

Abstract
Ca2+ signaling plays important roles during both axonal and dendritic growth. Yet whether and how Ca2+ rises may trigger and contribute to the development of long-range cortical connections remains mostly unknown. Here, we demonstrate that two separate limbs of the Ca2+/calmodulin-dependent protein kinase kinase (CaMKK)–CaMKI cascades, CaMKK–CaMKIα and CaMKK–CaMKIγ, critically coordinate axonal and dendritic morphogenesis of cortical neurons, respectively. The axon-specific morphological phenotype required a diffuse cytoplasmic localization and a strikingly α-isoform-specific kinase activity of CaMKI. Unexpectedly, treatment with muscimol, a GABAA receptor agonist, selectively stimulated elongation of axons but not of dendrites, and the CaMKK–CaMKIα cascade critically mediated this axonogenic effect. Consistent with these findings, during early brain development, in vivo knockdown of CaMKIα significantly impaired the terminal axonal extension and thereby perturbed the refinement of the interhemispheric callosal projections into the contralateral cortices. Our findings thus indicate a novel role for the GABA-driven CaMKK–CaMKIα cascade as a mechanism critical for accurate cortical axon pathfinding, an essential process that may contribute to fine-tuning the formation of interhemispheric connectivity during the perinatal development of the CNS.
Introduction
The formation of cortical neural circuits requires precisely controlled development of axons and dendrites. Although the molecular mechanisms underlying axon guidance in the CNS have been intensively studied (Tessier-Lavigne and Goodman, 1996; Dickson, 2002), the intracellular signaling and cytoskeletal remodeling mechanisms implicated in the precise extension and targeting of axonal arbors still remain mostly unsolved.
Ca2+ plays a central role in the regulation of neuronal morphogenesis. It is believed that there is an elevated optimal range for the intracellular Ca2+ concentration that supports maximal neurite outgrowth in various types of neurons (Kater et al., 1988; Gomez and Zheng, 2006). Ca2+/calmodulin-dependent protein kinases (CaMKs), a major Ca2+-dependent kinase family, are good candidates as potential downstream effectors of calcium elevation in neurons (Soderling and Stull, 2001; Hudmon and Schulman, 2002). Although the essential role of CaMKII subfamily members in neuronal plasticity has been shown, much less is known about the function of the CaMKI/IV subfamily, which forms several distinct kinase cascades downstream of CaMK kinase α (CaMKKα) and/or CaMKKβ (Soderling, 1999; Hook and Means, 2001; Hudmon and Schulman, 2002; Bito and Takemoto-Kimura, 2003). The CaMKI family includes four isoforms: α (Nairn and Greengard, 1987), β/Pnck (Yokokura et al., 1997), γ/CL3 (Takemoto-Kimura et al., 2003), and δ/CKLiK (Ishikawa et al., 2003). Recently, a few reports from several laboratories, including ours, have started to suggest, in vitro, that CaMKI activity may participate in the regulation of neuronal morphology such as growth cone motility (Wayman et al., 2004), neurite outgrowth (Schmitt et al., 2004; Uboha et al., 2007), activity-dependent growth of dendrites (Wayman et al., 2006; Takemoto-Kimura et al., 2007), and stabilization of spines (Saneyoshi et al., 2008). However, evidence based on materials from genetically engineered animals is still scarce, and in vivo validation of such findings is still much awaited. Furthermore, despite the heavy expression of all CaMKI isoforms in the developing forebrain, there is yet little information as to what kinds of endogenous activity or extracellular ligands may influence the activity of CaMKI, during a perinatal period when only spontaneous Ca2+ transients are generated, and when synaptic activity-driven Ca2+-mobilization is still missing.
We previously reported that a dendritic raft-anchored CaMK, CaMKIγ/CL3, plays an essential role in dendritic growth downstream of BDNF (Takemoto-Kimura et al., 2007). However, the exact context in which other CaMKI isoforms might contribute to neuronal morphogenesis remained obscure.
Here, we show genetic and pharmacogenetic evidence that demonstrates that two separate limbs of CaMKK–CaMKI cascades, CaMKK–CaMKIα and CaMKK–CaMKIγ, critically coordinate axonal and dendritic morphogenesis of immature cortical neurons, respectively. Furthermore, we found that activation of GABAA receptors promoted axonal growth via the CaMKK–CaMKIα pathway. During perinatal brain development, in vivo knockdown of CaMKIα significantly impaired the terminal elongation of callosal axon projections in the somatosensory cortex. Together, our data suggest that a GABA-driven CaMK cascade may play a critical role in activity-regulated refinement of cortical axon wiring.
Materials and Methods
Construction of expression plasmids and RNA interference vectors.
For RNA interference (RNAi) experiments, short hairpin RNA (shRNA) vectors, coexpressing mRFP1 as a morphological tracer, were constructed essentially as described previously (Takemoto-Kimura et al., 2007). To create pSUPER-shCaMKIα and pSUPER-shCaMKIα#2, two complementary 60 bp oligonucleotides carrying sense and antisense sequences for CATTGTAGCCCTGGATGAC (19 bp; corresponding to nucleotides 231-249 of mouse CaMKIα) and antisense and sense sequences for GATCAAGCACCCCAACATT (19 bp; corresponding to nucleotides 216-234 of mouse CaMKIα), respectively, were subcloned into the pSuper+mRFP1 plasmid backbone. pSUPER-shNega was generated similarly except that an artificial 19-mer sequence (ATCCGCGCGATAGTACGTA) was used as a target as described previously (Takemoto-Kimura et al., 2007). This sequence was based on a commercially available negative control small interfering RNA sequence (B-Bridge International), and we confirmed that it had no significant identity to any known mammalian gene based on a BLAST (basic local alignment search tool) search. Silent mutations were introduced into the shRNA target sequence of enhanced green fluorescent protein (EGFP)-tagged wild-type (WT) and mutant CaMKIα cDNAs to generate shRNA-resistant constructs (pEGFP-CaMKIαres and related constructs). Short hairpin RNA interference vectors against CaMKIα, CaMKIγ/CL3, and CaMKIV [shCaMKIα (this study); shCaMKIγ/CL3 and shCaMKIV (Takemoto-Kimura et al., 2007)] selectively suppressed expression of GFP-CaMKIα, GFP-CaMKIγ/CL3, GFP-CaMKIV, respectively (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). An antibody against CaMKIV (BD Biosciences Transduction Laboratories) also confirmed these results. The potency of the knockdown was estimated to be ∼70–80%, based on the reduction of overexpressed GFP-tagged proteins in Western blot analyses (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). In keeping with this, and consistent with a transfection efficiency of >50% in our electroporation, we also detected a target-specific decrease of 40∼50% in the amount of endogenous mRNA using a real-time PCR system (LightCycler 1.5; Roche Diagnostics) (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material).
Rat CaMKIα cDNA was inserted into pEGFPC1 vector (Clontech) to generate pEGFP-CaMKIα (Takemoto-Kimura et al., 2003). The expression vector for a constitutively active form, pEGFP-CaMKIαCA (286IHQS to 286EDDD; F307A) was created from pEGFP-CaMKIα by site-directed mutagenesis. Similarly, a point mutation was introduced to generate pEGFP-CaMKIαK49A. pCAG-EGFP-CaMKIγ/CL3 was as described previously (Takemoto-Kimura et al., 2007). CaMKKβ wild-type and V269F cDNA (Tokumitsu et al., 2003) (a kind gift from Dr. Hiroshi Tokumitsu, Kagawa University, Kagawa, Japan) was subcloned into pEGFPC3. Mouse CaMKIβ and CaMKIδ cDNAs were obtained from the German RZPD gene collection and RIKEN Genomic Science Center, respectively, and inserted into pEGFPC1 vector to generate pEGFP-CaMKIβ and pEGFP-CaMKIδ. All constructs were verified by sequencing.
