

Abstract
Triplex-forming oligonucleotides (TFOs) can bind to polypurine•polypyrimidine tracts in DNA and as a consequence, perturb normal functioning of a targeted gene. The effectiveness of such anti-gene TFOs can potentially be enhanced by covalent attachment of the TFO to its DNA target. Here we report that attachment of N-7-platinated guanine nucleosides to the 3′- and/or 5′-ends of oligopyrimidine TFOs enables these TFOs to form highly stable adducts with target DNA deoxyguanosines or deoxyadenosines that are adjacent to the TFO binding site. Such adduct formation stably anchors the TFO to its target. Depending on the sequences adjacent to the TFO binding site, adduct formation can occur on either strand of the DNA. Adduct formation by 3′,5′ bis platinated TFOs can result in formation of an interstrand cross-link between both strands of the DNA duplex. Formation of the adducts, which could be reversed by treatment with sodium cyanide, was dependent upon the ability of the TFO to bind to DNA and appeared to occur at a rate slower than that at which the TFO bound to the DNA duplex. The extent of adduct formation at 37°C by platinated deoxyribo-TFOs diminished as the pH was increased from 6.5 to 7.4. In contrast high levels (~86%) of adduct formation by platinated 2′-O-methylribo-TFOs were observed at both pH 6.5 and pH 7.4. Platinated 2′-O-methylribo-TFOs were also shown to bind to plasmid DNA and inhibit transcription in vitro, and to inhibit plasmid replication in E. coli cells. These results suggest that platinum-conjugated TFOs may be good candidates for use as anti-gene agents.
Over the past two decades, there has been considerable interest in developing oligonucleotides that can specifically inhibit gene expression. Two approaches that have received much attention are antisense oligonucleotides and siRNA. Both approaches target the RNA product of the expressed gene and ultimately result in destruction of this target. Because the RNA products are continuously produced when the gene is transcribed, the antisense oligonucleotide or siRNA must be present constantly in order to prevent their translation into protein.
An alternative approach that directly targets and inhibits gene expression utilizes triplex-forming oligonucleotides (TFOs). These oligonucleotides are designed to bind to double-stranded DNA at specific regions within the gene (1-3). As a consequence of this binding, transcription of the gene is inhibited or mutations are induced that result in aberrant expression of the gene. The potential advantages of the antigene strategy are the relatively few copies that need to be targeted and the possibility that targeting can permanently disable gene expression.
Triplex forming oligonucleotides interact via the major groove by binding to polypurine tracts within the DNA target (1-7). Oligopurine TFOs utilize reverse-Hoogsteen hydrogen bonds to form A•A-G and G•G-C triads and are oriented antiparallel to the polypurine tract. Oligopyrimidine TFOs utilize Hoogsteen hydrogen bonds to form T•A-T and C+•G-C triads and are oriented parallel to the polypurine tract. In this case the N-3 of the cytosine of the C+•G-C triad must be protonated in order to form a stable Hoogsteen interaction and consequently triplex stability diminishes with increasing pH.
Although TFOs can transiently block DNA transcription (8-11), inhibition of transcription can be enhanced considerably by linking the TFO covalently to the transcribed strand (12-15). Furthermore, covalent attachment of TFOs can provoke DNA repair leading to mutations in the targeted gene (16-19). For these reasons, efforts have been made to conjugate TFOs with functional groups that would enable the TFO to bind to its DNA target in an irreversible manner. Alkylating groups such as bromoacetamide (20, 21) and chlorambucil (22, 23) have been used successfully to covalently attach a TFO to DNA. In these cases alkylation occurs at the N-7 of a guanine adjacent to the TFO binding site. However, the alkylated guanine is prone to depurination, which would lead to release of the TFO. TFOs have also been conjugated with derivatives of psoralen, a photoreactive molecule that intercalates into DNA and can form cyclobutane adducts with thymines upon irradiation with long wavelength UV light (15, 24-28).
An alternative approach utilizes the ability of the cancer chemotherapeutic drug cisplatin to cross-link DNA. Cisplatin or cis-diamminedichloroplatinum(II) (cis-DDP) reacts primarily with the N-7 of purine bases to form a highly stable coordination complex (29-31). The primary target of cisplatin is guanine, and cisplatin can form both intrastrand and interstrand cross-links in DNA. Platinum compounds have been employed in several ways to cross-link TFOs to duplex DNA including tethering cisplatin to the 5′-end of a TFO (32, 33), targeting an interruption in the polypurine tract with a transplatin modified cytosine residue (34,35), and using transplatin monoadducts of guanine and cytosine to react within the Hoogsteen triad (36, 37).
In the latter approach, a transplatin modified dG, placed at the 5′-end of a TFO efficiently cross-linked to a guanine in the purine strand of the target DNA sequence. In contrast, a transplatin-modified dG placed at an internal position in the TFO was too destabilizing to allow efficient cross-linking. Cross-linking was found to be highly dependent upon pH, with virtually no cross-linking observed at pH 7.
In this report, we describe the binding and cross-linking properties of oligopyrimidine TFOs that have 5′-transPt-dG (Pt-TFO), 3′-transPt-dG (TFO-Pt) and bis-3′,5′-transPt-dG (Pt-TFO-Pt) modifications. We show that the platinum of a Pt-TFO can react with purines in either the purine or pyrimidine strand of a DNA target duplex, and that it preferentially reacts with directly opposed G or A residues in the duplex. We also show that the platinum of a TFO-Pt, which can form adducts with purines in either strand of the duplex target, reacts more efficiently than the platinum of a Pt-TFO. Furthermore, the doubly-modified TFO, Pt-TFO-Pt, can react with both strands of the duplex to form an interstrand cross-link. Finally we show that TFO-Pt and Pt-TFO-Pt that consist of 2′-O-methylribonucleotides cross-link efficiently with their DNA target at physiological temperature and pH and can be used to disrupt in vitro transcription and prevent replication in E. coli of a plasmid containing a binding site for the TFO.
EXPERIMENTAL PROCEDURES
Protected 2′-deoxy- and 2′-O-methylribonucleoside-3′-O-bis(diisopropylamino)-β-cyanoethylphosphoramidites, 2′-deoxy- and 2′-O-methylribonucleoside derivatized controlled pore glass supports (CPG) were purchased from Glen Research Inc. (Sterling, VA). 2′-O-methylribo-5-methylcytosine phosphoramidite and 2′-O-methylribo-5-methyluridine derivatized CPG were purchased from ChemGenes Corp. (Ashland, MA). The base protecting groups were benzoyl (A, C) or isobutryl (G). 4,5-Dicyanoimidazole, 5-ethylthiotetrazole, Cap Mix A, Cap Mix B and oxidizing solution were purchased from EMD Chemicals (Gibbstown, NJ). Phosphoramidite solutions were prepared using synthesis-grade acetonitrile that was dried and stored over calcium hydride. A platinum solution was prepared by incubating 5 mM trans-diamminedichloroplatinum(II) (trans-DDP) with 10 mM silver nitrate overnight at 37°C. The resulting precipitate was pelleted by centrifugation, and the supernatant containing trans-diamminediaquaplatinum(II) dinitrate was stored in the dark at room temperature. MALDI-TOF mass spectrometry analysis was performed at the AB Mass Spectrometry/Proteomics Facility at The Johns Hopkins School of Medicine, with support from the National Center for Research Resources Shared Instrumentation, Grant 1S10-RR14702.
