

Summary
IL-10 is an anti-inflammatory mediator, important in limiting immunopathology. Its impact is influenced both by the timing and localization of its release. Here we show that NK cells rapidly express IL-10 during acute infection with the rapidly disseminating pathogens Toxoplasma gondii, Listeria monocytogenes or Yersinia pestis. Direct IL-12 signals proved necessary and sufficient for NK induction of IL-10. NK cells from T. gondii-infected mice inhibited dendritic cell release of IL-12 in an IL-10-dependent manner and NK cell depletion resulted in elevated serum IL-12. Together these data suggest an innate, negative feedback loop, in which IL-12 limits its own production by eliciting IL-10 from NK cells. In contrast to the systemic pathogens, NK cell IL-10 was not elicited by locally restricted infection with influenza virus or with a Y. pestis strain attenuated to prevent dissemination. Thus, systemic infections uniquely engage NK cells in an IL-10-mediated immunoregulatory circuit that functions to alleviate inflammation during sepsis.
Introduction
Surviving infection requires a precise balance between the pro-inflammatory immune response needed to eliminate the pathogen, and anti-inflammatory signals essential to limit collateral damage to the host. Sepsis is a clinical example of the consequence when this balance is lost: the widespread immune activation associated with uncontrolled dissemination of a pathogen can result in systemic inflammation, multi-organ failure and death (Cohen, 2002). IL-10 is a potent immunosuppressive cytokine that provides one mechanism of counter-regulation and in diverse models of infection, IL-10 is critical for the maintenance of immune balance (Couper et al., 2008). During acute toxoplasmosis, for example, rapid production of pro-inflammatory interferon (IFN)-γ is essential to control the parasite (Suzuki et al., 1988) but IL-10-deficient mice die with increased intestinal and liver pathology, despite a reduced parasite burden (Gazzinelli et al., 1996; Suzuki et al., 2000).
Several cellular sources of IL-10 have been identified and in infectious diseases attention has focused on CD4+ T cells in lymphoid organs (Anderson et al., 2007; Jankovic et al., 2007; Li and Flavell, 2008; Roers et al., 2004). These cells, however, release cytokines only during the adaptive phase of immune responses, concomitant with the expression of potent effector functions. Potential sources of earlier IL-10 production include B cells and innate cells such as macrophages and dendritic cells (Corinti et al., 2001; Fillatreau et al., 2002; Katakura et al., 2004). Even NK cells, which are best known for their innate release of IFN-γ, can be stimulated to secrete IL-10 in vitro (Chakir et al., 2001; Deniz et al., 2008; Mehrotra et al., 1998) and, in both humans and mice, this activity has been associated with immunosuppression (De Maria et al., 2007; Maroof et al., 2008). However, the relative contribution of NK cells to the expression of IL-10 and the factors which elicit NK cell-derived IL-10 in vivo are still unknown.
To examine the sources of IL-10 during infection with diverse pathogens, we analyzed its expression in vivo using both wild-type mice and IL-10 reporter animals. We reveal that NK cells are the first and the most frequent IL-10-expressing population induced in non-lymphoid tissues during acute, systemic infection. NK cell release of IL-10 is elicited by IL-12, and it acts via negative feedback to inhibit the pathogen-stimulated production of IL-12 by dendritic cells. This regulatory circuit is specific to systemic inflammation and is not engaged by the limited IL-12 signaling that occurs during localized infections. NK cells therefore appear to offer an IL-10-mediated, immunosuppressive capacity that is rapidly activated in situations of systemic insult. Thus, the dissemination of a pathogen directs qualitative changes in the cellular response mounted by the infected host.
Results
NK cells express IL-10 upon infection with diverse pathogens
To identify the sources of IL-10 during acute infection, we infected bicistronic IL-10-GFP reporter mice, designated Vert-X (Madan et al., 2009; Sun et al., 2009), with the rapidly disseminating, protozoan pathogen T. gondii. Consistent with data obtained in this and other IL-10 reporter mice (Kamanaka et al., 2006; Maynard et al., 2007), and elsewhere (Jankovic et al., 2007; Stumhofer et al., 2007), reporter expression was low or absent in sham-infected or naïve animals (Figure 1A, B and data not shown) and was detected in CD4+ T cells one week after infection with T. gondii (Figure 1A). IL-10 expression was also apparent in CD8+ T cells, CD19+ B cells, and NK T cells (NK1.1+TCRβ+) (Figure 1A). Most strikingly, 75% ± 11 (mean ± SD) of NK cells (NK1.1+TCRβ-) in the liver and 84% ± 6 in the blood also acquired an IL-10-GFP+ phenotype (Figure 1A, B, C, F). Similar data was obtained after intraperitoneal infection with T. gondii (Figure S1). NK1.1+GFP+ cells were present in all analyzed tissues and, although rare in the spleen and mesenteric lymph node, were the predominant IL-10+ population in non-lymphoid organs (Figure 1B). The majority of NK1.1+GFP+ cells did not stain for TCRβ (Figure 1C) or CD3ε (data not shown), identifying them as NK cells rather than NK T cells or conventional T cells expressing NK1.1 upon activation (Slifka et al., 2000). RT-PCR analysis of WT C57BL/6 and Rag-/- non-reporter mice confirmed the contribution of innate immune cells to the expression of IL-10 during infection (Figure S2). Indeed NK cells sorted from T. gondii- but not sham-infected WT C57BL/6 mice showed clear induction of IL-10 in NK cells, both by RT-PCR (Figure 1D) and by IL-10 protein production upon ex vivo stimulation (Figure 1E). Kinetic studies in Vert-X mice revealed that NK cell IL-10 expression was initiated between 3 and 4 days after infection, preceding the induction of IL-10 in CD4+ T cells (Figure 1F). IL-10+ NK cells were more abundant than IL-10+ CD4+ cells in the blood and liver of T. gondii-infected mice, while their numbers were similar in the spleen (Figure 1G). Thus, NK cells represented an early IL-10-expressing population during toxoplasmosis, and were the predominant IL-10+ population in non-lymphoid tissues at the peak of the acute inflammatory response.
Figure 1. NK cells are the first and predominant IL-10 expressing population in acute toxoplasmosis.
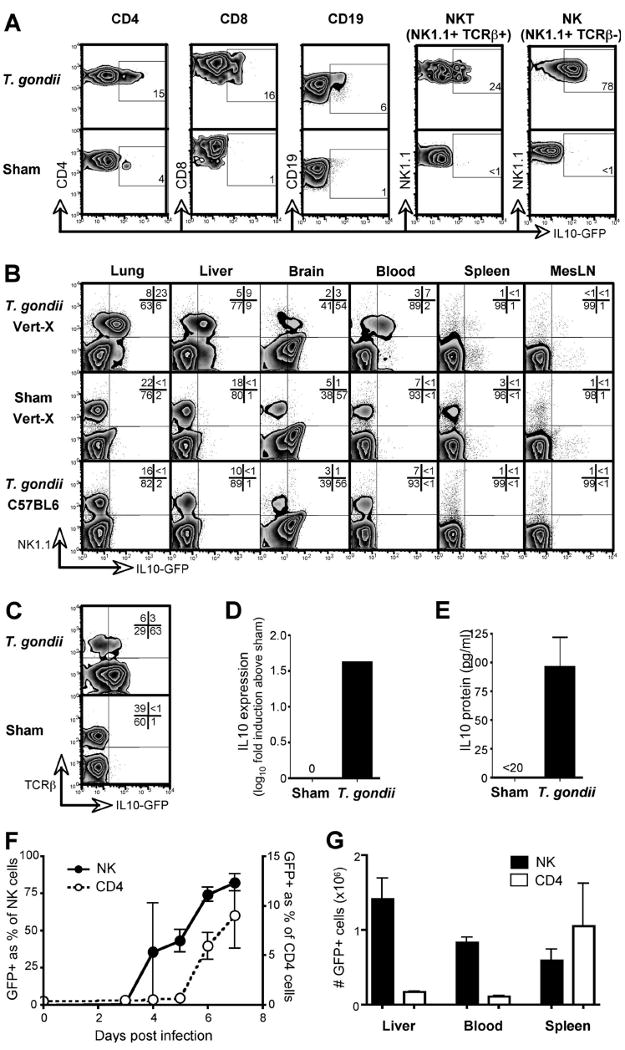
Vert-X IL-10 reporter mice were infected with T. gondii and, 7 days later, the indicated lymphocyte populations analyzed by flow cytometry. Sham-infected Vert-X mice were analyzed for comparison. (A) Liver cells were stained to distinguish lymphocyte subsets. Numbers indicate the percentage of GFP positive cells within each population, rounded to the nearest full digit. (B) Lymphocytes from various tissues were assessed for NK1.1 and IL-10. Infected non-reporter mice were included to control for autofluorescence. (C) NK1.1-gated cells were stained with TCRβ to discriminate NK and NK T cells. (D) NK cells (NK1.1+TCRβ-) were sorted from the livers of T. gondii-infected non-reporter mice. IL-10 expression was determined by RT-PCR and is shown as fold induction over sham-infected controls. (E) Cells were isolated as in (D) and IL-10 protein production was measured after stimulation with PMA and ionomycin. Error bars indicate SD of triplicate cultures. (F) Vert-X mice were bled at various times post-infection and GFP expression within the indicated populations was quantified by flow cytometry. Data depict the mean of three mice ± SD. (G) Vert-X mice were infected with T. gondii and, on day 7, the total numbers of GFP+ NK1.1+ TCRβ- (NK) and GFP+ CD4+ (CD4) cells calculated per organ or per ml of blood. Data depict the mean of three mice ± SD. All panels are representative of two or more independent experiments.