Gene targeting, neuronal culture, and pharmacology.
All animal experiments in this study were performed in accordance with regulations and guidelines for the care and use of the experimental animals of the University of Tokyo, and approved by the institutional review committee of University of Tokyo Graduate School of Medicine.
CaMKKα-knock-out (KO) mice were described previously (Blaeser et al., 2006). CaMKKβ-KO mice were produced similarly by deleting exon 2 (where the ATG starts) through exon 6 of the CaMKKβ gene. A detailed characterization of CaMKKβ-KO mice will be described elsewhere (F. Blaeser and T. A. Chatila, unpublished observations). CaMKKα- and CaMKKβ-KO mice were crossed to produce CaMKKα/β-double knock-out (DKO) mice. The targeting strategy of CaMKIγ/CL3-KO mice was as described previously (Takemoto- Kimura et al., 2007).
Dissociated cortical neurons were prepared and cultured from embryonic day 19 (E19) Sprague Dawley rats or E17 C57BL/6 mice (wild-type as well as mutant mice), essentially as described previously (Takemoto-Kimura et al., 2007). In brief, dissected cortices were incubated for 10 min with 10 mg/ml trypsin type XI (Sigma-Aldrich) plus 0.5 mg/ml DNase I type IV (Sigma-Aldrich) at room temperature and mechanically dissociated in Hanks solution, pH 7.4 (Sigma-Aldrich), with 0.5 mg/ml DNase I type IV and 12 mm MgSO4. Cortical neurons were transfected immediately after dissociation by electroporation using a Nucleofector (Amaxa Biosystems), plated onto poly-l-lysine-coated 12 mm coverslips (BD Biosciences), poly-d-lysine-coated glass-bottom dishes (MatTek) or six-well dish (BD Biosciences), and maintained in minimum essential medium (Invitrogen) containing 5 g/L glucose, 0.2 g/L NaHCO3, 0.1 g/L transferrin (Calbiochem), 2 mm GlutaMAX-I (Invitrogen), 25 μg/ml insulin (Sigma-Aldrich), B-27 supplement (Invitrogen), and 10% fetal bovine serum. Cultures were maintained in 5% CO2 at 37°C.
For inhibition and stimulation experiments, 2-[N-(2-hydroxyethyl)-N-(4-methoxybenzenesulfonyl)] amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN-93) (Calbiochem), 1,8-naphthoylene benzimidazole-3-carboxylic acid (STO-609) (Tocris Bioscience), mevastatin (Wako), muscimol (Tocris Bioscience), or BDNF [generously provided by Dainippon Sumitomo Pharma Co., Ltd. (Osaka, Japan) by courtesy of Dr. Chikao Nakayama] were added to the medium of cultured neurons expressing mRFP1 at 6 h after plating at the final concentrations of 10 μm (KN-93), 2.6 μm (STO-609), 10 μm (mevastatin), 1 μm (muscimol), and 50 ng/ml (BDNF), respectively. Bath application was performed by dissolving the reagents in one-half volume of the conditioned culture medium and by mixing this gently with the remaining one-half of the original medium in the dish. No medium change was done onward until fixation.
Immunocytochemistry, morphometric analyses, and visualization of raft-targeted proteins.
For morphometric analysis, cortical neurons were transfected immediately after dissociation by electroporation using Nucleofector and plated onto 12 mm poly-l-lysine-coated coverslips at the density of 5 × 105 cells (rats) or 7.5 × 105 cells (mice) per coverslip in 24-well plates. Dissociated cultures of rat and mouse cortical neurons and all measurements (axonal and dendritic length, axonal tip numbers) were performed at 2 d in vitro (DIV) essentially as described previously (Takemoto-Kimura et al., 2007). Images of neuronal morphologies were captured based on immunoreactivities against GFP, mRFP1, or mCherry, using the Olympus BX51 microscopy system with a 20× objective. Dendrites and axons were identified by standard morphological criteria as described previously (Takemoto-Kimura et al., 2007), and only neurons that possessed one clearly classifiable axon and one or more dendrites were analyzed. All quantitative analyses were performed by an observer blinded to the identity of the transfected constructs, genotypes of mice, or treated drugs.
Immunostaining was performed as described previously (Bito et al., 1996; Nonaka et al., 2006; Takemoto-Kimura et al., 2007). A rabbit anti-DsRed antibody (Takara) was used for quantitative morphometric analyses of RNAi, rescue, and forced expression experiments, and a rat anti-GFP antibody (Nacalai Tesque) was used to detect coexpressed constructs. An anti-GM130 antibody (BD Biosciences Transduction Laboratories) was used as a Golgi marker. As secondary antibodies, Alexa 488-, Alexa 594-conjugated anti-mouse, anti-rabbit, and anti-rat IgG antibodies (Invitrogen) were used. Fluorescent images were taken by a confocal laser microscopy system (LSM 510META-V3.2; Carl Zeiss) built on an inverted microscope (Axiovert 200M; Carl Zeiss) with the 40× objective [Plan-Neofluar 40×/numerical aperture (NA) 1.3, oil; Carl Zeiss] or using a CCD camera-based imaging analysis system (an Olympus BX51 equipped with a DP-70 camera). Visualization of raft-targeted proteins was performed as described previously (Takemoto-Kimura et al., 2007).
Western blot analysis.
For Western blot analysis, cortical neurons were transfected with pSUPER-shNega or pSUPER-shCaMKIα by electroporation using a Nucleofector and plated at a density of 5 × 106 cells in a six-well dish. At 2 DIV, the cells were lysed in lysis buffer containing 50 mm Tris-HCl, pH 6.8, 2% SDS, and 10% glycerol. A rabbit anti-CaMKIα antibody (Uezu et al., 2002) was used (a kind gift from Drs. Kohji Fukunaga and Jiro Kasahara, Tohoku University, Sendai, Japan). Chemiluminescence detection was performed using horseradish peroxidase-conjugated anti-rabbit IgG and ECL Plus reagent (GE Healthcare).
Calcium imaging.
Fluorescent calcium imaging was performed essentially as described previously (Furuyashiki et al., 2002; Takemoto-Kimura et al., 2007). Twenty-four hours after plating, cortical neurons on glass-bottom dishes were loaded with Fluo-4 AM (2.5 μm; Dojindo Laboratories) for 30 min at room temperature. After wash, cells were incubated at 37°C in a stage CO2 chamber (Tokai Hit Co., Ltd.) equipped on an LSM 510 META (Carl Zeiss). After baseline recording, a medium containing 20× muscimol (final concentration, 1 μm) was gently bath-applied. Fluorescence changes in the cell bodies of individual cells were analyzed using MetaMorph or ImageJ software, and data are expressed as ΔF/Fo.
In utero electroporation, data acquisition, and quantification of the terminal arborization of callosal axons.
In utero electroporation was performed as described previously (Mizuno et al., 2007). Equal amount of pSUPER-vectors (2 μg/μl) and pCAG-EGFP (2 μg/μl) were mixed together with the dye Fast Green (0.05%; Wako) for injection into the lateral ventricle. The postnatal brains [postnatal day 16 (P16)] were fixed by transcardial perfusion of 4% PFA in 0.1 m phosphate buffer followed by overnight immersive fixation in 4% PFA in PBS and then transferred to 30% sucrose in PBS for 1–2 d at 4°C. Serial coronal brain sections were prepared at 50 μm thickness by a cryostat (HM560; Microm), and every one section out of four was immunostained. Sections were permeabilized in 0.3% Triton X-100 in PBS, and then blocked in 5% normal goat serum, 1% BSA, and 0.3% Triton X-100 in PBS followed by fluorescent immunostaining of EGFP. Sections were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) (Invitrogen). Quantitative analyses were performed and compared using the utmost posterior section of the stained sets that included the corpus callosum.
