

Abstract
Bmi1 is a polycomb group proto-oncogene that has been implicated in multiple tumor types. However, its role in HCC development has not been well studied. In this manuscript, we report that Bmi1 is over-expressed in human HCC samples. When Bmi1 expression is knocked down in human HCC cell lines, it significantly inhibits cell proliferation and perturbs cell cycle regulation. T o investigate the role of Bmi1 in promoting liver cancer development in vivo, we stably expressed Bmi1 and/or an activated form of Ras (RasV12) in mouse liver. We found while Bmi1 or RasV12 alone is not sufficient to promote liver cancer development, co-expression of Bmi1 and RasV12 promotes HCC formation in mice. Tumors induced by Bmi1/RasV12 resemble human HCC by deregulation of genes involved in cell proliferation, apoptosis, and angiogenesis. Intriguingly, we found no evidence that Bmi1 regulates Ink4A/Arf expression in both in vitro and in vivo systems of liver tumor development. In summary, our study demonstrates that Bmi1 can cooperate with other oncogenic signals to promote hepatic carcinogenesis in vivo. Yet Bmi1 functions independent of Ink4A/Arf repression in liver cancer development.
INTRODUCTION
Bmi1, a member of the mammalian polycomb group of multimeric transcriptional repressors, is involved in the regulation of development, stem cell self-renewal, cell cycle, and senescence (1). Bmi1 was first identified as a c-myc cooperating oncogene in murine B-cell lymphomas (2). Subsequent studies have revealed that Bmi1 is required by both normal and leukemic hematopoietic stem cells to maintain their proliferative capacity (3, 4). In addition, Bmi1 has been shown to be important for self-renewal of neural stem cells (5), and its expression is essential for the tumorigenicity of MycN induced neuroblastoma (6). Studies have found that Bmi1 induces telomerase activity and subsequently immortalizes mammary epithelial cells (7). Perhaps the most prominent link between Bmi1 and tumor development is its inhibition of the Ink4A/Arf locus, which results in the regulation of cell senescence and proliferation (8, 9).
Deregulation of Bmi1 expression has been reported in multiple tumor types, including non-small cell lung carcinoma, colon carcinoma, medulloblastoma, metastatic melanoma, and nasopharyngeal carcinoma (10–14). Up-regulation of Bmi1 in human hepatocellular carcinoma (HCC) has also been reported (15, 16). In a recent study, Chiba et al showed that silencing Bmi1 expression decreased the side population (SP) cells in HCC cell lines (17). These SP subpopulation cells are considered to harbor cancer stem cell like properties (18). However, the exact role of Bmi1 during HCC pathogenesis remains unclear. There are currently no in vivo models, which demonstrate that Bmi1 functions as an oncogene and directly contributes to HCC pathogenesis.
In this paper, we describe that Bmi1 is over-expressed in human HCC samples. Bmi1 expression is also required for HCC cell proliferation in vitro. Notably, we established a novel mouse model for Bmi1 and demonstrate that Bmi1 can cooperate with activated Ras signaling to promote hepatic carcinogenesis in vivo. However, expression analysis suggests that Bmi1 functions independent of its ability to repress Ink4A/Arf tumor suppressor genes. Our data therefore provide solid evidence for a functional role of Bmi1 in liver cancer pathogenesis.
RESULTS
Bmi1 is over-expressed in human HCC samples
In our previous studies, we used genomic approaches, including cDNA microarray and array based comparative genomic hybridization, to characterize molecular variations in human HCC (19–21). We identified 703 genes, which are highly expressed in human HCC (21). One of these up-regulated genes is Bmi1. From this microarray study, Bmi1 expression is up-regulated in human HCC compared with non-tumor liver tissues (p=2×10−6, after Bonferoni correction) (Fig. 1A). To verify this observation, we performed real-time RT-PCR analysis for Bmi1 expression in an independent liver tumor sample set which have not been previously assayed in microarray studies. Again, we observed up-regulation of Bmi1 in HCC samples (p<0.001, Fig. 1B). In two recent studies, the over-expression of Bmi1 in human HCC samples was demonstrated at protein levels (15, 16).
Figure One.

Bmi1 expression and its correlation with Ink4A/Arf expression in human HCC samples. (A) Bmi1 expression in non-tumor liver and HCC samples assayed by cDNA microarrays; (B) Bmi1 expression in an independent liver tissues set assayed by real-time RT-PCR; (C) Correlation between Bmi1 and Ink4A/Arf expression in human HCC samples assayed by cDNA microarrays; (D) and (E) Correlation between Bmi1 and p14Arf (D) or p16Ink4A (E) expression in human HCC samples assayed using real-time RT-PCR.
Since p16Ink4A and p14Arf have been considered to be major targets of Bmi1 during tumor development, we investigated whether there is any correlation between Bmi1 and Ink4A/Arf expression in human HCC. On the cDNA microarrays, there was one probe corresponding to the C-terminal sequences of Ink4A/Arf, which hybridized to both p16Ink4A and p14Arf. Our analysis of this microarray data found no correlation between Bmi1 and total Ink4A/Arf expression (R=−0.094) (Fig. 1C). We next assayed the expression of Bmi1, p16Ink4A, and p14Arf individually using real-time RT-PCR in 19 human HCC and 4 non-tumor liver tissues. Again, we found no correlation between the expression values of Bmi1 and p16Ink4A or p14Arf (Fig. 1D and 1E).
