

Abstract
BACKGROUND
Dominantly inherited Creutzfeldt-Jakob disease (CJD) comprises 5–15% of all CJD cases. The E200K mutation in the prion protein (PrP) gene (PRNP) is the most frequent cause of familial CJD. Co-existent amyloid-beta (Aβ) pathology has been reported in some transmissible spongiform encephalopathies but not in familial CJD with the E200K mutation.
OBJECTIVE
To characterize a CJD family in which Aβ pathology co-distributes with spongiform degeneration.
DESIGN
Clinicopathological and molecular study of a family with CJD with the E200K-129M haplotype.
SETTING
Alzheimer’s disease research center
MAIN OUTCOME MEASURES
Clinical, biochemical, and neuropathological observations of 2 generations of a family.
RESULTS
In this kindred, three autopsied individuals showed pathological changes typical for this haplotype: spongiform degeneration, gliosis, neuronal loss, and PrP deposition. Moreover, two of these cases (ages 57 and 63) showed numerous Aβ plaques co-distributed with the spongiform degeneration. APOE genotyping in 2 cases revealed that Aβ plaques were present in the APOE4 carrier but not in the APOE4 noncarrier. Two additional individuals exhibited incomplete penetrance as they had no clinical evidence of CJD at death after age 80 and yet had affected siblings and children.
CONCLUSION
This is the first description of Aβ pathology in familial CJD with the E200K mutation. The co-distribution of plaques and CJD-associated changes suggests that PrP plays a central role in Aβ formation and that Aβ pathology and prion disease likely influence each other. The kindred described here provides support that PrPE200K may also result in increased Aβ deposition.
Introduction
Creutzfeldt-Jakob disease (CJD) is the most common human prion disease. Its clinical course is characterized by rapid onset and progression of dementia, myoclonus, and cerebellar, visual, pyramidal, and extrapyramidal dysfunction and invariably culminates in death, usually within a few months of onset 1–4. Spongiform degeneration, gliosis, and neuronal loss are the major histopathological hallmarks of the disease. Deposition of the scrapie prion protein (PrPSc), the pathological conformational isoform of the normal cellular glycoprotein PrP (PrPC), occurs in the brains of affected individuals 2.
Sporadic CJD has a worldwide incidence of approximately one person per million per year 5. Dominantly inherited familial CJD represents 5–15% of all CJD cases 6. Several mutations have been identified in the prion protein gene (PRNP). The E200K mutation results in a nonconservative substitution of lysine for glutamate at codon 200 2, 7 and accounts for more than 70% of familial CJD 8. Disease phenotype, duration, and age at onset are further influenced by the methionine (M)/valine (V) polymorphism at codon 129 in PRNP 2–4, 9. The prevalence of E200K mutation is especially high among those of Slovakian descent, including ancestors who migrated to Hungary 2, 8, 10, 11.
We report here a non-Jewish family of Hungarian heritage with the E200K-129M haplotype with five affected individuals in two successive generations who developed ataxia and a rapidly progressive dementia. This kindred illustrates several novel and atypical features of CJD with respect to clinical presentation, disease duration, genetics, and pathology in which two distinct misfolded proteins co-exist.
Report of Cases
Family History
This family emigrated from Hungary (I-1, I-2, II-1) to the US between 1902 and 1910. Five adult individuals in two successive generations developed ataxic gait followed by rapidly progressive dementia. The proband (II-10), a niece (III-4), and a nephew (III-5) were clinically examined by one of us (JCM) and another niece (III-8) was followed by a local neurologist between 1985 and 2006 (Figure 1). Cases II-10, III-4, and III-8 had postmortem examinations.
Figure 1.

Pedigree of E200K kindred with five affected individuals across two generations. II-2 and II-4 were asymptomatic at death and represent two separate instances of incomplete penetrance in a single kindred. Individual members of the kindred are represented as triangles to maintain anonymity. Arrow, proband; crossbar, deceased; black triangle, affected; asterisk, autopsy.
Patient II-10
Two months before her 63rd birthday, this woman developed an inability to walk and progressive memory decline with an inability to recognize family members. Examination six months after symptom onset revealed temporal disorientation, impaired learning and memory, and ataxic gait. Head computerized tomography (CT) scan revealed left frontal focal atrophy, later confirmed on magnetic resonance imaging (MRI). Neither diffusion weighted MRI nor cerebrospinal fluid (CSF) levels of the 14–3–3 protein were obtained. An electroencephalogram (EEG) was abnormal, showing frontal diffuse slowing with random sharp waves. The patient died at age 63, approximately 11 months after the onset of illness.