Confocal images were taken (LSM 510META-V3.2; Carl Zeiss) with a 10× objective (Plan-Neofluar 10×/NA 0.3; air; Carl Zeiss) with 10 μm optical sectioning. Z projection images taken at 512 × 512 pixels were acquired by average projection mode and background was subtracted, and the intensity was normalized by maximal intensity in the white matter. For one-dimensional fluorescence intensity profile analysis in Figure 7C, a rectangular zone (nominal width set at 100 pixels) was drawn along the vertical axis from the pial surface to the white matter, and the average pixel intensity projected onto the vertical axis was calculated. To quantify the impairment of the cortical wiring in Figure 7D, average intensity in a rectangle region (100 pixels in width) in the cortex was divided by that in the white matter. Three to five pups were used for quantification. All calculations were performed using MetaMorph software (version 7; Molecular Devices).
Figure 7.

Knockdown of CaMKIα impairs terminal extension of callosal axons in vivo. A, A control coronal section was obtained near the posterior end of the corpus callosum, from a P16 pup electroporated in utero with pSUPER-shNega and pCAG-EGFP on E15.5. The somatodendritic regions of layer II/III neurons were strongly labeled (asterisks) in the somatosensory cortex, from which callosal axons projected toward the contralateral cortical areas at the S1/S2 border region (arrowhead). Scale bar, 1 mm. CC, Corpus callosum; Hi, hippocampus; Th, thalamus. B, Terminal extension of callosal axons into the contralateral cortical layers was severely disrupted in CaMKIα-knockdown neurons (shKIα), although axons were able to reach the white matter (WM) beneath S1/S2 area (arrowhead). Scale bar, 1 mm. C, Axonal extension and terminal branch arborization were strongly impaired in layers II/III in CaMKIα-knockdown neurons, as illustrated by the magnified images of GFP marker showing the total axonal volumes present in the cortical layers (in pseudocolor), or by a one-dimensional fluorescence intensity profile analysis. Scale bar, 200 μm. D, Quantification of the cortical wiring defect caused by an aberrant terminal axon extension in the cortex. Two independent RNAi constructs (shKIα and shKIα #2) gave similar results. **p < 0.01 (one-way ANOVA with Tukey's test comparison with shNega).
Statistical analyses.
Statistical analyses were run separately for axonal and dendritic datasets throughout our study, while using scattered diagrams of paired data of axonal and dendritic lengths (“orthogonal plots”). Statistical analyses were performed using Prism 4.0 (GraphPad Software). Student's t test was used for comparisons of two groups. One- or two-way ANOVA with post hoc Tukey–Kramer or Bonferroni's test was used for factorial analysis among more than three groups. Kolmogorov–Smirnov test was applied to Figure 1, C and D. All data are shown as mean ± SEM, unless otherwise mentioned, and shaded regions in orthogonal plots graphically depict the zone of mean ± 2SEM on both axes to facilitate the evaluation of the phenotypes.
Figure 1.
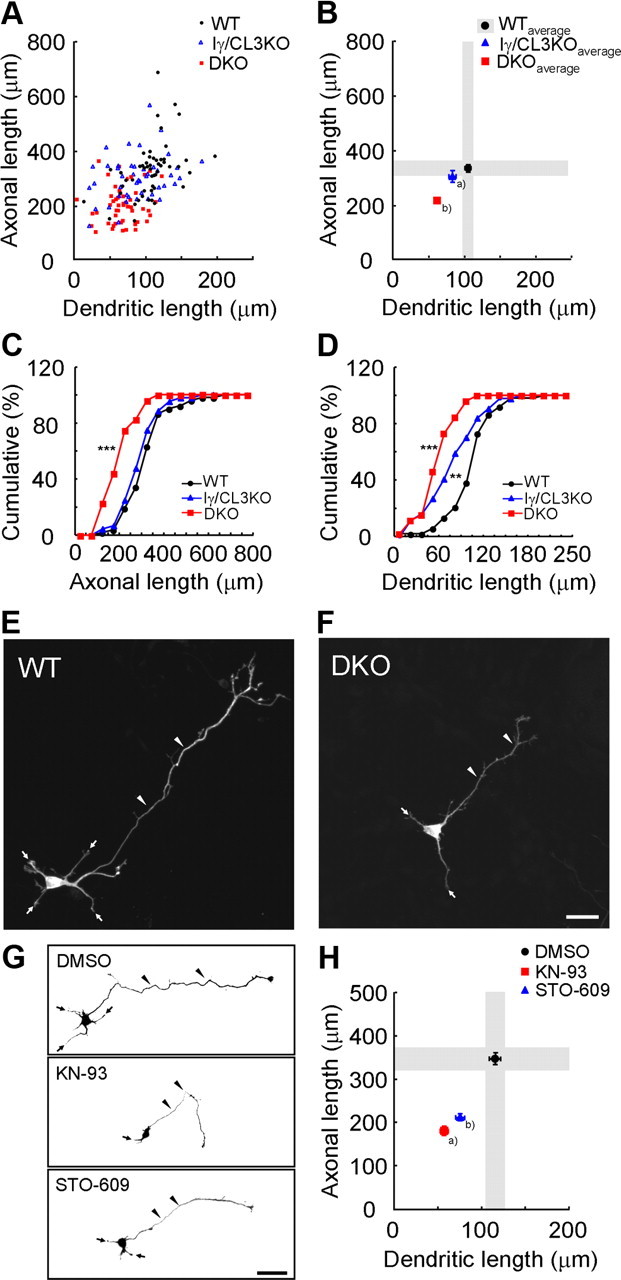
CaMKK-dependent CaMK cascades control cortical axonal and dendritic growth. A, B, A scattered plot (orthogonal plot) of data points (A) and averages (B) for both axonal and dendritic lengths obtained of individual neurons. Black circles, Wild type (WT). Blue triangles, Iγ/CL3 knock-out (Iγ/CL3 KO). Red squares, CaMKKα/β-double knock-out (DKO). Number of neurons: WT, n = 52; Iγ/CL3 KO, n = 44; DKO, n = 52. aDendrite, p < 0.01; bAxon, p < 0.001; dendrite, p < 0.001 (one-way ANOVA with Tukey's test comparison with WT). C, D, Cumulative probability analysis for total axonal length (C) and total dendritic length (D) in neurons from WT, DKO, and Iγ/CL3 KO mice. Number of neurons: WT, n = 52; DKO, n = 52; Iγ/CL3 KO, n = 44. **p < 0.01; ***p < 0.001, Kolmogorov–Smirnov test comparison with WT. E, F, Cortical neurons (2 d in vitro) from CaMKKα/β-DKO mice (F) showed impaired growth of axons (arrowheads) and dendrites (arrows) compared with neurons from WT mice (E). Scale bar, 25 μm. G, Treatment with KN-93, a general CaMK inhibitor, and STO-609, a blocker of CaMKKα/β, the upstream kinases of all CaMKI/IV, from 6 to 48 h after plating impaired both axonal (arrowheads) and dendritic (arrows) growth. Scale bar, 50 μm. H, An orthogonal plot shows a quantitative analysis of axonal and dendritic morphometric parameters from each neuron. Number of neurons: DMSO, n = 48; KN-93, n = 43; STO-609, n = 48. a,bAxon, p < 0.001; dendrite, p < 0.001 (one-way ANOVA with Tukey's test comparison with DMSO).