Altogether, these data demonstrate that Bmi1 is up-regulated in human HCC, suggesting that Bmi1 may play a role in HCC pathogenesis. However, Bmi1 expression does not appear to be correlated with the expression of Ink4A/Arf tumor suppressor genes in human HCC samples.
Stable shRNA-mediated knockdown of Bmi1 inhibits cancer cell growth in vitro
Our expression analysis suggests a potential role for Bmi1 during liver tumorigenesis. We therefore decided to study the functional significance of Bmi1 in hepatocarcinogenesis. We found that Bmi1 protein is highly expressed in human HCC cell lines (data not shown). To investigate whether Bmi1 is required during liver cancer development, we stably knocked down its expression using lentiviral shRNA in human HCC cell lines. To better study the relationship of Bmi1 expression and genetic alternations in human HCCs, we chose three HCC cell lines (SK-Hep1, Huh7, and Hep3B) with different genetic variations in AFP, p53, p16Ink4A, and p14Arf (Supplementary Table One).
We infected these cells with lentivirus encoding empty vector pLKO.1, a vector with scrambled shRNA (SC/pLKO.1) or vectors against Bmi1, Bmi1/pLKO.1. Because similar results were obtained using pLKO.1 or SC/pLKO.1 as controls (data not shown), only the data with pLKO.1 are shown here. To exclude non-specific RNAi-mediated effects, we tested two shRNA constructs which target different regions of the Bmi1 sequence (Bmi1/pLKO.1 #1 and Bmi1/pLKO.1 #2). We found that both Bmi1/pLKO.1 vectors efficiently silenced Bmi1 expression in human HCC cell lines, as confirmed by both real-time RT-PCR and Western blotting (Fig. 2A and B, Supplementary Fig. 1 ). Therefore, only data from the Bmi1/pLKO.1 #1 studies are shown.
Figure Two.

Down-regulation of Bmi1 and its effects on its target gene expression in human HIC cells. (A) Real-time RT-PCR analysis of Bmi1, p14Arf, and p16Ink4A in pLKO.1 or Bmi1/pLKO.1 infected HCC cells; (B) Protein expression of Bmi1 and p16 in pLKO.1 or Bmi1/pLKO.1 infected HCC cells. Actin was used as loading control.
We found that despite different genetic backgrounds and Ink4A/Arf status, silencing of Bmi1 inhibits growth of all three HCC cell lines (Fig. 3A and Supplementary Fig. 1). Furthermore, the expression of cell cycle genes, such as Cdc2, Cdc20, and Bub1 (22) are significantly down-regulated in Bmi1/pLKO.1 infected HCC cells (Fig. 3B and Supplementary Fig. 2). In addition, BrdU labeling revealed a decreased proliferative rate (Fig. 3C and Supplementary Fig. 3), whereas activated caspase 3 assay showed a slight increase in apoptosis when Bmi1 expression is silenced (data not shown). Finally, cell cycle analysis suggests that loss of Bmi1 perturbs cell cycle regulation and leads to G2/M accumulation (Fig. 3D, Supplementary Fig. 4 and Supplementary Table Two). There is also an increase of sub-G1 phase cells in Bmi1/pLKO.1 infected cells, providing further support of increased cell apoptosis when Bmi1 expression is inhibited (Fig. 3D, Supplementary Fig. 4 and Supplementary Table Two).
Figure Three.

Silencing Bmi1 expression leads to decreased cell growth in human HCC cells. (A) Cell growth assays in three HCC cell lines infected with lentivirus encoding pLKO.1 or Bmi1/pLKO.1. Cell numbers were counted at 3 days post infection; (B) Down-regulation of cell cycle genes: Mad2, Cdc2, Cdc20, and Bub1 in Bmi1/pLKO.1 infected Huh7 cells; (C) BrdU labeling assay showing a decrease of proliferation in Bmi1/pLKO.1 infected Hep3B cells; (D) Representative images of cell cycle distribution of Hep3B cells infected with pLKO.1 or Bmi1/pLKO.1 measured by PI staining and FACS analysis. The percentage of cells in each cell cycle phase is listed on the right panel. **: p <0.01; *: p <0.05.
In summary, the studies support that Bmi1 expression is required for in vitro growth of human HCC cell lines.
Loss of Bmi1 does not lead to significant increased expression of p16Ink4A or p14Arf in HCC cell lines
One of the major mechanisms of Bmi1 induced tumor development is its function as a potent inhibitor of CDKN2A which encodes two major proteins: p16Ink4A and p14Arf (p19Arf in mice) (8, 23). We first determined whether Bmi1 knockdown affects the mRNA expression of CDKN2A genes using real-time RT-PCR. SK-Hep1 cells have a deletion of Ink4A/Arf locus, whereas Huh7 cells have strong promoter methylation of p16Ink4A. Therefore, p16Ink4A expression is virtually undetectable in these two cell lines, regardless of whether Bmi1 is down-regulated (Fig. 2A). In addition, the loss of Bmi1 expression in transfected Hep3B cells does not appear to affect the expression of p16Ink4A. Likewise, we found that silencing Bmi1 expression does not lead to up-regulation of p14Arf in Huh7 and Hep3B cells, while p14Arf expression is absent in SK-Hep1 cells (Fig. 2A). We next assayed p16Ink4A protein expression in these HCC cell lines (Fig. 2B). Consistent with real-time RT-PCR results, p16Ink4A is undetectable in SK-Hep1 and Huh7 cells, whereas there is little change of p16Ink4A protein levels in Bmi1/pLKO.1 infected cell Hep3B cells (Fig. 2B).
