

Abstract
Tissue factor/coagulation factor VII (fVII) complex formation on the surface of cancer cells plays important roles in cancer biology such as cell migration and invasion, angiogenesis, and anti-apoptotic effects. We recently found that various cancer cells ectopically synthesize fVII, resulting in activation of cell motility and invasion. Here, we characterized mechanisms of hepatic and ectopic fVII (FVII) gene expression to identify molecular targets enabling selective inhibition of the ectopic expression. Unlike hepatic expression, HNF-4 binding to the promoter is not required for ectopic FVII expression, although Sp1 binding is essential. Furthermore, we found novel nuclear targets of basal hepatocytic and ectopic FVII expression. Notably, histone acetyltransferases p300 and CBP are exclusively recruited to the promoter region of the FVII gene specifically in breast cancer cells. We further show that curcumin, a dietary compound, can selectively inhibit ectopic fVII expression by targeting p300/CBP activity. These results suggest a strategy to inhibit ectopic fVII-induced tumor progression without impairment of the physiological hemostatic process.
Keywords: breast cancer, coagulation factor VII, curcumin, ectopic synthesis, histone acetyltransferase
Introduction
Produced in the liver, blood coagulation factor VII (fVII) is a key component of the extrinsic coagulation cascade(1). Tissue factor (TF) is an integral membrane protein widely expressed in normal and cancer cells, and it is the cellular receptor for fVII.(1) Upon formation of the TF/fVII complex, fVII converts to the active form of fVII (fVIIa) and initiates the downstream coagulation cascade. The TF/fVIIa complex forms because of the presence of fVII in exudated blood plasma, and it has been shown to stimulate many aspects of cancer progression, including cell growth,(2) motility,(3, 4) invasion,(3, 5) angiogenesis,(6) and anti-apoptotic effects(7, 8) via activation of the coagulation cascade and/or protease-activated receptors. Thus the TF-fVIIa pathway is an attractive target for cancer therapy, but concerns remain that blockade of this pathway may also impair normal hemostasis and therefore cause bleeding.(2)
We recently reported that various nonhepatic cancer cells ectopically express the FVII gene and that inhibition of the TF/fVII complex on the cell surface reduces cell motility and invasion.(9) We further showed that the ectopic expression is prominent in breast cancer cell lines. (9) Regarding molecular mechanisms, previous in vitro analyses have revealed that the transcription factors hepatocyte nuclear factor-4 (HNF-4) and Sp1 play crucial roles in hepatocytic expression of the human FVII gene(10-12); however, the regulators involved in ectopic FVII expression have not been defined. Determination of the molecular mechanisms of ectopic FVII expression may yield a method to block ectopic fVII synthesis selectively in cancer cells without loss of fVII synthesis by the liver. In the present study, we investigated hepatocytic and ectopic fVII expression in breast cancer cells to evaluate the epigenetic mechanisms on expression. We found that in cancer cells, unlike hepatocytes, HNF-4 is dispensable for FVII expression. p300 and CBP are selectively recruited to the active FVII promoter in breast cancer cells, but in hepatocytes, recruited HATs were heterogeneous. Furthermore, we show that HAT recruitment can be targeted for specific inhibition of ectopic fVII synthesis.
Results
HNF-4 is not required for ectopic FVII gene expression
To elucidate the mechanism of ectopic fVII expression in breast cancer cells, we used various cell lines with different FVII gene expression levels. YMB-1 and MDA-MB-453 (hereafter 453) cells were breast cancer cells with high FVII expression levels. T98G, MDA-MB-231 (hereafter 231), and OVSAYO cells are glioblastoma, breast cancer, and ovarian cancer cells, respectively, with very low FVII expression. Hepatoma cell lines, HepG2 and HUH6 clone 5 (hereafter HUH), as well as primary cultures of human hepatocytes (hNHeps) were used as controls for expression of FVII in liver cells.
We first performed nucleotide sequencing and quantitative real-time PCR of the FVII 5’ region in tumor cells. This region was not mutated or amplified in the high fVII-expressing YMB-1 cells (data not shown). We next tested whether HNF-4 is expressed in cancer cells that ectopically express the FVII gene. Western blotting showed that, in contrast to HepG2 (9), YMB-1, 453, OVSAYO, and T98G cells did not express HNF-4 (Fig. 1A). Chromatin immunoprecipitaton (ChIP) analysis revealed that, unlike HepG2 and HUH, the FVII promoter region was not occupied by HNF-4 in YMB-1 cells (Fig. 1B), excluding the possibility that trace HNF-4 bound to the promoter and caused ectopic fVII expression.
Figure 1. Ectopic activation of FVII promoter does not require HNF-4 binding in cancer cells.