Patient III-4
At age 61, this woman developed progressive difficulty with walking and balance and began repeating statements and stories. Five months later, she had difficulty taking medications correctly, preparing meals, managing household finances, and identifying family members. Eight months after onset of symptoms, the patient was no longer ambulatory and had no spontaneous conversation. She required full care for dressing and grooming and was incontinent. CSF was sampled and was positive for 14–3–3 protein. A brain MRI with diffusion imaging revealed restricted diffusion signal abnormalities involving the periventricular white matter, bilateral basal ganglia, and thalamus as previously described in individuals with E200K-129M 12, 13. An EEG was abnormal with excessive generalized diffuse slowing but without periodic activity. The patient died at age 62, 13 months after her initial presentation.
Patient III-5
At age 52, this man presented with gait and balance difficulty and reduced spontaneous speech. Examination revealed hesitant speech with errors in auditory comprehension and unsteady gait. Four months after symptom onset, he was noted to have no spontaneous conversation, poor recall, temporal and geographic disorientation, and wide-based unsteady gait. No involuntary movements were observed. An EEG was abnormal with mild generalized slowing without periodic waveforms. CSF was sampled and was positive for 14–3–3 protein. MRI revealed moderate diffuse cortical atrophy. He died at age 54, approximately 17 months after onset of gait difficulty.
Patient III-8
The patient came to medical attention six months after symptom onset at age 56. The history indicated deteriorating memory function with inability to carry out accustomed activities and language dysfunction. The examination was notable for paucity of speech and difficulty in auditory comprehension. Hypertonicity and bradykinesia were noted in the right upper and lower extremities and the gait was both apraxic and ataxic. The EEG was abnormal with moderate generalized slowing (left posterior hemispheric predominance) without periodic waveforms. The CSF 14–3–3 protein assay was positive. MRI revealed asymmetric increased diffusion in the cortex of the left frontal and posterior parietal lobes and the basal ganglia (Figures 2A and 2B). Subsequently, she became mute and developed action myoclonus. She died at age 57, approximately seven months after the onset of illness.
Figure 2.
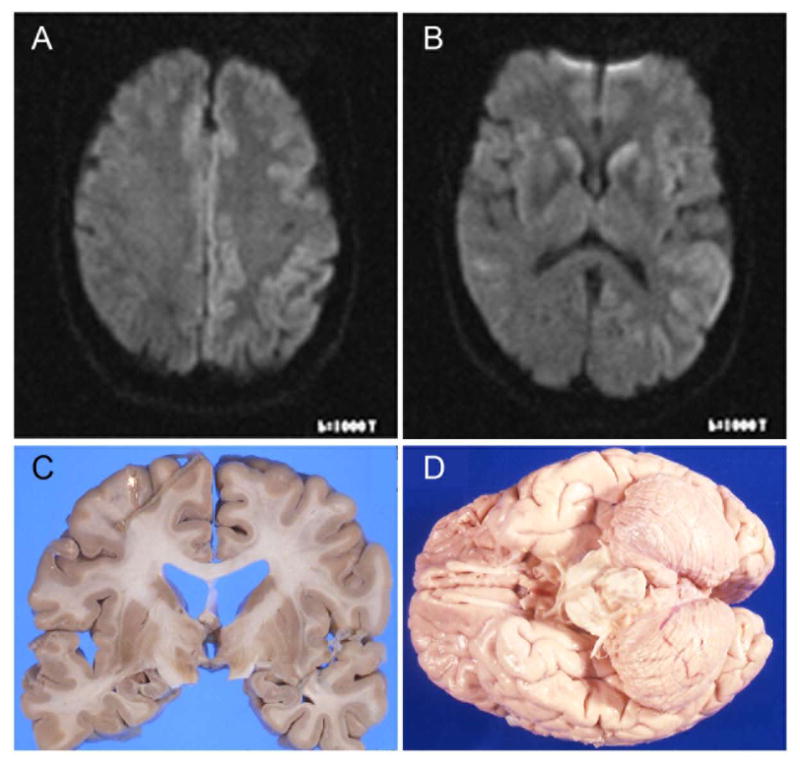
A & B. Cerebral magnetic resonance images for patient III-8. On diffusion weighted images, diffusion restriction seen in: left frontal and parietal cortices (A) and caudate (B). C & D: Postmortem macroscopic evaluation of proband (II-10). Prominent atrophy of the cerebral hemispheres (C) or of the cerebellum (D) is absent.
Patient II-7
A brother (II-7) of the proband (II-10) died at age 60. History from relatives indicated that he had a rapidly progressive dementia with similar features as the proband.
Patient II-2
A sister (II-2) of the proband (II-10) died at age 81 after a stroke. Prior to death there were no known motor, coordination, or cognitive deficits. Two of her children (III-4 and III-5) developed CJD.
Patient II-4
Another sister (II-4) of the proband (II-10) died at age 82 from sepsis. Prior to death there were no known motor or coordination deficits or cognitive decline. One of her children (III-8) developed CJD.
Methods
Neuropathology
Paraffin sections from formalin-fixed blocks of cerebral cortex were obtained from three family members who came to autopsy (II-10, III-4, and III-8). These were processed by using a standard battery of histological stains including hematoxylin and eosin, Luxol Fast Blue, Periodic Acid Schiff, Bielschowsky (silver) to evaluate spongiosis, gliosis, and neuronal loss.