In summary, our data showed that Bmi1 is required for HCC cell proliferation; however the effect of Bmi1 in promoting HCC cell growth is independent of Ink4A/Arf status.
Overexpression of Bmi1 cooperates with activated Ras to induce HCC in mice
We next determined whether Bmi1 can function as an oncogene by establishing a mouse model. We reasoned that it is unlikely Bmi1 alone is sufficient to induce liver cancer formation in vivo. Therefore, we searched for other signaling pathways that may be able to cooperate with Bmi1 to promote hepatic carcinogenesis. We chose activated Ras as the second signal, based on studies which have shown that Bmi1 is capable of cooperating with activated Ras to transform cells in vitro (24, 25). In addition, Ras/MAPK signaling is known to be activated in all human HCC samples (26). Therefore it represents a critical genetic alteration present in human HCC. Furthermore, studies from our and other labs have found that activated Ras alone is not sufficient to induce HCC formation in mice (27, 28).
We applied hydrodynamic transfection to stably express Bmi1 (with c-terminal V5 tag) and/or an activated form of N-ras (RasV12) into mouse hepatocytes. These animals were then monitored and sacrificed at specific time points or when moribund. We found that while over-expression of RasV12 (n=15) or Bmi1 (n=5) alone was not sufficient to promote liver tumor development, the co-expression of Bmi1 and RasV12 induced liver tumors in 78.6% (11/14) of the mice between 15 to 30 weeks post injection (Fig. 4A). Tumors tend to be multifocal, sometimes with over 100 tumor nodules scattered around the entire liver (Fig. 4B and data not shown).
Figure Four.

Bmi1 cooperates with activated Ras (RasV12) to promote hepatic carcinogenesis in vivo. (A) Tumor development incident curves in mice. The cumulative hazard represents the relative probability of tumor development in each condition; (B) Representative gross image of liver tumors induced by Bmi1/RasV12. Arrows indicate visible tumor nodules; (C) Quantitative RT-PCR analysis of AFP expression in normal liver and Bmi1/RasV12 tumor samples; (D) H&E staining of non-tumor liver (NT) and HCC (T) induced by Bmi1/RasV12 (upper). Immunohistochemical staining with anti-V5 antibody showing staining of V5-tagged Bmi1 in HCC cells in a tumor nodule, with sporadic staining in non-tumor liver tissues (middle). Insets: expanded view showing specific nuclear staining of Bmi1 in non-tumor liver or HCC cells. Immunohistochemical staining of phospho-ERK in both NT and T samples (lower); (E) Western blot analysis of N-Ras, phospho-ERK, and ERK expression. Actin was used as loading control.
Histological examination of liver tumor samples induced by Bmi1/RasV12 showed that tumors consisted of neoplastic cells with frequent trabecular disorganization, which are characteristic of HCC (Fig. 4D). In most cases, the tumor cells appear to be well differentiated. Real-time RT-PCR analysis revealed high expression of HCC specific marker α-fetoprotein (AFP) (Fig. 4C), further confirming the tumors to be of hepatocellular origin.
We next examined the tumor nodules for expression of injected Bmi1 (with a c-terminal V5 tag) and RasV12. Using anti-V5 antibody we observed that all tumor cells showed positive nuclear staining of Bmi1 (Fig. 4D). Sporadic expression of Bmi1 was also detected in the hepatocytes of surrounding non-tumor liver. RasV12 is indicated by elevated protein levels (Fig. 4E). Since activated Ras is a potent inducer of MAPK signaling, we investigated the activity of RasV12 by assaying for the presence of phospho-ERK. Both western blot and immunohistochemical analyses detected strong expression of phospho-ERK in the tumors (Fig. 4D & E).
Altogether, these results support that Bmi1 and RasV12 can cooperate to induce HCC in vivo.
Molecular characterization of Bmi1/RasV12 induced HCC
We then investigated the molecular features of Bmi1/RasV12 induced tumors in order to determine whether these traits resemble phenotypes observed in human HCC. We first assayed for cell proliferation in Bmi1/RasV12 tumor samples. Our detection of proliferative marker, Ki67, suggested the tumor cells to be highly proliferative (Fig. 5A). We also observed increased expression of cyclin E1 in liver tumor samples, while there is little variability in the expression of cell cycle regulator, cyclin D1 (Fig. 5B). In addition, we found that these tumors exhibited elevated levels of cell cycle inhibitor p21 (Fig. 5B), which is likely to be a feedback response to the activated Ras signaling. Furthermore, anti-apoptotic marker survivin and cell-cell adhesion marker E-cadherin were also found to be up-regulated in liver tumor samples (Fig. 5B). The up-regulation of E-cadherin is consistent with well-differentiated tumor histology and is frequently observed in certain mouse models of HCC (29, 30). The occurrence of angiogenesis during liver carcinogenesis can be distinguished by the expression of endothelial markers, like PODXL1 (31). Although PODXL1 is not typically expressed by normal liver sinusoidal endothelial cells, this marker is frequently present in the endothelial cells of liver tumors (31). Therefore, we analyzed our samples for PODXL1 and observed that only endothelial cells within tumor nodules stained positive for this marker (Fig. 5A). Furthermore, these HCC samples also highly expressed angiogenic factor Ang2 (Fig. 5B).
Figure Five.
