

Abstract
Chemical tags for live cell imaging are emerging as viable alternatives to the fluorescent proteins for labeling proteins with small molecule probes. Among reported chemical tags, trimethoprim (TMP)-tag stands out for having sufficient cell permeability and selectivity to allow imaging of intracellular proteins. TMP-tag provides a non-covalent label in which the protein of interest is fused to E. coli dihydrofolate reductase (DHFR) and then labeled with a cell permeable TMP-probe heterodimer. To complement the utility of the non-covalent TMP-tag, we sought to render the TMP-tag covalent for applications such as single-molecule tracking and pulse-chase labeling that would benefit from a more permanent modification. Based on the long-standing use of proximity-induced reactivity for irreversible inhibitor design and its more recent application to in vitro chemical biology tools, we designed an eDHFR variant with a unique Cys residue positioned to react with an acrylamide electrophile installed on the TMP-probe label. In vitro experiments show that the eDHFR:Leu28Cys nucleophile reacts rapidly and quantitatively with the TMP-acrylamide-probe. Most significantly, the balance in reactivity provided by the acrylamide electrophile allows intracellular proteins tagged with eDHFR:Leu28Cys to be labeled with a TMP-acrylamide-fluorescein heterotrimer in live cells with minimal background. Thus, the TMP electrophile described here can be used immediately as a covalent chemical tag in live cells. Moreover, proximity-induced reactivity is shown to be sufficiently selective for use in a living cell, suggesting a general approach for the development of orthogonal covalent chemical tags from existing non-covalent ligand-protein pairs.
Introduction
The fluorescent proteins (FPs) have emerged as invaluable tools for tagging and visualizing proteins in vivo (1-3). FPs allow individual proteins to be selectively tagged in the complex environment of a living cell by simple genetic encoding. Naturally occurring and engineered FPs have been exploited to improve the brightness, photostability, expression, and spectral range of the tags (3). To complement the utility of the FPs and to facilitate growing efforts to carry out demanding biophysical measurements in live cells, chemical tags have been developed over the past decade that combine the selectivity provided by genetic encoding with a modular small molecule probe (4-6). To date, the protein-based chemical tags have been most successful in achieving sufficient selectivity to enable high signal-to-noise imaging of intracellular proteins (7-12).
We have previously demonstrated that the high-affinity interaction between E. coli dihydrofolate reductase (eDHFR) and trimethoprim (TMP) can be used to selectively label proteins with fluorescent tags in vivo (7, 8). A plasmid encoding a fusion between eDHFR and the protein of interest can be constructed and then transfected into live cells using standard methods in molecular biology. Then, the eDHFR fusion protein can be selectively labeled by non-covalent binding to a cell-permeable TMP-fluorophore heterodimer. Because eDHFR has a molecular weight (18 kD) two-thirds that of GFP and is a stable, monomeric protein, eDHFR is an attractive protein chemical tag. TMP has high selectivity for eDHFR over mammalian DHFRs, and thus eDHFR-tagged proteins can be labeled in wild-type mammalian cell backgrounds with high selectivity.
The straightforward synthesis of TMP derivatives provides facile access to a variety of TMP-fluorophores. TMP has excellent cell permeability properties, reflecting its use clinically as an antibiotic. Optimized TMP-green and -red tags that improve the cell permeability of the fluorescein chromophore heterodimer have been reported that allow efficient labeling of intracellular proteins (8). Thus, TMP-tag is one of the few chemical tags that allows intracellular proteins to be labeled in live cells with high-resolution.
To complement the utility of our non-covalent TMP-tag, we sought to design a covalent TMP-tag that would retain the cell permeability of TMP but provide a more permanent modification for single-molecule tracking, pulse-chase labeling and other applications. Proximity-induced reactivity appealed to us as a potentially general method for converting a non-covalent protein tag to a covalent one. It is now well established that irreversible enzyme inhibitors can be created by appending a functional group on the inhibitor that forms a covalent bond with an amino acid inside or outside the ligand-binding pocket (13-16). More recently broad-specificity suicide substrates have been used to clone enzymes based on their reaction class, e.g. proteases, using cell lysates (17). Individual enzymes have been labeled using selective inhibitors modified with appropriate electrophiles using proximity-induced reactivity in cell lysates. Finally, there have been efforts to design chemical tags for live-cell imaging based on proximity-induced reactivity (18, 19). Significantly, Belshaw and co-workers demonstrated that with optimization of the electrophile and the position of the protein nucleophile, proximity-induced labeling could be extremely rapid, with 50% labeling occurring in times as short as 15 minutes. To date, however, proximity-induced chemical tags have not shown sufficient selectivity to enable high-resolution imaging in live cells (18, 19).
Here we evaluate the potential of proximity-induced reactivity for engineering covalent tags suitable for in vivo imaging by generating a covalent TMP-tag. First, acrylamide and Cys were selected as the functional groups for modification of TMP and eDHFR. Using molecular modeling programs, a heterotrimeric TMP-acrylamide-probe was designed that could react with the unique Cys installed on the surface of eDHFR and the TMP analog was subsequently synthesized. Then, the system was characterized in vitro and in vivo in order to determine the efficiency of labeling. Finally, the utility of the covalent pair as an imaging tag was assessed, testing its ability to selectively label fusion proteins in vivo. Not only does this work provide a new covalent tag for live cell imaging, but also it demonstrates that proximity-induced labeling can provide the balance in reactivity needed for selective protein modification in live cells.