Synthesis of oligonucleotides
The sequences of the oligonucleotides used in this report can be found in Table 1 and Figures 3-5 and 8. These oligonucleotides were synthesized on an ABI DNA synthesizer (model 3400), using phosphoramidite chemistry and standard automated procedures. The phosphoramidite solutions were made up to a concentration of 0.15 M and 5-ethylthiotetrazole was used as the activating agent. Coupling times were 120 s for 2′-deoxyribonucleoside phosphoramidites and 360 s for 2′-O-methylribonucleoside phosphoramidites. The oligonucleotides were deprotected and removed from the solid support by incubating the CPG in a solution containing 300 μL of concentrated ammonium hydroxide and 100 μL of 95% ethanol for 3.5 hrs at 55°C. The supernatant from this reaction was evaporated to dryness and the residue dissolved in 200 μL of 50% aqueous acetonitrile for storage at 4°C. The crude oligonucleotides were purified by strong anion exchange HPLC on a Dionex DNAPac PA 100-4 5 250 mm column (Dionex Corp., Sunnyvale, CA), using a 30 min linear gradient of 0 to 0.5 M sodium chloride in a solution containing 100 mM Tris (pH 7.8) and 10% acetonitrile at a flow rate of 1.0 mL/min. The purified oligonucleotides were desalted on a C-18 Clarity Tube (Phenomenex, Inc., Torrance, CA). The concentration of purified oligomers was determined from their UV absorbance at 260 nm, using calculated extinction coefficients based on nearest neighbor approximations (38). The compositions of the oligonucleotides were confirmed by MALDI-TOF mass spectrometry, as shown in Table 1.
Table 1.
Platinated TFOs and their target DNA duplex.
| Oligonucleotidea | Abr. | Tm °Cb | Calc. mass | Obs. mass |
|---|---|---|---|---|
| d–GTTCTTCTTTTCCTTTCT | 1d | 51 | 5430.92 | 5431.64 |
| d–TTCTTCTTTTCCTTTCTG | 2d | 46 | 5430.92 | 5433.50 |
| d–GTTCTTCTTTTCCTTTCTG | 3d | 49 | 5759.97 | 5760.45 |
| mr–TTCTTCTTTTCCTTTCTG | 2mr | 72 | 5971.32 | 5971.70 |
| mr–GTTCTTCTTTTCCTTTCTG | 3mr | 71 | 6330.47 | 6332.39 |
| d–(Pt–G)TTCTTCTTTTCCTTTCT | t1d | ND | 5659.92 | 5656.13 |
| d–TTCTTCTTTTCCTTTCT(Pt–G) | t2d | ND | 5659.92 | 5658.37 |
| d–(Pt–G)TTCTTCTTTTCCTTTCT(Pt–G) | t3d | ND | 6218.07 | 6213.79 |
| mr–TTCTTCTTTTCCTTTCT(Pt–G) | t2mr | ND | 6200.42 | 6198.84 |
| mr–(Pt–G)TTCTTCTTTTCCTTTCT(Pt–G) | t3mr | ND | 6788.67 | 6785.19 |
| d–CCGAAGAAGAAAAGGAAAGAGCC GGCTTCTTCTTTTCCTTTCTCGG–d |
4R 4Y |
65c | 7154.31 6926.07 |
7155.18 6926.12 |
d, 2′-deoxyribo; mr, 2′-O-methylribo; C, 5-methylcytosine; Pt-G, N7-trans-diamminediaquaplatinum(II) derivative of guanine; and Bold, TFO binding site.
Triplex melting temperatures were determined at pH 6.5 in triplex buffer containing 100 mM MOPS, 100 mM sodium acetate, and 2.5 mM magnesium acetate. ND, not determined.
Melting temperature of the target duplex at pH 6.5.
Figure 3. Adduct formation between platinated TFOs and DNA duplexes.

(a) Sequences of the DNA duplexes. Bases shown in bold lie outside the TFO binding site. (b) Percent adduct formation between platinated TFOs t1d (dark blue), t2d (medium blue), or t3d (light blue) and the purine or pyrimidine strands of each of the DNA duplexes. Solutions containing 1 μM 32P-labeled duplex and 2 μM platinated TFO were incubated in triplex buffer at 37°C for 48 hrs and then analyzed by PAGE on a 20% denaturing gel. The amount of adduct formed was quantitated by phosphorimaging the gel.
Figure 5. Sequence dependent interactions between platinated TFOs and DNA duplexes.

(a) Sequence of the scrambled duplex, SRY. (b) Solutions containing 1 μM 32P-labeled duplex SRY or 1 μM 32P-labeled duplex 4RY and 2 μM platinated TFO were incubated in triplex buffer at 37°C for 48 hrs and then analyzed by PAGE on a 20% denaturing gel. (c) Solutions containing 1 μM 32P-labeled duplex 4RY, 2 μM platinated TFO and increasing concentrations of competitor non-platinated TFO were incubated in triplex buffer at 37°C for 48 hrs and then analyzed by PAGE on a 20% denaturing gel. The percentage of adduct formed by each platinated TFO versus competitor concentration was determined.
Figure 8. Interaction of platinated TFO with plasmid DNA.

(a) Sequence of duplex insert 5RY. The TFO binding site is indicated by the double underline; the guanine target for reaction with platinated TFO 2tmr is underlined; EcoRI and HindIII complementary overhangs are indicated by heavy underlines; and the BglII restriction site is indicated by the arrows. (b) Reaction of 1 μM 5RY with 0 μM (lane 1), 2 μM (lane 2), 4 μM (lane 3) or 10 μM t2mr in buffer containing 50 mM HEPES, 100 mM sodium chloride, 5 mM magnesium chloride, pH 7.5 incubated at 37°C for 24 hrs. The 5Y strand was labeled with 32P and the products of the reaction were analyzed on a 15% denaturing polyacrylamide gel. (c) Reaction of 500 ng of p-GEM-TFO with 0 μM TFO (lane 1), 10 μM 2mr (lane 2), 10 μM t2mr (lane 3) or 0 μM TFO (lane 4) in buffer containing 50 mM HEPES, 100 mM sodium chloride, 5 mM magnesium chloride, pH 7.5 incubated at 37°C for 72 hrs. Following the reaction, the plasmids were digested with 10 units (lanes 1-3) or 0 units (lane 4) of BglII and the digests were analyzed by electrophoresis on a 0.8% agarose gel. Lane M is a 1 kb DNA ladder.
Preparation of trans-DDP modified TFOs
The N7s of the terminal guanines of the TFOs were platinated by dissolving the dried oligomer in an aqueous solution of 144 μM trans-diamminediaquaplatinum(II) dinitrate to give a final oligomer concentration of 0.2 nM. The solution was incubated at 37°C for 2-3 hrs. The platinated TFOs were then purified by SAX-HPLC as described above and their compositions were determined by MALDI-TOF mass spectrometry as shown in Table 1.