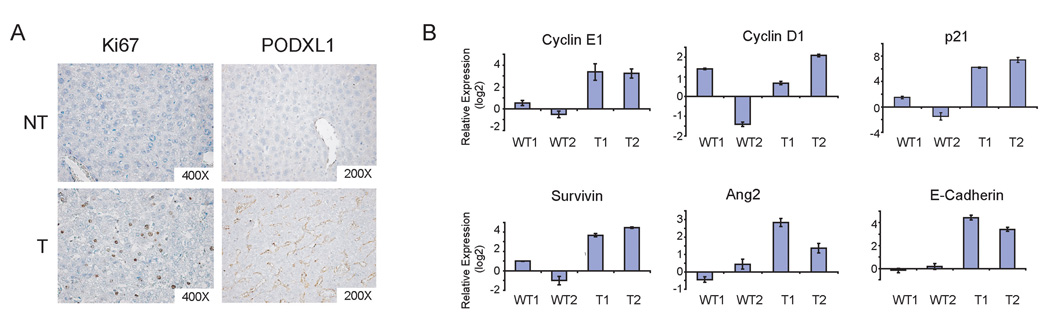
Characterization of liver tumors induced by Bmi1/RasV12. (A) Immunohistochemical staining of Ki67 and PODXL1 in non-tumor liver (NT) and tumor (T) tissues; (B) Quantitative RT-PCR analysis of genes in two wildtype liver tissues and two Bmi1/Ras liver tumors. In all cases, the expression values of the two wildtype samples were averaged, set to 1 and used to normalize liver tumor samples.
Overall, our data suggests that Bmi1/RasV12 expressing tumors resemble a subset of human HCC characterized by the deregulation of factors involved in proliferation, apoptosis, and angiogenesis.
Up-regulation of p16Ink4A/p19Arf expression in Bmi1/RasV12 induced liver tumors
Bmi1 has been shown to cooperate with RasV12 to transform MEF cells via inhibition of Ink4A/Arf locus (24). We therefore investigated whether this regulation is a mechanism by which Bmi1 and activated Ras promote tumorigenesis in vivo. We examined the expressions of p16Ink4A and p19Arf in our samples by quantitative RT-PCR. We have used multiple primers against p16Ink4A and p19ARF, and we found that in all cases, both p16Ink4A and p19ARF can only be detected after more than 30 cycles of PCR, indicating that p16Ink4A and p19ARF are expressed at very low levels in normal liver tissues. In contrast, p16Ink4A expression is up-regulated ~5 fold and p19Arf expression is up-regulated ~50 fold in Bmi1/RasV12 tumor samples (Fig. 6).
Figure Six.

Up-regulation of p16Ink4A and p19Arf in liver tumor samples induced by Bmi1/RasV12. The relative expression of p16Ink4A and p19Arf of two wildtype liver tissues (WT) and two tumor samples (T) is shown.
Regulation of Ink4A/Arf expression by RasV12 and Bmi1 in mouse hepatocytes
The up-regulation of p16Ink4A and p19Arf in Bmi1/RasV12 tumor samples is quite surprising since Bmi1 is a known inhibitor of Ink4A/Arf locus. One of the possibilities is that Ras is a potent inducer of p16Ink4A and p19Arf. The up-regulation of Ink4A/Arf may be why activated Ras alone is not sufficient to induce HCC formation in vivo. It is possible that the partial inhibition of Ras-induced Ink4A/Arf expression by Bmi1 is what eventually leads to hepatic carcinogenesis. If this is the case, it is likely that Bmi1/RasV12 tumor cells may have somewhat elevated expression of p16Ink4A and p19Arf compared to normal liver. However, this hypothesis is only possible if RasV12 can strongly induce p16Ink4A and p19Arf expression in hepatocytes. We therefore investigated the regulation of p16Ink4A and p19Arf by RasV12 or Bmi1 in normal mouse hepatocytes.
First, we generated adenovirus encoding activated Ras. Adenoviral infection has been shown to be able to transfect 100% of mouse hepatocytes at MOI of 10 (32). We infected mouse primary hepatocytes with either control adenovirus encoding EGFP (AD-EGFP), or activated Ras (AD-RasV12-HA). Western blot analysis demonstrated the expression of RasV12 in infected cells (Fig. 7A). Using quantitative RT-PCR, we found that p16Ink4A expression is repressed, whereas p19ARF expression remains unchanged in primary mouse hepatocytes after AD-RasV12-HA infection (Fig. 7A). Next we transfected primary mouse hepatocytes with plasmids encoding activated N-Ras (RasV12-HA) and/or Bmi1 (Bmi1-V5) using Targefect-Hepatocyte transfection reagents, which have a transfection efficiency of 50% for primary hepatocytes. The expression of RasV12 and Bmi1 are indicated by western blot analysis (Fig. 7B). We found that while the expression of p16Ink4A is down-regulated by RasV12 transfection, there is very little change in the expression of p19Arf. Bmi1 up-regulates p16Ink4A expression and inhibits p19Arf expression in this condition (Fig. 7B). Co-transfection with Bmi1 and RasV12 showed similar impact on p16Ink4A while having little effect on p19Arf (Fig. 7B). Next, we assayed p16Ink4A protein expression in these mouse hepatocytes. We found that p16Ink4A protein is undetectable in primary mouse hepatocytes in all these conditions (Fig. 7A).
Figure Seven.