(A) Western blot analysis of HNF-4 expression in cancer cells. β-actin was also examined as the protein-loading control. (B) ChIP analysis of HNF-4 binding in cancer cells. The black bar shows a PCR-amplified region within the FVII 5’ promoter. Hatched and open circles are indicative of previously identified Sp1 and HNF-4 binding sites, respectively. A bent arrow is indicative of the position of the major transcription start site identified in a hepatocyte.(10) I designates an input PCR control using DNA prepared from sonicated chromatin without immunoprecipitation. (C) Luciferase constructs used for the deletion analysis of FVII-promoter activity in cancer cells. Hatched and open circles are indicative of Sp1 and HNF-4 binding sites, respectively. An open triangle designates a minimum 6 base pairs (ACTTTG)(10) deletion from the HNF-4 binding site. A bent arrow is indicative of the major transcription start site.(10) Numbering is from the translation start site. (D) The relative promoter activities of deletion constructs at 24 h post-transfection are shown for each cell line as percentages of the −400/+1 construct in HepG2 cells. Cancer cells were co-transfected with a control vector expressing Renilla luciferase to correct variations in transfection efficiency in each experiment. Columns, mean (three independent experiments (hereafter n = 3)); Bars, SD. *P < 0.05.
The HNF-4 binding site is dispensable, and the Sp1 binding site is essential for ectopic FVII gene expression
To determine the regulatory regions responsible for ectopic FVII expression, we next performed luciferase reporter gene assays. A FVII promoter fragment (Fig. 1C, −400/+1) derived from MCAS cells(9), in which FVII is not expressed and site-directed mutants were fused to the pGL4.10 vector (Fig. 1C). Constructs were transfected into various cancer cells with different endogenous FVII expression levels. Luciferase activities in nonhepatic cell extracts were compared with those in a positive control cell line, HepG2.(10, 11) The promoter activity of construct −400/+1 in HepG2 cells was set to 100% (10, 11), and activities of YMB-1 and 453 cells were approximately 90% and 50%, respectively, of HepG2 cells (Fig. 1D). The relative levels of promoter activities were comparable to endogenous fVII mRNA levels in these cells (data not shown), suggesting that the −400/+1 region contains all necessary promoter elements to study ectopic fVII transcription. Promoter activities in very low fVII-expressing cells were less than 5% of the activity in HepG2 cells (Fig. 1D). Truncated reporter construct −400/-212 or −400/-111, which lacked Sp1 and HNF-4 binding sites, showed reduced promoter activities in HepG2, YMB-1, and 453 cells (Fig. 1D), indicating contributions of the deleted regions to fVII transcriptional activation. Experiments with constructs −111/-83 or −114/+1Δ, which lacked or were mutated in the HNF-4 site, revealed that luciferase activities were decreased in HepG2 and HUH cells (75% and 87% decreases, respectively) (Fig. 1D) compared with the −114/+1 construct, confirming that HNF-4 binding is critical for hepatocytic fVII expression. In contrast, experiments with these constructs revealed that absence of the HNF-4 binding site slightly (10% decrease) and moderately (35% decrease) impair promoter activities compared with the −114/+1 construct in YMB-1 and 453 cells, respectvely (Fig. 1D), suggesting that the HNF-4 binding site was largely dispensable for ectopic FVII expression and that the Sp1 site was sufficient to induce ectopic FVII activation. This conclusion was also supported by data showing that deletion of the Sp1 site in the −84/+1 construct significantly diminished promoter activities in the YMB-1 and 453 cells (Fig. 1C, D).
Sp1, Egr-1, USF, and CREB bind to the active FVII gene promoter chromatin
Reporter gene analysis revealed that transcription factors other than Sp1 may be involved in ectopic FVII gene regulation. Thus, we next investigated additional transcription factors associated with structurally active FVII promoter. We used TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCHJ.html), which is based on the TRANSFAC database, to predict novel transcription factor binding sites at a threshold score of 80 and found some within the 5’-region investigated by the reporter assay. USF, known to share binding sites with c-Myc, (13) was predicted to bind regions (Fig. 2A). Additional Sp1 and HNF-4 binding sites were also predicted within these regions (Fig. 2A). Egr-1 overlaps with the GC-rich elements of Sp1 binding sites (14) found in the FVII promoter region. CREB, which is activated in response to increased cyclic AMP concentrations and binds to CRE elements, (15) was also predicted to bind (Fig. 2A).
Figure 2. Sp1, Egr-1, USF-1, and CREB associate with the FVII promoter chromatin.

(A) The putative binding sites for transcription factors harbored in the proximal FVII promoter. Circles and a bent arrow are previously identified Sp1 and HNF-4 binding sites and transcription start site, respectively. (B) Western blot analysis of transcription factors that were suspected to regulate FVII gene expression in cancer cells. β-actin level was also examined as the protein-loading control. (C) ChIP analysis of transcription factors. ChIP was performed for the FVII promoter in cancer cells using normal rabbit IgG or polyclonal antibody of each transcription factor. PCR primers used were as described in Fig. 1. Data were also quantitatively estimated by qPCR. Fold enrichment of immunoprecipitated template DNA for each transcription factor against background (templates precipitated with IgG) is shown for each cell line. Columns, mean; Bars, SD (n = 3).
Western blotting analysis showed that the cellular content of these transcription factors was comparable in the cell lines tested (Fig 2B). ChIP analysis revealed that Sp1, Egr-1, and CREB bound to the FVII promoter in HepG2 and YMB-1 cells, whereas these factors were found associated only at low levels with the FVII promoter in OVSAYO and T98G cells (Fig 2C). We did not detect significant c-Myc binding to the FVII promoter, although the positive control experiment showed that c-Myc binds to the VEGF promoter region in OVSAYO cells (Fig. S1). A similar binding pattern of these transcription factors was observed in hnHeps (Fig. 2C). siRNA transfection followed by quantitative RT-PCR and immunoblotting analyses revealed that Egr-1 and USF-1 could downregulate and CREB could upregulate ectopic and hepatocytic FVII expressions (Fig. S2).