Thermal denaturation experiments
A solution of 1 μM 4RY duplex and 1 μM TFO was prepared in triplex buffer (pH 6.5), which contained 100 mM MOPS, 100 mM sodium acetate, and 2.5 mM magnesium acetate. The solution was heated to 90°C for 10 min, allowed to cool slowly to room temperature and incubated overnight at 4°C to anneal the triplex. The absorbance at 260 nm was monitored using a Cary 3E spectrophotometer as the solution was heated from 5°C to 80°C at a rate of 0.4°C/min. The melting temperature was determined by plotting the first derivative of the absorbance vs. temperature profile and is accurate to ±1°C.
Determination of binding of platinum-modified TFOs to DNA
The 5′-hydroxyl group of the pyrimidine strand of the target DNA duplex was labeled using, polynucleotide kinase and γ-[32P]-ATP at a specific activity of 10 Ci/mmol. The labeled strand was mixed with an equimolar amount of unlabeled complementary strand, and the solution was evaporated to dryness. The residue was dissolved in 100 μL of triplex buffer to a final duplex concentration of 1 μM. The solution was heated to 90°C for 10 min, and then slowly cooled to room temperature. Solutions containing 100 pmol of platinated TFO were evaporated to dryness, the residue dissolved in 10 μL of the labeled duplex solution and the solution incubated overnight at 4°C. A2 μL aliquot of 50% glycerol was added to each sample, and the samples were electrophoresed on a 20% native polyacrylamide gel at 100 V and 4°C in triplex buffer. The gel was imaged using a Fuji FLA-7000 phosphorimager.
Reaction of platinum-modified TFOs with DNA
1 μM [32P]-labeled duplex with a specific activity of 10 Ci/mmol was prepared as described above. Solutions containing 10-200 pmol of platinated TFO were evaporated to dryness and dissolved in 10 μL of the labeled duplex solution to a final concentration of 1-20 μM TFO. Each reaction mixture was incubated at 37°C and quenched with 7 μL of formamide gel loading buffer. The samples were electrophoresed on a 20% denaturing polyacrylamide gel at 800 V. The gel was imaged by a Fuji FLA-7000 phosphorimager and radioactivity in each band was quantitated using ImageGuage software.
Pt-TFO cross-link reversal by cyanide treatment
Platinum modified TFO (t1d, t2d or t3d) was cross-linked to the target duplex using the procedure described above. The slower migrating band, which corresponds to the cross-linked product, was excised from the denaturing gel and the gel slice was extracted by shaking in 20% acetonitrile solution containing 0.1 M ammonium acetate buffer (pH 6.25) for 48 hrs at 37°C. The DNA was precipitated in 100% ethanol overnight at −20°C, and then centrifuged for 20 min at 4°C. The pellet was resuspended in 70% ethanol and centrifuged for 10 min at 4°C. The pellet was dried under vacuum and dissolved in either 20 μL of water or 20 μL of 200 mM sodium cyanide. Samples were incubated at 37°C for 48 hr and electrophoresed on a 20% denaturing polyacrylamide gel at 800 V. The gel was imaged by a Fuji FLA-7000 phosphorimager and radioactivity in each band was quantitated using ImageGuage software.
TFO binding kinetics
Triplex formation results in a decrease in absorbance at 260 nm. This phenomenon has been employed previously to estimate the rate of third strand association (39, 40). Separate solutions of 2 μM t1d and 2 μM 4RY were prepared in triplex buffer (pH 6.5). The duplex solution (4RY) was annealed and stored overnight at 4°C. 500 μL of each solution was equilibrated at 37°C and the solutions were quickly, but thoroughly mixed in a 1 mL quartz cuvette to give a final concentration of 1 μM. The absorbance at 260 nm was immediately monitored using a Cary 3E spectrophotometer (1 data point/3 sec) over the course of 20,000 sec (5.56 hrs) at 37°C. The kon value was calculated using the Varian software supplied with the spectrophotometer using second order kinetics. The reported kon value is an average of four independent experiments. The t1/2 of binding was calculated using the equation t1/2 = 1/(kon*[duplex]0).
Construction of plasmid p-GEM-TFO
Plasmid pGEM-3Z (Promega, Madison WI) was cut with EcoRI and HindIII and the resulting linear DNA was purified by agarose gel electrophoresis. The HindIII restriction site is immediately adjacent to a T7 promoter sequence in the plasmid. One equivalent of the linearized DNA was ligated with 6 equivalents of duplex 5RY, whose sequence is shown in Figure 8a, using T4 DNA ligase. The ligated DNA was then transformed into DH5α E. coli cells; the transformed cells were plated on LB agar plates supplemented with ampicilin, bromo-chloro-indolyl-galactopyranoside (X-gal) and isopropyl-β-D-thioglactopyranoside(IPTG); and white colonies were selected. Individual colonies were grown in LB medium; plasmid DNA was isolated using a QIAprep Spin Miniprep kit (Qiagen Sciences); and the DNA was sequenced to confirm the presence of the 5RY insert. The insert is oriented such that the 5Y strand of the duplex is part of the plasmid strand that can be transcribed by T7 RNA polymerase.
Reaction of platinum-modified TFO 2tmr with p-GEM-TFO
Solutions (10 μL) containing 500 ng of pGEM-TFO and either 0 μM TFO, 10 μM non-platinated TFO 2mr or 10 μM platinated TFO t2mr in buffer composed of 50 mM HEPES, 100 mM sodium chloride, 5 mM magnesium chloride, pH 7.5, were incubated for 72 hrs at 37°C. A 0.5 μL aliquot was removed from each reaction and each aliquot was incubated in 9.5 μL of a solution containing 100 mM sodium chloride, 50 mM Tris-hydrochloride, 10 mM magnesium chloride, 1 mM dithiothreitol, pH 7.9, and 10 units of BglII for 2 hrs at 37°C. A 5 μL aliquot of each digest was mixed with 5 μL of 20% glycerol loading buffer and the solutions were electrophoresed on a 0.8% agarose gel containing Syber Green dye. The gel was scanned on a Fuji FLA-7000 imager.
Transcription of p-GEM-TFO DNA
pGEM-TFO treated with 0 μM TFO, 10 μM non-platinated TFO 2mr or 10 μM platinated TFO t2mr as described above was incubated with HindIII for 2 hrs at 37°C. Examination by agarose gel electrophoresis showed that all three plasmid preparations were completely linearized. Transcription was carried out using the Riboprobe System T-7 kit supplied by Promega. Each reaction contained between 1.5 to 11.6 ng of linearized plasmid DNA, 9.8 mM dithiothreitol, 40 units of RNasin inhibitor, 0.49 mM each of rATP/rCTP/rUTP, 11.8 μM rGTP, 50 μCi of α-32- P-rGTP and 19 units of T7 RNA polymerase in 20.4 μL of transcription buffer that consisted of 40 mM Tris hydrochloride (pH 7.9), 10 mM sodium chloride, 6 mM magnesium chloride, 2 mM spermidine, 0.05% Tween. The reactions were incubated for 1 hr at 37°C; passed through a Sephadex G-25 Micro-Spin column (GE Healthcare) and then electrophoresed on a 15% denaturing polyacrylamide gel at 800 V. The gel was phosphorimaged, the 42 nucleotide run-off products were quantified from the phosphorimage and the amount of product was normalized to the amount of plasmid DNA used in the reaction.