Regulation of p16Ink4A and p19Arf expression by RasV12 and/or Bmi1 in primary mouse hepatocytes. (A) Primary mouse hepatocytes were infected with adenovirus encoding EGFP or RasV12-HA. Western blotting shows the expression of HA-tagged RasV12 (left), and real-time RT-PCR shows the quantification of p16Ink4A and p19Arf expression in adenoviral infected primary hepatocytes (right); (B) Primary mouse hepatocytes were transfected with plasmids encoding EGFP, RasV12-HA and/or Bmi1-V5. Western blotting shows the expression of HA-tagged RasV12, V5-tagged Bmi1 and p16Ink4A (left). Arrow shows the ectopically expressed Bmi1-V5 that migrates higher than endogenous Bmi1 on gel. IMR90 cell lysate was used as a positive control for p16Ink4A protein expression. Quantification of p16Ink4A and p19Arf mRNA expression in transfected primary hepatocytes was performed using real-time RT-PCR (right). **: p <0.01; *: p <0.05.
Ras has been shown to induce cell senescence in certain cell types (33, 34), but the induction of cell senescence in hepatocytes by Ras has not been reported. It is possible that Bmi1 cooperates with RasV12 to promote HCC pathogenesis by overcoming Ras mediated induction of senescence. We therefore investigated whether RasV12 or Bmi1 regulates cell senescence in primary hepatocytes. We transfected primary mouse hepatocytes with EGFP, RasV12, Bmi1, or RasV12 and Bmi1, and assayed for cell senescence. We found no evidence that either RasV12 or Bmi1 can modulate senescent status in primary hepatocytes (Supplementary Fig. 5 and Supplementary Table Three).
In summary, our data do not support activation of Ink4A/Arf by Ras or inhibition of Ink4A/Arf by Bmi1 overexpression in hepatocytes. The experiments therefore indicate that Bmi1 cooperates with RasV12 to promote HCC pathogenesis in an Ink4A/Arf independent manner.
DISCUSSION
There is increasing evidence supporting Bmi1 as an important oncogene in tumor development. Up-regulation of Bmi1 expression has been reported in multiple tumor types. Studies also showed that Bmi1 expression is required for in vitro cell proliferation in Ewing Sarcoma, lung cancer, and medulloblastoma cells (35–37), whereas overexpression of Bmi1 enhances cell survival in epidermis (38) and prostate cancer cells (39). Using Bmi1 knockout mice, studies demonstrated that Bmi1 expression is required for the tumorigenesis of leukemia and lung cancer in vivo (4, 40). However, there is still little evidence whether Bmi1 overexpression can directly contribute to carcinogenesis, especially in solid tumors. Proper mouse models need to be established to address this critical question. In our current study, we showed that Bmi1 is over-expressed in human HCC samples and required for HCC cell growth in vitro. More importantly, we established a novel mouse model which demonstrates that Bmi1 can cooperate with activated Ras to promote HCC pathogenesis in mice. Our study therefore provides pivotal data supporting Bmi1 as an oncogene and its role in hepatic carcinogenesis.
In this study, we used activated Ras to mimic the activation of Ras/MAPK pathway and in combination with Bmi1 to induce HCC in our mouse model. Although there is ubiquitous activation of Ras/MAPK signaling in human HCC, Ras mutations are, in fact, very rare (41, 42). De-regulation of other factors, including tumor suppressor genes Spry2 and RASSF1, overexpression of H-Ras, as well as over-activation of receptor tyrosine kinases, such as EGFR and c-Met, have been implicated in human HCC, all of which result in up-regulation of this pathway (26). Therefore, combination of these genetic alternations with Bmi1 over-expression in mouse models will provide additional in vivo models to mimic human HCC pathogenesis. Interestingly, we assayed the expressions of H-Ras and RASSF1A in human HCC samples, and found there to be no correlation between their expressions and Bmi1 expression (Supplementary Fig. 6). Clearly, future experiments will be needed to determine whether the expression of other factors involved in the activation of Ras/MAPK pathways, such as c-Met or EGFR, are associated with up-regulation of Bmi1 expression during HCC pathogenesis.
An important implication of our study is that the tumorigenicity of Bmi1 during HCC pathogenesis is independent of its ability to repress Ink4A/Arf expression. First, we showed there is no correlation between the expressions of Bmi1 and p16Ink4A or p14Arf in human HCC samples. Secondly, we found that the down-regulation of Bmi1 inhibits HCC cell growth independent of Ink4A/Arf status. Finally, we showed that in mouse tumor cells induced by Bmi1/RasV12, there is no down-regulation of p16Ink4A or p19Arf expression. Consistent with our observation, several recently published reports have revealed an Ink4A/Arf independent role for Bmi1 during tumor pathogenesis. Bruggeman et al, for instance, showed that Bmi1 controls mouse glioma development in an Ink4a/Arf-independent manner (43). Bmi1 knockdown significantly inhibits cell growth in both wildtype and p16Ink4A null Ewing Sarcoma (35), and medulloblastoma cell lines (37). Thus, although inhibition of Ink4A/Arf tumor suppressor gene expression has been widely considered to be the key mechanism of the oncogenic activity of Bmi1, more recent data suggest a critical role of an Ink4A/Arf independent mechanism for Bmi1 during carcinogenesis.