In all cases, immunohistochemistry was carried out on deparaffinized, rehydrated, formic acid pretreated sections. For standard and double PrP and Aβ immunostaining, sections were treated with hydrochloric acid and microwaved for antigen retrieval. For PrP immunostaining, sections were probed with the monoclonal antibody 3F4 (1:3,000;14), incubated with polymer/HRP (EnVision G/2 double stain system rabbit/mouse; Dako, Carpinteria, CA), and visualized with diaminobenzidine tetrahydrochloride 3, 15. For Aβ immunostaining, sections were probed with 4G8 (1:3,000;14) monoclonal antibody, incubated with polymer AP, and visualized with either fast blue BB salt hemi (zinc chloride) salt (Sigma, St. Louis, MO) or permanent red substrate (EnVision; Dako, Carpinteria, CA). For ApoE immunohistochemistry, sections were pretreated with citrate buffer and hydrogen peroxide. Sections were incubated with ApoE4 (1:15000; MBL, Woburn, MA) monoclonal antibody.
PRNP Genotype Determination
Genomic DNA was extracted from frozen brain tissue obtained from cases III-4 and III-8 at autopsy 16. PRNP coding region was amplified and sequenced as previously described 3, 4, 17. Sequence analysis confirmed the presence of the E200K mutation and excluded other mutations. Codon 129 genotype was determined by digestion of amplified DNA with Nsp I restriction endonuclease 4. Genomic DNA from patient II-10 could not be obtained due to a lack of archival frozen tissue.
APOE Genotype Determination
APOE genotyping was performed using an ABI Real Time TaqMan® SNP Genotyping assay. Briefly, genomic DNA was used for allelic determination of SNP 112 (rs 429358; E4 allele) and SNP 158 (rs 7412; E2 allele) APOE gene. Both SNP assays were done separately in two plates for the same samples and the results were combined for genotypes. 50 ng of DNA was combined with 1x final concentration of universal TaqMan® PCR master mix (Applied Biosystems, Foster City, CA) with 0.5x final concentration of primers for SNP 112 and SNP 158. Primers for both SNPs were tagged with VIC® and FAM™ fluorescent dyes. Real time PCR was performed and results were tabulated independently for each SNP. Genotypes were combined for APOE genotype of an individual.
Brain homogenates preparation and PK digestion
Brain tissue homogenates (10% w/v) from frozen brain tissue were prepared in lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% NP–40, 0.5% sodium deoxycholate, 100 mM Tris, pH 8.0). For proteinase K (PK; Sigma, St. Louis, MO) digestion of PrP, brain homogenates were incubated with 100 μg/ml of PK for 1 h at 37°C. Digestion was stopped by adding PMSF at a final concentration of 3 mM.
Western blot
PK-treated and –untreated samples were boiled in an equal volume of 2x sodium dodecyl sulfate (SDS) sample buffer (6% SDS, 5% β-mercaptoethanol, 20% glycerol, 4 mM EDTA, 125 mM Tris–HCl, pH 6.8) for 10 min. Proteins were separated by 15% Tris–HCl gels (BioRad, Hercules, CA). Proteins were transferred from gels to polyvinylidene fluoride (PVDF) membrane (Immobilon-P; Millipore) at 70 V for 2 h at room temperature. Membranes were incubated with blocking buffer (5% non-fat milk in Tris-buffered saline Tween–20 (TBS-T)) for 1 h, then probed with 3F4 (1:40000) to human PrP residues 109–112 14. Finally membranes were incubated with a horseradish peroxidase-conjugated goat anti-mouse antibody (1:3000) for 1 h. The PrP bands were visualized on Kodak film by the ECL Plus (GE Healthcare, Piscataway, NJ) as described by the manufacturer. Western blots were analyzed by Image Acquisition and Analysis software LabWorks 4.0 (UVP Inc., Upland, CA).
Results
Neuropathologic Findings
For cases II-10, III-4, and III-8, gross examination revealed normal weight for each brain (range 1300–1430 g). The cerebral hemispheres appeared full, with normal sulcal and gyral patterns (Figure 2C). The cerebellum was grossly normal in these cases (Figure 2D). In all cases, histochemical examination revealed the pathognomonic findings for CJD with spongiform degeneration, neuronal loss, and gliosis throughout the hippocampus, neocortex, basal ganglia, and thalamus (Figure 3A). There was absent to minimal spongiform change noted in the cerebellum for two cases (II-10 and III-8). In III-4, the cerebellum showed a moderate degree of spongiosis in the molecular layer and atrophy and gliosis in the dentate nucleus and the cortex, especially at the level of the vermis.
Figure 3.