To determine whether other pathogens besides T. gondii also elicit NK cell IL-10, Vert-X mice were inoculated with two distinct, rapidly disseminating bacteria, and their NK cells analyzed 4 days later (Figure 2A). After infection with the gram-positive bacterium Listeria monocytogenes, 79% ± 11 of blood NK cells became GFP-positive. Likewise, infection with the gram-negative bacterium Yersinia pestis elicited IL-10 expression in 35% ± 21 of NK cells. Sham-infected mice showed 0.5% ± 0.5 and 0.3% ± 0.1 GFP+ NK cells, respectively. These data suggested that NK cell-derived IL-10 may be a common feature of the immune response to multiple pathogens that rapidly disperse throughout the host. To test whether systemic, sterile inflammation would also elicit NK cell IL-10, we injected Vert-X mice i.v. with either CpG or LPS. Indeed systemic CpG triggered IL-10 expression in 40% ± 8 of NK cells and LPS elicited IL-10 in 42% ± 3 of NK cells, compared to 0.2% ± 0.05 and 0.2% ± 0.07 in mice given PBS (Figure 2B). Thus systemic inflammation appeared sufficient to generate IL-10+ NK cells, independently of an initiating pathogen.
Figure 2. NK cell IL-10 is elicited by systemic inflammations.
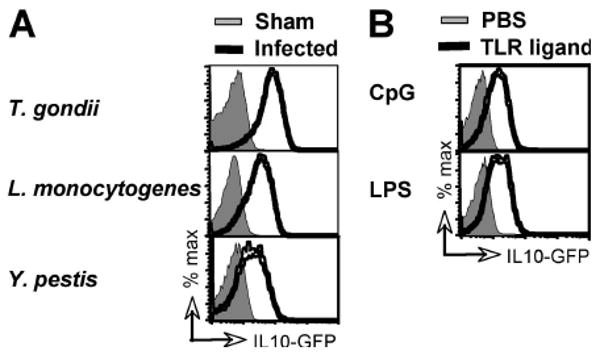
(A) Vert-X mice were infected with T. gondii, L. monocytogenes, or Y. pestis and analyzed by flow cytometry 7, 4, and 4 days later, respectively, together with sham-infected controls. (B) CpG or LPS was given to Vert-X mice i.v. in PBS and recipients were analyzed 48 h later. In both panels, data shown are gated on NK cells from peripheral blood and are representative of 3-5 mice per group in at least two independent experiments.
Systemic inflammation elicits IL-10 in pre-existing, circulating NK cells
Although mature NK cells typically emerge from the bone marrow into the circulation, a recent study by Di Santo and colleagues revealed a novel, thymic pathway of NK cell development (Vosshenrich et al., 2006). Thymus-derived NK cells were distinguished by their considerable cytokine production, a feature they share with the CD56brightCD16neg NK cell subset in humans which can produce significant amounts of IL-10 (Cooper et al., 2001; Vosshenrich et al., 2006). In mice, thymus-derived NK cells are characterized by the expression of CD127, a marker not found on NK cells developing in the bone marrow (Vosshenrich et al., 2006). Consistent with this observation, CD127+ NK cells were readily detectable in the thymi of both naïve mice and those acutely infected with T. gondii, but were infrequent in other tissues (Figure 3A). However, GFP+ NK cells in T. gondii-infected mice were exclusively CD127neg in all tissues, including the thymus. Thus, IL-10+ NK cells appear to be bone-marrow derived and not to originate in the thymus.
Figure 3. Circulating NK cells initiate IL-10 expression in response to infection.
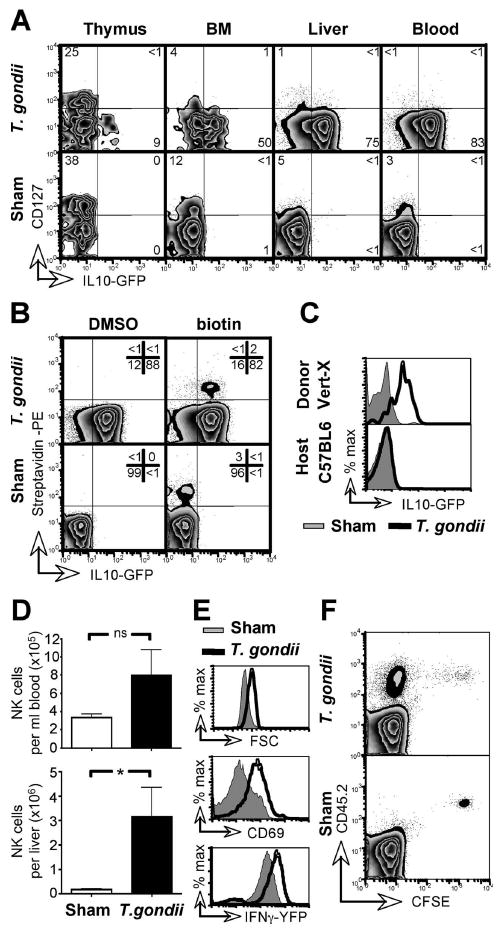
(A) Vert-X mice were T. gondii- or sham-infected and, 7 days later, NK cells from the indicated organs were analyzed by flow cytometry. Numbers indicate the percentage of cells within quadrants and were rounded to full digits. (B) Vert-X mice were injected i.v. with NHS-biotin in DMSO, or with DMSO alone, and infected with T. gondii 12 h later. On day 7 PBL were stained with streptavidin and analyzed by flow cytometry. Data are gated on NK cells. (C) PBL from naïve, CD45.1+ Vert-X mice were transferred i.v. into CD45.2+ C57BL/6 mice 12 h before T. gondii infection. PBL from recipient mice were harvested 7 days later and GFP expression quantified in NK cells from both the transferred (Vert-X) and host (C57BL/6 non-reporter) populations. (D) C57BL/6 mice were infected with T. gondii and NK cells enumerated in the indicated organs 7 days later. Error bars indicate SD from four mice per group. (E) Mice were infected as in (C) and blood-borne NK cells were analyzed by flow cytometry for forward scatter and CD69. Yeti IFN-γ-reporter mice were similarly infected and blood-borne NK cells assessed for YFP expression. (F) PBL from naïve, CD45.2+ C57BL/6 mice were labeled with CFSE and transferred i.v. into congenic CD45.1+ mice 12 h before T. gondii infection. PBL were harvested and CFSE dilution analyzed within the NK cell population 7 days later. All panels are representative of at least two independent experiments with 3-4 mice per group. *, P < 0.05
While the absence of CD127 on IL-10+ NK cells suggested that they originate in the bone marrow, it was not clear whether they emerge as an inherently IL-10+ subset, or whether preexisting, mature NK cells acquire IL-10 expression during toxoplasmosis. To test this, blood-borne cells were surface biotinylated in vivo prior to infection, by i.v. injection of a transiently reactive NHS-biotin moiety (Mullarky et al., 2005). 7 days later NK cells were analyzed to assess their biotinylation, indicating their presence in the blood before infection, and IL-10 expression (Figure 3B). 85% ± 6 of the biotinylated NK cells initiated GFP expression in response to T. gondii infection, whereas only 1% ± 0.04 of labeled NK cells became GFP+ in sham-infected animals. The frequency and extent of IL-10 expression within the biotinylated NK cells were equivalent to those of their non-labeled counterparts in the same animal, demonstrating that the surface biotinylation did not interfere with IL-10 induction. Together these results suggested that NK cells in the circulation prior to infection acquire IL-10 expression during toxoplasmosis. To confirm this, we adoptively transferred blood-borne, GFP- lymphocytes from naïve Vert-X mice into CD45 disparate hosts and infected the recipients with T. gondii. Donor NK cells became 70% ± 5 GFP+ in T. gondii-infected, but not sham-infected hosts (2% ± 2) (Figure 3C). Together these results demonstrate that infection with T. gondii induces IL-10 expression in mature, circulating NK cells.