Results and Discussion
Design of a Covalent TMP-Tag
We envisioned converting our previously reported non-covalent TMP-tag into a covalent tag by incorporating appropriate reactive groups on TMP and eDHFR that, when held in proximity, would react to form a covalent bond (Figure 1). Key to this design was the choice of reactive groups that balance low reactivity towards endogenous cellular molecules with efficient proximity-induced reactivity when TMP binds to eDHFR. Many combinations of amino acids and electrophiles have been published for the design of irreversible inhibitors (14). For the covalent TMP-tag, the choice was limited to pairs in which the reactive group on the protein was a natural amino acid in order to retain genetic encoding. Therefore, Cys and acrylamide were chosen as the reactive pair. Ligands containing acrylamide have been designed that can irreversibly label target proteins after one minute of incubation when reactive groups are optimally positioned in the binding pocket (15). At the same time, acrylamide is a very selective electrophile, having low background reactivity with cellular proteins (15, 20).
Figure 1.
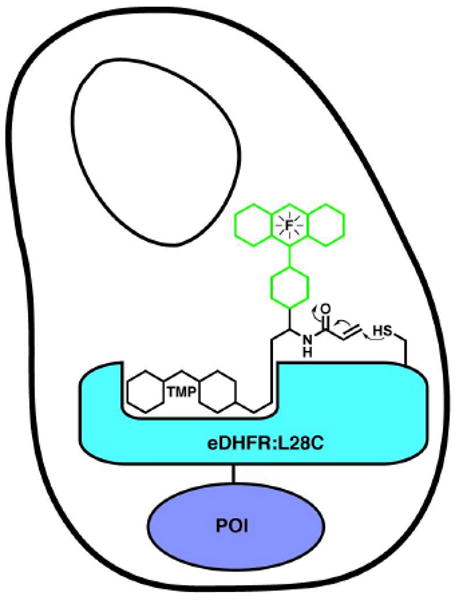
A covalent TMP-tag. We sought to convert our non-covalent trimethoprim (TMP) E. coli dihydrofolate reductase (DHFR) chemical tag into a covalent tag using proximity-induced reactivity. An acrylamide electrophile was installed on the trimethoprim-fluorophore (TMP-F) label. Then molecular modeling was used to design a Cys nucleophile on the surface of eDHFR optimally positioned to react with the acrylamide electrophile when TMP bound to the active-site of eDHFR. A covalent TMP-tag would be advantageous for pulse-chase experiments, single-molecule imaging and other applications that benefit from a more permanent chemical modification.
With selection of the reactive pair, the next step was to design an eDHFR:Cys variant and a corresponding acrylamide TMP electrophile (A-TMP). There are no high-resolution structures of TMP bound in the active site of eDHFR available; however, there is a structure of TMP bound to the highly homologous L. casei DHFR (21). Therefore, a structural alignment of the E. coli (22) and L. casei enzymes was used to create a model of TMP bound to eDHFR. Proximity-accelerated reactivity can be achieved by reacting an electrophilic group on a ligand with a nucleophilic residue on the protein that is located on the inside or outside of the binding pocket. Reaction with a residue on the outside of the pocket is more general and straightforward since it does not interfere with ligand binding interactions. Previous work from Belshaw and co-workers (18) showed that the shortest alkylation rates were achieved with residues in closest proximity to the binding pocket. Therefore, the residue Leu28 was selected for mutation to Cys because it is located just outside of the binding pocket and has a solvent accessible side chain. In the model of TMP bound to eDHFR, the 4′-OH group of TMP is located near the opening of the binding pocket, close to the protein surface. Leu28 is also located along the protein surface. Accordingly, the required linker length was approximated as the arc, ℓ=2πRsinθ, where R is the radius of gyration and θ is the angle between the 4′-OH and Leu28, using the center of the protein as the vertex. After determining an estimate for the linker, the molecular modeling program MacroModel (23) was used to produce energy-minimized structures of TMP analogs containing varying linker lengths. The linker lengths of these analogs were calculated as the distance from the 4′-OH to the reactive β-carbon of the acrylamide, and it was established that a 21-atom linker would be sufficient to span the length of ℓ. The chosen analog was then superimposed onto TMP in the crystal structure using PyMOL (24) in order to verify the design (Figure 2).
Figure 2.
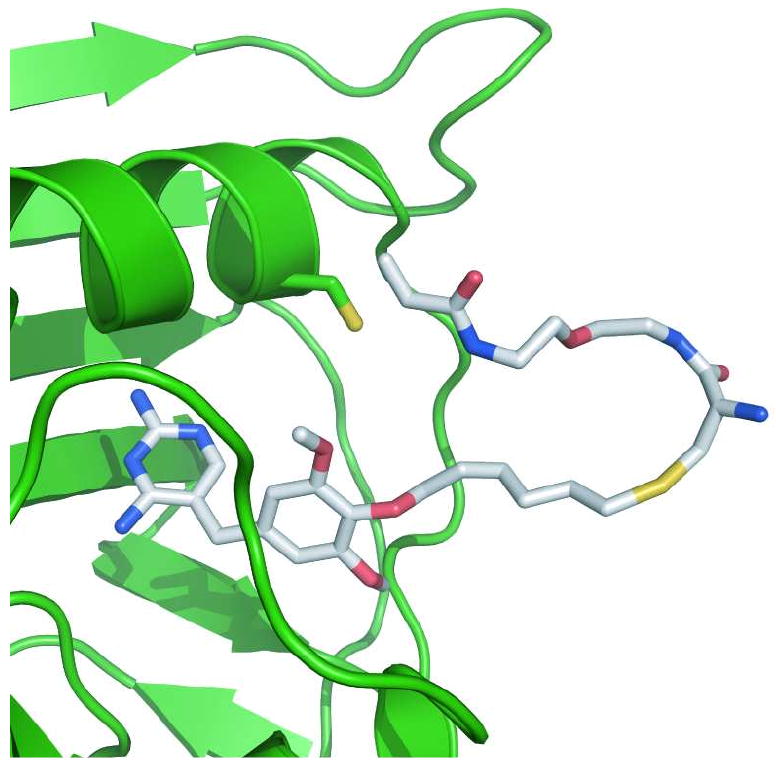
The design of a covalent TMP-DHFR pair. Cartoon of a TMP-acrylamide heterodimer bound to eDHFR, with an engineered Leu28Cys in position to react with the acrylamide electrophile. The eDHFR protein is represented in green as a ribbon diagram, with the Cys28 residue shown using the stick representation. The TMP-acrylamide heterodimer is also represented as sticks with coloring based on elements. Since no structure has been solved of TMP bound to eDHFR, the model was created by structural alignment of eDHFR (22) with the L. casei DHFR (21). The A-TMP structure was created in MacroModel (23) and then superimposed on TMP in the eDHFR model. The graphic was prepared using PyMOL (24).