Results
CaMKK pathway regulates axonal and dendritic growth during early stages of cortical development
We previously reported that a dendritic raft-anchored CaMKIγ/CL3 (Takemoto-Kimura et al., 2003) plays an essential role in dendritic growth downstream of BDNF during the morphological maturation of cortical neurons (Takemoto-Kimura et al., 2007). As four distinct CaMKI isoforms (α, β, γ/CL3, and δ) and CaMKIV are activated by upstream CaMKKs α and β, we sought to test how potently neuritogenesis was disturbed in cultured cortical neurons generated from CaMKKα/β-DKO mice. To specifically identify the genotype contribution to either axonal or dendritic growth, we orthogonally plotted the dendritic length (i.e., total length of all dendritic processes) and axonal length (i.e., total length of all axonal processes including branches) for each GFP-expressing cortical neuron blindly chosen from multiple fields of view (Fig. 1A,B; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Although cortical neurons from CaMKIγ/CL3 knock-out mice revealed a strikingly dendrite-specific deficit (Fig. 1A–D), we found that both axons and dendrites were significantly shortened in cortical neurons from DKO mice, compared with neurons from WT mice (Fig. 1A–F). Exposure to KN-93, which blocks all CaMK species (CaMKII, CaMKI, CaMKIV, and CaMKK), also reduced both total axonal and dendritic lengths (Fig. 1G,H). Specific blockade of the CaMKKs, using STO-609, a selective inhibitor for CaMKKs (Tokumitsu et al., 2002), resulted in a quantitatively similar impairment (Fig. 1G,H). Together, these genetic and pharmacological experiments clearly demonstrated that CaMKK-mediated CaMK cascades played critical roles both in axonogenesis and dendritogenesis of immature cortical neurons, consistent with a previous work on other cell types (Wayman et al., 2004). Furthermore, our data pointed to the presence of a selective CaMKK–CaMK cascade that strongly supported cortical axonal growth in a manner that was distinct from the dendritic contribution of CaMKIγ.
Suppression of CaMKIα expression specifically impairs axonal but not dendritic growth
To identify which of CaMKI or CaMKIV isoform(s) was involved in regulation of the axonal growth, we designed several short hairpin-type pSUPER vectors that were targeted to specific isoforms of the CaMKI/IV subfamily members. In this RNAi experiment, we also coexpressed a PGK promoter-driven mRFP1 as a morphological tracer. In a control experiment, polarized cortical neurons grown for 48 h typically grew 5∼6 dendrites and a single axon. Knockdown using an shCaMKIα vector was prominent enough such that even an overexpressed GFP-CaMKIα became barely detectable 48 h after transfection, whereas the control mRFP1 expression level remained unchanged (Fig. 2A). Strong suppression of endogenous CaMKIα expression in shCaMKIα-transfected neurons was also demonstrated by Western blot analysis using an anti-CaMKIα antibody (Fig. 2B). A lack of cross-knockdown effects across α-, γ-CaMKI isoforms and CaMKIV was verified (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 2.

Knockdown of CaMKIα specifically impairs axonal but not dendritic growth. A, Efficient downregulation of exogenous GFP-CaMKIα was achieved by a CaMKIα-targeted shRNA vector (shCaMKIα), but not by a control vector (shNega), in rat cortical neurons. The mRFP1 expression, which was driven by a dual promoter in a pSUPER+mRFP1 vector, remained unchanged. Scale bar, 50 μm. B, Knockdown of endogenous CaMKIα was evaluated by Western blot analysis using an anti-CaMKIα antibody. Rat cortical neurons were transfected with pSUPER-shNega or pSUPER-shCaMKIα by electroporation, and the cells were lysed at 2 DIV. shCaMKIα suppressed endogenous CaMKIα, whereas the control mRFP1 expression level remained unchanged. C, D, shCaMKIα-expressing rat cortical neurons (shCaMKIα) (D) showed impaired axonal growth (arrowheads), whereas the dendritic morphology was spared (arrows) compared with neurons from shNega-expressing rat cortical neurons (shNega) (C). Scale bar, 25 μm. E, An orthogonal plot of averaged data; n = 15 for all groups. aAxon, p < 0.001. bDendrite, p < 0.001 (one-way ANOVA with Tukey's test comparison with shNega). Scale bar, 25 μm. F, Introduction of shCaMKIα-resistant wild-type GFP-CaMKIα (WTres) successfully rescued the axonal defect elicited by shCaMKIα. In contrast, an shCaMKIα-resistant kinase-inactive GFP-CaMKIα (a K49Ares point mutant) was unable to rescue the shCaMKIα phenotype. n = 15 for all groups. a,bAxon, p < 0.001 (one-way ANOVA with Tukey's test comparison with shNega + mock).
Under these conditions, shCaMKIα-treated neurons showed unchanged dendritic growth but had a markedly shorter axon (Fig. 2C,D). Under the same conditions, in contrast, CaMKIγ/CL3 knockdown specifically blocked dendritic, but not axonal, outgrowth (Fig. 2E) (Takemoto-Kimura et al., 2007), whereas CaMKIV knockdown had no effect (Fig. 2E). The striking specificity in the axonal phenotype of CaMKIα was replicated even when axonal growth was measured under conditions in which shCaMKIα-transfected neurons were kept in suspension culture for an extended period (48 h) before plating, to ensure a maximized knockdown efficiency (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The impairment in axonal growth observed in CaMKIα-diminished neurons was rescued by expression of an shCaMKIα-resistant WT-CaMKIα (WTres), but not by that of an shCaMKIα-resistant kinase-inactive CaMKIα (K49Ares), demonstrating the requirement of the kinase activity of CaMKIα (Fig. 2F). Together, our results strongly implicated the CaMKK–CaMKIα cascade as a critical player in the control of cortical axonal growth.
Two separate CaMKK–CaMKI cascades control cortical axonal and dendritic growth
In keeping with this robust selectivity in the knockdown experiments, forced expression of either one of the four CaMKI isoforms revealed that total axonal length was stimulated only by an increase in CaMKIα, whereas dendrite growth was promoted only by CaMKIγ/CL3 expression (Fig. 3A,B). No change in primary axon number was detected in CaMKIα-overexpressing neurons, suggesting that CaMKIα did not act on axon specification per se (data not shown). Most critically, expression of a constitutively active CaMKIα (CaMKIαCA), was sufficient to rescue the axonal deficit, but without altering dendritic atrophy, in cortical neurons from DKO mice (Fig. 3C) or in WT neurons treated with STO-609 (Fig. 3D). However, forced expression of CaMKIαWT, which enzymatically remains inactive in the absence of CaMKK activity, had no effect in either of these backgrounds (Fig. 3C,D). In a parallel experiment, both axonal and dendritic defects in WT neurons treated with STO-609 were rescued by transfection of a STO-609-resistant CaMKKβ V269F mutant (Tokumitsu et al., 2003) (Fig. 3E).
Figure 3.