Infection with T. gondii was also associated with an increase in number of NK cells in the blood and liver (Figure 3D), suggesting that the induction of IL-10 may be concurrent with cell proliferation. Indeed, NK cells in T. gondii-infected mice displayed an activated phenotype with increased FSC profile, upregulation of the acute activation marker CD69, and enhanced expression of IFN-γ as indicated by YFP fluorescence in Yeti reporter mice (Mayer et al., 2005; Stetson et al., 2003) (Figure 3E). Given that essentially all NK cells upregulate IFN-γ (Figure 3E) and that the bulk population is IL-10-GFP+ (Figure 1, 2), the majority of NK cells must express both cytokines. To determine whether circulating NK cells proliferate upon infection, we adoptively transferred CFSE-labeled PBL from naïve CD45.2 donors into naïve CD45.1 hosts and infected the recipient mice with T. gondii. As shown in Figure 3F, NK cells divided markedly in T. gondii-infected but not sham-infected hosts, as revealed by the loss of CFSE fluorescence. Taken together, these data show that the induction of IL-10 in pre-existing, circulating NK cells is associated with activation and extensive proliferation.
IL-12 is necessary and sufficient to induce NK cell IL-10
Systemic infections with both protozoa and bacteria generate robust IL-12 responses in their host (Gazzinelli et al., 1994; Tripp et al., 1994) and, since IL-12 can elicit expression of IL-10 by NK cells in vitro (Grant et al., 2008), we hypothesized that NK cell IL-10 in vivo might also be driven by IL-12. Indeed, direct ex vivo analysis 5 days after infection with T. gondii revealed that STAT4, the signaling adapter of the IL-12/23 receptor complex, was phosphorylated in virtually all NK1.1+ cells, demonstrating actively engaged signaling (Figure 4A) (Wu et al., 2000). This in vivo activation of STAT4 was IL-12p40-dependent, because it was completely abolished by the administration of a neutralizing anti-IL-12p40 antibody (Figure 4A). To test whether IL-12p40 was necessary for the induction of NK cell IL-10, Vert-X mice were treated with anti-IL-12p40 during T. gondii infection and analyzed after 1 week. Neutralization of IL-12p40 abrogated the induction of IL-10 in NK cells (only 5% ± 2 of blood NK cells were GFP+ in anti-p40 treated mice, compared to 81% ± 7 in isotype treated controls) (Figure 4B). Because the IL-12p40 subunit is shared by IL-12 and IL-23 (Hunter, 2005), these experiments did not discriminate which of these cytokines is required to induce NK cell IL-10. We therefore analyzed IL-12Rβ2-deficient NK cells, which are selectively non-responsive to IL-12 (Wu et al., 2000). To distinguish direct effects of IL-12 on NK cells from global abnormalities caused by the complete absence of IL-12 responsiveness, we generated mixed bone marrow chimeras reconstituted with equal parts of IL-12Rβ2-/- and CD45.1 congenic WT bone marrow. T. gondii-infected chimeras displayed STAT4 phosphorylation in WT but not IL-12Rβ2-/- NK cells (Figure 4C), demonstrating both that IL-12 signaling was selectively disrupted and that no other cytokine triggered the phosphorylation of STAT4 in IL-12Rβ2-/- cells. Importantly, in contrast to the WT cells sorted from the same T. gondii-infected animal, IL-12Rβ2-/- NK cells failed to induce IL-10 (Figure 4D). These data show in vivo that direct IL-12 signals are necessary for the induction of IL-10 in NK cells.
Figure 4. IL-12 is necessary and sufficient to elicit IL-10 production by NK cells.
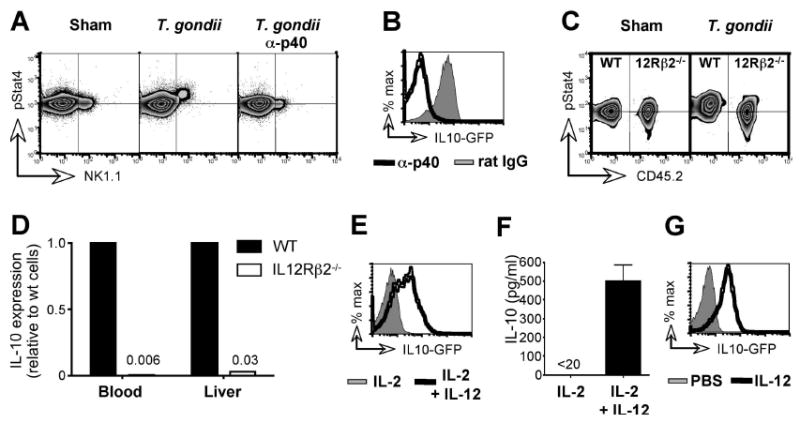
(A) C57BL/6 mice received anti-IL-12p40 or an isotype mAb on days -1 and 3 and were T. gondii- or sham-infected on day 0. On day 4 PBL were stained directly ex vivo for NK1.1 and phosphorylated Stat4. Horizontal guidelines indicate the mode fluorescence intensity of pStat4 staining in sham-infected animals. (B) Vert-X mice were treated as in (A). NK cells in the blood were analyzed for GFP on day 7. (C) Mixed bone marrow chimeras were generated by reconstituting of CD45.1+ recipients with equal parts of CD45.1+ WT and CD45.2+ IL-12Rβ2-/- BM. Reconstituted mice were T. gondii- or sham-infected and 4 days later PBL were stained as in (A). (D) WT and IL-12Rβ2-/- NK cells from the blood and liver of infected chimeras were FACS sorted on day 7 and analyzed for IL-10 by RT-PCR. Transcript levels in WT cells were set to 1. (E) PBL from naïve Vert-X mice were cultured for 48 h in the presence of IL-2 alone or IL-2 and IL-12. GFP expression by NK cells was determined by flow cytometry. (F) NK cells were FACS sorted from the livers of C57BL/6 mice and cultured as in (E). Culture supernatants were harvested 48 h later and analyzed for IL-10 protein. Error bars indicate SD of triplicate cultures. (G) Naïve Vert-X mice were injected with recombinant IL-12 i.v. and blood was analyzed 48 h later. Data are gated on NK cells. All panels are representative of 3-5 mice per group and at least two independent experiments.