Synthesis of the Heterotrimeric TMP
The synthetic design of the covalent TMP-tag, A-TMP-fluorescein (1), was planned around a tri-branched core for installation of (1) the TMP ligand, (2) the fluorophore and (3) the acrylamide electrophile (Scheme 1). Cysteine was used for the branched core because it has three chemically unique sites of attachment, minimizing the need for protecting groups. The route was also designed to introduce the probe in the last step, allowing for straightforward derivatization with a variety of probes.
Scheme 1.
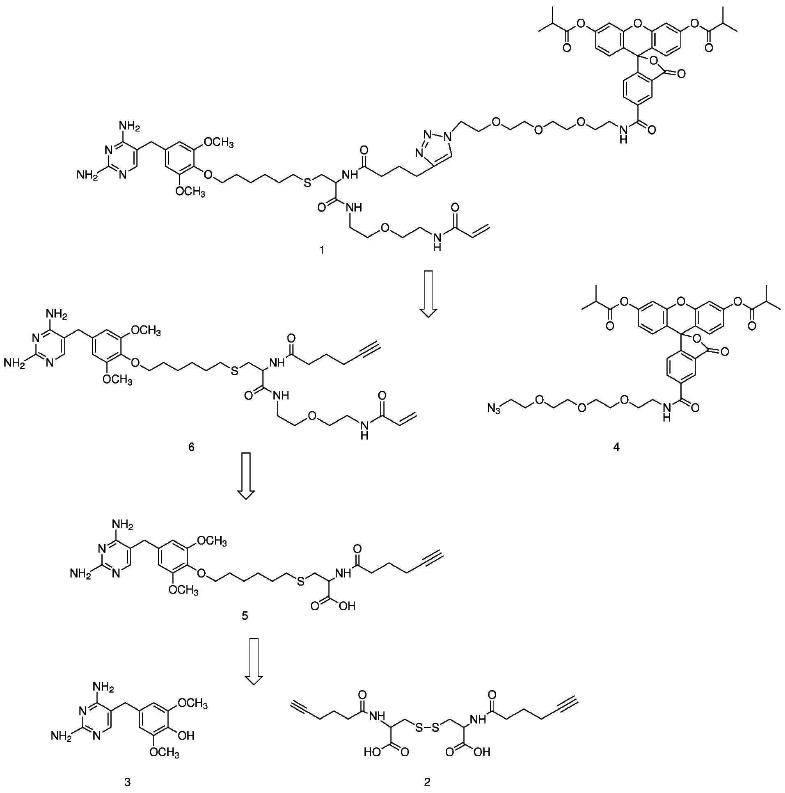
Retrosynthetic analysis of TMP-fluorophore-acrylamide heterotrimer.
The first step of the synthesis was activation of 5-hexynoic acid as the succinimidyl ester (S2, Supplementary Scheme S3). Two equivalents of the activated alkyne were then selectively coupled to the two amines of the oxidized di-cysteine using Schotten-Bauman conditions producing compound 2. The cysteine dimer had been selected as the precursor for the core in order to initially protect the nucleophilic thiol.
Derivatization of TMP takes advantage of the fact that substitution at the 4′-methoxy position does not significantly disrupt binding of TMP to eDHFR (25). The 4′-methoxy of TMP was selectively hydrolyzed to a hydroxyl to generate TMP-OH (3), as previously described (26). Based on the modeling results, we hypothesized that a six-carbon linker would prevent the branching position from interfering with binding. Thus, TMP-OH was alkylated with commercially available 1,6-dibromohexane to produce TMP-linker-bromide (S1, Supplementary Scheme S1), with a second electrophile available for coupling to the cysteine core.
The succinimidyl ester of 6-carboxyfluorescein was coupled to the amino group of 11-azido-3,6,9-trioxaundecan-1-amine, derivatizing the fluorophore with an azido group for coupling to the alkyne of the cysteine core via a copper-catalyzed 1,3-dipolar cycloaddition reaction known as “Click chemistry”. To improve the membrane permeability of the final molecule, the hydroxyl groups of the crude product were selectively protected by reaction with isobutyric anhydride in pyridine producing compound 4 (8).
The synthesis of the final product from the cysteine core was completed in four steps. First, the disulfide bond of compound 2 was reduced to the thiol using n-tributylphosphine and then immediately alkylated with TMP-linker-bromide (S1). Second, 1,5-diamino-3-oxapentane was coupled to the free carboxylic acid using standard peptide coupling conditions to produce S3 (Supplementary Scheme S3).