We further tested expression of estrogen receptor α (ERα) and progesterone receptor (PR) in breast cancer cells because these receptors activate some gene promoters. (16, 17) Western blotting analysis showed that these receptors were not detected in the tested cells (Fig. S1). HIF-2α (HIF2), which associates with ectopic FVII induction during hypoxia, (9) was not expressed in YMB-1 cells under normoxia (Fig. S1).
Recruitment of heterogeneous histone acetyltransferases to the acetylated FVII promoter chromatin
Our data indicate that transcription factors described above are shared between hepatic and breast cancer cells and, therefore, cannot be targeted for selective inhibition of ectopic FVII expression. Histone acetylation within a gene promoter is an important step for activating transcription (18) by providing open chromatin structure accessible for transcription factors. We next tested the acetylation status of histone H4 in the FVII gene promoter region. ChIP analysis using an anti-histone H4 antibody revealed that histone H4 was highly acetylated in HepG2, YMB-1, and 453 cells but was poorly acetylated in low fVII-expressing OVSAYO and T98G cells (Fig 3A), consistent with the binding pattern of transcription factors (Fig. 2C).
Figure 3. Histone acetylation and histone acetyltransferases recruited to the FVII promoter in hepatocytes and breast cancer cells.
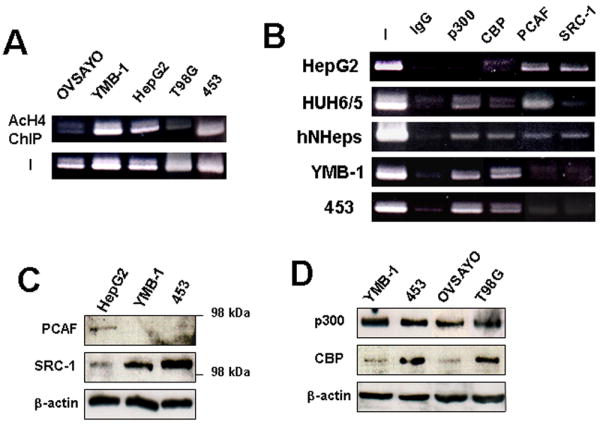
(A) ChIP analysis of histone H4 acetylation in the FVII 5’ region in cancer cells. (B) ChIP analysis of HATs associated with the FVII promoter in cancer cells. (C) Western blotting analysis of PCAF and SRC-1 expression in cancer cells. (D) Western blotting analysis of p300 and CBP expression in FVII-expressing and nonexpressing cancer cells.
HNF-4 directly interacts with the coactivator HATs, p300/CBP (19, 20), and/or SRC-1 (19, 20), resulting in the formation of open chromatin structure with acetylated histones to make the promoter region accessible to other transactivators. In addition, another HAT, p300/CBP-associated factor (PCAF) was also reported to be involved in the activation of HNF-4 responsive genes (21); however, the manner in which HATs associate with a specific gene promoter is poorly understood. Thus we next investigated whether p300, CBP, SRC-1, and PCAF associated with hepatic or ectopically activated FVII promoters. ChIP analysis revealed that PCAF and SRC-1 associate with the FVII promoter in HepG2 cells, although p300 and CBP did not bind (Fig. 3B). On the other hand, p300 and PCAF were recruited to the promoter region in HUH cells (Fig. 4B). All the HATs tested were recruited to the FVII promoter of hNHeps. These results demonstrate that the FVII promoter is differentially targeted by multiple HAT activities depending on the cell type. In the case of YMB-1 and 453 cells, only p300 and CBP associated with the FVII promoter region (Fig. 3B). Western blotting indicated that breast cancer cells do not express PCAF (Fig. 4C). On the other hand, SRC-1 was expressed, but it did not associate with the FVII promoter in breast cancer cells (Fig. 3C). Furthermore, ectopic FVII expression was not the result of overexpression of p300 and CBP because western blotting showed that the cellular content of these HATs in cancer cells did not correlate with FVII expression level (Fig. 3D).
Figure 4. Selective inhibition of ectopic FVII gene expression by curcumin.