To test whether IL-12 is also sufficient to elicit NK cell-derived IL-10, lymphocytes from naïve Vert-X mice were cultured with IL-2 in the absence or presence of IL-12 (Figure 4E). The addition of IL-12 alone was sufficient to induce the expression of IL-10 in NK cells and NK cells sorted from naive C57BL/6 mice released significant amounts of IL-10 only when stimulated by the addition of IL-12 (Figure 4F), a finding consistent with recently published data (Grant et al., 2008). Moreover, systemic i.v. administration of recombinant IL-12 to naïve Vert-X mice efficiently triggered IL-10 expression in NK cells (70% ± 3 of blood NK cells became GFP+, compared to 0.3% ± 0.04 in PBS controls) (Figure 4G). Together these data formally demonstrate that direct IL-12 signals are both necessary and sufficient to induce NK cells to produce IL-10 in vivo.
NK cell IL-10 inhibits IL-12 production by DC
Since the anti-inflammatory action of IL-10 is thought to be largely mediated by its ability to inhibit the production of IL-12 by antigen presenting cells (Moore et al., 2001), these data suggested that a negative feedback loop could exist in which IL-12 limits its own production by eliciting IL-10 from NK cells. Indeed, when NK cells were isolated from T. gondii-infected C57BL/6 mice and mixed with LPS-stimulated, IL-10-/- DCs, such that the only potential source of IL-10 in these cultures was the NK cells, their net effect was to significantly inhibit IL-12 production (Figure 5A). This suppression of DC function was dependent on IL-10, since the inclusion of an IL-10R-blocking antibody instead enabled the NK cells to strongly potentiate IL-12 release (Figure 5A). Interestingly, NK cell-derived IL-10 was functional even at a concentration undetectable in the culture supernatants; it became directly measurable only when cytokine consumption was prevented by the IL-10R-blocking antibody. Considerable quantities of IFN-γ were also detected in the co-cultures, particularly when LPS was used to trigger DC-derived IL-12 and thereby augment NK stimulation (Figure 5A). These results suggest that activated NK cells release both IL-10 and IFN-γ, and that the selective inhibition of only IL-10 enables the remaining IFN-γ to enhance DC activation. Together these data reveal that the net function of IL-10+ NK cells is to limit the release of pro-inflammatory IL-12.
Figure 5. NK- cell IL-10 limits IL-12 production by DC.
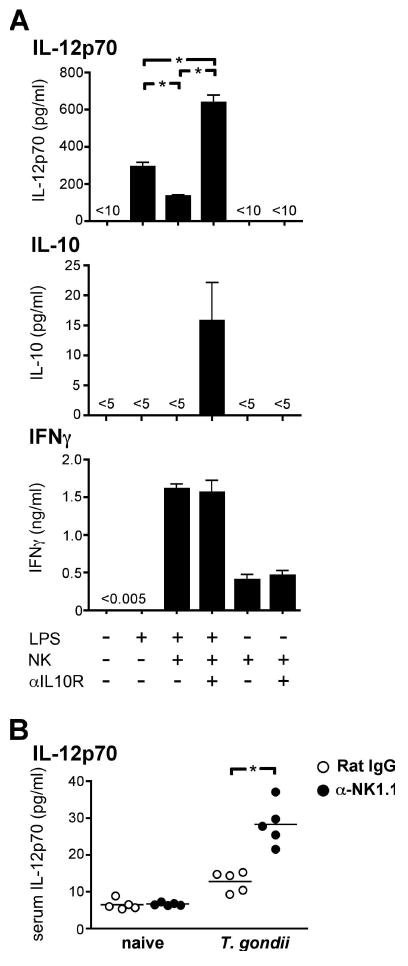
(A) Wild type NK cells were sorted from the livers of C57BL/6 mice infected with T. gondii 7 d earlier, and cultured with IL-10-/- DCs. LPS and either anti-IL-10R or an isotype control antibody were included as indicated. Culture supernatants were harvested after 16 h and analyzed by cytometric cytokine bead array. Error bars indicate SD of triplicate cultures. (B) C57BL/6 mice received anti-NK1.1 or an isotype mAb on days -1 and 3 and were T. gondii- or sham-infected on day 0. Serum was collected on day 5 and analyzed for IL-12 by cytometric bead array. Both panels are representative of two independent experiments. *, P < 0.001
To test whether NK cells exhibit the same immunosuppressive activity in vivo during systemic infection, we depleted NK cells during acute toxoplasmosis in C57BL/6 mice. Indeed, NK cell-depleted animals showed significantly higher concentrations of serum IL-12 compared with isotype-treated controls (Figure 5B), suggesting that NK cells can act to inhibit infection-induced systemic inflammation. Their immunosuppressive function appeared similarly effective to that of CD4+ T cells, since the depletion of either population led to equivalent increases in serum IL-12 and IFN-γ in T. gondii-infected mice (Figure S3). Neither NK nor CD4+ T cells, when singly depleted, could recapitulate the blockade of IL-10 function achieved by treatment with an anti-IL-10R antibody (Figure S3) and together these data suggest that, while neither population is the only source, both NK cells and CD4+ T cells contribute IL-10 to the immune response elicited by T. gondii infection.
NK cell IL-10 is triggered by systemic availability of IL-12
Our earlier experiments indicated that NK cell-derived IL-10 was driven by systemic inflammation associated with diverse, rapidly disseminating pathogens. To investigate whether localized infection would also elicit NK cell IL-10, we challenged Vert-X mice with the respiratory influenza virus. In contrast to the systemic pathogens T. gondii and Y. pestis, influenza virus did not induce IL-10 expression in NK cells (Figure 6A). This was true in both the blood and in the infected tissues (BAL and lung), at days 3, 5 and 10 after infection, and at infectious doses ranging from 300 to 30,000 EID50 (Figure 6A and data not shown). One possible explanation for these data is that, in contrast to systemic inflammation, local infection does not elicit sufficient IL-12 to induce NK cell IL-10. This hypothesis was consistent with the absence of ex vivo STAT4 phosphorylation in NK cells during influenza virus infection (Figure 6B). To test whether increased availability of IL-12 could initiate NK cell expression of IL-10, we infected Vert-X mice with influenza virus and, 5 days later, injected rIL-12 i.v. As expected (Fig. 4G), this treatment resulted in phosphorylation of Stat4 in essentially all NK cells in both the blood and BAL, at an equivalent level to that achieved by rIL-12 administered to naïve mice (Figure 6B). Importantly, the injection of IL-12 into influenza virus-infected mice also triggered IL-10 expression by NK cells in all tissues examined, including the blood, BAL and lung (82% ± 3 of NK cells in the blood, 70% ± 8 in the BAL and 75% ± 3 in lung were GFP+ after rIL-12 injection, compared to 0.3% ± 0.04, 1.0% ± 0.3 and 0.2% ± 0.1 in infected mice given PBS) (Figure 6C). These data show that, during infection with influenza virus, NK cells retain the capacity to express IL-10 in response to IL-12 and will do so when sufficient IL-12 is available
Figure 6. NK cell IL-10 is elicited by systemic and not local inflammation.