Third, the succinimidyl ester of acrylamide was introduced to the free amine of the linker producing compound 6, and completing the 21-atom linker between TMP and the β-carbon of the acrylamide. Last, the azide of compound 4 was coupled to the alkyne of compound 6 using Click chemistry, giving compound 1, a heterotrimeric A-TMP-fluorescein (A-TMP-Fl), from three components in seven linear steps in 0.8 percent overall yield from 5-hexynoic acid, the longest linear route.
For preliminary in vitro studies, a biotin derivative was also synthesized, A-TMP-biotin (A-TMP-B). The detailed synthesis of this TMP heterotrimer will be subsequently reported in a separate publication (1H-NMR, Supplementary Figure S2).
In Vitro Characterization of Covalent Labeling
To verify that the designed eDHFR:L28C/A-TMP pair underwent efficient proximity-induced covalent labeling, the pair was first characterized in vitro. Using purified protein, A-TMP was shown to label eDHFR:L28C covalently, with the reaction 50% complete after 50 minutes.
First, an E. coli expression vector encoding the eDHFR:L28C variant was constructed, and then the eDHFR:L28C protein was overexpressed and purified. eDHFR has two naturally occurring cysteines, Cys85 and Cys152. While these two Cys are buried in the protein structure, they were removed to ensure there would be no interference with the cysteine labeling reaction or with protein folding and purification. Both Cys85 and Cys152 were mutated to serine, a conservative mutation structurally and electronically. Finally, the reactive cysteine was introduced with a L28C mutation. All point mutations in eDHFR were made using Stratagene's QuikChange Site-Directed Mutagenesis Kit. Primers were designed not only to encode the mutated codon but also to introduce a new unique restriction site for analysis. After each round of site-directed mutagenesis, successful mutants were identified by colony PCR and restriction digestion. The presence of only the desired mutations was confirmed by sequencing the full coding region. Even though this variant has three point mutations C85S, C152S, and L28C, for clarity the protein variant is referred to as eDHFR:L28C throughout this manuscript. A variant containing the C85S and C152S mutations, but lacking the L28C mutation, was used as a control and is referred to as eDHFR:−2C. The mutated eDHFR genes were encoded in vectors containing the T7 promoter and had His6 tags. The proteins were overexpressed via the T7 promoter using standard conditions and purified using Ni-NTA spin columns. Both proteins were judged to be >95% pure by Coomassie staining of a SDS-PAGE gel.
To verify that the designed pair would react efficiently to form a covalent bond, purified eDHFR:L28C was reacted with A-TMP-B and then analyzed by SDS-PAGE and Western analysis (Figure 3, panel a). The labeling reaction conditions were based on precedent reported by Belshaw and co-workers when the same nucleophile-electrophile pair was used (20). Purified eDHFR:L28C at a 5 μM concentration was incubated with 10 μM A-TMP-B in phosphate buffered saline (PBS), pH7 with 1 mM reduced glutathione for three hours at 37°C. As a control, A-TMP-B was incubated under the same conditions with eDHFR:−2C to confirm that covalent labeling was dependent on the L28C mutation. This result proves that eDHFR:L28C is covalently labeled by A-TMP-B.
Figure 3.
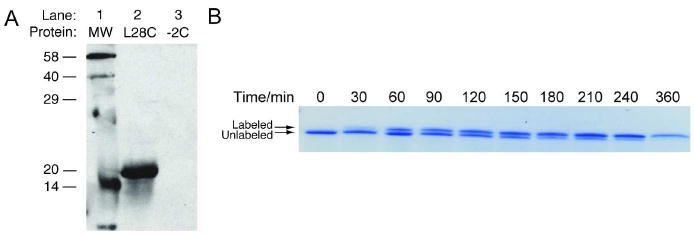
Covalent labeling with A-TMP-tag in vitro. To demonstrate the rate and efficiency of covalent labeling, A-TMP-biotin was incubated with purified eDHFR and then analyzed by sodium dodecyl sulfate (SDS)-PAGE. a) Western analysis of in vitro covalent labeling reaction. Purified eDHFR at a concentration of 5 μM was incubated with 10 μM A-TMP-biotin in phosphate buffered saline (PBS) with 1 mM glutathione at 37°C for three hours. A biotinylated protein ladder was run in Lane 1. Lanes 2, 5 μM purified eDHFR:L28C incubated with 10 μM A-TMP-biotin; Lane 3, 5 μM eDHFR:−2C incubated with 10 μM A-TMP-biotin. b) Determination of the rate of covalent labeling. Purified eDHFR:L28C was labeled under the same reaction conditions in a. At various time points, aliquots were quenched with 6× SDS. The reaction was analyzed by SDS-PAGE and Coomassie staining and it was determined that 50% labeling occurs in approximately 50 minutes and that the reaction is near quantitative at the end.
Having shown that the labeling reaction worked, next, the rate and efficiency of labeling was determined. An alkylation reaction was set up using the same conditions as described above, and aliquots were quenched at various time points by the addition of 6× SDS. Initial experiments revealed that alkylation of eDHFR:L28C by A-TMP-B produces a gel shift, and thus the rate could be analyzed simply by analysis of a Coomassie stained SDS-PAGE gel (Figure 3, panel b). The progress of the reaction was quantified by densitometry analysis of the gel using the program Image-J (27). The time required for 50% alkylation of eDHFR:L28C was calculated to be 50 minutes from the pseudo first-order rate constant for the appearance of the alkylated product. Covalent labeling of eDHFR:L28C after three hours of incubation was calculated to be >95%.