Colony formation by E.coli transformed with p-GEM-TFO
Samples (150 pg each) of pGEM-TFO treated with 0 μM TFO, 10 μM non-platinated TFO 2mr or 10 μM platinated TFO t2mr as described above were electroporated at 1.8 kv into electrocompetent DH5α E. coli cells in 40 μL of GYT medium that contained 1.25 mg/mL yeast extract, 2.5 mg/mL tryptone and 10% glycerol. The cells were then immediately diluted with 1.0 mL of SOC medium that contained 20 mg/mL Bacto-tryptone, 5 mg/mL Bacto-yeast extract, 8.6 mM sodium chloride and 2.5 mM potassium chloride, and then incubated with shaking at 37°C for 1 hr. The cells were then spread onto LB plates supplemented with ampicilin and the plates were incubated overnight at 37°C. Two separate experiments were carried out in triplicate and the number of colonies formed was counted.
RESULTS AND DISCUSSION
Synthesis and Characterization of platinated TFOs
Previous studies by Leng and coworkers had examined the ability of oligodeoxyribopyrimidine TFOs with a 5′-transPt-dG modification to form adducts with DNA (36, 37). We have extended these studies to include oligodeoxyribopyrimidine and oligo-2′-O-methylribopyrimidine TFOs that contain 5′-, 3′- and 5′,3′-transPt-dG modifications. The sequences of these TFOs and the site targeted for triplex formation are shown in Table 1. The target duplex sequence (4RY) serves as a model for triplex formation with five cytosine residues within a 17 bp long polypurine tract, and is similar to a previously studied purine target sequence found in the coding region of the polymerase gene of HIV-1 (34,35). Each of the TFOs consisted of either 2′-deoxyribonucleotides or 2′-O-methylribonucleotides and contained 5-methylcytosine (C) and 5-methyluracil (T) bases. One or both ends of the TFO contained guanine nucleosides that were platinated to give the monofunctional N-7 trans-[Pt(NH3)2(G)(H2O)]+ adduct. Platination of the TFOs was carried out by incubating the TFO in a solution containing trans-diamminediaquaplatinum(II) dinitrate at 37°C. Under these conditions, platination occurs preferentially at the N-7 of the guanine of the TFO (36). Because addition of the platinum group adds a positive charge to the oligonucleotide, the platinated TFO can be separated from non platinated TFO by strong anion exchange HPLC (see Figure S1). The compositions of the platinated TFOs were confirmed by MALDI-TOF mass spectrometry analysis (see Table 1). Additional peaks observed in the HPLC chromatograph (see Figures S1 and S2) were isolated and identified by MALDI-TOF mass spectrometry as multiply platinated species. The overall yields of the purified and desalted Pt-TFOs were approximately 20-30%.
The TFOs were tested for their ability to bind to a DNA target sequence, 4RY, by measuring the melting temperatures of the triplexes formed by the non-platinated TFOs at pH 6.5, as shown in Table 1. The TFOs that consisted of deoxyribonucleotides, 1d, 2d, and 3d, formed relatively stable triplexes whose third strands dissociated at approximately 50°C, a temperature lower than the melting temperature of duplex 4RY, 65°C. We and others have shown previously that incorporation of 2′-O-methylribonucleotides can stabilize TFO binding (40-45). In accord with these findings, the TFOs containing 2′-O-methylribonucleotides, 2mr and 3mr, formed triplexes that melted with a single transition at greater than 70°C. Representative melting curves can be found in Figure S3. Melting temperatures for the corresponding platinum modified TFOs were not determined because these TFOs were expected to form adducts with the DNA target. However, a previous study has shown that modification of a guanine at the end of a polypyrimidine TFO with a non reactive platinum group resulted in a slight stabilization of a triplex formed by this TFO (37). Most likely this stabilization results from introduction of the positively charged platinum group. Based on this result we expected that the platinated TFOs shown in Table 1 would form triplexes whose stabilities are comparable to those of the parent unmodified TFOs.
Adduct formation between platinated TFOs and DNA duplexes
We examined the interactions of the platinated TFOs by electrophoretic mobility shift assays on native polyacrylamide gels. Each of the platinated deoxyribo-TFOs was incubated with DNA target duplex, 4RY, at 4°C, and the solution was then subjected to electrophoresis on a native polyacrylamide gel run at 4°C to prevent dissociation of the triplex due to heating of the gel. As shown in Figure 1a, the mobility of 4RY decreased in the presence of Pt-TFO, t1d, TFO-Pt, t2d, or Pt-TFO-Pt, t3d, a result consistent with triplex formation.
Figure 1. Interactions of platinated TFOs with DNA duplex 4RY.

(a) Solutions containing 10 μM platinated TFO and 1 μM duplex 4RY, whose purine strand was labeled with 32P-phosphate, were incubated at 4°C in triplex buffer that contained 100 mM MOPS, pH 6.5, 100 mM sodium acetate, 2.5 mM magnesium acetate, and were then subjected to electrophoresis on a 20% native polyacrylamide run at 4°C and 100 V. (b) Solutions containing platinated TFO and 1 μM duplex 4RY, whose purine strand was labeled with 32P-phosphate (shown in red), were incubated at 37°C for 48 hrs in triplex buffer and were then subjected to electrophoresis on a 20% denaturing polyacrylamide gel. Percent adduct formation is shown below the corresponding lane (% X-L). (c) Solutions containing platinated TFO and 1 μM duplex 4RY, whose pyrimidine strand was labeled with 32P-phosphate (shown in red), were incubated at 37°C for 48 hrs in triplex buffer and were then subjected to electrophoresis on a 20% denaturing polyacrylamide gel. Percent adduct formation is shown below the corresponding lane (% X-L).
The platinated TFOs were then tested for their ability to form adducts with each strand of the target duplex. Increasing concentrations of the platinated TFOs were incubated at 37°C with duplex 4RY whose purine or pyrimidine strand was labeled with 32P phosphate. The reaction mixtures were then subjected to electrophoresis on a denaturing polyacrylamide gel. As shown in Figure 1b, incubation of the target with Pt-TFO t1d resulted in 50% adduct formation with the purine strand, whereas only 5% adduct formation was observed with TFO-Pt t2d. Approximately 50% adduct formation was observed when Pt-TFO-Pt t3d was incubated with purine strand labeled 4RY. In this case, however, the electrophoretic mobility of the adduct was significantly slower than that observed for the adducts formed by either t1d or t2d. This slower mobility suggested that t3d had formed adducts with both strands of the duplex. Reaction of Pt-TFO t1d with pyrimidine-labeled 4RY resulted in approximately 10% adduct formation, as shown in Figure 1c, whereas approximately 90% adduct formation was observed with TFO-Pt t2d. A similar level of adduct formation was also observed with Pt-TFO-Pt t3d. As seen in Figure 1c, t3d formed two types of adducts, one whose mobility was similar to that of adducts formed by t1d and t2d, and a slower mobility adduct. This latter adduct, which represents approximately 35 - 40% of the total adduct formation, is most likely the TFO cross-linked to both strands of the duplex. It migrates more slowly on the denaturing gel because the three strands are linked together as indicated schematically in Figure 1c. These results show that adduct formation can occur when the transPt-dG is placed at either end of the TFO and that either strand of the duplex can be targeted. These results also suggest that cross-linking is strand specific and possibly sequence specific depending on the location, 5′ or 3′, of the platinated-G in the TFO.