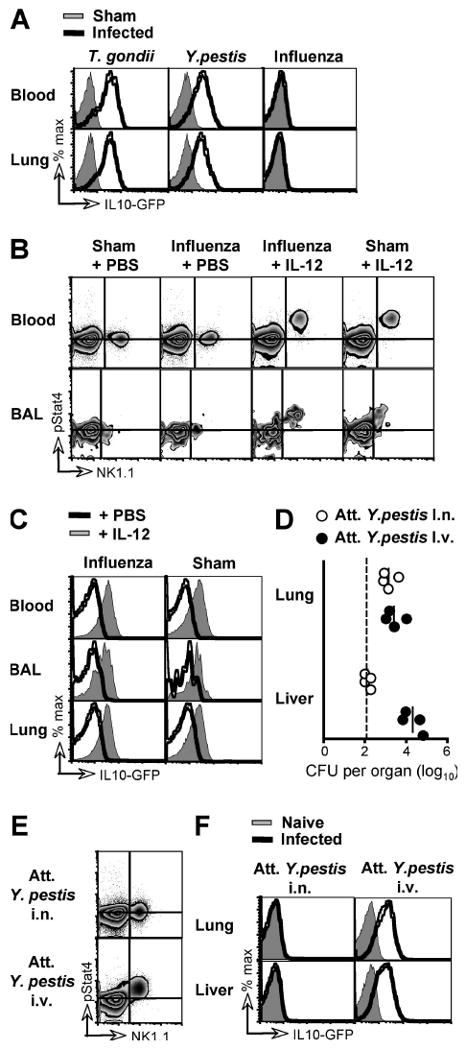
(A) Vert-X mice were infected with T. gondii, Y. pestis or influenza virus and analyzed by flow cytometry 7, 4, and 5 days later, respectively, together with sham-infected controls. Data shown are gated on NK cells from the indicated organs. (B) C57BL/6 mice were either sham-infected or infected with influenza virus i.n. and, 5 days later, BAL cells and PBL stained directly ex vivo for NK1.1 and phosphorylated Stat4. Some mice received recombinant IL-12 by i.v. injection 2 h before sacrifice. Horizontal guidelines indicate the mode fluorescence intensity of pStat4 staining in PBL and BAL lymphocytes from sham-infected animals, respectively. (C) Vert-X mice were either sham-infected or infected with influenza virus and, 3 days later, injected i.v. with either PBS or recombinant IL-12. Cells were analyzed by flow cytometry after a further 48 h. Data shown are gated on NK cells. (D) Vert-X mice were infected with 3×106 CFU of the attenuated Kim10+caf1- strain of Y. pestis by either i.n. or i.v. administration. 4 days later, the indicated organs were ground and plated to assess bacterial burden. Dashed line denotes limit of detection. (E) C57BL/6 mice were infected as in (D) and 2 days later, PBL were stained directly ex vivo for NK1.1 and phosphorylated Stat4, as in (B). (F) Vert-X mice were infected as in (D) and analyzed by flow cytometry 4 days later. Data in all panels are representative of 3-5 mice per group in at least two independent experiments.
To confirm that the absence of IL-12-driven IL-10 expression in NK cells during influenza was due to the local containment of the infection rather than a unique feature of viral pathogens, we sought an experimental system in which the same microbe could be used in either a local or systemic challenge. In contrast to the parental wild-type Y. pestis, the attenuated Kim10+caf1- strain (Philipovskiy and Smiley, 2007) fails to disseminate from the lung when inoculated i.n. but establishes systemic infection when delivered i.v. (Figure 6D). Intranasal infection with this attenuated Y. pestis did not engage STAT4 signaling in circulating NK cells, whereas systemic administration of an identical dose of the same attenuated bacterium stimulated robust STAT4 phosphorylation in the bulk NK cell population (Figure 6E). Moreover, i.n. infection did not elicit IL-10 in NK cells, even in the lung, and despite a locally equivalent bacterial burden to that achieved by disseminated infection following i.v. challenge (0.3% ± 0.1 of lung NK cells and 0.2% ± 0.1 in the liver were GFP+ 4 days after i.n. inoculation, compared to 59% ± 2 and 53% ± 2 after systemic delivery; Figure 6F). The lack of NK cell-derived IL-10 reflected the localized infection rather than the intranasal route of pathogen encounter, since the rapidly disseminating, parental Y. pestis strain did induce IL-10 expression in NK cells after i.n. delivery (Figure 6A). Together these results suggest that the levels of IL-12 that induce NK cell-derived IL-10 are a unique feature of systemic infection, demonstrating that the dissemination of a pathogen can change the cellular and cytokine composition of the host response.
Discussion
IL-10 has a critical role in dampening immune responses and preventing immunopathology, largely due to its ability to mitigate IL-12 (Anderson et al., 2007; Belkaid, 2007; Corinti et al., 2001; Gazzinelli et al., 1996; Jankovic et al., 2007; Suzuki et al., 2000). In this study we have identified NK cells as important mediators of this regulatory circuit: during disseminated infections, NK cells rapidly express IL-10 in response to systemic IL-12, and thereby limit further production of IL-12. The release of IL-10 by NK cells has been previously observed in vitro (Grant et al., 2008; Mehrotra et al., 1998) and, recently, during the late stages of murine visceral leishmaniasis (Maroof et al., 2008). In contrast, we demonstrate here that IL-10+ NK cells are an early and dominant feature of acute systemic inflammatory responses, but are not elicited by localized infections with influenza virus or a Y. pestis bacterium attenuated to prevent its dissemination from the site of infection. The activation of an NK cell-mediated immunosuppressive loop therefore appears to be a unique characteristic of systemic inflammation, and we propose that NK cells provide rapid and widespread IL-10 release to counter situations of dangerous escalation in pro-inflammatory cytokines.
Expression of IL-10 by NK cells was triggered in vivo by three rapidly disseminating pathogens, T. gondii, L. monocytogenes and Y. pestis. The acute, systemic inflammatory response generated by these infections is characteristic of sepsis, where the balance between IL-10 and IL-12 can be critical for survival (Jansen et al., 1996). NK cells have been reported to exacerbate disease in some murine models of sepsis (Barkhausen et al., 2008; Etogo et al., 2008), an effect attributed to their production of IFN-γ. However there is also evidence that transgenic expression of IL-15, a NK cell growth factor, protects mice from septic shock (Hiromatsu et al., 2003). Our data suggest that NK cells can simultaneously express both IFN-γ and IL-10 (Figure 1, 3, 5; (Chakir et al., 2001) and, while other reports have illustrated the ability of NK cells from naïve mice to potentiate a Th1 response (Goldszmid et al., 2007; Guan et al., 2007; Martin-Fontecha et al., 2004), our data demonstrate that during acute systemic infection, the net outcome of the co-production of IL-10 and IFN-γ by NK cells is immune regulation (Figure 5). The concurrent expression of both IFN-γ and IL-10 by NK cells parallels that of highly activated Th1 cells (Anderson et al., 2007; Jankovic et al., 2007), and indeed the production of IL-10 by human NK cell clones has also been proposed to be a late stage of terminal differentiation (Loza and Perussia, 2001).
One of the most striking findings in our study is the majority of NK cells which rapidly express IL-10 during systemic inflammation. We show that IL-10 expression is initiated in pre-existing, circulating NK cells, and that direct IL-12 signals are both necessary and sufficient for its induction. This extends a recent report that IL-12 can initiate the production of IL-10 by murine NK cells cultured in vitro (Grant et al., 2008). Intriguingly, IL-12 is paramount in driving Th1-polarized immune responses (Gazzinelli et al., 1994; Magram et al., 1996) and yet localized, respiratory infection with the influenza virus did not elicit NK cell IL-10, despite clear Th1 induction (Mayer et al., 2005). NK cells were not refractory to IL-12 stimulation during influenza infection and indeed systemic administration of exogenous IL-12 elicited IL-10+ NK cells even in the context of viral infection (Figure 6). This might suggest that the concentration of IL-12 induced by influenza infection, although sufficient to polarize a Th1 response, remains below a threshold required to activate NK cell IL-10 expression. Alternatively, the location of IL-12 released during influenza may be tightly controlled, perhaps restricted to the synapse between APC and cognate T cell, and inaccessible to NK cells.
Direct interactions between NK cells and DC have been described both within the lymph node (Bajenoff et al., 2006) and in peripheral tissues (Moretta, 2002). The distribution of IL-10+ NK cells in predominantly non-lymphoid tissues (Figure 1B) positions them at a site where DC can be conditioned prior to their migration into the lymph node. IL-10 has potent effects on both DC and macrophages, inhibiting their expression of MHC and costimulatory molecules and suppressing their production of pro-inflammatory cytokines, including IL-12 (Figure 5) (Li and Flavell, 2008; Moore et al., 2001), but the impact of IL-10 weakens as the APC mature (Moore et al., 2001; Perona-Wright et al., 2007). Thus the localization and kinetics of the NK cell IL-10 response grants these cells the potential to influence the adaptive immune response from the point of its initiation. This timescale may be particularly important in sepsis, where fatality can precede the adaptive immune response (Cohen, 2002).