Finally, we demonstrated that, as would be expected for a covalent tag based on high-affinity binding, the labeling reaction could be carried out at stoichiometric concentrations of the fluorophore, even at low protein concentrations, without any significant decrease in reaction rate. Currently, the majority of chemical tags are based on enzymatic modification with a fluorescent substrate. For the reaction rate of the enzyme-catalyzed reaction to be maximal, the fluorophore concentration needs to be near or above the substrate KM, typically at a μM to mM concentration. Because in vivo many proteins are at concentrations below the KM, the fluorophore must be used in large excess over the protein, resulting in high background fluorescence from unreacted fluorophore. In contrast, tags like TMP-tag, based on high-affinity binding, need only be applied near or above the KD, typically in the low nM range. Therefore, most proteins in the cell can be labeled using stoichiometric concentrations of fluorophore, minimizing background noise due to unbound fluorophore. First, the A-TMP labeling reaction was modeled using COPASI (28) (see Supporting Information for additional details). The model predicts that the time for 50% labeling remains 50 minutes down to 100 nM protein concentration; the time for 50% labeling at 10 nM protein concentration is 72 minutes and at 1 nM protein concentration is 150 minutes. To verify the model and to characterize the reaction rate and endpoint dependence on reactant concentrations, the kinetics were determined with eDHFR:L28C and A-TMP-B both at a 1 μM concentration (Figure S3). The results confirmed that, as predicted by the model, 50% labeling still occurred in ca. 50 minutes. These results suggest that chemical tags based on high-affinity binding should minimize background fluorescence because the fluorophore can be used at low, stoichiometric concentrations.
In Vivo Characterization of Covalent Labeling
Having confirmed that A-TMP efficiently labeled eDHFR:L28C covalently in vitro, the covalent eDHFR:L28C/A-TMP pair next was evaluated in vivo. A heterotrimeric A-TMP-Fl was used to both characterize the in vivo covalent labeling reaction and to visualize intracellular labeling of a nucleus-targeted eDHFR:L28C fusion protein. A-TMP-Fl was shown to label the fusion protein covalently and with high signal-to-noise using in-gel fluorescence and confocal microscopy. There was no evidence of cytotoxicity, as the cells labeled with A-TMP-Fl showed no loss of viability or any other phenotypic variations compared with mock treated control cells.
To test the utility of the A-TMP-tag for live cell imaging of intracellular proteins, wild-type fibroblasts expressing a nucleus-targeted eDHFR:L28C-red fluorescent protein (RFP) fusion was labeled with A-TMP-Fl (Figure 4, panel a). An eDHFR:L28C-RFP fusion protein was constructed so that cells positively transfected with eDHFR:L28C could be identified by excitation of RFP. Fibroblast cells transiently transfected with the DNA encoding nucleus-targeted eDHFR:L28C-RFP were incubated with 1 μM of A-TMP-Fl for ten minutes. The cells were then washed twice and imaged by confocal fluorescent microscopy. Significantly, all transfected cells show distinct nuclear staining with no significant background labeling in the cytosol or in untransfected cells, demonstrating that the A-TMP-tag has sufficient selectivity to label intracellular proteins with high signal-to-noise in live cells.
Figure 4.
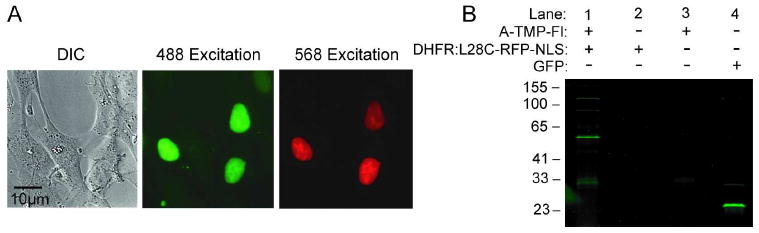
Covalent labeling with A-TMP-tag in vivo. To demonstrate the specificity of covalent labeling in living cells, A-TMP-fluorescein was used to label wild type fibroblasts expressing a nucleus-targeted eDHFR:L28C-RFP fusion protein. a) Live cell imaging of intracellular proteins using the covalent A-TMP-tag. A nucleus-targeted eDHFR:L28C-RFP fusion protein was labeled with A-TMP-fluorescein (A-TMP-Fl) in wild type fibroblasts. Cells transiently transfected with vector encoding the nucleus-targeted eDHFR:L28C-RFP fusion were incubated with 1 μM A-TMP-Fl in phosphate buffered saline (PBS) for ten minutes, washed twice with PBS, incubated for one hour, and then imaged using live cell, confocal microscopy. Confocal micrographs: left column, differential image contrast; middle column, excitation of fluorescein at 488 nm; right column, excitation of RFP at 568 nm. b) In-gel fluorescence scanning analysis of in vivo covalent labeling reaction. Cells were nucleofected 48 hours prior to labeling experiment. The cells were then washed with PBS and incubated for 3 hours in 4 mL of complete DMEM either with or without 1 μM A-TMP-Fl. The cells were washed twice with PBS, trypsinized, and pelleted. The supernatant was removed, and the cells were lysed and the lysate was analyzed by sodium dodecyl sulfate (SDS)-PAGE and in-gel fluorescence scanning, exciting with a 488 nm laser. Lane 1, cells transiently transfected with nucleus-targeted eDHFR:L28C-RFP and incubated with A-TMP-Fl; Lane 2, cells transiently transfected with nucleus-targeted eDHFR:L28C-RFP but not incubated with A-TMP-Fl; Lane 3, untransfected cells incubated with A-TMP-Fl; Lane 4, a positive control of cells transiently transfected with a cytostolic GFP vector. These results show A-TMP-Fl reacts covalently and selectively with eDHFR:L28C in vivo.