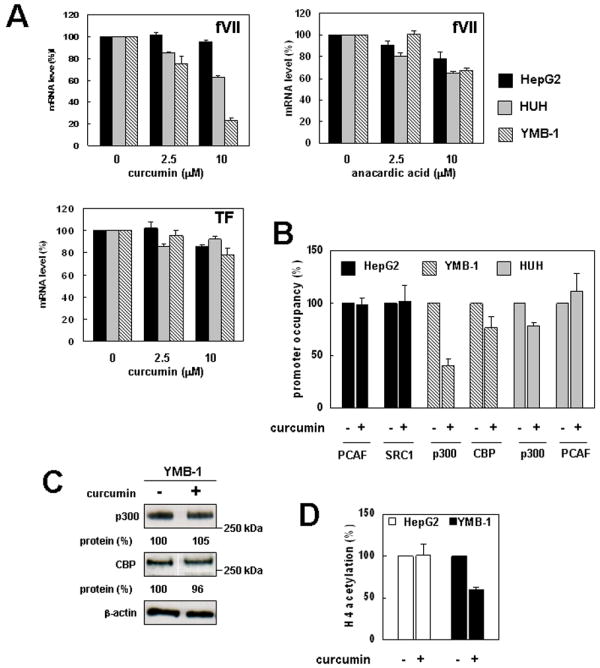
(A) Effect of HAT inhibitors on fVII mRNA expression level in cancer cells. Cancer cells were cultured with or without HAT inhibitors (curcumin or anacardic acid) for 24 h, then mRNA levels (fVII and TF) were analyzed by real-time PCR. fVII mRNA expression was normalized to the 18S ribosomal RNA level. (B) Effect of curcumin on FVII promoter occupancy by HATs in cancer cells. Quantitative ChIP analysis was performed as previously described. Relative level of occupancy is shown for each cell line as percentages of the curcumin (-) experiment. Columns, mean; Bars, SD (n = 3). (C) Western blot analysis of p300 and CBP in cell lysate of YMB-1 cells cultured with or without curcumin. Relative band intensities were densitometrically determined using ImageJ software (http://rsb.info.nih.gov/ij/). (D) Quantitative ChIP analysis of acetylation level of histone H4 in the FVII promoter in cancer cells cultured in the presence or absence of curcumin. Relative level of H4 acetylation is shown for each cell line as percentages of the curcumin (-) experiment. Columns, mean; Bars, SD (n = 3).
Selective inhibition of ectopic fVII synthesis by targeting p300/CBP activity
Given that the ectopically activated FVII promoter was devoid of PCAF and SRC-1, inhibiting p300/CBP activity was predicted to suppress ectopic FVII expression possibly without reducing fVII production in hepatocytes, which recruit alternative factors to the FVII promoter. Among reported natural HAT inhibitors (22, 23), only curcumin exhibits a p300/CBP-selective inhibitory effect (23). Thus, we next tested whether curcumin could inhibit ectopic FVII expression, while not affecting hepatic synthesis. HepG2, HUH, and YMB-1 cells were cultured with 2.5 or 10 μM of curcumin for 24 h, then total RNA was extracted. We confirmed that these concentrations were nontoxic to all cell types used in this experiment by determining the number of viable cells using trypan-blue exclusion (data not shown) and cell proliferation analyses (Fig. S3). Quantitative RT-PCR analysis revealed that, as expected, the fVII mRNA level was unchanged in HepG2 cells (Fig. 4A). The mRNA level in HUH cells was moderately (less than 40%) decreased when cells were treated with 10-μM curcumin. In contrast, the mRNA level was diminished in a dose-dependent manner in YMB-1 cells, and the decrease was more than 70% when cells were treated with 10-μM curcumin (Fig. 4A). On the other hand, experiments using the same concentrations of anacardic acid (22), a natural p300 and PCAF inhibitor, did not selectively inhibit ectopic FVII expression (Fig. 4A). We confirmed that, as previously reported (24, 25), curcumin did not significantly influence basal TF mRNA level in cancer cells (Fig. 4A).
To elucidate the inhibitory mechanism of curcumin, we next examined the effect of curcumin on the promoter occupancy of HATs in cancer cells. ChIP analysis with cells treated with 10-μM curcumin for 24 h revealed that binding of p300 and CBP was decreased in YMB-1 cells, whereas PCAF and SRC-1 occupancies were unchanged in HepG2 cells (Fig. 4B). In addition, the impairment of promoter binding in YMB-1 cells was preferential for p300 rather than CBP. Total protein levels of p300 and CBP were unchanged when cells were treated with curcumin (Fig. 4C). Thus, curcumin reduced promoter occupancy by these factors. As expected, the promoter occupancy by HATs was less compromised in HUH cells by curcumin treatment (Fig. 4B). ChIP analysis revealed that the acetylation level of histone H4 in the FVII promoter was reduced after curcumin treatment in YMB-1 cells, whereas they were unchanged in HepG2 cells (Fig. 4D), suggesting that inhibition of p300/CBP-mediated histone acetylation contributed to repression of ectopic fVII expression. In addition, RT-PCR analysis revealed that treatment with 500 nM trichostatin (a natural histone deacetylase inhibitor) in T98G and OVSAYO cells resulted in an increase in fVII mRNA level (Fig. S3), consistent with the concept that histone deacetylation is an important mechanism for maintaining very low FVII expression in nonhepatic cells.
Furthermore, we examined whether curcumin inhibited ectopic fVII synthesis at the protein level in cancer cells without impairments of fVII production from hepatocytes. HepG2 and HUH cells were cultured with or without 10 μM of curcumin for 24 h. The amount of fVII secreted in cell-culture media was determined by commercially available fVII detection system (Fig. 5A). The fVII secretion from HepG2 cells was unchanged after curcumin treatment (Fig. 5A). fVII production from HUH cells was reduced ca. 20% (79±7.6% relative to vehicle-treated cells), consistent with mRNA levels (Fig. 4A & 5A). Unexpectedly, unlike HepG2 cell, YMB-1 cells did not secrete fVII in cell-culture media (Fig. 5B). However, western blot analysis revealed that fVII is detectable in YMB-1 cell lysate, suggesting that ectopically synthesized fVII functions in an autocrine manner. The expression level was decreased by 10-μM-curcumin treatment (Fig. 5B). Furthermore, based on the fXa generation assay, ectopic fVII synthesis (fVII present at exterior cell membrane) was diminished ca. 60% (43±1.6% relative to vehicle-treated cells) when YMB-1 cells were cultured with 10-μM curcumin for 24 h (Fig. 5C).