The dominance of the IL-10+ phenotype within the NK population differs from the lower frequency within T cell subsets (Figure 1A), and presumably reflects the ability of NK cells to respond to cytokine alone, whereas T cells also require recognition of cognate antigen. A recent report demonstrated that WT CD4+ T cells were sufficient to rescue Rag-/-IL-10-/- mice from lethal toxoplasmosis (Jankovic et al., 2007); in contrast, we reveal here that NK cells are a prominent source of IL-10 in immunologically intact animals, providing IL-10 before antigen-specific sources are available. Multiple cell types express IL-10 during immune-driven inflammatory responses (Figure 1A, S3;(Moore et al., 2001)). The contribution of each one reflects both their distinct temporal and spatial distributions and the different stimuli that elicit their IL-10. Given the active engagement of the IL-12/STAT4 pathway in essentially all circulating NK cells during acute, systemic infection (Figure 4, 6), and the sufficiency of IL-12 signals alone to trigger the release of IL-10 protein (Figure 4F), it is likely that the majority of GFP+ NK cells that we observe are secreting IL-10 in vivo. The activation of the IL-12/STAT4 pathway for the induction of NK cell-derived IL-10 is conserved between humans and mice, because IL-12 also triggers the production of IL-10 by purified human NK cells in vitro (Akuffo et al., 1999; Cooper et al., 2001; Mehrotra et al., 1998; Peritt et al., 1998). Indeed, a STAT4 binding site, CNS+3.10, was recently identified in the il10 gene and is conserved between these species (Grant et al., 2008). Septic patients often exhibit simultaneously increased plasma concentrations of IL-10 and IL-12, a finding that has been proposed as a diagnostic criterion (Sherwin et al., 2008).
Therapeutically, the ability of IL-12 to elicit IL-10 from NK cells has additional consequence, suggesting that the administration of IL-12 or inflammatory adjuvants could counter-intuitively elicit immunosuppression. Indeed, IL-12-treated cancer patients have been reported to display elevated serum IL-10 levels (Portielje et al., 2003). Collectively these findings underscore the clinical importance of understanding the IL-12:IL-10 balance. Here we have shown that NK cells are a rapidly available and abundant source of IL-10 upon IL-12 stimulation, thus defining a novel mechanism of immune regulation active during acute, systemic inflammatory disease.
Experimental Procedures
Mice
Vert-X (C57BL/6 IL-10/eGFP) mice (Madan et al., 2009), Yeti (C57BL/6 IFN-γ-eYFP) (Stetson et al., 2003), C57BL/6, CD45.1+ congenic (B6.SJL-PtprcaPep3b/BoyJ), Rag1-/- (B6.129S7-Rag1tm1Mom/J), IL-12Rβ2-/- (B6.129S1-Il12rb2tm1Jm/J) and IL-10-/- (B6.129P2-Il10tm1Cgn/J) mice were bred and housed in the animal facility of the Trudeau Institute. Animals were kept under specific pathogen-free conditions in filter top cages and were used at 6-12 weeks of age. All experimental procedures were approved by the Institutional Animal Care and Use Committee.
Mixed bone marrow chimeras
CD45.1+ C57BL/6 mice were lethally irradiated (950 rads) and reconstituted with 1 × 107 bone marrow cells, comprising equal parts CD45.1+ WT and CD45.2+ IL-12Rβ2-/- bone marrow. Chimeras were allowed to reconstitute for 6-8wk before infection.
Infections, TLR challenge and IL-12 administration
Mice were infected with 10 cysts of ME49 Toxoplasma gondii by gavage, 1 × 105 CFU Listeria monocytogenes (strain EGD) by i.p. injection, 2 × 105 CFU Yersinia pestis (strain KIM D27) by i.n. instillation, or 300 EID50 of the A/HK-×31 (×31, H3N2) influenza A virus i.n., as described previously (Johnson et al., 2003; Mayer et al., 2005; Mullarky et al., 2005; Parent et al., 2005). The attenuated Yersinia pestis strain Kim10+caf1- (Philipovskiy and Smiley, 2007) was injected either i.n. or i.v., as indicated, at a dose of 3 × 106 CFU per mouse. Sham-infected animals received either uninfected brain, when controls for T. gondii infection, or PBS by the appropriate route. T. gondii – infected mice were analyzed 7 days post-infection; those infected with L. monocytogenes or Y. pestis were analyzed on day 4. Both CpG (ODN1826 (InvivoGen)) and LPS (from E. coli B4:111 (Sigma)) were administered i.v. at a dose of 50 μg/mouse, and recombinant mouse IL-12p70 (PeproTech) was injected i.v. at a dose of 1 μg/mouse. Controls received PBS alone and mice were analyzed 48 h post injection.
Tissue sampling and flow cytometry
Single cell suspensions were prepared from blood, spleen, mesenteric LN, bone marrow and thymus by mechanical disruption and red cell lysis, where needed. Liver lymphocytes, lungs and brains were perfused, digested for 40 min with collagenase IV (100 U/ml) and DNaseI (10 U/ml) and lymphocytes enriched at the interphase of a discontinuous 60/40 Percoll gradient. The following mAbs were used for flow cytometry: CD3ε (clone 145-2C11), CD4 (RM4-5), CD8α (53-6.7), CD11c (HL3), CD19 (ID3), CD45.1 (A20), CD45.2 (104), CD69 (H1.273), CD127 (A7R34), I-Ab (28-16-8S), NK1.1 (PK136) and TCRβ (H57-597). NK cells were identified as NK1.1+ and either CD3- or TCRβ-. Intracellular staining for phopshorylated Stat4 was performed on PBL or BAL cells immediately after red cell lysis using PhosFlow reagents (BD Biosciences) according to the manufacturer's instructions.
In vivo biotinylation
Labelling of circulating lymphocytes was achieved by intravenous injection of 1.2 mg of sulfo-N-hydroxysuccinimide-biotin (Calbiochem) dissolved in 10% DMSO and given as two doses separated by 2 h (Mullarky et al., 2005). Mice were infected 12 h later and biotinylated cells were identified by staining with streptavidin-PE.
In vivo cytokine neutralization
0.75 mg per mouse of rat anti-mouse IL-12/23 p40 antibody (clone C17.8,), anti-IL-10R antibody (clone 1B1.3A) or an IgG isotype control (HPRN), was administered i.p. 12 h before and 3 d after infection.
Cell transfer
Peripheral blood lymphocytes were harvested from naïve CD45.1+ Vert-X or CD45.2+ C57BL/6 mice and 2-3 × 106 cells were transferred i.v. into CD45 disparate WT hosts. Recipients were infected 12 h later. Where indicated, cells were labelled with CFSE (3 μM) for 10 min at 37°C prior to transfer.
Cell sorting, in vitro activation and cytokine quantification
NK cells were purified from T. gondii- or sham-infected livers using a FACSVantage cell sorter. Resultant cells were >95% NK1.1+TCRβ- and were used immediately for RNA extraction and RT-PCR, or cultured for 48 h at 1 × 106/ml in the presence of IL-2 (300 U/ml), IL-2 and IL-12 (5 ng/ml), or phorbol 12-myristate 13-acetate (PMA, 50 ng/ml) and ionomycin (1 μM). Cytokines in culture supernatants were quantified with the Mouse Inflammation Cytometric Bead Array kit (BD PharMingen) according to the manufacturer's instructions. Unfractionated PBL from naïve Vert-X mice were similarly cultured with IL-2 ± IL-12 and their GFP expression measured 48 h later by FACS, as detailed above.
RT-PCR
RNA extraction and quantitative real-time RT-PCR were performed as described (Johnson et al., 2003; Mohrs et al., 2005).
Dendritic cell and NK cell co-culture
DCs were generated from IL-10-/- BM in the presence of recombinant GM-CSF (Peprotech), as described (MacDonald et al., 2001), and were >90% CD11c+MHCII+. DCs were harvested on day 10 of culture and replated at 1 × 106 DC/ml in the presence or absence of LPS (100 ng/ml, from E.coli 011:B4, Sigma), an equal number of NK cells sort-purified from T. gondii-infected livers (see above), anti-IL10R antibody (40 μg/ml, clone 1B1.3A) or a rat IgG isotype control (HPRN). Culture supernatants were collected at 16 h and cytokines measured by cytometric bead array, as above.