To determine the rate and efficiency of labeling in vivo, live cells were incubated with A-TMP-Fl, and the covalent labeling reaction was examined by SDS-PAGE and in-gel fluorescence. NIH Swiss 3T3 fibroblasts transiently transfected with nucleus-targeted eDHFR:L28C-RFP were incubated at 37°C for 3 hours in DMEM complete media. After incubation, each set of cells was lysed and evaluated by SDS-PAGE and in-gel fluorescence scanning (Figure 4, panel b). Lanes 1 and 2 contain the lysate of cells transfected with the eDHFR:L28C-RFP fusion that were incubated with and without 1 μM A-TMP-Fl, respectively. Lane 3 contains the lysate of untransfected cells that were incubated in media containing 1 μM A-TMP-Fl, thus testing for background labeling of endogenous cellular proteins. Cells transfected with a cytostolic GFP vector were used as a positive control for detection (Lane 4). In Lane 1, the only major band is the expected 52 kDa eDHFR:L28C-RFP fusion protein; confirming the identity of this band, it emits in both the green and red fluorescence channels, and it is directly observed as a gel shift in the time course experiment (Supplementary Figure S5). The in vivo time course experiment also demonstrates that covalent labeling reaction is robust, with >95% labeling occurring after two hours. Interestingly, there are a few minor higher and lower molecular weight bands in the lysate of the transfected cells (Lane 1) (see Supporting Information for a table of the relative intensities of the bands). The minor lower molecular weight bands also appear in the untransfected cell lysate (Lane 3) and grow in over time (Figure S5); therefore, we conclude that these bands reflect slow background labeling, which it may be possible to minimize through further optimization of the labeling conditions. The minor higher molecular weight bands only occur in the lysate of the transfected cells (Lane 1) and also are observed in the red channel (Figure S5), suggesting that these bands are most likely either degradation products or adducts of the labeled eDHFR:L28C-RFP fusion proteins. Currently, we are investigating the identity and origin of these three minor bands. The absence of any major bands in Lane 3 confirms that there is no significant background labeling of endogenous intracellular proteins, demonstrating that the covalent labeling reaction is highly selective. Together, these results establish that proximity-induced reactivity produces a highly specific, covalent modification verifying the utility of the eDHFR:L28C/A-TMP pair as a chemical tag for live cell labeling experiments, and more broadly, the utility of proximity-induced reactivity for the development of new chemical tags.
To determine the approximate time required for covalent labeling in vivo, the live cell labeling experiment was repeated, incubating fibroblasts transiently transfected with nucleus-targeted eDHFR:L28C-RFP with 1 μM A-TMP-Fl for various lengths of time and subsequently analyzing the reaction by SDS-PAGE and in-gel fluorescence scanning (Supplementary Figure S5). Since covalent labeling produced a discernable gel shift, detection of red fluorescence enabled the determination of both the time course and the extent of covalent labeling. The progress of the reaction was quantified by densitometry analysis of the gel using the program Image-J (27). The gel established that the in vivo covalent labeling reaction is >95% complete after 2 hours, slightly faster than that measured in vitro. Thus, the in-gel fluorescence analysis demonstrates that the A-TMP-Fl labeling reaction is (1) covalent, (2) near quantitative, (3) 50% complete in less than 1 hour, and (4) exhibits no significant background labeling of endogenous proteins.
Discussion
Together these results establish that A-TMP-tag is a robust covalent chemical tag for live cell imaging. The reaction between eDHFR:L28C and A-TMP is rapid, with in vitro kinetics showing that 50% covalent labeling occurs in 50 minutes, but at the same time specific, allowing an intracellular protein to be imaged in a living cell with high signal-to-noise. eDHFR is an 18 kD, stable, monomeric protein. Thus, eDHFR should be a suitable label for any protein that can be imaged with FPs and may even offer advantages for proteins that are perturbed by the slightly larger FPs (ca. 27 kD) (1) or by complications from oligomerization of certain FPs (3). The protocol for labeling with A-TMP-tag is straightforward—the A-TMP-fluorophore is cell permeable and is simply added to the cell media immediately prior to imaging, with no additional reagents or cell-permeabilization procedures required that might affect cell viability. As expected given that TMP has high selectivity for bacterial DHFRs and is used clinically as an antibiotic and given that E. coli DHFR is not endogenous to mammalian cells, expression of eDHFR tagged proteins and addition of A-TMP-fluorophores show no signs of cytotoxicity or interference with endogenous biological pathways. Complimentary to our non-covalent TMP-tag, the covalent A-TMP-tag reported here should be advantageous for pull-down, pulse-chase, and other applications that require a more permanent chemical modification. With growing interest in single-molecule imaging of live cells (29) and evidence that super-resolution imaging methods can break the diffraction barrier for molecular imaging of cells (30-33), covalent chemical tags are attractive because they have the potential to allow proteins to be labeled in live cells with small molecule fluorophores with high photon flux.