Figure 5. Curcumin inhibits ectopic fVII production at the protein level.
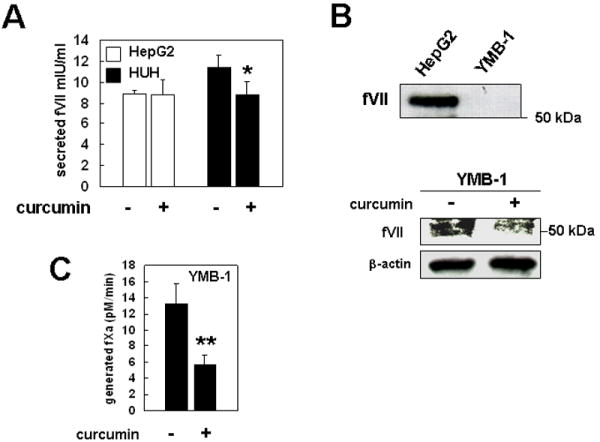
(A) fVII secreted in cell-culture supernatant from hepatic cancer cells cultured with or without curcumin was measured by a chromogenic activity assay based on fXa generation activity. Columns, mean; Bars, SD (n = 3). (B) Unlike hepatic cell, ectopically produced fVII is not secreted in cell-culture media (upper). Conditioned media for each cell-type (2 × 106 cells) were prepared for immunoblotting. Detection of ectopically expressed fVII in whole cell lysate of YMB-1 cells and its inhibition by curcumin (lower). (C) A procoagulant activity on YMB-1 cells cultured in the presence or absence of curcumin was measured by fXa generation assay. Columns, mean; Bars, SD (n = 3). **P < 0.005 different from secreted fVII level from HUH cells (*).
Ectopic expression of fVII in breast cancer tissues
In addition to ectopic synthesis by tumor cells, fVII may accumulate in the tumor stroma because of extravasation from hyperpermeable vessels (26) or synthesis by macrophages (27, 28). fVII transcripts in breast cancer tissues were determined by quantitative RT-PCR analysis using total RNA extracted from 30 Formalin-fixed, paraffin-embedded (FFPE) primary breast cancer specimens. Forty-seven percent (14 of 30 samples) ectopically expressed fVII mRNA (Fig. 6A). Expression levels in breast cancer tissues were comparable or even higher than those of normal liver. The normal breast tissue examined contained a barely detectable amount of fVII mRNA. Immunohistochemical analyses confirmed fVII protein synthesis in breast cancer tissues, specifically in the tumors cells but not in adjacent noncancerous areas of the mammary glands (Fig. 6B). These data demonstrate that ectopic fVII expression is common in breast cancer tissues.
Figure 6. Detection of ectopic fVII expression from surgical specimens of primary breast cancer tissues.
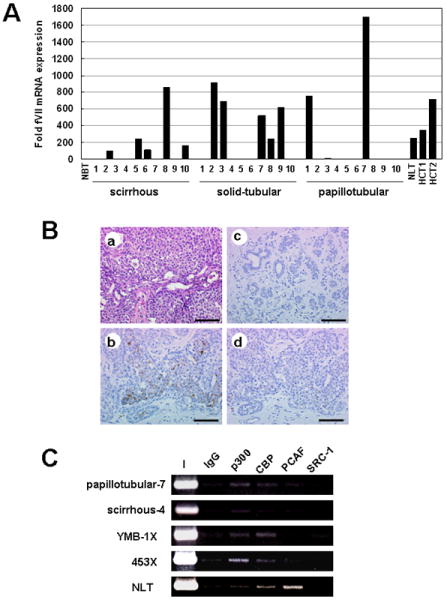
(A) Detection of fVII mRNA from FFPE breast cancer tissues by qRT-PCR analysis. Data were normalized as previously described. NBT, NLT, and HCT are indicative of normal breast, normal liver tissues, and the non-neoplastic part of FFPE hepatectomy-specimens, respectively. (B) Immunohistochemistry of fVII. From a representative case (corresponds to case no. 2 of solid-tubular carcinomas), hematoxylin-eosin staining (a) and immunostainings with anti-fVII were presented. Cancer cells stained with anti-fVII (b), but noncancerous mammary glands on the same section showed no signal (c). To demonstrate specificity of the antibody, the antibody was preincubated with fVII, and these controls showed a marked reduction in signal on cancer cells (d). Scale bars, 100 μm. (C) ChIP analysis of HATs binding to FVII promoter in tissue samples. X is indicative of xenograft sample.
HATs associated with FVII promoter in normal hepatic and breast cancer tissues
To examine the binding pattern of HATs in vivo, we last performed ChIP using surgically removed tissue samples. ChIP analysis revealed that p300 and CBP bind to the FVII promoter in fVII expressing but not in nonexpressing breast cancer tissues (Fig. 6C). Experiments using mouse xenograft samples of YMB-1 and 453 cells showed that binding pattern of HATs observed in these cells in vitro was conserved in vivo (Fig. 6C). In normal hepatic tissue, we found that PCAF preferentially binds to the FVII promoter as in the case of cultured hepatocytes (Fig. 3B and 6C).