Clearly, the next step in the characterization of molecular mechanisms of Bmi1 is to identify novel targets and/or pathways regulated by Bmi1 during HCC pathogenesis, and investigate how they cooperate with activated Ras/MAPK signaling to induce liver cancer formation. Some potential targets of Bmi1 have been identified in human cancer cell lines. For example, hTert is thought to be a major target in Bmi1 induced immortalization of mammary epithelial cells (7). NID1, a gene related to cell adhesion has been implicated as a Bmi1 target in Ewing Sarcoma cells (35). Signaling molecules including BMP5, TGF-β2, and Notch2 have been found to be regulated by Bmi1 in medulloblastoma cell lines (37). It would be of interest to determine whether the expression of these factors is modulated by Bmi1 during HCC pathogenesis. A recent study indicated that the loss of Bmi1 results in the increase of reactive oxygen species and subsequent stimulation of the DNA damage response pathway (44). Activation of the DNA damage response pathway has been found to be an important barrier to tumorigenesis. In our recent studies, we observed up-regulation in the mRNA of p53 and ATM genes in Bmi1/RasV12 induced liver tumor samples (Lee SA, unpublished observation). Clearly, it would be important to further characterize the expression of these genes in tumor samples at protein levels. Analysis of the regulation of this pathway by Bmi1 in liver may identify an additional function for Bmi1 during the development of HCC.
MATERIALS AND METHODS
Human tissue samples and RNA preparation
Samples of tumor and non-tumor liver tissues were collected from liver resections at The University of Hong Kong. Tissues were frozen in liquid nitrogen within half an hour after they were resected. Total RNA was extracted using Trizol (Invitrogen). This study was approved by the Ethics Committee of the University of Hong Kong and the Internal Review Boards from UCSF.
Constructs and reagents
Two shRNA constructs targeting Bmi1: Bmi1/pLKO.1 #1 (TRCN0000020155, NM_005180.5-693s1c1) and Bmi1/pLKO.1 #2 (TRCN0000020156, NM_005180.5-1061s1c1), used to silence Bmi1 expression were obtained from OpenBiosystems (Huntsville, AL). Control pLKO.1 (empty vector) or SC/PLKO.1 (with a scrambled sequence) plasmids were obtained from Addgene (Cambridge, MA). The hyperactive sleeping beauty construct (pCMV/SB) by Dr. Mark Kay of Stanford University; and pCaggs-RasV12 by Dr. David Largaespada of University of Minnesota. The pT3-EF1α vector containing duplicated inverted repeats (IR) for sleeping beauty mediated integration and EF1α promoter (pT3-EF1α) used for hydrodynamic injection was described by Tward A et al (45). Human Bmi1 (with a C-terminal V5 tag) was cloned into pT3-EF1α via the Gateway PCR cloning strategy (Invitrogen, Carlsbad, CA). All plasmids were purified using the Endotoxin free Maxi prep kit (Sigma, St. Louis, MO) before injecting into mice.
Cell culture, lentiviral infection, cell proliferation, BrdU labeling, caspase-3 activity and cell cycle assays
All human HCC cell lines were purchased from ATCC except Huh7, which were kindly provided by Dr. Ben Yen of UCSF. The cells are cultured in DMEM plus 10% fetal bovine serum. Lentivirus was generated and used to infect HCC cells. Three days post infection, cells were expanded and selected with 1µg/ml puromycin for 3 days and harvested for protein or RNA analysis. To assay cell proliferation rate, equal number of cells were seeded in 6 well plates and counted 3 to 4 days post seeding. Cell cycle analysis was performed by flow cytometry after propidium iodide (PI) staining, and the results were analyzed using FlowJo. BrdU labeling was as described (46) and Caspase-3 activity was measured using Caspase-Glo3/7 Assay kit (Promega, Madison, WI).
Hepatocyte isolation, transfection, adenovirus infection, and cell senescence assay
Primary hepatocyte isolation was performed using standard collagenase perfusion method as described (47). The hepatocytes were transfected with plasmids using Targefect-Hepatocyte reagents (Targeting Systems, El Cajon, CA) per manufacturer’s instruction. Ad-H-RasV12 was kindly provided by Dr. Judy Meinkoth of the University of Pennsylvania (48). Adenovirus was amplified and titered by Vector Biolabs (Philadelphia, PA) and used to infect primary hepatocytes at 50 MOI. Hepatocytes were harvested 30 hours post transfection or infection. Hepatocyte senescence was determined using senescence β-galactosidase staining kit (Cell Signaling Technology, Beverly, MA).
Mouse hydrodynamic transfection and monitoring
Wildtype FVB/N mice were used in this study. The hydrodynamic transfection procedure are as described previously (45). The injected mice were monitored weekly and sacrificed between 14 to 30 weeks post injection. All mice were housed, fed, and treated in accordance with protocols approved by the committee for animal research at the University of California, San Francisco.
Histology and immunohistochemistry
Animals were euthanized and their livers were removed and rinsed in PBS. Samples collected from the livers were either frozen in dry ice for RNA and protein extraction or fixed overnight in freshly prepared cold 4% paraformaldehyde. Fixed tissue samples were embedded in paraffin. Five micron sections were placed on slides and stained with hematoxylin and eosin. Immunohistochemistry was performed as described (28). Antibodies and dilutions were as follows: anti-V5, 1:1000 (Invitrogen); anti-phospho-ERK, 1:100 (Cell Signalling Technology); anti-PODXL1, 1:200 (Applied Genomics, Burlingame, CA); and anti-Ki67, 1:150 (Lab vision, Fremont, CA).