A-TMP-tag can now be used in conjunction with other covalent chemical tags for multicolor tagging applications. While several different chemical tags have been reported, few can actually label intracellular proteins in living cells with high specificity and hence good signal-to-noise. For multi-color labeling of intracellular proteins, there is clear evidence in the published literature that TMP-tag and SNAP-tag provide high signal-to-noise labeling for intracellular proteins. TMP-tag provides a non-covalent label based on binding of TMP by eDHFR (7, 8). Johnsson and co-workers developed SNAP-tag, a labeling method based on the human DNA repair protein O6-alkylguanine-DNA alylktransferase (AGT) (10), where an evolved AGT variant is specifically labeled in vivo by O6-benzylguanine-probe heterodimers (34). HaloTag (12), an irreversible dehalogenase variant developed by Promega, CLIP-tag (11), the newly reported orthogonal variant of SNAP-tag, and now our covalent A-TMP-tag all show promise for providing additional orthogonal tags for labeling intracellular proteins, but require further experimental vetting. Tsien's original tetracysteine tag suffers from background labeling of Cys-rich proteins but remains the only short peptide tag for labeling intracellular proteins (9). For multi-color labeling of extracellular membrane proteins, biotin ligase (35) and phosphopantetheinyl transferase (36, 37) additionally can be used, and provide short peptide tags. An important next step for the chemical tagging field will be the optimization of a set of orthogonal chemical tags that can be used simultaneously to study the dynamic interactions of individual proteins in different biological pathways in the complex environment of the cell.
Proximity-induced reactivity provides an additional strategy for engineering covalent chemical tags in vivo—beyond developing entirely new ligand-receptor or substrate-enzyme pairs or creating an orthogonal pair with both high activity and selectivity by directed evolution. Enzyme-mediated chemical tags in theory are attractive because of their rapid chemical modification of the tagged protein. However, given that for in vivo experiments the rate-limiting step is the time required for the small molecule probe to enter the cell (typically ≥1 hour), in practice there likely will be no temporal advantage to enzyme-mediated tagging compared to proximity-induced covalent modification. In its first embodiment here, the covalent A-TMP-tag covalent labels 50% of purified eDHFR:L28C in vitro by 50 min—already on the order of typical protocols for labeling proteins in vivo with chemical tags. Given that Belshaw and co-workers (18) showed that a similar exo-reaction could be optimized to give 50% labeling within 15 min and that endo suicide substrates show 50% reaction as fast as 1 min (15), the rate for labeling with the A-TMP-tag presumably can be further improved by optimization of the orientation of the Cys nucleophile and acrylamide electrophile, or even directed evolution. While proximity-induced reactivity has long been exploited in the field of organic chemistry and a few manuscripts have explored its use as a tool for chemical biology, here we establish that proximity-induced reactivity has sufficient selectivity for specific labeling in a live cell.
Here we have developed a new covalent chemical tag for in vivo imaging of intracellular proteins, and, for the first time, we have established that proximity-induced reactivity provides the specificity required for challenging cellular applications, such as high-resolution imaging. While demonstrated here with TMP and eDHFR, the same acrylamide electrophile and Cys nucleophile presumably can be readily extended to convert other chemical dimerizers, such as SLF and FKBP12* or dexamethasone and the glucocorticoid receptor, into covalent chemical tags. Future development of additional orthogonal reactions and the extension to engineering applications beyond chemical tags should begin to elucidate the full potential of classical reactions in organic chemistry for synthetic biology.
Methods
Synthetic Methods
Synthetic procedures and characterization of each compound are provided in the Supporting Information.
Vector Construction
Details of vector construction are provided in the Supporting Information.
In-Gel Fluorescence Scanning
Procedures for in-gel fluorescence scanning are provided in the Supporting Information.
Protein Purification
The resulting eDHFR:L28C plasmid was expressed in BL21(DE3)pLysS cells (Invitrogen). Cells were grown at 37°C to an OD600 of 0.6 and then, induced with 0.4 mM IPTG for three hours and purified using nickel-NTA spin columns (Qiagen). The protein was dialyzed three times in phosphate buffered saline (PBS) at 4°C, snap froze and stored at −80°C.
In Vitro Alkylation Reactions
Purified eDHFR:L28C (5 μM) was incubated with A-TMP-B (10 μM) in PBS with reduced glutathione (1 mM) at 37°C. At selected time points, aliquots (30 μL) were removed from the reaction mixture, quenched with 6× SDS. Samples from the in vitro alkylation experiments were run on Criterion 15% Tris-HCl gels (BioRad) for 55 min at 200 V. Bands were quantified by densitometry analysis of or Coomassie stained gels by Image-J.
Live Cell Imaging
The A-TMP-Fl dye (1 μM) was added to Ringers solution, applied to cells for 10 minutes and washed twice with ringers containing 10% (v/v) FBS. Images were obtained using a FV500 confocal microscope (Olympus) using a 60× (1.4 NA) objective. Floview Tif images were processed with ImageJ UCSD Plugins.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM-071754). S. Gallagher was supported by a National Defense Science and Engineering Graduate fellowship. We would like to acknowledge K. Lemberg and L. Brown for experimental assistance.
Footnotes
Supporting Information Available: This material is free via the Internet.