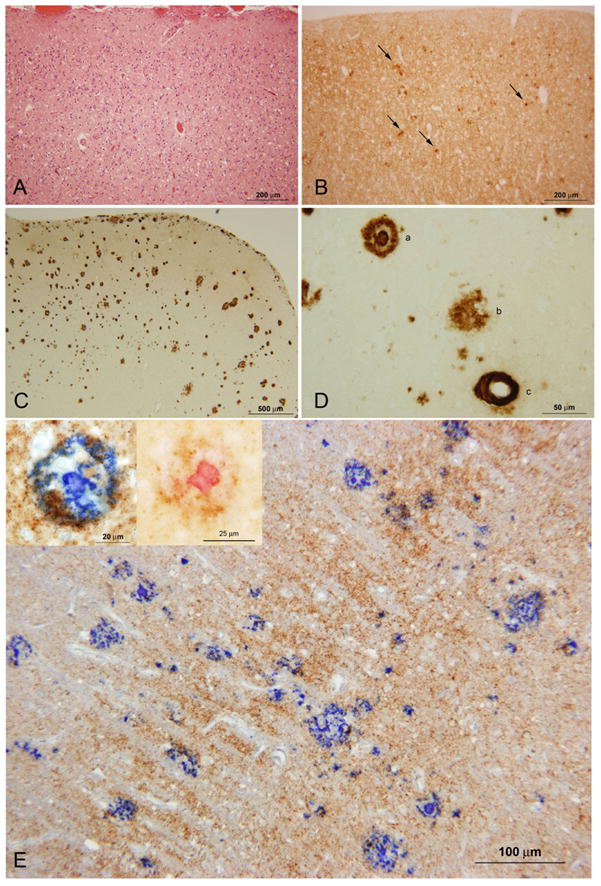
Histology and immunohistochemistry of case III-8. A: fine spongiform degeneration and gliosis in the occipital cortex; B: diffuse PrP and clusters of PrP aggregates (arrows) were detectable throughout the cortex; C: the same cortical region shows Aβ plaques; D: higher magnification shows the extracellular Aβ deposits, represented by a ring-with-core plaque (a), diffuse plaque (b), and cerebral amyloid angiopathy (c). E: double immunostaining for PrP and Aβ. The Aβ plaques are stained in blue whereas PrP stains in brown. The insets on the top left corner show a higher magnification of the Aβ plaques that were stained with fast blue (left) and permanent red (right); PrP stains in brown. Monoclonal antibodies 3F4 and 4G8 were used to label PrP and Aβ, respectively. The scale bar is indicated at the bottom of each figure.
Prion protein deposition, primarily in diffuse synaptic form and to a lesser extent in clusters of PrP aggregates (Figure 3B), was evident throughout the brain with sparing of the cerebellum in two subjects (II-10 and III-8). In case III-4, PrP deposition was present in the cerebellum in granule and plaque-like structures. Moreover, in two cases (II-10 and III-8) there was a population of Aβ plaques that co-distributed with spongiform degeneration in the hippocampus, neocortex, basal ganglia, thalamus, but not in the cerebellum (Figure 3C–E). Double immunostaining revealed co-distribution of Aβ plaques with PrP immunostaining in every region examined where Aβ and PrP abnormal immunostaining occurred (Figure 3E).
Western blot analysis of frozen brain tissue samples from patients III-4 and III-8 was conducted by the National Prion Disease Pathology Surveillance Center and demonstrated the presence of the abnormal, PK-resistant prion protein (PrPSc) characterized by the under-representation of the unglycosylated PrPSc (21 kDa) consistent with that of E200K familial CJD (Figure 4) 4.
Figure 4.

Western blot analysis. Brain homogenates obtained from the frontal cortex were either PK-untreated (lanes 1–2 and 7) or treated with PK (lanes 3–6 and 8) and probed with 3F4. The PK-resistant PrPSc from patients III-4, III-8 and the unrelated E200K-129M case (lanes 3 and 4, and 8 respectively) shows electrophoretic mobility similar to, but PrP glycoform ratio different from, PrPSc type 1 sCJD. T1: PrPSc type 1; T2: PrPSc type 2; Molecular weight masses are expressed in kilodalton (kDa) and are indicated on the right side of the figure.
Genetic Findings
Genetic analysis of patients III-4 and III-8 confirmed both the presence of a known familial CJD mutation, E200K, as well as the codon 129 genotype. The haplotype for these two representative individuals was E200K-129M while the genotype at codon 129 of the normal allele was 129V and 129M for III-4 and III-8, respectively. Genotype for patient II-10 was not available. Given the Aβ burden demonstrated by immunohistochemistry, APOE genotype was also determined. APOE genotype for patient III-4 was ε2/ε3 and for patient III-8 was ε3/ε4. APOE genotype for patient II-10 was inconclusive when assessed by immunohistochemistry using an ApoE4 specific antibody.
Two individuals (II-2 and II-4) who were likely E200K mutation carriers exhibited incomplete penetrance as they had no cognitive or motor deficits prior to death, yet each had affected siblings and one or more affected children.