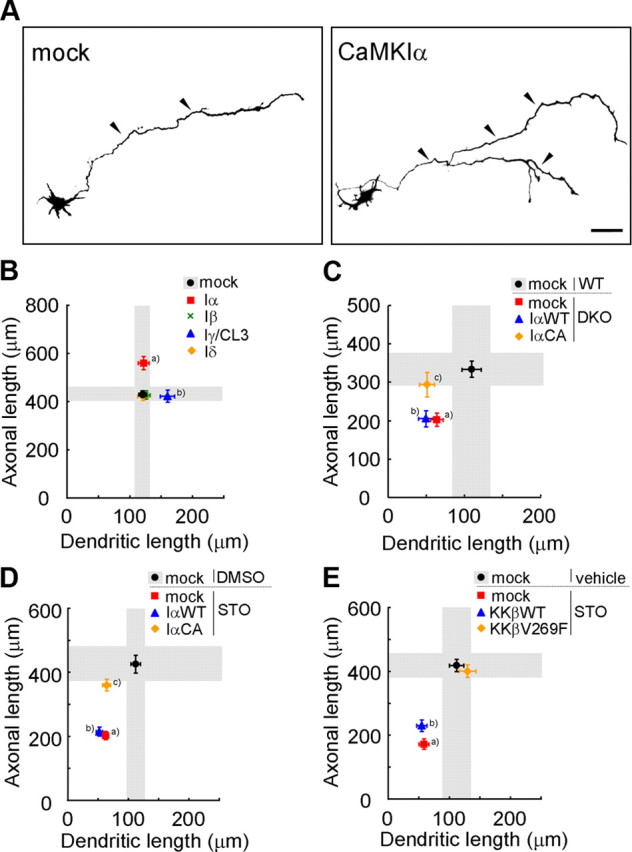
A specific role for a CaMKK–CaMKIα cascade in promoting axonal growth in cortical neurons. A, Representative images of rat cortical neurons transfected with GFP-CaMKIα. Scale bar, 50 μm. B, Overexpression of CaMKIα and CaMKIγ facilitated axonal and dendritic growth, respectively. n = 15 for all groups. aAxon, p < 0.001; bdendrite, p < 0.05 (one-way ANOVA with Tukey's test comparison with mock). C, The axonal growth defect in DKO mice was selectively rescued by coexpression of a constitutively active CaMKIα (CaMKIαCA), but not by a wild-type CaMKIα (CaMKIαWT); the dendritic growth defect was left unaltered; n = 15 for all groups. aAxon, p < 0.01; dendrite, p < 0.05; baxon, p < 0.01; dendrite, p < 0.001; cdendrite, p < 0.01 (one-way ANOVA with Tukey's test comparison with WT plus mock). D, Only the axonal growth defects caused by STO-609 treatment were rescued by expression of a constitutively active CaMKIα (CaMKIαCA), but not of a wild-type CaMKIα (CaMKIαWT). Dendritic growth defects remained unchanged; n = 15 for all groups. a,bAxon, p < 0.001; dendrite, p < 0.001; cDendrite, p < 0.001 (one-way ANOVA with Tukey's test comparison with DMSO plus mock). E, Both axonal and dendritic growth defects caused by STO-609 treatment were rescued by introducing an STO-609-resistant CaMKKβ mutant (V269F), but not a CaMKKβ-WT. n = 15 for all groups. a,bAxon, p < 0.001; dendrite, p < 0.01 (one-way ANOVA with Tukey's test comparison with vehicle plus mock).
Together, these data strongly implicated the CaMKK–CaMKIα and CaMKK–CaMKIγ cascades as parallel pathways acting independently in the promotion of axonal and dendrite growth, respectively, in cultured cortical neurons.
Both localization and kinase specificity of CaMKIα play important roles in CaMKIα-dependent axonal growth
Our data, so far, suggested that the axonogenic action of CaMKIα manifested in a manner that was completely orthogonal and independent to the dendritogenic effect mediated by CaMKIγ/CL3, despite a high degree of structural identity (71% amino acid identity in the catalytic domain sequences). What then discriminated the distinct function of these two kinases?
To identify the molecular determinants involved in axonogenic and dendritogenic selectivity of the CaMKK–CaMKI cascades, we generated CaMKIα/γ chimeras such that each kinase domain was paired with either cytosolic or Golgi/raft localization signals in the C terminus (Fig. 4A) (Takemoto-Kimura et al., 2007). We then tested their potencies to rescue the defect caused by knockdown of endogenous CaMKIα. As expected, forced expression of an shCaMKIα-resistant WT-CaMKIα (Iαres) rescued the axonal impairment in CaMKIα knockdown neurons (Fig. 4B). The WT-CaMKIγ/CL3 (Iγ), however, promoted dendritic growth without showing any effect on axonal deficit. CaMKIα is believed to be freely diffusible. However, the C-terminal region of CaMKIγ/CL3 is lipidified by prenylation and palmitoylation, targeting it preferentially into lipid rafts, which are highly abundant in dendrites and in Golgi (Takemoto-Kimura et al., 2007) (Fig. 4A; supplemental Fig. 4, available at www.jneurosci.org as supplemental material). A dendritic raft-targeted mutant of CaMKIα, GFP-CaMKIα+Cterm (Iαraft-res), was unable to rescue the axonal impairment in CaMKIα knockdown neurons (Fig. 4B). A cytoplasmic, raft-excluded mutant of CaMKIγ/CL3, namely CaMKIγ/ CL3ΔCterm (Iγcyto), had no ability, either (Fig. 4B), contrary to our expectations. Thus, surprisingly, CaMKI-mediated selectivity of neurite growth might not be simply determined by the localization of a CaMKIα or CaMKIγ in or out of the membrane rafts.
Figure 4.

Functional segregation of CaMKK–CaMKIα and CaMKK–CaMKIγ cascades. A, The domain structures and subcellular localizations of CaMKIα (Iα), CaMKIγ/CL3 (Iγ), and their chimeras. GFP-CaMKIα (Iα), CaMKIγ/CL3 (Iγ), and their chimeras distribution detected by anti-GFP immunostaining showed colocalization with a Golgi marker, GM130. GFP-Iγ and Iαraft-res signals were also enriched within Golgi (arrowheads). Single representative confocal sections are shown for Golgi localization. GFP-Iγ and -Iαraft-res fluorescence was retained after detergent treatment in a punctate manner in 2 DIV cortical neurons along the dendrites, demonstrating a sizable portion of detergent-resistant GFP-Iγ and Iαraft-res in the dendritic rafts. Line scans of pixel fluorescence, performed within a chosen field of a 15 μm dendritic segment. Scale bars: right, 50 μm; middle, 5 μm; left, 100 μm. B, Neither Iγ, Iαraft-res, nor Iγcyto were able to rescue the axonal phenotype caused by knockdown of CaMKIα; n = 15 for all groups. a,b,cAxon, p < 0.001; daxon, p < 0.001, dendrite, p < 0.01 (one-way ANOVA with Tukey's test comparison with shNega plus mock). C, Neither Iα, Iαraft, nor Iγcyto-res were able to rescue the dendritic phenotype caused by knockdown of CaMKIγ/CL3; n = 15 for all groups. a,c,dDendrite, p < 0.001; baxon, p < 0.001, dendrite, p < 0.001 (one-way ANOVA with Tukey's test comparison with shNega plus mock). D, Overexpression of CaMKIα, specifically increased axon length in cortical neurons; n = 15 for all groups; n = 15 for all groups. aAxon, p < 0.05; bdendrite, p < 0.001 (one-way ANOVA with Tukey's test comparison with mock).
To further confirm this, the chimeras were expressed in the background of CaMKIγ/CL3-knockdown neurons. Expression of an RNAi-resistant WT-CaMKIγ/CL3 (Iγres) rescued the dendritic impairment, whereas WT-CaMKIα (Iα) promoted axonal growth without an effect on dendrite impairment (Fig. 4C). Again, however, neither a freely diffusible CaMKIγ/CL3ΔCterm (Iγcyto-res), nor a raft-targeted CaMKIα, CaMKIα+Cterm (Iαraft), had any effect (Fig. 4C). Furthermore, forced expression of CaMKIα, CaMKIγ, and CaMKIα/γ chimeras in a naive background revealed that total axonal length was stimulated only by an increase in CaMKIα (Fig. 4D). Together, these data provide strong functional evidence in support of the notion that CaMKK–CaMKIα and CaMKK–CaMKIγ are not duplicative mechanisms with simply altered targeting of downstream kinases, but are genuinely segregated cascades that are divergent at the level of kinase substrate specificity.