References
- 1.Giepmans BNG, Adams SR, Ellisman MH, Tsien RY. Review - The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 2.Tsien RY. The green fluorescent protein. Annual Review of Biochemistry. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 3.Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J Cell Sci. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- 4.O'Hare HM, Johnsson K, Gautier A. Chemical probes shed light on protein function. Curr Opin Struct Biol. 2007;17:488–494. doi: 10.1016/j.sbi.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Miller LW, Cornish VW. Selective chemical labeling of proteins in living cells. Curr Opin Chem Biol. 2005;9:56–61. doi: 10.1016/j.cbpa.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Chen I, Ting AY. Site-specific labeling of proteins with small molecules in live cells. Curr Opin Biotechnol. 2005;16:35–40. doi: 10.1016/j.copbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Miller LW, Cai Y, Sheetz MP, Cornish VW. In vivo protein labeling with trimethoprim conjugates: a flexible chemical tag. Nat Methods. 2005;2:255–257. doi: 10.1038/nmeth749. [DOI] [PubMed] [Google Scholar]
- 8.Calloway NT, Choob M, Sanz A, Sheetz MP, Miller LW, Cornish VW. Optimized fluorescent trimethoprim derivatives for in vivo protein labeling. Chembiochem. 2007;8:767–774. doi: 10.1002/cbic.200600414. [DOI] [PubMed] [Google Scholar]
- 9.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 10.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 11.Gautier A, Juillerat A, Heinis C, Correa IR, Jr, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. ACS Chem Biol. Vol. 3. 2008. HaloTag: a novel protein labeling technology for cell imaging and protein analysis; pp. 373–382. [DOI] [PubMed] [Google Scholar]
- 13.Baker BR. The Case for Irreversible Inhibitors as Anticancer Agents, an Essay. Cancer Chemoth Rep. 1959:1–10. [Google Scholar]
- 14.Powers JC, Asgian JL, Ekici OD, James KE. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 15.Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, Smaill JB, Trumpp-Kallmeyer S, Dobrusin EM. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci U S A. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chmura AJ, Orton MS, Meares CF. Antibodies with infinite affinity. Proc Natl Acad Sci U S A. 2001;98:8480–8484. doi: 10.1073/pnas.151260298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 18.Krusemark CJ, Belshaw PJ. Covalent labelling of fusion proteins in live cells via an engineered receptor-ligand pair. Org Biomol Chem. 2007;5:2201–2204. doi: 10.1039/b705185a. [DOI] [PubMed] [Google Scholar]
- 19.Nonaka H, Tsukiji S, Ojida A, Hamachi I. Non-enzymatic covalent protein labeling using a reactive tag. J Am Chem Soc. 2007;129:15777–15779. doi: 10.1021/ja074176d. [DOI] [PubMed] [Google Scholar]
- 20.Levitsky K, Boersma MD, Ciolli CJ, Belshaw PJ. Exo-mechanism proximity-accelerated alkylations: investigations of linkers, electrophiles and surface mutations in engineered cyclophilin-cyclosporin systems. Chem Bio Chem. 2005;6:890–899. doi: 10.1002/cbic.200400383. [DOI] [PubMed] [Google Scholar]
- 21.Polshakov VI, Smirnov EG, Birdsall B, Kelly G, Feeney J. NMR-based solution structure of the complex of Lactobacillus casei dihydrofolate reductase with trimethoprim and NADPH. J Biomol NMR. 2002;24:67–70. doi: 10.1023/a:1020659713373. [DOI] [PubMed] [Google Scholar]
- 22.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 23.MacroModel. version 9.1. Schrodinger, LLC; New York, NY: 2005. [Google Scholar]
- 24.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 25.Roth B, Aig E, Rauckman BS, Strelitz JZ, Phillips AP, Ferone R, Bushby SR, Sigel CW. 2,4-Diamino-5-benzylpyrimidines and analogues as antibacterial agents. 5. 3′,5′-Dimethoxy-4′-substituted-benzyl analogues of trimethoprim. J Med Chem. 1981;24:933–941. doi: 10.1021/jm00140a005. [DOI] [PubMed] [Google Scholar]
- 26.Brossi A, Grunberg E, Hoffer M, Teitel S. Synthesis and chemotherapeutic activity of two metabolites of trimethoprim. J Med Chem. 1971;14:58–59. doi: 10.1021/jm00283a017. [DOI] [PubMed] [Google Scholar]
- 27.Rasband WS. ImageJ. U. S. National Institutes of Health; Bethesda: 19972007. [Google Scholar]
- 28.Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. COPASI--a COmplex PAthway SImulator. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- 29.Xie XS, Yu J, Yang WY. Living cells as test tubes. Science. 2006;312:228–230. doi: 10.1126/science.1127566. [DOI] [PubMed] [Google Scholar]
- 30.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 31.Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. Proc Natl Acad Sci U S A. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bates M, Huang B, Dempsey GT, Zhuang X. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007;317:1749–1753. doi: 10.1126/science.1146598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinis C, Schmitt S, Kindermann M, Godin G, Johnsson K. ACS Chem Biol. Vol. 1. 2006. Evolving the substrate specificity of O6-alkylguanine-DNA alkyltransferase through loop insertion for applications in molecular imaging; pp. 575–584. [DOI] [PubMed] [Google Scholar]
- 35.Chen I, Howarth M, Lin W, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 36.George N, Pick H, Vogel H, Johnsson N, Johnsson K. Specific labeling of cell surface proteins with chemically diverse compounds. J Am Chem Soc. 2004;126:8896–8897. doi: 10.1021/ja048396s. [DOI] [PubMed] [Google Scholar]
- 37.Yin J, Liu F, Li X, Walsh CT. Labeling proteins with small molecules by site-specific posttranslational modification. J Am Chem Soc. 2004;126:7754–7755. doi: 10.1021/ja047749k. [DOI] [PubMed] [Google Scholar]
- 38.Miller LW, Sable J, Goelet P, Sheetz MP, Cornish VW. Methotrexate conjugates: a molecular in vivo protein tag. Angew Chem Int Ed Engl. 2004;43:1672–1675. doi: 10.1002/anie.200352852. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.