Discussion
In the present study, we demonstrate that the FVII promoter is epigenetically activated without HNF-4 binding in breast cancer cells, whereas Sp1 binding is essential for both hepatic and cancer cell expression. The mechanism described here is different from the previously identified pathway of hypoxia-inducible fVII expression in ovarian cancer, which involved HIF2-mediated transcriptional activation. Indeed, we did not find evidence that hypoxia induces ectopic fVII synthesis in a limited number of breast cancer cell lines (Fig. S4), indicating cancer-type-specific mechanisms for the induction of fVII expression.
Our ChIP analyses revealed that, in addition to previously identified transcription factor binding, Egr-1, USF-1, and CREB are components of hepatic and ectopically activated FVII promoter chromatin. We showed that the FVII gene is negatively regulated by these transcription factors. Consequently, we suggest that Sp1, Egr-1, USF, and CREB are recruited to active chromatin and regulate hepatic and ectopic FVII gene expression to maintain proper steady-state levels of transcripts.
HNF-4 activates target-gene promoters in cooperation with p300, CBP, and/or SRC-1 (19). Our experiments showed that several HATs are recruited to the FVII gene promoter (p300, CBP, and SRC-1), with some variations depending on the hepatocyte cell line studied. In contrast, PCAF was found invariably with the FVII promoter in all tested hepatocytes. In ectopically fVII-synthesizing cells, p300 and CBP were predominantly recruited to the FVII promoter region, and an activating transcription factor must replace HNF-4. An Sp1 element is crucial for ectopic FVII expression, and we consider it likely that Sp1 is the crucial transactivator, based on several reports demonstrating promoter activation through interaction between Sp1 and p300 (29-31).
Curcumin is a dietary compound, and its application to cancer therapy based on its antiproliferative- and apoptosis-inducing effects has been widely studied (32). Our data demonstrate that curcumin-sensitive p300/CBP-specific activity selectively suppresses ectopic fVII expression. Our data indicate that p300/CBP stimulate ectopic transcription, at least in part, by histone acetylation. Curcumin selectively reduced ectopic fVII synthesis, but not hepatic FVII expression that is supported by heterogeneous HAT recruitment. This concept may be generally true for breast cancer cells because fVII transcription was also suppressed in 453 cells via p300 inhibition by curcumin (Fig. S5). These data provide new evidence that the repertoire of cell-type-specific HAT recruitment can be exploited as strategy for selective transcriptional repression.
Recent advances suggest that HATs could be an attractive target for treating cancer (33-35). In addition, curcumin inhibits various chromatin components and reduces TF induction via inhibition of transcription factors NF-κB, AP-1, and Egr-1, potentially resulting in anti-tumor effects (24, 25). In fact, recent preclinical trials have revealed that curcumin analogs effectively suppress human breast tumor growth(36, 37). Because we previously demonstrated that ectopic fVII synthesis activates motility and invasion of cancer cells (9) and because TF-fVIIa-PAR2 signaling is essential for breast cancer growth (2) and development (38), we suggest that reduction of constitutive ectopic fVII synthesis through inhibition of p300/CBP activity may add a new approach to the growing list of anti-cancer strategies. Further studies of clinical association of ectopic fVII expression and animal studies with fVII-expressing breast cancer cells will provide answers to this issue.
Materials and methods
Cells, tissue specimens, RNA, and cell culture
Most human cancer cell lines were as previously described.(9) The MDA-MB-231 cell line was obtained from the Japanese Collection of Research Bioresources (Osaka, Japan). hNHeps-Human hepatocytes were obtained from Lonza (Walkersville, MD, USA). Total RNA from normal human tissues was obtained from Ambion (Austin, TX, USA). All cells were cultured as previously described.(9) Total RNAs from formalin-fixed, paraffin-embedded (FFPE) specimens of 30 breast cancer tissues and 2 normal liver tissues were extracted with RecoverAll Nucleic Acid Isolation Kit (Ambion). FFPE tissues from surgically removed specimens were all prepared from patients of the Kanagawa Cancer Center Hospital under written agreements in the study, which was approved by the institutional review board.
Immunohistochemistry
Immunohistochemistry for fVII on FFPE tissue sections using 5 μg/ml of anti-VII polyclonal rabbit antibody(26) was generally done as previously described.(26) To demonstrate the specificity of the staining, a control immunohistochemistry was also done with 5 μg/ml of purified fVII antigen added to the antibody solution.
Quantitative RT-PCR analysis
We determined fVII and TF mRNA levels by RT-PCR as previously described.(9) As an internal standard, the expression level of 18S ribosomal RNA was determined using One Step SYBR RT-PCR Kit (Takara, Tokyo, Japan) and QuantiTect Primer Assay (Qiagen, Valencia, CA, USA). TF expression was determined with the oligomers described in “Supplemental Materials.”
Plasmid construction and luciferase reporter gene assay
A PCR-amplified fragment of the FVII 5’ region and its deletion mutants were inserted into pGL4.10 (Promega, Madison, WI, USA) at the NheI/HindIII site. PCR was performed with genomic DNA of MCAS cells.(9) A mutant construct without the minimal HNF-4 binding site was prepared using the QuikChange Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Luciferase assays were performed using the Dual-luciferase Reporter Assay System (Promega). The pRL-TK vector (Promega) was co-transfected to correct variations in transfection efficiency. Luciferase activities were measured using a Wallac 1420 ARVO MX (PerkinElmer, Turku, Finland).