It seemed likely that the transPt-dG group of the TFO platinates the guanine residues of 4RY adjacent to the TFO binding site. It has been shown that cyanide is an extremely strong ligand for platinum and that adding cyanide to a Pt-G adduct will displace the platinum from guanine (46-50). The t1d-4R, t2d-4Y, and t3d-4Y adducts were isolated from the gel and were treated with 200 mM sodium cyanide. The products of these reactions were then analyzed on a denaturing gel. As shown in Figure 2, the mobilities of the treated adducts reverted to those of the labeled strand alone. This result is consistent with the formation of a platinum adduct between the platinated TFO and the labeled target strand.
Figure 2. Reversal of platinated TFO adducts by treatment with cyanide.
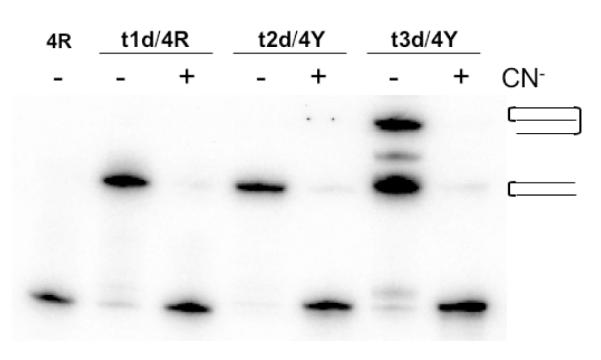
Adducts formed by reaction of the platinated TFOs with duplex 4RY were isolated and treated with either water or 200 mM sodium cyanide. The reaction mixtures were then subjected to electrophoresis on a 20% denaturing polyacrylamide gel. The labeled strand of duplex 4RY is indicated at the top of the gel.
In order to better understand the base preference of adduct formation, a series of duplexes were synthesized in which a set of two identical base pairs, which could serve as sites for platination, were placed at each end of the duplex. The sequences of the four duplexes containing the four possible combinations of the base pairs are shown in Figure 3a. The purine- or pyrimidine-rich strand of each duplex was labeled with 32P, the labeled duplexes were reacted with the platinated TFOs at 37°C and the products analyzed by denaturing PAGE. The results are summarized in Figure 3b. As expected, significant adduct formation was observed with duplexes whose labeled strand contained guanine (GY or GR) or adenine (AY or AR) residues at the ends of the duplex. It is likely that adduct formation occurred as a result of platination of the N-7 of these bases. Very little adduct formation was observed when either cytosines or thymines were placed at the ends of the labeled strands, a result consistent with the known reactivity of these bases with platinum. We were surprised to find that adduct formation occurred equally well on either the pyrimidine or purine strand of the duplex when guanines or adenines were the target of platination. In a previous study of a 5′-transPt-dG modified TFO, no adduct formation with the pyrimidine strand was observed (36). However, in this case the pyrimidine strand of the target duplex contained only pyrimidines at the platination site.
The duplexes shown in Figure 3a contain two potential sites for platination: one base from the end of the triplex binding site (n-1) and two bases from the end of the triplex binding site (n-2). In order to better characterize the precise site of platination for each of the platinated TFOs, we employed another set of duplexes (shown in Figure 4a) that have a single G residue at either the n-1 or the n-2 position. The purine or pyrimidine strand of each duplex was 5′-end labeled with a 32P phosphate group and reactions were carried out with each of the platinated TFOs as described above. As shown in Figure 4b, a clear pattern of adduct formation emerged for each of the platinated TFOs. Preferential adduct formation by the Pt-TFO, t1d, was observed with duplexes that contained guanine at the n-1 position in either the purine or pyrimidine strand of the duplex. In contrast, adduct formation by the TFO-Pt, t2d, occurred to the greatest extent with the duplex whose pyrimidine strand contained a guanine at position n-2, and to lesser extents with the duplexes that contained an n-2 guanine in the purine strand or an n-1 guanine in the pyrimidine strand. As expected, the bis-platinated Pt-TFO-Pt, t3d, formed adducts equally well with the pyrimidine and purine strands of both duplexes.
Figure 4. Adduct formation between platinated TFOs and DNA duplexes.
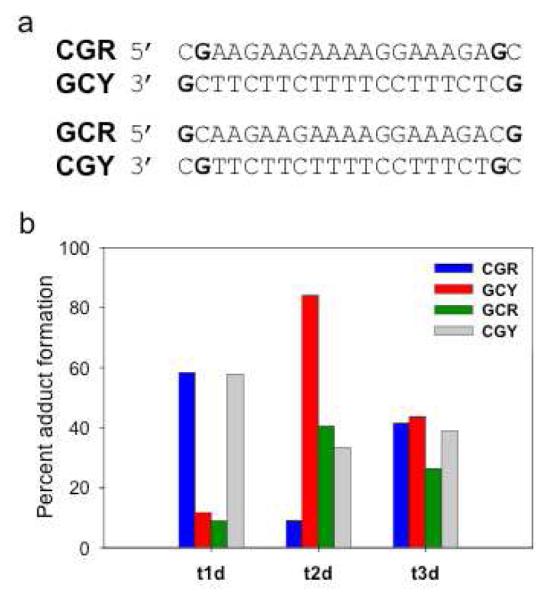
(a) Sequences of DNA duplexes. Potential sites of platination are shown in bold. (b) Percent adduct formation between platinated TFOs t1d, t2d or t3d and the purine or pyrimidine strands of the DNA duplexes. Solutions containing 1 μM 32P-labeled duplex and 2 μM platinated TFO were incubated in triplex buffer at 37°C for 48 hrs and then analyzed by PAGE on a 20% denaturing gel. The amount of cross-linking was quantitated by phosphorimaging the gel.
The experiments described above show that platinated TFOs can form adducts with DNA duplexes that contain guanines or adenines adjacent to the TFO binding site in either strand of the duplex. These platinated TFOs retained approximately 60-90% of their initial activity after storage for 1.5 months in aqueous 50% acetonitrile at 4°C. These results suggest that it should be possible to design stable platinated TFOs that can target a rather broad range of sequences in DNA.
Specificity of adduct formation
Experiments were carried out using a “scrambled” DNA duplex to ensure that adduct formation by the platinated TFOs was dependent upon the ability of the TFO to bind to its target. The sequence of this scrambled duplex, SRY, is shown in Figure 5a. It contains the same number of CG and TA base pairs as DNA duplex 4RY (see Table 1) and has the same terminal sequences adjacent to the TFO binding site. Each of the platinated TFOs was incubated at 37°C with duplex SRY, whose purine or pyrimidine strand was labeled with a 5′-32P-phosphate group and the products of the reaction were analyzed by denaturing PAGE. Control reactions were run using 32P-labeled 4RY as a target duplex. As shown in Figure 5b, the platinated TFOs formed adducts with either the purine or pyrimidine strand of the 4RY. In contrast, very little adduct formation with either the SR or SY strand of the scrambled duplex was observed. In a second experiment, increasing concentrations of non-platinated TFO were added to reaction mixtures containing the platinated TFOs and DNA duplex 4RY. The amount of adduct formation was determined by denaturing PAGE. The results are summarized in Figure 5c. A significant decrease in adduct formation by each of the platinated TFOs was observed in the presence of an equimolar amount of competitor non-platinated TFO (2 μM) and adduct formation was completely inhibited at higher competitor concentrations.