In vivo cell depletions
0.75 mg per mouse of rat anti-mouse NK1.1 antibody (clone PK136), anti-CD4 (clone Gk1.5) or an IgG isotype control (HPRN), was administered i.p. 12 h before and 3 d after T. gondii infection. Serum was collected on day 5 and cytokines measured by cytometric bead array, as above.
Statistical analysis
All data are presented as the means ± s.d. unless otherwise indicated. Numerical data were analyzed for statistical significance using Student's unpaired t-test with Prism software (GraphPad). P values <0.05 were considered statistically significant.
Supplementary Material
Supplemetary Figure 1. NK cell expression of IL-10 is unaffected by route of T. gondii infection. Vert-X IL-10 reporter mice were infected with T. gondii either by gavage (p.o.) or by intraperitoneal injection (i.p.). 7 days later, lymphocytes from various tissues were assessed for NK1.1 and IL-10 by flow cytometry. Naive Vert-X mice were analyzed for comparison. Numbers indicate the percentage of cells within each quadrant, rounded to the nearest full digit.
Supplementary Figure 2. IL-10 induction is maintained in Rag-deficient animals. WT and Rag2-/- mice were infected with T. gondii and the indicated organs harvested 1, 4 and 7 days later. IL-10 expression was measured by real time RT-PCR and is shown as the logarithm of the fold induction relative to that of sham-infected controls. Data depict the mean of five mice ± SD and are representative of four independent experiments.
Supplemetary Figure 3. NK cells and CD4+ cells mediate similar suppression of pro-inflammatory serum cytokines. C57BL/6 mice received either anti-NK1.1 (PK136), anti-CD4 (GK1.5), anti-IL-10R (1B1.3A) or an isotype mAb (HRPN) on days -1 and 3 and were T. gondii- or sham-infected on day 0. Serum was collected on day 5 and analyzed for both IL-12 and IFN-γ by cytometric bead array.
Acknowledgments
We thank Ron LaCourse and Brandon Sells for cell sorting, Paula Lanthier for technical help, Jacob Kohlmeier for critical comments and Robert Brubaker, James Bliska and Robert North for provision of bacterial strains. This work was supported by funds from Trudeau Institute and the National Institutes of Health grants AI072296 (M.M.), AI061577 (S.T.S.), AI61587 (L.L.J.), and AI057992 (C.L.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akuffo H, Alexis A, Eidsmo L, Saed A, Nylen S, Maasho K. Natural killer cells in cross-regulation of IL-12 by IL-10 in Leishmania antigen-stimulated blood donor cells. Clin Exp Immunol. 1999;117:529–534. doi: 10.1046/j.1365-2249.1999.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4+CD25-Foxp3- Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med. 2007;204:285–297. doi: 10.1084/jem.20061886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenoff M, Breart B, Huang AY, Qi H, Cazareth J, Braud VM, Germain RN, Glaichenhaus N. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J Exp Med. 2006;203:619–631. doi: 10.1084/jem.20051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkhausen T, Frerker C, Putz C, Pape HC, Krettek C, van Griensven M. Depletion of NK cells in a murine polytrauma model is associated with improved outcome and a modulation of the inflammatory response. Shock. 2008;30:401–410. doi: 10.1097/SHK.0b013e31816e2cda. [DOI] [PubMed] [Google Scholar]
- Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol. 2007;7:875–888. doi: 10.1038/nri2189. [DOI] [PubMed] [Google Scholar]
- Chakir H, Lemay AM, Webb JR. Cytokine expression by murine DX5+ cells in response to IL-12, IL-18, or the combination of IL-12 and IL-18. Cell Immunol. 2001;212:71–81. doi: 10.1006/cimm.2001.1844. [DOI] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, Carson WE, Caligiuri MA. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97:3146–3151. doi: 10.1182/blood.v97.10.3146. [DOI] [PubMed] [Google Scholar]
- Corinti S, Albanesi C, la Sala A, Pastore S, Girolomoni G. Regulatory activity of autocrine IL-10 on dendritic cell functions. J Immunol. 2001;166:4312–4318. doi: 10.4049/jimmunol.166.7.4312. [DOI] [PubMed] [Google Scholar]
- Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- De Maria A, Fogli M, Mazza S, Basso M, Picciotto A, Costa P, Congia S, Mingari MC, Moretta L. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol. 2007;37:445–455. doi: 10.1002/eji.200635989. [DOI] [PubMed] [Google Scholar]
- Deniz G, Erten G, Kucuksezer UC, Kocacik D, Karagiannidis C, Aktas E, Akdis CA, Akdis M. Regulatory NK cells suppress antigen-specific T cell responses. J Immunol. 2008;180:850–857. doi: 10.4049/jimmunol.180.2.850. [DOI] [PubMed] [Google Scholar]
- Etogo AO, Nunez J, Lin CY, Toliver-Kinsky TE, Sherwood ER. NK but not CD1-restricted NKT cells facilitate systemic inflammation during polymicrobial intra-abdominal sepsis. J Immunol. 2008;180:6334–6345. doi: 10.4049/jimmunol.180.9.6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, Sher A. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J Immunol. 1994;153:2533–2543. [PubMed] [Google Scholar]
- Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- Goldszmid RS, Bafica A, Jankovic D, Feng CG, Caspar P, Winkler-Pickett R, Trinchieri G, Sher A. TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-gamma production. J Exp Med. 2007;204:2591–2602. doi: 10.1084/jem.20070634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant LR, Yao ZJ, Hedrich CM, Wang F, Moorthy A, Wilson K, Ranatunga D, Bream JH. Stat4-dependent, T-bet-independent regulation of IL-10 in NK cells. Genes Immun. 2008;9:316–327. doi: 10.1038/gene.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Moretto M, Bzik DJ, Gigley J, Khan IA. NK cells enhance dendritic cell response against parasite antigens via NKG2D pathway. J Immunol. 2007;179:590–596. doi: 10.4049/jimmunol.179.1.590. [DOI] [PubMed] [Google Scholar]
- Hiromatsu T, Yajima T, Matsuguchi T, Nishimura H, Wajjwalku W, Arai T, Nimura Y, Yoshikai Y. Overexpression of interleukin-15 protects against Escherichia coli-induced shock accompanied by inhibition of tumor necrosis factor-alpha-induced apoptosis. J Infect Dis. 2003;187:1442–1451. doi: 10.1086/374643. [DOI] [PubMed] [Google Scholar]
- Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5:521–531. doi: 10.1038/nri1648. [DOI] [PubMed] [Google Scholar]
- Jankovic D, Kullberg MC, Feng CG, Goldszmid RS, Collazo CM, Wilson M, Wynn TA, Kamanaka M, Flavell RA, Sher A. Conventional T-bet+Foxp3- Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J Exp Med. 2007;204:273–283. doi: 10.1084/jem.20062175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen PM, van der Pouw Kraan TC, de Jong IW, van Mierlo G, Wijdenes J, Chang AA, Aarden LA, Taylor FB, Jr, Hack CE. Release of interleukin-12 in experimental Escherichia coli septic shock in baboons: relation to plasma levels of interleukin-10 and interferon-gamma. Blood. 1996;87:5144–5151. [PubMed] [Google Scholar]
- Johnson LL, Berggren KN, Szaba FM, Chen W, Smiley ST. Fibrin-mediated protection against infection-stimulated immunopathology. J Exp Med. 2003;197:801–806. doi: 10.1084/jem.20021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamanaka M, Kim ST, Wan YY, Sutterwala FS, Lara-Tejero M, Galan JE, Harhaj E, Flavell RA. Expression of Interleukin-10 in Intestinal Lymphocytes Detected by an Interleukin-10 Reporter Knockin tiger Mouse. Immunity. 2006;25:941–952. doi: 10.1016/j.immuni.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Katakura T, Miyazaki M, Kobayashi M, Herndon DN, Suzuki F. CCL17 and IL-10 as effectors that enable alternatively activated macrophages to inhibit the generation of classically activated macrophages. J Immunol. 2004;172:1407–1413. doi: 10.4049/jimmunol.172.3.1407. [DOI] [PubMed] [Google Scholar]