Comment
We report here a familial CJD kindred of Hungarian ancestry with the E200K-129M haplotype which features five affected individuals in two successive generations. Distinctive aspects of this kindred include the type and distribution of CJD and Aβ pathological findings and the apparent incomplete penetrance. The unusual pathological finding in this family was the presence of numerous Aβ plaques in two of the three cases (II-10 and III-8) suggesting that Aβ deposition is linked with CJD pathophysiology.
Cortical Aβ plaques are observed in Alzheimer’s disease (AD) and less commonly in clinically normal older adults 18, 19. However, in this kindred our patients were all under the age of 65. The Aβ plaques were unaccompanied by notable neurofibrillary pathology as would be characteristic of AD. The Aβ burden was not restricted to cortical areas and was also found in subcortical regions such as basal ganglia. There have been reports of Aβ plaques in other transmissible spongiform encephalopathies such as sporadic and familial Gerstmann-Sträussler syndrome (GSS) 20, iatrogenic CJD, older sporadic CJD cases 21–33, and in a case from a PRNP gene insertion kindred 34. However, to our knowledge there have been no reports of Aβ pathology among familial CJD with E200K mutation.
Neither of the two cases with Aβ plaques had a history of traumatic brain injury which has been proposed to result in increased Aβ deposition 35. APP and PS1 genes were not screened for mutations since review of the literature did not provide conclusive evidence of these mutations coexisting with PRNP mutations and contributing to CJD pathology 36–38. APOE genotyping was pursued since there is a strong association of the ε4 allele and increased Aβ deposition 25, 39. Patient III-4, who lacked Aβ pathology, had a ε2/ε3 genotype; whereas, patient III-8, who had considerable Aβ burden had ε3/ε4 genotype. APOE genotype for patient II-10 could not be established due to limited archival clinical material.
The remarkable co-distribution of plaques and CJD-associated changes in cases II-10 and III-8 suggests that this Aβ deposition is associated with CJD pathophysiology rather than an independent process. For instance, all three cases that came to autopsy had evidence of spongiform changes and gliosis which could be present in a region in the absence of Aβ plaques; however, the converse was not the case. This suggests that spongiform change is an upstream event and is required for Aβ deposition. Several lines of evidence indicate that the PRNP genotype and/or PrPC have an influence on the AD phenotype and that Aβ and PrPC metabolisms are interconnected: 1) the codon 129 methionine/valine polymorphism of the PRNP gene is a risk factor for AD 40–43; 2) the occurrence of mature Aβ plaques has been reported in two familial forms of prion disease (34; present study); 3) Aβ induced synaptic dysfunction is mediated through its binding to PrPC44; 4) the formation of Aβ is increased in scrapie-infected mice as well as in the presence of PRNP pathogenic mutations 41 z; and 5) depletion of PrPC or the presence of disease-associated mutant PrP in mouse N2a cells results in failed β-secretase inhibition with resultant increase in Aβ levels 42. Taken together, these findings suggest that PrPC plays a central role in Aβ formation and that Aβ pathology and prion disease likely influence each other. The kindred described here provides support that PrPE200K may also result in increased Aβ deposition. The impact of E200K may be modulated by the codon 129 status on the normal allele 40, 42, 43, 45. For instance it is known that PrP deposition patterns are different for 129MM versus 129MV 2, 46. In the case of III-4 the codon 129 status was MV and may have resulted in differential PrP deposition and therefore the lack of downstream Aβ deposition 42. Also, III-4 APOE status was ε2/ε3 which may have further decreased the likelihood of Aβ deposition. Recognition of this possibility may encourage other investigators to assess potential Aβ deposition in cases with classic CJD pathology.
This kindred also demonstrates a novel genetic feature of incomplete penetrance. Patients II-2 and II-4 had no motor and cognitive impairment prior to death from acute illnesses at ages 81 and 82 and yet each had two affected siblings and had one or more affected children. Since the E200K mutation is dominantly inherited, these two individuals likely were mutation carriers without CJD features because of incomplete penetrance. Ninety-six percent of E200K mutation carriers develop the clinical CJD phenotype if they live past the age of 80 5 except for those with Slovakian heritage (such as this kindred), where penetrance may only be 59% 11, 47.
Another distinctive feature of this kindred is the clinicopathological presentation. Presenting clinical features of E200K-129M typically include cognitive abnormalities in up to 83% of patients and cerebellar signs in up to 55% 2. These initial features are followed by development of dementia in all patients, cerebellar signs in 79%, and myoclonus in 73% during the course of the disease 2. The clinical presentation in this family also features ataxia followed by dementia. Classically the ataxia seen in E200K-129M is attributed to severe spongiform degeneration, gliosis, and neuronal loss in the cerebellum. However, in this kindred the cerebellum was spared with only moderate involvement in one case (III-4), a finding consistent with its E200K-129M haplotype 2. Alternately, the gait disturbance observed in this family could be attributable to involvement of the spinal cord instead of the cerebellum. Cases of an amyotrophic form of CJD have been reported; however, in those instances motor signs consistent with motor neuron disease were observed 48–51. Spinal cords were not available for analysis in this family. In typical E200K-129M, the mean age of onset is 58 years with the mean duration of 6 months 2, 52. While the age of onset in this family ranged from 52 to 62, the duration of illness was longer as it ranged from 7 to 17 months 11. Other typical CJD features absent in this family were positive sharp waves on EEG and prominent brain atrophy 2, 52.