Taken together, the results described above demonstrate that adduct formation between the platinated TFOs and DNA duplex 4RY is driven by the ability of the TFO to bind specifically to this target and not by non-sequence-specific platination of the DNA duplex. This behavior suggests that it should be possible to design platinated TFOs that can react with their designated DNA targets in a specific manner with little or no off-target effects.
Kinetics of TFO binding and adduct formation
The kinetics of triplex formation were determined by observing the decrease in absorbance upon mixing TFO 1d with DNA duplex 4RY at 37°C as shown in Figure 6a. The average rate constant, kon, for the association of the TFO with the duplex is 654.51 s−1M−1 as determined from four separate experiments. This translates to a half-life of triplex formation of 25.5 minutes. This value can be compared with those of Xodo, who found a kon of 2557 M−1s−1 for a 18-mer deoxy TFO that was assayed at 26.1°C (39) and Puri et. al., observed a kon value of approximately 600 M−1s−1 for a 17-mer 2′-O-methylribo TFO at 25°C (40).
Figure 6. Kinetics of triplex adduct formation.

(a) Equal volumes of solutions containing 1 μM TFO 1d and DNA duplex 4RY in triplex buffer were mixed and the absorbance at 260 nm was recorded as a function of time at 37°C. (b) Solutions containing 1 μM 32P-labeled duplex 4RY and 2 μM t2d were incubated in triplex buffer at 37°C for various lengths of time and analyzed by PAGE on a 20% denaturing gel. The percent adduct formation was determined as a function of time.
The kinetics of adduct formation between the platinated TFO t2d and DNA duplex 4RY were determined. In these experiments, 1 μM 4RY was incubated with 2 μM platinated TFO, t2d, at 37°C and the extent of adduct formation at various times was assayed by denaturing PAGE. Figure 6b shows the averaged results from three independent experiments. The platinated TFO achieves maximal adduct formation after 10-12 hours and the t1/2 of adduct formation is approximately 3 hours. This result suggests that adduct formation occurs at a rate significantly slower than that at which the TFO binds to its target. It appears then that platination is the rate limiting step in adduct formation. These observations are consistent with the specificity of adduct formation described above and further support the idea that TFO binding precedes platination of the DNA target.
Effect of pH on adduct formation
The experiments described above were carried out at pH 6.5, a pH that favors protonation of the 5-methylcytosines in the TFO and thus enhances the stability of the triplex. A 2-5 fold decrease in adduct formation was observed when the reactions were carried out at pH 7.4 (see Figure 7a). As demonstrated in Table 1, triplex stability can be increased significantly by replacing deoxyribonucleotides in the TFO with 2′-O-methylribonucleotides. This change significantly increases the amount of adduct formation as shown in Figure 7b. The 3′-platinated mr-TFO, t2mr, and 3′,5′-bis-platinated mr-TFO, t3mr, both give extensive adduct formation with duplex 4RY at both pH 6.5 and pH 7.4. These results suggest that platinated 2′-O-methyribo-TFOs could be designed that would function effectively under physiological conditions.
Figure 7. Effect of pH on adduct formation by platinated deoxyribo- and 2′-O-methylribo-TFOs.

(a) Solutions containing 1 μM duplex 4RY and 2 μM platinated deoxyribo-TFOs t1d, t2d, or t3d in pH 6.5 or pH 7.4 triplex buffer were incubated at 37°C for 48 hrs and then subjected to 20% denaturing PAGE. (b) Solutions containing 1 μM duplex 4RY and 2 μM platinated 2′-O-methylribo-TFOs t2mr or t3mr in pH 6.5 or 7.4 triplex buffer at 37°C for 48 hrs and then subjected to 20% denaturing PAGE. The percent adduct formation as determined by phosphorimaging is shown below each lane. The purine strand of duplex 4RY is labeled in each case.
Interactions with plasmid DNA
The interaction of 3′-platinated mr-TFO t2mr with a purine tract in the supercoiled form of plasmid p-GEM-TFO was studied. This plasmid was constructed by inserting duplex 5RY, whose sequence is shown in Figure 8a, into the EcoRI and HindIII restriction sites of the polylinker region of p-GEM-3Z. Duplex 5RY, whose purine tract is identical in sequence to that of duplex 4RY, contains a unique BglII restriction site that is not otherwise found in p-GEM-3Z. The underlined guanine located 10 nucleotides from the 5′-end of 5Y serves as a target site for platination by TFO t2mr.
Binding experiments were carried out at pH 7.5 and 37°C to measure the extent of adduct formation between t2mr and duplex 5RY, whose pyrimidine-rich strand was labeled with 32P. As shown in Figure 8b, addition of increasing concentrations of t2mr resulted in the formation of a new product whose mobility on a denaturing polyacrylamide gel was consistent with formation of an adduct between t2mr and strand 5Y of the duplex. Quantification of the phosphorimage of the gel showed that 79% of the duplex was converted to adduct after incubation with 10 μM t2mr for 24 hrs. The cross-linked duplex was not hydrolyzed when treated with BglII (data not shown). This result demonstrates that triplex and adduct formation by t2mr obstructs the recognition site of the restriction enzyme.
Plasmid p-GEM-TFO was incubated with 10 μM t2mr or 10 μM non-platinated TFO 2mr at 37°C. The plasmid DNA was then treated with BglII and the resulting digests were analyzed by agarose gel electrophoresis. As shown in Figure 8c, supercoiled plasmid alone (lane 1) or supercoiled plasmid that had been incubated with 2mr (lane 2) was completely converted by BglII to linear DNA. In contrast, 84% of the plasmid that had been incubated with t2mr was resistant to digestion by BglII and remained in the supercoiled form (lane 3). These results show that platinated TFO t2mr can bind to and react with the 5RY insert in the supercoiled plasmid under essentially physiological conditions.
The site on the plasmid into which duplex 5RY was inserted is flanked by promoter sequences for T-7 and Sp6 RNA polymerases. The insert is oriented such that the strand containing the 5Y sequence would be used as a template for transcription by T-7 RNA polymerase. Run-off transcription experiments were carried out to determine if transcription is inhibited when t2mr is bound to the plasmid. Plasmid that had been incubated with t2mr as described above was linearized by digestion with BglII. The linearized plasmid was then transcribed using T-7 RNA polymerase and the amount of run-off transcript, which is 42 nucleotides in length, was compared to that obtained from untreated p-GEM-TFO or plasmid treated with 2mr. The non-platinated TFO, 2mr, inhibited transcription approximately 25%, whereas transcription was inhibited approximately 95% by the platinated TFO, t2mr.
In addition to inhibiting transcription, the platinated TFO would also be expected to interfere with plasmid replication. To test this, 2mr- or t2mr -treated p-GEM-TFO, which contains an ampicilin resistance gene, was electroporated into wild type DH5α E. coli cells and the electroporated cells were spread on LB agar plates that contained ampicilin. The number of colonies formed by cells electroporated with 2mr was reduced 67% relative to cells electroporated with untreated pGEM-TFO. An even greater amount of inhibition of colony formation, 84%, was observed for the cells electroporated with t2mr -treated p-GEM-TFO.