GABA is one of the physiological ligand acting upstream of CaMKIα to promote axonal growth during early stages of cortical development
The biological significance of this specificity could be demonstrated if the physiological signal triggering the axonogenic effect of CaMKIα was identified. To this end, we searched for a potential extracellular ligand that induced intracellular calcium elevation and potently stimulated axonal growth. We found that muscimol, a GABAA receptor agonist with a known excitatory action during perinatal development (Owens et al., 1996; Represa and Ben-Ari, 2005), specifically promoted elongation of axons, but not of dendrites, in cultured cortical neurons (Fig. 5A,B). Under the same conditions, we confirmed that BDNF had a complementary growth effect mostly selective for dendrites (Takemoto- Kimura et al., 2007).
Figure 5.

Muscimol, a GABAA receptor agonist, specifically stimulates elongation of axons in cultured cortical neurons. A, B, Representative images (A) and ensemble data (B) of immature cortical neurons treated with either muscimol (a GABAA receptor agonist) or BDNF. Muscimol significantly promoted axonal growth (arrowheads). In contrast, BDNF had no effect on axons but mainly affected dendrites. Scale bar, 50 μm. n = 15 for all groups. aAxon, p < 0.001; bdendrite, p < 0.01 (one-way ANOVA with Tukey's test comparison with vehicle).
To test the extent of requirement for GABA, we added bicuculline, a GABAA receptor antagonist, in the medium and found that axonal growth was rather selectively impaired (Fig. 6A). Furthermore, we confirmed that muscimol application triggered a strong Ca2+ influx in our cortical neurons (Fig. 6B,C). In keeping with this, forced expression of KCC2, a neuronal K+/Cl− cotransporter that lowers intracellular Cl− concentration, and that is upregulated during development to convert the GABA action from excitation to inhibition (Rivera et al., 1999), impaired both constitutive and muscimol-stimulated axonal growth (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Pharmacological blockade of all CaM kinases using KN-93 (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), or of CaMKK using STO-609 (Fig. 6D), completely blocked the axonogenic muscimol effect. CaMKIα RNAi (shIα), but not CaMKIγ/CL3 RNAi (shIγ), selectively impaired muscimol-stimulated axonal growth (Fig. 6E), and this effect was rescued by coexpressing an shCaMKIα-resistant CaMKIα WT (IαWTres), but not CaMKIγ/CL3 WT (IγWT) (Fig. 6F). Thus, a CaMKK–CaMKIα cascade may critically mediate GABAA-stimulated axon outgrowth during the early development of a cortical neuron.
Figure 6.

Activation of GABAA receptors promotes axonal growth via the CaMKK–CaMKIα pathway in immature cortical neurons. A, Bicuculline, a GABAA receptor antagonist, blocked axonal growth; n = 15 for all groups. aAxon, p < 0.001 (Student's t test comparison with vehicle). B, Embryonic cortical neurons (1 DIV) were loaded with a calcium indicator, Fluo-4 AM, and calcium responses were measured by time-lapse imaging. A green fluorescence image was overlaid on a differential interference contrast image. The colored boxes indicate the location of cells shown in C. Scale bar, 50 μm. C, Representative calcium responses in individual cells after muscimol administration. Three different types of calcium responses were revealed (green, blue, and red). An averaged response from 10 cells in a microscopic field is revealed in black. D, Both basal and muscimol-stimulated axonal growths were suppressed with STO-609, a specific blocker of CaMKKα/β; n = 15 for all groups. Axon: two-way ANOVA, muscimol effect, F(1,56) = 14.38, p = 0.0004; drug effect, F(1,56) = 225.63, p < 0.0001; muscimol × drug, F(1,56) = 15.79, p = 0.0002. Dendrite: two-way ANOVA, muscimol effect, F(1,56) = 0.39, p = 0.5336; drug effect, F(1,56) = 105.76, p < 0.0001; muscimol × drug, F(1,56) = 0.13, p = 0.7199. a,bAxon, p < 0.001 (comparison with vehicle plus DMSO); caxon, p < 0.001 (comparison with muscimol plus DMSO); n.s. (comparison with vehicle plus STO). E, CaMKIα knockdown quantitatively inhibited axonal growth induced by muscimol treatment, to an extent similar to that obtained with STO-609; n = 15 for all groups. Axon: two-way ANOVA, muscimol effect, F(1,84) = 66.44, p < 0.0001; RNAi effect, F(2,84) = 168.04, p < 0.0001; muscimol × RNAi, F(2,84) = 19.96, p < 0.0001. Dendrite: two-way ANOVA, muscimol effect, F(1,84) = 0.23, p = 0.6305; RNAi effect, F(2.84) = 61.58, p < 0.0001; muscimol × RNAi, F(2.84) = 0.01, p = 0.9888. a,bAxon, p < 0.001 (comparison with vehicle plus shNega); caxon, p < 0.001 (comparison with muscimol plus shNega); n.s. (comparison with vehicle plus shIα); daxon, p < 0.001 (comparison with vehicle plus shIγ); n.s. (comparison with muscimol plus shNega). F, Introduction of shCaMKIα-resistant wild-type GFP-CaMKIα (WTres) specifically rescued the suppression of muscimol-induced axonal growth triggered by knockdown of CaMKIα; n = 15 for all groups. aAxon, p < 0.001; dendrite, n.s.; baxon, n.s.; dendrite, p < 0.001 (one-way ANOVA with Tukey's test comparison with muscimol plus shIα plus mock); caxon, n.s.; dendrite, p < 0.001 (t test comparison with muscimol plus shIγ plus mock).
Contribution of CaMKIα in fine-tuning axonal pathfinding in vivo
We finally tested the in vivo relevance of these findings by investigating the function of CaMKIα during activity-dependent cortical wiring in vivo. The callosal axons that originate from layer II/III pyramidal neurons of the somatosensory cortex are known to elongate and target themselves to the border between the S1 and S2 areas of the contralateral cortex, where they suddenly turn and grow into the cortical layers and develop their terminal branches mainly at layers II–III and V. Previous reports demonstrated that reduction of neuronal excitability by overexpression of an inwardly rectifying potassium channel, Kir2.1, impaired such layer-specific development of the terminal branches in the visual cortex (Mizuno et al., 2007) and in the somatosensory cortex (Wang et al., 2007). Furthermore, premature elimination of excitatory GABA drive by forced expression of KCC2 or knockdown of NKCC1 in newly born cortical neurons dramatically perturbed the morphological maturation of the dendrites (Cancedda et al., 2007; Wang and Kriegstein, 2008) or of the terminal callosal axon branches (H. Mizuno, T. Hirano, and Y. Tagawa, unpublished data).
If the morphogenetic effect of excitatory GABA required Ca2+ signaling, could the CaMKK–CaMKIα pathway perhaps mediate activity-dependent control of callosal axonal extension? To test this, CaMKIα was knocked down in the somatosensory layer II/III neurons by in utero electroporation at E15.5, and an effect on axonal growth was examined. At P16, control neurons terminated their axons into a restricted region (border of S1/S2 area) in the contralateral cortex and extensively developed their terminal branches into layers II/III and V (Fig. 7A). CaMKIα-knockdown neurons extended interhemispheric axonal projections in the white matter, suggesting the CaMKIα may not be absolutely required for midline crossing and progression of axon fibers (Fig. 7B). However, their terminal axonal extension into the cortical layers was severely diminished, especially in layers II/III (Fig. 7C,D). These results indicate a developmentally critical role of CaMKIα in activity-dependent regulation of cortical connectivity in vivo.