- Li MO, Flavell RA. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity. 2008;28:468–476. doi: 10.1016/j.immuni.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Loza MJ, Perussia B. Final steps of natural killer cell maturation: a model for type 1-type 2 differentiation? Nat Immunol. 2001;2:917–924. doi: 10.1038/ni1001-917. [DOI] [PubMed] [Google Scholar]
- MacDonald AS, Straw AD, Bauman B, Pearce EJ. CD8- dendritic cell activation status plays an integral role in influencing Th2 response development. J Immunol. 2001;167:1982–1988. doi: 10.4049/jimmunol.167.4.1982. [DOI] [PubMed] [Google Scholar]
- Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, Yogev N, Gu Y, Khodoun M, Hildeman D, et al. Nonredundant Roles for B Cell-Derived IL-10 in Immune Counter-Regulation. J Immunol. 2009 doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Posttranscriptional regulation of il10 gene expression allows natural killer cells to express immunoregulatory function. Immunity. 2008;29:295–305. doi: 10.1016/j.immuni.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A, Sallusto F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–1265. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- Mayer KD, Mohrs K, Crowe SR, Johnson LL, Rhyne P, Woodland DL, Mohrs M. The functional heterogeneity of type 1 effector T cells in response to infection is related to the potential for IFN-{gamma} production. J Immunol. 2005;174:7732–7739. doi: 10.4049/jimmunol.174.12.7732. [DOI] [PubMed] [Google Scholar]
- Maynard CL, Harrington LE, Janowski KM, Oliver JR, Zindl CL, Rudensky AY, Weaver CT. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- Mehrotra PT, Donnelly RP, Wong S, Kanegane H, Geremew A, Mostowski HS, Furuke K, Siegel JP, Bloom ET. Production of IL-10 by human natural killer cells stimulated with IL-2 and/or IL-12. J Immunol. 1998;160:2637–2644. [PubMed] [Google Scholar]
- Mohrs K, Harris DP, Lund FE, Mohrs M. Systemic dissemination and persistence of th2 and type 2 cells in response to infection with a strictly enteric nematode parasite. J Immunol. 2005;175:5306–5313. doi: 10.4049/jimmunol.175.8.5306. [DOI] [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Moretta A. Natural killer cells and dendritic cells: rendezvous in abused tissues. Nat Rev Immunol. 2002;2:957–964. doi: 10.1038/nri956. [DOI] [PubMed] [Google Scholar]
- Mullarky IK, Szaba FM, Berggren KN, Parent MA, Kummer LW, Chen W, Johnson LL, Smiley ST. Infection-stimulated fibrin deposition controls hemorrhage and limits hepatic bacterial growth during listeriosis. Infect Immun. 2005;73:3888–3895. doi: 10.1128/IAI.73.7.3888-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent MA, Berggren KN, Kummer LW, Wilhelm LB, Szaba FM, Mullarky IK, Smiley ST. Cell-mediated protection against pulmonary Yersinia pestis infection. Infect Immun. 2005;73:7304–7310. doi: 10.1128/IAI.73.11.7304-7310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peritt D, Robertson S, Gri G, Showe L, Aste-Amezaga M, Trinchieri G. Differentiation of human NK cells into NK1 and NK2 subsets. J Immunol. 1998;161:5821–5824. [PubMed] [Google Scholar]
- Perona-Wright G, Anderton SM, Howie SE, Gray D. IL-10 permits transient activation of dendritic cells to tolerize T cells and protect from central nervous system autoimmune disease. Int Immunol. 2007;19:1123–1134. doi: 10.1093/intimm/dxm084. [DOI] [PubMed] [Google Scholar]
- Philipovskiy AV, Smiley ST. Vaccination with live Yersinia pestis primes CD4 and CD8 T cells that synergistically protect against lethal pulmonary Y. pestis infection. Infect Immun. 2007;75:878–885. doi: 10.1128/IAI.01529-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portielje JE, Lamers CH, Kruit WH, Sparreboom A, Bolhuis RL, Stoter G, Huber C, Gratama JW. Repeated administrations of interleukin (IL)-12 are associated with persistently elevated plasma levels of IL-10 and declining IFN-gamma, tumor necrosis factor-alpha, IL-6, and IL-8 responses. Clin Cancer Res. 2003;9:76–83. [PubMed] [Google Scholar]
- Roers A, Siewe L, Strittmatter E, Deckert M, Schluter D, Stenzel W, Gruber AD, Krieg T, Rajewsky K, Muller W. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004;200:1289–1297. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin C, Broadbent R, Young S, Worth J, McCaffrey F, Medlicott NJ, Reith D. Utility of Interleukin-12 and Interleukin-10 in Comparison with Other Cytokines and Acute-Phase Reactants in the Diagnosis of Neonatal Sepsis. Am J Perinatol. 2008 doi: 10.1055/s-0028-1090585. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Pagarigan RR, Whitton JL. NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J Immunol. 2000;164:2009–2015. doi: 10.4049/jimmunol.164.4.2009. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, Kronenberg M, Locksley RM. Constitutive Cytokine mRNAs Mark Natural Killer (NK) and NK T Cells Poised for Rapid Effector Function. J Exp Med. 2003;198:1069–1076. doi: 10.1084/jem.20030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O'Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute Influenza virus infection by producing IL-10. Nat Med. 2009;15:277–284. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science. 1988;240:516–518. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Sher A, Yap G, Park D, Neyer LE, Liesenfeld O, Fort M, Kang H, Gufwoli E. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J Immunol. 2000;164:5375–5382. doi: 10.4049/jimmunol.164.10.5375. [DOI] [PubMed] [Google Scholar]
- Tripp CS, Gately MK, Hakimi J, Ling P, Unanue ER. Neutralization of IL-12 decreases resistance to Listeria in SCID and C.B-17 mice. Reversal by IFN-gamma. J Immunol. 1994;152:1883–1887. [PubMed] [Google Scholar]
- Vosshenrich CA, Garcia-Ojeda ME, Samson-Villeger SI, Pasqualetto V, Enault L, Richard-Le Goff O, Corcuff E, Guy-Grand D, Rocha B, Cumano A, et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol. 2006;7:1217–1224. doi: 10.1038/ni1395. [DOI] [PubMed] [Google Scholar]
- Wu C, Wang X, Gadina M, O'Shea JJ, Presky DH, Magram J. IL-12 receptor beta 2 (IL-12R beta 2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J Immunol. 2000;165:6221–6228. doi: 10.4049/jimmunol.165.11.6221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemetary Figure 1. NK cell expression of IL-10 is unaffected by route of T. gondii infection. Vert-X IL-10 reporter mice were infected with T. gondii either by gavage (p.o.) or by intraperitoneal injection (i.p.). 7 days later, lymphocytes from various tissues were assessed for NK1.1 and IL-10 by flow cytometry. Naive Vert-X mice were analyzed for comparison. Numbers indicate the percentage of cells within each quadrant, rounded to the nearest full digit.
Supplementary Figure 2. IL-10 induction is maintained in Rag-deficient animals. WT and Rag2-/- mice were infected with T. gondii and the indicated organs harvested 1, 4 and 7 days later. IL-10 expression was measured by real time RT-PCR and is shown as the logarithm of the fold induction relative to that of sham-infected controls. Data depict the mean of five mice ± SD and are representative of four independent experiments.
Supplemetary Figure 3. NK cells and CD4+ cells mediate similar suppression of pro-inflammatory serum cytokines. C57BL/6 mice received either anti-NK1.1 (PK136), anti-CD4 (GK1.5), anti-IL-10R (1B1.3A) or an isotype mAb (HRPN) on days -1 and 3 and were T. gondii- or sham-infected on day 0. Serum was collected on day 5 and analyzed for both IL-12 and IFN-γ by cytometric bead array.