Preparation of lysates and Western blotting
Liver tissues or cell lines were lysed in M-PER mammalian protein extraction buffer (Pierce, Rockford, IL) plus proteinase inhibitor cocktail (Roche, Palo Alto, CA) and Halt phosphotase inhibitor cocktail (Pierce). Protein content of the lysate was quantified using the BCA protein assay (Pierce). Western blotting was performed as described (45). Antibodies were used as follows: anti-Bmi1, 1:1000 (Millipore, Billerica, MA) or 1:1000 (Invitrogen); anti-phospho-ERK, 1:1000; anti-ERK, 1:1000 (Cell Signaling Technology); anti-actin, 1:1000 (Sigma, St. Louis, MO); anti-V5, 1:5000 (Invitrogen); anti-NRas, 1:1000; anti-p16, 1:1000; and anti-HA, 1:1000 (Santa Cruz Biotechnology, Santa Cruz, CA)
Real-time RT-PCR
Total RNA was extracted from frozen liver tissues using Trizol (Invitrogen) and digested with DNase I to remove any genomic DNA contamination. Sybergreen based real-time RT-PCR was carried out as described (21), and rRNA was used as internal control. Transcript quantification was performed in triplicate for every sample and reported relative to rRNA. The primer pairs used are listed in Supplementary Table Four.
Statistical analysis
The Pearson’s correlation coefficient (R) was used to determine the correlations between gene expression values and p-value was determined using SPSS statistical program. Student’s t test was used to evaluate statistical significance among experimental groups. Values of P < 0.05 were considered to be significant.
Supplementary Material
ACKNOWLEDGEMENT
We would like to thank Sandra Huling of the UCSF Liver Center Morphology Core for histology support; Ali Naqvi, Theresa Canavan and Chris Her of the Cell and Tissue Biology Core for isolating mouse hepatocytes; Ali Brincat and Xiaoyin Wang of Sandler Lentivirus Core for lentivirus production; and Bill Hyun for FACS analysis. We would like to thank Drs. David Largaespada, Mark Kay, Judy Meinkoth and Ben Yen for reagents used in the study. This work is supported by NIH grants R21CA131625 and R01CA136606 to X.C, R01CA094150 to G.D., and P30DK026743 to the UCSF liver center.
Abbreviations
- HCC
hepatocellular carcinoma.
Footnotes
Conflict of Interest Statement:
The authors declare no conflict of interest.
REFERENCE
- 1.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118(4):409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mumyc transgenic mice. Cell. 1991;65(5):753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- 3.Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 4.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 5.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425(6961):962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui H, Hu B, Li T, et al. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am J Pathol. 2007;170(4):1370–1378. doi: 10.2353/ajpath.2007.060754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dimri GP, Martinez JL, Jacobs JJ, et al. The Bmi-1 oncogene induces telomerase activity and immortalizes human mammary epithelial cells. Cancer Res. 2002;62(16):4736–4745. [PubMed] [Google Scholar]
- 8.Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113(2):175–179. doi: 10.1172/JCI20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 10.Leung C, Lingbeek M, Shakhova O, et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428(6980):337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 11.Vonlanthen S, Heighway J, Altermatt HJ, et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer. 2001;84(10):1372–1376. doi: 10.1054/bjoc.2001.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JH, Yoon SY, Kim CN, et al. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer Lett. 2004;203(2):217–224. doi: 10.1016/j.canlet.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Mihic-Probst D, Kuster A, Kilgus S, et al. Consistent expression of the stem cell renewal factor BMI-1 in primary and metastatic melanoma. Int J Cancer. 2007;121(8):1764–1770. doi: 10.1002/ijc.22891. [DOI] [PubMed] [Google Scholar]
- 14.Sacchi S, Federico M, Dastoli G, et al. Treatment of B-cell non-Hodgkin's lymphoma with anti CD 20 monoclonal antibody Rituximab. Crit Rev Oncol Hematol. 2001;37(1):13–25. doi: 10.1016/s1040-8428(00)00069-x. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Pan K, Zhang HK, et al. Increased polycomb-group oncogene Bmi-1 expression correlates with poor prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134(5):535–541. doi: 10.1007/s00432-007-0316-8. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki M, Ikeda H, Itatsu K, et al. The overexpression of polycomb group proteins Bmi1 and EZH2 is associated with the progression and aggressive biological behavior of hepatocellular carcinoma. Lab Invest. 2008 doi: 10.1038/labinvest.2008.52. [DOI] [PubMed] [Google Scholar]
- 17.Chiba T, Miyagi S, Saraya A, et al. The polycomb gene product BMI1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res. 2008;68(19):7742–7749. doi: 10.1158/0008-5472.CAN-07-5882. [DOI] [PubMed] [Google Scholar]
- 18.Chiba T, Kita K, Zheng YW, et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006;44(1):240–251. doi: 10.1002/hep.21227. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Cheung ST, So S, et al. Gene expression patterns in human liver cancers. Mol Biol Cell. 2002;13(6):1929–1939. doi: 10.1091/mbc.02-02-0023.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patil MA, Gutgemann I, Zhang J, et al. Array-based comparative genomic hybridization reveals recurrent chromosomal aberrations and Jab1 as a potential target for 8q gain in hepatocellular carcinoma. Carcinogenesis. 2005;26(12):2050–2057. doi: 10.1093/carcin/bgi178. [DOI] [PubMed] [Google Scholar]