Discussion
Differential control of cortical axonogenesis and dendritogenesis by activation of CaMKIα and CaMKIγ/CL3
In a previous work (Takemoto-Kimura et al., 2007), we showed that a lipid-modified CaM kinase CaMKIγ/CL3 (a membrane-anchored CaMKI isoform) was directed to the dendrites on raft insertion and could potently promote early dendritic development, with little effect on axon outgrowth, in cultured cortical neurons. In striking contrast to CaMKIγ/CL3, we here demonstrate that a cytosolic sister kinase, CaMKIα, has a complementary role: it has little role in dendritogenesis, but is necessary and sufficient to promote axonogenesis in the same preparation. Additionally, our present work established that CaMKIα regulates axonal extension in vivo. Additional rigorous quantitative studies are awaited to establish the potential role of other CaMKK–CaMK signaling pathways in cortical neuritogenesis in general.
How can such specificity of axonal/dendritic growth be regulated by two separate yet structurally resembling kinases lying downstream of the same CaMKKs? The chimeric kinase experiments (Fig. 4) strongly suggested that the diverging kinase substrate specificities and the dissimilarity in subcellular localization (cytosol vs dendritic rafts) might provide a basis for the strikingly differential effect of CaMKIα and CaMKIγ/CL3 during axonal and dendritic development. In support of this functional segregation between the two distinct CaMKK–CaMKI cascades, we identified an extracellular ligand, GABA, which specifically stimulated axonal growth via CaMKIα (this study), whereas BDNF selectively promoted dendritic growth via CaMKIγ (Takemoto-Kimura et al., 2007), during an early developmental stage of cortical neurons.
In principle, BDNF could rather selectively act on dendrites in part because of the strong affinity of the active TrkB receptor to lipid rafts (Suzuki et al., 2004), which are enriched on dendrites. At this point, however, how GABA stimuli could possibly generate an axon-specific effect remains rather unclear, although preliminary Ca2+ imaging experiments indicated that GABAA stimulation might trigger growth cone-localized Ca2+ transients (S. Kamijo, H. Fujii, S. Takemoto-Kimura, and H. Bito, unpublished data). It is noteworthy that many potential in vitro substrates of CaMKIα have previously been associated with axonal or presynaptic functions. These include synapsin I (Nairn and Greengard, 1987), Numb and Numbl (Tokumitsu et al., 2005), microtubule affinity regulating kinase 2 (MARK2/Par-1b) (Uboha et al., 2007), and β-Pak-interacting exchange factor (βPIX) (Saneyoshi et al., 2008). Although some of these known substrates of CaMKI may potentially underlie a part of early axonal growth, additional work is clearly needed to fully elucidate how an axonogenic substrate may be activated via phosphorylation by CaMKIα.
A pivotal role for a GABA-driven CaMKK–CaMKIα cascade in controlling axonal morphogenesis during early development
In this work, we identified a crucial role for GABA in controlling cortical axon outgrowth during early development via a CaMKK–CaMKIα cascade. In immature cortical neurons, what is the mechanism by which GABA can stimulate axonal development in a CaMKI-dependent manner? Recent studies showed that GABAA receptors activation has potent excitatory effects in immature, but not in mature, neurons (Ben-Ari et al., 2007). The excitatory action of GABA was demonstrated to be caused by a high basal Cl− concentration in immature neurons, because of a high amount of the Na+–K+–2Cl− cotransporter (NKCC1) which favors Cl− influx, whereas the K+–Cl− cotransporter (KCC2) primarily responsible for Cl− efflux is still low in expression (Payne et al., 2003). Because of elevated intracellular Cl− concentration in immature neurons, GABAA receptors activation thus induces depolarization (Ben-Ari et al., 2007), thereby likely triggering the opening of voltage-gated Ca2+ channels, which then generates enough Ca2+ influx leading to CaMKK–CaMKIα activation.
During early development, it is now known that GABA controls a variety of biological processes. Our work has only addressed the significance of the CaMKK–CaMKIα cascade in GABA-mediated cortical axonogenesis during the perinatal period. Whether other GABA-regulated processes may also be mediated by CaMKIα clearly remains to be investigated. For instance, the process of cortical migration has also been reported to be regulated by GABA through signal transduction pathways involving Ca2+, both in vitro (Behar et al., 1996, 1998, 2000) and in vivo (Heck et al., 2007). Interestingly, treatment with calmidazolium, an inhibitor of calmodulin, reduces the migration rate in cerebellar granule cells (Kumada and Komuro, 2004). Additional studies are needed to determine whether the CaMKK–CaMKIα cascade may play additional roles in such developmental processes as well.
A CaMKK–CaMKIα pathway may regulate fine-sculpting of cortical wiring
We here established the critical importance of an axonogenic GABA–CaMKK–CaMKIα pathway during early development in vitro. Moreover, this study indicated that CaMKIα regulated activity-dependent extension of terminal cortical axons in vivo. Interestingly, premature elimination of excitatory GABA action by forced expression of KCC2 in newly born cortical neurons dramatically perturbed the morphological maturation of the dendrites (Cancedda et al., 2007) or of the terminal callosal axon branches (H. Mizuno, T. Hirano, and Y. Tagawa, unpublished data). Our present findings thus uncover an unexpected role of the CaMKK–CaMKIα cascade as one key mechanism in GABA-driven activity-dependent regulation of cortical connectivity. More studies are needed to establish whether and how other Ca2+-mobilizing signals (e.g., BDNF) may spatially and temporally interact and perhaps cooperate with such an axonogenic GABA–CaMKK–CaMKIα pathway. Finally, our data lend support to the existence of a perinatal time window of structural refinement, during which spontaneous Ca2+ signaling regulated by trophic factors, guidance signals, and ambient neurotransmitters, such as BDNF or GABA, critically fine-tunes cortical connectivity, perhaps even before the receipt of the earliest sensory cues.
Footnotes
This work was supported in part by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (S.T.-K., Y.T., T.H., H.O., H.B.) and the Ministry of Health, Labour and Welfare of Japan (H.B.), 21st Century Center of Excellence (COE) and Global COE Programmes (H.B.), by a grant from the National Institutes of Health (T.A.C.), and by awards from the Astellas Foundation for Research on Metabolic Disorders (H.B., S.T.-K.), the Naito Foundation (S.T.-K.), the Cell Science Research Foundation, the Takeda Foundation, and the Toray Science Foundation (H.B.). N.A.-I. and M.N. were predoctoral and postdoctoral fellows funded by Japan Society for the Promotion of Science, respectively. We thank all members of the Bito laboratory for support and discussion, T. Soderling and G. Wayman for constructive comments on a previous version of this work, and M. Kano for initially providing access to a validated working stock of muscimol. We are grateful to H. Tokumitsu (Kagawa University) for CaMKK-β WT and V269F cDNA; to J. Nabekura (National Institute for Physiological Sciences, Aichi, Japan) and K. Nakayama (Showa University, Tokyo, Japan) for a KCC2 plasmid; to R. Y. Tsien (Howard Hughes Medical Institute and University of California, San Diego, La Jolla, CA) for mRFP1 and mCherry cDNAs; and to K. Fukunaga and J. Kasahara (Tohoku University) for a rabbit anti-CaMKIα antibody. BDNF was supplied by Dainippon Sumitomo Pharma Co., Ltd. through the courtesy of C. Nakayama and T. Ishiyama. We are also indebted to assistance from K. Saiki, Y. Kondo, and T. Kinbara.