Taken together, the experiments described above suggest that the platinated 2′-O-methylribo-TFO can bind effectively to supercoiled DNA, and that such binding is able to interfere with transcription and replication of plasmid DNA. Experiments are currently in progress to determine if platinated TFOs of this type can be used to inhibit gene expression in mammalian cells.
Supplementary Material
ACKNOWLEDGEMENTS
This research was supported by a grant from the National Institutes of Health, GM 057140 (PSM). The authors wish to thank Ms. Lynn Periera for carrying out the transcription experiments; the laboratory of Dr. Michael Matunis for providing electrocompetent DH5α cells; and Ms. Erica Hlavin for advice on transforming E. coli cells.
Footnotes
Supporting Information Available: SAX HPLC chromatograms of t2d (Figure S1), t3d (Figure S2) and representative thermal denaturation curves (Figure S3) are provided. This material is available free of charge via the Internet at http://pubs.acs.org/BC.
REFERENCES
- (1).Chan PP, Glazer PM. Triplex DNA: fundamentals, advances, and potential applications for gene therapy. J. Mol. Med. 1997;75:267–282. doi: 10.1007/s001090050112. [DOI] [PubMed] [Google Scholar]
- (2).Knauert MP, Glazer PM. Triplex forming oligonucleotides: sequence-specific tools for gene targeting. Hum. Mol. Genet. 2001;10:2243–2251. doi: 10.1093/hmg/10.20.2243. [DOI] [PubMed] [Google Scholar]
- (3).Vasquez KM, Glazer PM. Triplex-forming oligonucleotides: principles and applications. Q. Rev. Biophys. 2002;35:89–107. doi: 10.1017/s0033583502003773. [DOI] [PubMed] [Google Scholar]
- (4).Thuong NT, Helene C. Sequence specific recognition and modification of double-helical DNA by oligonucleotides. Angew. Chem. Int. Ed. Engl. 1993;32:666–690. [Google Scholar]
- (5).Radhakrishnan I, Patel DJ. DNA triplexes: solution structures, hydration sites, energetics, interactions, and function. Biochemistry. 1994;33:11405–11416. doi: 10.1021/bi00204a001. [DOI] [PubMed] [Google Scholar]
- (6).Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annu. Rev. Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- (7).Fox KR. Targeting DNA with triplexes. Curr. Med. Chem. 2000;7:17–37. doi: 10.2174/0929867003375506. [DOI] [PubMed] [Google Scholar]
- (8).Alunni-Fabbroni M, Manfioletti G, Manzini G, Xodo LE. Inhibition of T7 RNA polymerase transcription by phosphate and phosphorothioate triplex-forming oligonucleotides targeted to a R.Y site downstream from the promoter. Eur. J. Biochem. 1994;226:831–839. doi: 10.1111/j.1432-1033.1994.00831.x. [DOI] [PubMed] [Google Scholar]
- (9).Hacia JG, Dervan PB, Wold BJ. Inhibition of Klenow fragment DNA polymerase on double-helical templates by oligonucleotide-directed triple-helix formation. Biochemistry. 1994;33:6192–6200. doi: 10.1021/bi00186a019. [DOI] [PubMed] [Google Scholar]
- (10).Giovannangeli C, Perrouault L, Escude C, Gryaznov S, Helene C. Efficient inhibition of transcription elongation in vitro by oligonucleotide phosphoramidates targeted to proviral HIV DNA. J. Mol. Biol. 1996;261:386–398. doi: 10.1006/jmbi.1996.0471. [DOI] [PubMed] [Google Scholar]
- (11).Giovannangeli C, Helene C. Triplex-forming molecules for modulation of DNA information processing. Curr. Opin. Mol. Ther. 2000;2:288–296. [PubMed] [Google Scholar]
- (12).Degols G, Clarenc JP, Lebleu B, Leonetti JP. Reversible inhibition of gene expression by a psoralen functionalized triple helix forming oligonucleotide in intact cells. J. Biol. Chem. 1994;269:16933–16937. [PubMed] [Google Scholar]
- (13).Ebbinghaus SW, Fortinberry H, Gamper HB., Jr. Inhibition of transcription elongation in the HER-2/neu coding sequence by triplex-directed covalent modification of the template strand. Biochemistry. 1999;38:619–628. doi: 10.1021/bi980981g. [DOI] [PubMed] [Google Scholar]
- (14).Intody Z, Perkins BD, Wilson JH, Wensel TG. Blocking transcription of the human rhodopsin gene by triplex-mediated DNA photocrosslinking. Nucleic Acids Res. 2000;28:4283–4290. doi: 10.1093/nar/28.21.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Besch R, Marschall C, Schuh T, Giovannangeli C, Kammerbauer C, Degitz K. Triple helix-mediated inhibition of gene expression is increased by PUVA. J. Invest. Dermatol. 2004;122:1114–1120. doi: 10.1111/j.0022-202X.2004.22521.x. [DOI] [PubMed] [Google Scholar]
- (16).Wang G, Levy DD, Seidman MM, Glazer PM. Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol. Cell. Biol. 1995;15:1759–1768. doi: 10.1128/mcb.15.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang G, Seidman MM, Glazer PM. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science. 1996;271:802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- (18).Vasquez KM, Narayanan L, Glazer PM. Specific mutations induced by triplex-forming oligonucleotides in mice. Science. 2000;290:530–533. doi: 10.1126/science.290.5491.530. [DOI] [PubMed] [Google Scholar]
- (19).Faruqi AF, Datta HJ, Carroll D, Seidman MM, Glazer PM. Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol. Cell. Biol. 2000;20:990–1000. doi: 10.1128/mcb.20.3.990-1000.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Grant KB, Dervan PB. Sequence-specific alkylation and cleavage of DNA mediated by purine motif triple helix formation. Biochemistry. 1996;35:12313–12319. doi: 10.1021/bi9608469. [DOI] [PubMed] [Google Scholar]
- (21).Taylor MJ, Dervan PB. Kinetic analysis of sequence-specific alkylation of DNA by pyrimidine oligodeoxyribonucleotide-directed triple-helix formation. Bioconjug. Chem. 1997;8:354–364. doi: 10.1021/bc970035x. [DOI] [PubMed] [Google Scholar]
- (22).Kutyavin IV, Gamper HB, Gall AA, Meyer RB. Efficient, specific interstrand cross-linking of double-stranded DNA by a chlorambucil-modified triplex-forming oligonucleotide. J. Am. Chem. Soc. 1993;115:9303–9304. [Google Scholar]
- (23).Reed MW, Lukhtanov EA, Gorn V, Kutyavin IV, Gall AA, Wald A, Meyer RB. Synthesis and reactivity of aryl nitrogen mustard-oligodeoxyribonucleotide conjugates. Bioconjug. Chem. 1998;9:64–71. doi: 10.1021/bc970134a. [DOI] [PubMed] [Google Scholar]