This is the first description of Aβ plaque pathology in familial CJD with E200K mutation. The co-distribution of Aβ deposition and spongiform degeneration in this family lends credence to the idea that these two synaptic proteins, APP and PrP, may interact to result in disease.
Acknowledgments
Funding Support: This work was supported in part by grants P50 AG05681 and P01 AG03991 (JCM); T32 NS007205 (NG, RJP); P01 AG14359, CDC UR8/CCU515004, the Charles S. Britton Fund, and the CJD Foundation (PG).
Footnotes
Financial Disclosure: None reported
Author Contributions: Study concept and design: Ghoshal, Perrin, Josephson, Sun, and Morris. Acquisition of Data: Ghoshal, Cali, Perrin, Josephson, Sun, Gambetti, and Morris. Analysis and interpretation of data: Ghoshal, Cali, Perrin, Gambetti, and Morris. Drafting of the manuscript: Ghoshal, Josephson, Sun, and Morris. Critical revision of the manuscript for important intellectual content: Ghoshal, Cali, Perrin, Gambetti, and Morris. Obtained funding: Ghoshal, Perrin, Gambetti, Morris. Administrative, technical, and material support: Ghoshal, Cali, Perrin, and Morris. Study supervision: Gambetti and Morris.
Additional Contributions: C. Christopher Clark, MD and Stuart Weiss, MD provided clinical records; Richard Torack, MD, Arie Perry, MD, and Nigel Cairns, PhD, FRCPath provided neuropathological evaluation; Alison Goate, DPhil, Joanne Norton, RN, Sumi Chakraverty, MS, and Pam Millsap, RN provided genotyping assistance and pedigree development; Manuela Pastore, PhD conducted initial Western blots and determined glycoform status; Yvonne Cohen, BS and Diane Kofskey, BS provided technical assistance.
References
- 1.Brown P, Gibbs CJ, Jr, Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994 May;35(5):513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 2.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 3.Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996 Jun;39(6):767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 4.Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999 Aug;46(2):224–233. [PubMed] [Google Scholar]
- 5.Spudich S, Mastrianni JA, Wrensch M, et al. Complete penetrance of Creutzfeldt-Jakob disease in Libyan Jews carrying the E200K mutation in the prion protein gene. Mol Med. 1995 Sep;1(6):607–613. [PMC free article] [PubMed] [Google Scholar]
- 6.Hsiao K, Prusiner SB. Inherited human prion diseases. Neurology. 1990 Dec;40(12):1820–1827. doi: 10.1212/wnl.40.12.1820. [DOI] [PubMed] [Google Scholar]
- 7.Goldgaber D, Goldfarb LG, Brown P, et al. Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker’s syndrome. Exp Neurol. 1989 Nov;106(2):204–206. doi: 10.1016/0014-4886(89)90095-2. [DOI] [PubMed] [Google Scholar]
- 8.Lee HS, Sambuughin N, Cervenakova L, et al. Ancestral origins and worldwide rdistribution of the PRNP 200K mutation causing familial Creutzfeldt-Jakob disease. Am J Hum Genet. 1999 Apr;64(4):1063–1070. doi: 10.1086/302340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parchi P, Zou W, Wang W, et al. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A. 2000 Aug 29;97(18):10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacs GG, Laszlo L, Bakos A, et al. Increased incidence of genetic human prion disease in Hungary. Neurology. 2005 Nov 22;65(10):1666–1669. doi: 10.1212/01.wnl.0000184513.95290.80. [DOI] [PubMed] [Google Scholar]
- 11.Kovacs GG, Puopolo M, Ladogana A, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005 Nov;118(2):166–174. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- 12.Fulbright RK, Kingsley PB, Guo X, et al. The imaging appearance of Creutzfeldt-Jakob disease caused by the E200K mutation. Magn Reson Imaging. 2006 Nov;24(9):1121–1129. doi: 10.1016/j.mri.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Fulbright RK, Hoffmann C, Lee H, Pozamantir A, Chapman J, Prohovnik I. MR imaging of familial Creutzfeldt-Jakob disease: a blinded and controlled study. AJNR Am J Neuroradiol. 2008 Oct;29(9):1638–1643. doi: 10.3174/ajnr.A1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kascsak RJ, Rubenstein R, Merz PA, et al. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987 Dec;61(12):3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cali I, Castellani R, Yuan J, et al. Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain. 2006 Sep;129(Pt 9):2266–2277. doi: 10.1093/brain/awl224. [DOI] [PubMed] [Google Scholar]