References
- Behar TN, Li YX, Tran HT, Ma W, Dunlap V, Scott C, Barker JL. GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J Neurosci. 1996;16:1808–1818. doi: 10.1523/JNEUROSCI.16-05-01808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Schaffner AE, Scott CA, O'Connell C, Barker JL. Differential response of cortical plate and ventricular zone cells to GABA as a migration stimulus. J Neurosci. 1998;18:6378–6387. doi: 10.1523/JNEUROSCI.18-16-06378.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Schaffner AE, Scott CA, Greene CL, Barker JL. GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb Cortex. 2000;10:899–909. doi: 10.1093/cercor/10.9.899. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Bito H, Takemoto-Kimura S. Ca2+/CREB/CBP-dependent gene regulation: a shared mechanism critical in long-term synaptic plasticity and neuronal survival. Cell Calcium. 2003;34:425–430. doi: 10.1016/s0143-4160(03)00140-4. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Blaeser F, Sanders MJ, Truong N, Ko S, Wu LJ, Wozniak DF, Fanselow MS, Zhuo M, Chatila TA. Long-term memory deficits in pavlovian fear conditioning in Ca2+/calmodulin kinase kinase alpha-deficient mice. Mol Cell Biol. 2006;26:9105–9115. doi: 10.1128/MCB.01452-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298:1959–1964. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- Furuyashiki T, Arakawa Y, Takemoto-Kimura S, Bito H, Narumiya S. Multiple spatiotemporal modes of actin reorganization by NMDA receptors and voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A. 2002;99:14458–14463. doi: 10.1073/pnas.212148999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez TM, Zheng JQ. The molecular basis for calcium-dependent axon pathfinding. Nat Rev Neurosci. 2006;7:115–125. doi: 10.1038/nrn1844. [DOI] [PubMed] [Google Scholar]
- Heck N, Kilb W, Reiprich P, Kubota H, Furukawa T, Fukuda A, Luhmann HJ. GABA-A receptors regulate neocortical neuronal migration in vitro and in vivo. Cereb Cortex. 2007;17:138–148. doi: 10.1093/cercor/bhj135. [DOI] [PubMed] [Google Scholar]
- Hook SS, Means AR. Ca2+/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Tokumitsu H, Inuzuka H, Murata-Hori M, Hosoya H, Kobayashi R. Identification and characterization of novel components of a Ca2+/calmodulin-dependent protein kinase cascade in HeLa cells. FEBS Lett. 2003;550:57–63. doi: 10.1016/s0014-5793(03)00817-2. [DOI] [PubMed] [Google Scholar]
- Kater SB, Mattson MP, Cohan C, Connor J. Calcium regulation of the neuronal growth cone. Trends Neurosci. 1988;11:315–321. doi: 10.1016/0166-2236(88)90094-x. [DOI] [PubMed] [Google Scholar]
- Kumada T, Komuro H. Completion of neuronal migration regulated by loss of Ca2+ transients. Proc Natl Acad Sci U S A. 2004;101:8479–8484. doi: 10.1073/pnas.0401000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Hirano T, Tagawa Y. Evidence for activity-dependent cortical wiring: formation of interhemispheric connections in neonatal mouse visual cortex requires projection neuron activity. J Neurosci. 2007;27:6760–6770. doi: 10.1523/JNEUROSCI.1215-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairn AC, Greengard P. Purification and characterization of Ca2+/calmodulin-dependent protein kinase I from bovine brain. J Biol Chem. 1987;262:7273–7281. [PubMed] [Google Scholar]
- Nonaka M, Doi T, Fujiyoshi Y, Takemoto-Kimura S, Bito H. Essential contribution of the ligand-binding βB/βC loop of PDZ1 and PDZ2 in the regulation of postsynaptic clustering, scaffolding, and localization of postsynaptic density-95. J Neurosci. 2006;26:763–774. doi: 10.1523/JNEUROSCI.2489-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28:278–283. doi: 10.1016/j.tins.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, Soderling TR. Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron. 2008;57:94–107. doi: 10.1016/j.neuron.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt JM, Wayman GA, Nozaki N, Soderling TR. Calcium activation of ERK mediated by calmodulin kinase I. J Biol Chem. 2004;279:24064–24072. doi: 10.1074/jbc.M401501200. [DOI] [PubMed] [Google Scholar]
- Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Stull JT. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem Rev. 2001;101:2341–2352. doi: 10.1021/cr0002386. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto-Kimura S, Terai H, Takamoto M, Ohmae S, Kikumura S, Segi E, Arakawa Y, Furuyashiki T, Narumiya S, Bito H. Molecular cloning and characterization of CLICK-III/CaMKIgamma, a novel membrane-anchored neuronal Ca2+/calmodulin-dependent protein kinase (CaMK) J Biol Chem. 2003;278:18597–18605. doi: 10.1074/jbc.M300578200. [DOI] [PubMed] [Google Scholar]
- Takemoto-Kimura S, Ageta-Ishihara N, Nonaka M, Adachi-Morishima A, Mano T, Okamura M, Fujii H, Fuse T, Hoshino M, Suzuki S, Kojima M, Mishina M, Okuno H, Bito H. Regulation of dendritogenesis via a lipid-raft-associated Ca2+/calmodulin-dependent protein kinase CLICK-III/CaMKIgamma. Neuron. 2007;54:755–770. doi: 10.1016/j.neuron.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Ikeda M, Saji I, Kobayashi R. STO-609, a specific inhibitor of the Ca2+/calmodulin-dependent protein kinase kinase. J Biol Chem. 2002;277:15813–15818. doi: 10.1074/jbc.M201075200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Inuzuka H, Ishikawa Y, Kobayashi R. A single amino acid difference between alpha and beta Ca2+/calmodulin-dependent protein kinase kinase dictates sensitivity to the specific inhibitor, STO-609. J Biol Chem. 2003;278:10908–10913. doi: 10.1074/jbc.M213183200. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Hatano N, Inuzuka H, Sueyoshi Y, Yokokura S, Ichimura T, Nozaki N, Kobayashi R. Phosphorylation of Numb family proteins. Possible involvement of Ca2+/calmodulin-dependent protein kinases. J Biol Chem. 2005;280:35108–35118. doi: 10.1074/jbc.M503912200. [DOI] [PubMed] [Google Scholar]
- Uboha NV, Flajolet M, Nairn AC, Picciotto MR. A calcium- and calmodulin-dependent kinase Iα/microtubule affinity regulating kinase 2 signaling cascade mediates calcium-dependent neurite outgrowth. J Neurosci. 2007;27:4413–4423. doi: 10.1523/JNEUROSCI.0725-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezu A, Fukunaga K, Kasahara J, Miyamoto E. Activation of Ca2+/calmodulin-dependent protein kinase I in cultured rat hippocampal neurons. J Neurochem. 2002;82:585–593. doi: 10.1046/j.1471-4159.2002.00984.x. [DOI] [PubMed] [Google Scholar]
- Wang CL, Zhang L, Zhou Y, Zhou J, Yang XJ, Duan SM, Xiong ZQ, Ding YQ. Activity-dependent development of callosal projections in the somatosensory cortex. J Neurosci. 2007;27:11334–11342. doi: 10.1523/JNEUROSCI.3380-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci. 2008;28:5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Yokokura H, Terada O, Naito Y, Hidaka H. Isolation and comparison of rat cDNAs encoding Ca2+/calmodulin-dependent protein kinase I isoforms. Biochim Biophys Acta. 1997;1338:8–12. doi: 10.1016/s0167-4838(97)00004-6. [DOI] [PubMed] [Google Scholar]