- (24).Takasugi M, Guendouz A, Chassignol M, Decout JL, Lhomme J, Thuong NT, Helene C. Sequence-specific photo-induced cross-linking of the two strands of double-helical DNA by a psoralen covalently linked to a triple helix-forming oligonucleotide. Proc. Natl. Acad. Sci. U.S.A. 1991;88:5602–5606. doi: 10.1073/pnas.88.13.5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Giovannangeli C, Thuong NT, Helene C. Oligodeoxynucleotide-directed photo-induced cross-linking of HIV proviral DNA via triple-helix formation. Nucleic Acids Res. 1992;20:4275–4281. doi: 10.1093/nar/20.16.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Havre PA, Gunther EJ, Gasparro FP, Glazer PM. Targeted mutagenesis of DNA using triple helix-forming oligonucleotides linked to psoralen. Proc. Natl. Acad. Sci. U.S.A. 1993;90:7879–7883. doi: 10.1073/pnas.90.16.7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Guieysse AL, Praseuth D, Giovannangeli C, Asseline U, Helene C. Psoralen adducts induced by triplex-forming oligonucleotides are refractory to repair in HeLa cells. J. Mol. Biol. 2000;296:373–383. doi: 10.1006/jmbi.1999.3466. [DOI] [PubMed] [Google Scholar]
- (28).Ping YH, Rana TM. Mechanism of site-specific psoralen photoadducts formation in triplex DNA directed by psoralen-conjugated oligonucleotides. Biochemistry. 2005;44:2501–2509. doi: 10.1021/bi0488707. [DOI] [PubMed] [Google Scholar]
- (29).Connors TA, Roberts JJ. Platinum coordination complexes in cancer chemotherapy. Springer Verlag; Berlin: 1974. [Google Scholar]
- (30).Lippert B. Multiplicity of metal ion binding patterns to nucleobases. Coordination Chemistry Reviews. 2000;200:487–516. [Google Scholar]
- (31).Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog. Nucleic Acid Res. Mol. Biol. 2001;67:93–130. doi: 10.1016/s0079-6603(01)67026-0. [DOI] [PubMed] [Google Scholar]
- (32).Sharma SK, McLaughlin LW. Cross-linking of a DNA conjugate tethering a cis-bifunctional platinated complex to a target DNA duplex. J. Am. Chem. Soc. 2002;124:9658–9659. doi: 10.1021/ja020500n. [DOI] [PubMed] [Google Scholar]
- (33).Sharma SK, McLaughlin LW. Triplex mediated delivery of a platinum complex to a specific DNA target site. J. Inorg. Biochem. 2004;98:1570–1577. doi: 10.1016/j.jinorgbio.2004.05.001. [DOI] [PubMed] [Google Scholar]
- (34).Campbell MA, Mason TM, Miller PS. Interactions of platinum(II)-derivatized triplex-forming oligonucleotides with DNA. Can. J. Chem. 2007;85:241–248. [Google Scholar]
- (35).Campbell MA, Miller PS. Cross-linking to an interrupted polypurine sequence with a platinum-modified triplex-forming oligonucleotide. J. Biol. Inorg. Chem. 2009;14 doi: 10.1007/s00775-009-0499-3. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Colombier C, Lippert B, Leng M. Interstrand cross-linking reaction in triplexes containing a monofunctional transplatin-adduct. Nucleic Acids Res. 1996;24:4519–4524. doi: 10.1093/nar/24.22.4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bernal-Mendez E, Sun J. s., Gonzalez-Vilchez F, Leng M. Reactivity of transplatin-modified oligonucleotides in triple-helical DNA complexes. New J. Chem. 1998;22:1479–1483. [Google Scholar]
- (38).Puglisi JD, Tinoco I., Jr. Absorbance melting curves of RNA. Methods Enzymol. 1989;180:304–325. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]
- (39).Xodo LE. Kinetic analysis of triple-helix formation by pyrimidine oligodeoxynucleotides and duplex DNA. Eur. J. Biochem. 1995;228:918–926. doi: 10.1111/j.1432-1033.1995.tb20340.x. [DOI] [PubMed] [Google Scholar]
- (40).Puri N, Majumdar A, Cuenoud B, Miller PS, Seidman MM. Importance of clustered 2′-O-(2-aminoethyl) residues for the gene targeting activity of triple helix-forming oligonucleotides. Biochemistry. 2004;43:1343–1351. doi: 10.1021/bi035808l. [DOI] [PubMed] [Google Scholar]
- (41).Shimizu M, Konishi A, Shimada Y, Inoue H, Ohtsuka E. Oligo(2′-O-methyl)ribonucleotides. Effective probes for duplex DNA. FEBS Lett. 1992;302:155–158. doi: 10.1016/0014-5793(92)80428-j. [DOI] [PubMed] [Google Scholar]
- (42).Shimizu M, Koizumi T, Inoue H, Ohtsuka E. Effects of 5-methyl substitution in 2′-O-methyloligo-(pyrimidine)nucleotides on triple-helix formation. Bioorg. Med. Chem. Lett. 1994;4:1029–1032. [Google Scholar]
- (43).Wang S, Kool ET. Relative stabilities of triple helices composed of combinations of DNA, RNA and 2′-O-methyl-RNA backbones: chimeric circular oligonucleotides as probes. Nucleic Acids Res. 1995;23:1157–1164. doi: 10.1093/nar/23.7.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wang S, Kool ET. Origins of the large differences in stability of DNA and RNA helices: C-5 methyl and 2′-hydroxyl effects. Biochemistry. 1995;34:4125–4132. doi: 10.1021/bi00012a031. [DOI] [PubMed] [Google Scholar]
- (45).Ushijima K, Ishibashi T, Yamakawa H, Tsukahara S, Takai K, Maruyama T, Takaku H. Inhibition of restriction endonuclease cleavage by triple helix formation with RNA and 2′-O-methyl RNA oligonucleotides containing 8-oxo-adenosine in place of cytidine. Biochemistry. 1999;38:6570–6575. doi: 10.1021/bi982848u. [DOI] [PubMed] [Google Scholar]
- (46).Cassidy RA, Puri N, Miller PS. Effect of DNA target sequence on triplex formation by oligo-2′-deoxy- and 2′-O-methylribonucleotides. Nucleic Acids Res. 2003;31:4099–4108. doi: 10.1093/nar/gkg436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lippard SJ, Ushay HM, Merkel CM, Poirier MC. Use of antibodies to probe the stereochemistry of antitumor platinum drug binding to deoxyribonucleic acid. Biochemistry. 1983;22:5165–5168. [Google Scholar]
- (48).Raudaschl-Sieber G, Lippert B. Reaction of cyanide with Pt-nucleobase complexes: preparative, spectroscopic, and structural studies. Unexpected stability of Pt-thymine and Pt-uracil complexes. Inorg. Chem. 1985;24:2426–2432. [Google Scholar]
- (49).Anin MF, Leng M. Distortions induced in double-stranded oligonucleotides by the binding of cis- or trans-diammine dichloroplatinum(II) to the d(GTG) sequence. Nucleic Acids Res. 1990;18:4395–4400. doi: 10.1093/nar/18.15.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Blommaert FA, Saris CP. Detection of platinum-DNA adducts by 32P-postlabelling. Nucleic Acids Res. 1995;23:1300–1306. doi: 10.1093/nar/23.8.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.