- 16.Petersen RB, Tabaton M, Chen SG, et al. Familial progressive subcortical gliosis: presence of prions and linkage to chromosome 17. Neurology. 1995 Jun;45(6):1062–1067. doi: 10.1212/wnl.45.6.1062. [DOI] [PubMed] [Google Scholar]
- 17.Khorana S, Gagel RF, Cote GJ. Direct sequencing of PCR products in agarose gel slices. Nucleic Acids Res. 1994 Aug 25;22(16):3425–3426. doi: 10.1093/nar/22.16.3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999 Mar;45(3):358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 19.Morris JC, Storandt M, McKeel DW, Jr, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996 Mar;46(3):707–719. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- 20.Miyazono M, Kitamoto T, Iwaki T, Tateishi J. Colocalization of prion protein and beta protein in the same amyloid plaques in patients with Gerstmann-Straussler syndrome. Acta Neuropathol. 1992;83(4):333–339. doi: 10.1007/BF00713522. [DOI] [PubMed] [Google Scholar]
- 21.Barcikowska M, Kwiecinski H, Liberski PP, Kowalski J, Brown P, Gajdusek DC. Creutzfeldt-Jakob disease with Alzheimer-type A beta-reactive amyloid plaques. Histopathology. 1995 May;26(5):445–450. doi: 10.1111/j.1365-2559.1995.tb00252.x. [DOI] [PubMed] [Google Scholar]
- 22.Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998 Aug;96(2):116–122. doi: 10.1007/s004010050870. [DOI] [PubMed] [Google Scholar]
- 23.Vital A, Canron MH, Gil R, Hauw JJ, Vital C. A sporadic case of Creutzfeldt-Jakob disease with beta-amyloid deposits and alpha-synuclein inclusions. Neuropathology. 2007 Jun;27(3):273–277. doi: 10.1111/j.1440-1789.2007.00755.x. [DOI] [PubMed] [Google Scholar]
- 24.Masters CL, Gajdusek DC, Gibbs CJ., Jr The familial occurrence of Creutzfeldt-Jakob disease and Alzheimer’s disease. Brain. 1981 Sep;104(3):535–558. doi: 10.1093/brain/104.3.535. [DOI] [PubMed] [Google Scholar]
- 25.Debatin L, Streffer J, Geissen M, Matschke J, Aguzzi A, Glatzel M. Association between Deposition of Beta-Amyloid and Pathological Prion Protein in Sporadic Creutzfeldt-Jakob Disease. Neurodegener Dis. 2008 Mar 18;:1–8. doi: 10.1159/000121389. [DOI] [PubMed] [Google Scholar]
- 26.Yagishita S, Iwabuchi K, Amano N, Yokoi S. Further observation of Japanese Creutzfeldt-Jacob disease with widespread amyloid plaques. J Neurol. 1989 Mar;236(3):145–148. doi: 10.1007/BF00314329. [DOI] [PubMed] [Google Scholar]
- 27.Preusser M, Strobel T, Gelpi E, et al. Alzheimer-type neuropathology in a 28 year old patient with iatrogenic Creutzfeldt-Jakob disease after dural grafting. J Neurol Neurosurg Psychiatry. 2006 Mar;77(3):413–416. doi: 10.1136/jnnp.2005.070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuchiya K, Yagishita S, Ikeda K, et al. Coexistence of CJD and Alzheimer’s disease: an autopsy case showing typical clinical features of CJD. Neuropathology. 2004 Mar;24(1):46–55. doi: 10.1111/j.1440-1789.2003.00513.x. [DOI] [PubMed] [Google Scholar]
- 29.Powers JM, Liu Y, Hair LS, Kascsack RJ, Lewis LD, Levy LA. Concomitant Creutzfeldt-Jakob and Alzheimer diseases. Acta Neuropathol. 1991;83(1):95–98. doi: 10.1007/BF00294437. [DOI] [PubMed] [Google Scholar]
- 30.Gaches J, Supino-Viterbo V, Foncin JF. Association of Alzheimer’s disease and Creutzfeldt-Jakob’s disease (author’s transl) Acta Neurol Belg. 1977 Jul-Aug;77(4):202–212. [PubMed] [Google Scholar]
- 31.Muramoto T, Kitamoto T, Koga H, Tateishi J. The coexistence of Alzheimer’s disease and Creutzfeldt-Jakob disease in a patient with dementia of long duration. Acta Neuropathol. 1992;84(6):686–689. doi: 10.1007/BF00227747. [DOI] [PubMed] [Google Scholar]
- 32.Leuba G, Saini K, Savioz A, Charnay Y. Early-onset familial Alzheimer disease with coexisting beta-amyloid and prion pathology. JAMA. 2000 Apr 5;283(13):1689–1691. doi: 10.1001/jama.283.13.1689-a. [DOI] [PubMed] [Google Scholar]