Western blot analysis
We performed western blotting as previously described(9). Primary antibodies used are described in “Supplemental Materials”.
Chromatin immunoprecipitation analysis
Chromatin immunoprecipitation (ChIP) analyses were performed as previously described.(9) In the case of experiments with surgically removed human tissues or xenograft tumor samples, tissues were homogenized to give very small pieces, then subjected to formalin fixation as in the case of cultured cells. We used the antibodies described in the “Western blot analysis” section. The antibody for acetyl-histone H4 (06-866) was obtained from Upstate Biotechnology. ChIP data were also quantitatively estimated by real-time PCR as described in the “Quantitative RT-PCR analysis” section. The primers and probes used are described in “Supplemental Materials.”
Formation of tumors in immunodeficient mice
Animal experiments were performed under approved protocols of the institutional animal use and care committee. YMB-1 or MDA-MB-453 cells (106 cells/50 μl PBS) were injected into the mammary fat pad of 6-week-old female SCID mice (Charles River Japan, Inc., Yokohama, Japan). After 12 weeks, mice were sacrificed, and tumors were excised.
Treatment of cells with HAT and histone deacetylase inhibitors
Cells (6 × 105 cells), initially cultured for 16 h in 60-mm dishes, were treated with curcumin (Sigma), anacardic acid (Calbiochem, San Diego, CA, USA), or trichostatin (Wako, Tokyo, Japan) dissolved in ethanol as a concentrated stock solution. Cells were also mock-treated with an identical volume of ethanol for comparison.
fXa generation assay
The fXa generation assay was performed as previously described.(9)
Determination of fVII concentrations in cell-culture supernatants secreted from hepatic cancer cells
The fVII concentrations in cell-culture medium were determined using an AssaySense Human Factor VII Chromogenic Activity Assay Kit (Assay Pro, St. Charles, MO, USA). Briefly, 6.5 × 104 cells/333 μl medium were seeded in each well of a 24-well plate then cultured for 14 h. Curcumin or an identical volume of vehicle was added to the cells and further cultured for 24 h. fVII in cell-culture supernatant was captured by a monoclonal anti-human fVII antibody immobilized onto a microplate. fVII captured by antibody against human fVII was converted to its active form by TF treatment then subjected to determination of fXa generating activity.
Supplementary Material
Acknowledgments
We would like to thank the Smoking Research Foundation (Y. Miyagi) for their support. WR is supported by NIH grant HL60742. This work was partly supported by a Grant-in-Aid for the Encouragement of Basic Science and Technology from the Science and Technology Office of the Kanagawa Prefectural Government (S. Koizume), and by a grant from the Japanese Ministry of Education, Culture, Sports, Science and Technology (Y. Miyagi).
Footnotes
Disclosure of Conflict of Interests There are no conflicts of interest to declare.
References
- 1.Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53(4):505–18. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 2.Versteeg HH, Schaffner F, Kerver M, et al. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111(1):190–9. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006;66(1):307–14. doi: 10.1158/0008-5472.CAN-05-1735. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X, Bailly MA, Panetti TS, Cappello M, Konigsberg WH, Bromberg ME. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J Thromb Haemost. 2004;2(1):93–101. doi: 10.1111/j.1538-7836.2004.00545.x. [DOI] [PubMed] [Google Scholar]
- 5.Hjortoe GM, Petersen LC, Albrektsen T, et al. Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood. 2004;103(8):3029–37. doi: 10.1182/blood-2003-10-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uusitalo-Jarvinen H, Kurokawa T, Mueller BM, Andrade-Gordon P, Friedlander M, Ruf W. Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27(6):1456–62. doi: 10.1161/ATVBAHA.107.142539. [DOI] [PubMed] [Google Scholar]
- 7.Versteeg HH, Spek CA, Slofstra SH, Diks SH, Richel DJ, Peppelenbosch MP. FVIIa:TF induces cell survival via G12/G13-dependent Jak/STAT activation and BclXL production. Circ Res. 2004;94(8):1032–40. doi: 10.1161/01.RES.0000125625.18597.AD. [DOI] [PubMed] [Google Scholar]
- 8.Sorensen BB, Rao LV, Tornehave D, Gammeltoft S, Petersen LC. Antiapoptotic effect of coagulation factor VIIa. Blood. 2003;102(5):1708–15. doi: 10.1182/blood-2003-01-0157. [DOI] [PubMed] [Google Scholar]
- 9.Koizume S, Jin MS, Miyagi E, et al. Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res. 2006;66(19):9453–60. doi: 10.1158/0008-5472.CAN-06-1803. [DOI] [PubMed] [Google Scholar]
- 10.Pollak ES, Hung HL, Godin W, Overton GC, High KA. Functional characterization of the human factor VII 5’-flanking region. J Biol Chem. 1996;271(3):1738–47. doi: 10.1074/jbc.271.3.1738. [DOI] [PubMed] [Google Scholar]