- 21.Patil MA, Chua MS, Pan KH, et al. An integrated data analysis approach to characterize genes highly expressed in hepatocellular carcinoma. Oncogene. 2005;24(23):3737–3747. doi: 10.1038/sj.onc.1208479. [DOI] [PubMed] [Google Scholar]
- 22.Whitfield ML, Sherlock G, Saldanha AJ, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13(6):1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pardal R, Molofsky AV, He S, Morrison SJ. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb Symp Quant Biol. 2005;70:177–185. doi: 10.1101/sqb.2005.70.057. [DOI] [PubMed] [Google Scholar]
- 24.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397(6715):164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 25.Datta S, Hoenerhoff MJ, Bommi P, et al. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007;67(21):10286–10295. doi: 10.1158/0008-5472.CAN-07-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calvisi DF, Ladu S, Gorden A. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130(4):1117–1128. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 27.Harada N, Oshima H, Katoh M, Tamai Y, Oshima M, Taketo MM. Hepatocarcinogenesis in mice with beta-catenin and Ha-ras gene mutations. Cancer Res. 2004;64(1):48–54. doi: 10.1158/0008-5472.can-03-2123. [DOI] [PubMed] [Google Scholar]
- 28.Lee SA, Ho C, Roy R, et al. Integration of genomic analysis and in vivo transfection to identify sprouty 2 as a candidate tumor suppressor in liver cancer. Hepatology. 2008;47(4):1200–1210. doi: 10.1002/hep.22169. [DOI] [PubMed] [Google Scholar]
- 29.Calvisi DF, Ladu S, Conner EA, Factor VM, Thorgeirsson SS. Disregulation of E-cadherin in transgenic mouse models of liver cancer. Lab Invest. 2004;84(9):1137–1147. doi: 10.1038/labinvest.3700147. [DOI] [PubMed] [Google Scholar]
- 30.Wei Y, Van Nhieu JT, Prigent S, Srivatanakul P, Tiollais P, Buendia MA. Altered expression of E-cadherin in hepatocellular carcinoma: correlations with genetic alterations, beta-catenin expression, and clinical features. Hepatology. 2002;36(3):692–701. doi: 10.1053/jhep.2002.35342. [DOI] [PubMed] [Google Scholar]
- 31.Chen X, Higgins J, Cheung ST, et al. Novel endothelial cell markers in hepatocellular carcinoma. Mod Pathol. 2004 doi: 10.1038/modpathol.3800167. [DOI] [PubMed] [Google Scholar]
- 32.Prost S, Sheahan S, Rannie D, Harrison DJ. Adenovirus-mediated Cre deletion of floxed sequences in primary mouse cells is an efficient alternative for studies of gene deletion. Nucleic Acids Res. 2001;29(16):E80. doi: 10.1093/nar/29.16.e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 34.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998;12(19):3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douglas D, Hsu JH, Hung L, et al. BMI-1 promotes ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res. 2008;68(16):6507–6515. doi: 10.1158/0008-5472.CAN-07-6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Q, Su B, Liu D, et al. Antisense RNA-mediated suppression of Bmi-1 gene expression inhibits the proliferation of lung cancer cell line A549. Oligonucleotides. 2007;17(3):327–335. doi: 10.1089/oli.2007.0070. [DOI] [PubMed] [Google Scholar]
- 37.Wiederschain D, Chen L, Johnson B, et al. Contribution of polycomb homologues Bmi-1 and Mel-18 to medulloblastoma pathogenesis. Mol Cell Biol. 2007;27(13):4968–4979. doi: 10.1128/MCB.02244-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee K, Adhikary G, Balasubramanian S, et al. Expression of Bmi-1 in epidermis enhances cell survival by altering cell cycle regulatory protein expression and inhibiting apoptosis. J Invest Dermatol. 2008;128(1):9–17. doi: 10.1038/sj.jid.5700949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan C, He L, Kapoor A, et al. Bmi1 promotes prostate tumorigenesis via inhibiting p16(INK4A) and p14(ARF) expression. Biochim Biophys Acta. 2008;1782(11):642–648. doi: 10.1016/j.bbadis.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Dovey JS, Zacharek SJ, Kim CF, Lees JA. Bmi1 is critical for lung tumorigenesis and bronchioalveolar stem cell expansion. Proc Natl Acad Sci U S A. 2008;105(33):11857–11862. doi: 10.1073/pnas.0803574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsuda H, Hirohashi S, Shimosato Y, Ino Y, Yoshida T, Terada M. Low incidence of point mutation of c-Ki-ras and N-ras oncogenes in human hepatocellular carcinoma. Jpn J Cancer Res. 1989;80(3):196–199. doi: 10.1111/j.1349-7006.1989.tb02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Challen C, Guo K, Collier JD, Cavanagh D, Bassendine MF. Infrequent point mutations in codons 12 and 61 of ras oncogenes in human hepatocellular carcinomas. J Hepatol. 1992;14(2–3):342–346. doi: 10.1016/0168-8278(92)90181-n. [DOI] [PubMed] [Google Scholar]
- 43.Bruggeman SW, Hulsman D, Tanger E, et al. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell. 2007;12(4):328–341. doi: 10.1016/j.ccr.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 44.Liu J, Cao L, Chen J, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009 doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tward AD, Jones KD, Yant S, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0706578104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wojtowicz JM, Kee N. BrdU assay for neurogenesis in rodents. Nat Protoc. 2006;1(3):1399–1405. doi: 10.1038/nprot.2006.224. [DOI] [PubMed] [Google Scholar]
- 47.Nelsen CJ, Rickheim DG, Tucker MM, Hansen LK, Albrecht JH. Evidence that cyclin D1 mediates both growth and proliferation downstream of TOR in hepatocytes. J Biol Chem. 2003;278(6):3656–3663. doi: 10.1074/jbc.M209374200. [DOI] [PubMed] [Google Scholar]
- 48.Cheng G, Lewis AE, Meinkoth JL. Ras stimulates aberrant cell cycle progression and apoptosis in rat thyroid cells. Mol Endocrinol. 2003;17(3):450–459. doi: 10.1210/me.2002-0344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.