- 33.Wakabayashi K, Hinokuma K, Takahashi H, Seki K, Tanaka M, Ikuta F. Coexistence of Creutzfeldt-Jakob disease and senile dementia of the Alzheimer type. Neuropathology. 1995 December;15(3–4):122–126. [Google Scholar]
- 34.Collinge J, Brown J, Hardy J, et al. Inherited prion disease with 144 base pair gene insertion. 2. Clinical and pathological features. Brain. 1992 Jun;115 (Pt 3):687–710. doi: 10.1093/brain/115.3.687. [DOI] [PubMed] [Google Scholar]
- 35.Van Den Heuvel C, Thornton E, Vink R. Traumatic brain injury and Alzheimer’s disease: a review. Prog Brain Res. 2007;161:303–316. doi: 10.1016/S0079-6123(06)61021-2. [DOI] [PubMed] [Google Scholar]
- 36.El Hachimi KH, Cervenakova L, Brown P, et al. Mixed features of Alzheimer disease and Creutzfeldt-Jakob disease in a family with a presenilin 1 mutation in chromosome 14. Amyloid: Int J Exp Clin Invest. 1996;3:223–233. [Google Scholar]
- 37.Dermaut B, Cruts M, Backhovens H, et al. Familial Creutzfeldt-Jakob disease in a patient carrying both a presenilin 1 missense substitution and a prion protein gene insertion. J Neurol. 2000 May;247(5):364–368. doi: 10.1007/s004150050603. [DOI] [PubMed] [Google Scholar]
- 38.Perry RT, Go RC, Harrell LE, Acton RT. SSCP analysis and sequencing of the human prion protein gene (PRNP) detects two different 24 bp deletions in an atypical Alzheimer’s disease family. Am J Med Genet. 1995 Feb 27;60(1):12–18. doi: 10.1002/ajmg.1320600104. [DOI] [PubMed] [Google Scholar]
- 39.Schmechel DE, Saunders AM, Strittmatter WJ, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993 Oct 15;90(20):9649–9653. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dermaut B, Croes EA, Rademakers R, et al. PRNP Val129 homozygosity increases risk for early-onset Alzheimer’s disease. Ann Neurol. 2003 Mar;53(3):409–412. doi: 10.1002/ana.10507. [DOI] [PubMed] [Google Scholar]
- 41.Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008 Apr;88(2):673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 42.Parkin ET, Watt NT, Hussain I, et al. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc Natl Acad Sci U S A. 2007 Jun 26;104(26):11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riemenschneider M, Klopp N, Xiang W, et al. Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology. 2004 Jul 27;63(2):364–366. doi: 10.1212/01.wnl.0000130198.72589.69. [DOI] [PubMed] [Google Scholar]
- 44.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009 Feb 26;457(7233):1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puoti G, Rossi G, Giaccone G, et al. Polymorphism at codon 129 of PRNP affects the phenotypic expression of Creutzfeldt-Jakob disease linked to E200K mutation. Ann Neurol. 2000 Aug;48(2):269–270. [PubMed] [Google Scholar]
- 46.Mitrova E, Belay G. Creutzfeldt-Jakob disease with E200K mutation in Slovakia: characterization and development. Acta Virol. 2002;46(1):31–39. [PubMed] [Google Scholar]
- 47.Chapman J, Ben-Israel J, Goldhammer Y, Korczyn AD. The risk of developing Creutzfeldt-Jakob disease in subjects with the PRNP gene codon 200 point mutation. Neurology. 1994 Sep;44(9):1683–1686. doi: 10.1212/wnl.44.9.1683. [DOI] [PubMed] [Google Scholar]
- 48.Nowacki P, Kulczycki J, Narolewska A, Grzelec H. Amyotrophic form of Creutzfeldt-Jakob disease with rapid course in 82-year-old man. Folia Neuropathol. 2000;38(4):161–163. [PubMed] [Google Scholar]
- 49.Iwasaki Y, Yoshida M, Hashizume Y, Kitamoto T, Sobue G. Neuropathologic characteristics of spinal cord lesions in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. 2005 Nov;110(5):490–500. doi: 10.1007/s00401-005-1076-7. [DOI] [PubMed] [Google Scholar]
- 50.Allen IV, Dermott E, Connolly JH, Hurwitz LJ. A study of a patient with the amyotrophic form of Creutzfeldt-Jakob disease. Brain. 1971;94(4):715–724. doi: 10.1093/brain/94.4.715. [DOI] [PubMed] [Google Scholar]
- 51.Worrall BB, Rowland LP, Chin SS, Mastrianni JA. Amyotrophy in prion diseases. Arch Neurol. 2000 Jan;57(1):33–38. doi: 10.1001/archneur.57.1.33. [DOI] [PubMed] [Google Scholar]
- 52.Kahana E, Zilber N, Abraham M. Do Creutzfeldt-Jakob disease patients of Jewish Libyan origin have unique clinical features? Neurology. 1991 Sep;41(9):1390–1392. doi: 10.1212/wnl.41.9.1390. [DOI] [PubMed] [Google Scholar]