- 11.Erdmann D, Heim J. Orphan nuclear receptor HNF-4 binds to the human coagulation factor VII promoter. J Biol Chem. 1995;270(39):22988–96. doi: 10.1074/jbc.270.39.22988. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg D, Miao CH, Ho WT, Chung DW, Davie EW. Liver-specific expression of the human factor VII gene. Proc Natl Acad Sci U S A. 1995;92(26):12347–51. doi: 10.1073/pnas.92.26.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qyang Y, Luo X, Lu T, et al. Cell-type-dependent activity of the ubiquitous transcription factor USF in cellular proliferation and transcriptional activation. Mol Cell Biol. 1999;19(2):1508–17. doi: 10.1128/mcb.19.2.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rong Y, Hu F, Huang R, et al. Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res. 2006;66(14):7067–74. doi: 10.1158/0008-5472.CAN-06-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2(8):599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 16.Dong J, Tsai-Morris CH, Dufau ML. A novel estradiol/estrogen receptor alpha-dependent transcriptional mechanism controls expression of the human prolactin receptor. J Biol Chem. 2006;281(27):18825–36. doi: 10.1074/jbc.M512826200. [DOI] [PubMed] [Google Scholar]
- 17.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. Progesterone regulates transcription of the p21(WAF1) cyclin- dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem. 1998;273(17):10696–701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–19. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 19.Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell. 2006;24(2):233–43. doi: 10.1016/j.molcel.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14(13):1553–77. [PubMed] [Google Scholar]
- 21.Martinez-Jimenez CP, Gomez-Lechon MJ, Castell JV, Jover R. Underexpressed coactivators PGC1alpha and SRC1 impair hepatocyte nuclear factor 4 alpha function and promote dedifferentiation in human hepatoma cells. J Biol Chem. 2006;281(40):29840–9. doi: 10.1074/jbc.M604046200. [DOI] [PubMed] [Google Scholar]
- 22.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278(21):19134–40. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 23.Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279(49):51163–71. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 24.Bierhaus A, Zhang Y, Quehenberger P, et al. The dietary pigment curcumin reduces endothelial tissue factor gene expression by inhibiting binding of AP-1 to the DNA and activation of NF-kappa B. Thromb Haemost. 1997;77(4):772–82. [PubMed] [Google Scholar]
- 25.Pendurthi UR, Williams JT, Rao LV. Inhibition of tissue factor gene activation in cultured endothelial cells by curcumin. Suppression of activation of transcription factors Egr-1, AP-1, and NF-kappa B. Arterioscler Thromb Vasc Biol. 1997;17(12):3406–13. doi: 10.1161/01.atv.17.12.3406. [DOI] [PubMed] [Google Scholar]
- 26.Fischer EG, Riewald M, Huang HY, et al. Tumor cell adhesion and migration supported by interaction of a receptor-protease complex with its inhibitor. J Clin Invest. 1999;104(9):1213–21. doi: 10.1172/JCI7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chapman HA, Jr, Allen CL, Stone OL, Fair DS. Human alveolar macrophages synthesize factor VII in vitro. Possible role in interstitial lung disease. J Clin Invest. 1985;75(6):2030–7. doi: 10.1172/JCI111922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoffman M, Colina CM, McDonald AG, Arepally GM, Pedersen L, Monroe DM. Tissue factor around dermal vessels has bound factor VII in the absence of injury. J Thromb Haemost. 2007;5(7):1403–8. doi: 10.1111/j.1538-7836.2007.02576.x. [DOI] [PubMed] [Google Scholar]
- 29.Hung JJ, Wang YT, Chang WC. Sp1 deacetylation induced by phorbol ester recruits p300 to activate 12(S)-lipoxygenase gene transcription. Mol Cell Biol. 2006;26(5):1770–85. doi: 10.1128/MCB.26.5.1770-1785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang YN, Chang WC. Induction of disease-associated keratin 16 gene expression by epidermal growth factor is regulated through cooperation of transcription factors Sp1 and c-Jun. J Biol Chem. 2003;278(46):45848–57. doi: 10.1074/jbc.M302630200. [DOI] [PubMed] [Google Scholar]
- 31.Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG. Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Mol Cell. 2000;6(3):551–61. doi: 10.1016/s1097-2765(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 32.Sa G, Das T. Anti cancer effects of curcumin: cycle of life and death. Cell Div. 2008;3:14. doi: 10.1186/1747-1028-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sung B, Pandey MK, Ahn KS, et al. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood. 2008;111(10):4880–91. doi: 10.1182/blood-2007-10-117994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer NG, Chin SF, Ozdag H, et al. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci U S A. 2004;101(19):7386–91. doi: 10.1073/pnas.0401002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eliseeva ED, Valkov V, Jung M, Jung MO. Characterization of novel inhibitors of histone acetyltransferases. Mol Cancer Ther. 2007;6(9):2391–8. doi: 10.1158/1535-7163.MCT-07-0159. [DOI] [PubMed] [Google Scholar]
- 36.Adams BK, Ferstl EM, Davis MC, et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorganic & medicinal chemistry. 2004;12(14):3871–83. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Shoji M, Sun A, Kisiel W, et al. Targeting tissue factor-expressing tumor angiogenesis and tumors with EF24 conjugated to factor VIIa. Journal of drug targeting. 2008;16(3):185–97. doi: 10.1080/10611860801890093. [DOI] [PubMed] [Google Scholar]
- 38.Versteeg HH, Schaffner F, Kerver M, et al. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res. 2008;68(17):7219–27. doi: 10.1158/0008-5472.CAN-08-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.