

Abstract
Nasal-type natural killer (NK) cell lymphoma is an infrequent aggressive malignant disease with very poor prognosis. We aimed to explore the possible role of the transcription factor STAT3 in the pathophysiology of this malignancy, as it was involved in oncogenesis and chemoresistance. For this, we established and characterized a continuous interleukin 2-dependent NK cell line (MEC04) from a patient with a fatal nasal-type NK cell lymphoma. Cells harbored poor cytotoxic activity against K562 cells, and spontaneously secreted interferon-γ, IL-10 and vascular-endothelium growth factor in vitro. STAT3 was phosphorylated in Y705 dimerization residue in MEC04 cells and restricted to the nucleus. Y705 STAT3 phosphorylation involved JAK2, since exposure of cells to AG490 inhibitor inhibited Y705 STAT3 phosphorylation. By using recombinant transducible TAT-STAT3β (βisoform), TAT-STAT3Y705F (a STAT3 protein mutated on Y705 residue which prevents STAT3 dimerization), and peptides inhibiting specifically STAT3 dimerization, we inhibited STAT3 phosphorylation and cell growth, with cell death induction. Finally, STAT3 was phosphorylated in Y705 residue in the nuclei of lymphoma cells in 8/9 patients with nasal-type NK/T cell lymphoma and in YT, another NK cell line. Our results suggest that STAT3 protein has a major role in the oncogenic process of nasal-type NK cell lymphomas, and may represent a promising therapeutical target.
Keywords: Animals; Extracellular Signal-Regulated MAP Kinases; physiology; Female; Humans; Interferon-gamma; biosynthesis; Janus Kinase 2; physiology; Killer Cells, Natural; pathology; Lymphoma, T-Cell; etiology; genetics; immunology; pathology; Male; Mice; Mice, SCID; Middle Aged; Nose Neoplasms; etiology; genetics; immunology; pathology; Phosphorylation; STAT3 Transcription Factor; antagonists & inhibitors; physiology; bcl-X Protein; physiology
INTRODUCTION
Natural killer (NK)/T cell lymphomas have been recently recognized as a distinct lymphoma subtype within the WHO classification 1,2. Clinically, it is a highly aggressive disease that typically presents as destructive lesions localized in the nasal cavity, the nasopharynx, paranasal sinuses or palate, and shows a predilection for Asians, Mexicans and South Americans. Nasal NK/T cell lymphomas derive putatively from natural killer (NK) cells or more rarely from a subset of γδ or αβ cytotoxic T cells and show a striking association with EBV 3. This latter is demonstrated by the presence of EBV genomes in the neoplastic cells using in situ hybridization with probes directed against EBERs and a type 2 latency with variable expression of the latent membrane protein 1 (LMP-1) and absence of EBNA-2 4. End-stage disease may also be accompanied by a leukemic phase and may involve other extranodal sites such as liver, spleen, bone marrow, central nervous system, lung or skin. Pathological features of nasal-type NK/T cell lymphomas are typically characterized by prominent tissue necrosis, as a consequence of angiocentric growth pattern and vascular damage with angioinvasion of the wall of vessels by lymphoma cells which show a high range of cytological appearance and frequent apoptotic figures. Nasal-type NK/T cell lymphomas are commonly complicated by hemophagocytic syndrome, which generally heralds death. This disorder is characterized by a poor outcome with only 39% and 49% 5-year event free survival and overall survival, respectively, due to chemoresistance 5. Few putative oncogenic mechanisms were reported. These involve mutations of genes regulating apoptosis such as Fas and p53 6–10, as well as expression of expression of P-glycoproteins 11. Moreover, a low expression level of the granzyme B protease inhibitor 9 (PI9) was associated with a worse prognosis and cell dedifferentiation, suggesting a role for this protein in the pathophysiology of these malignancies 5. However, the mechanisms involved in the pathogenesis and resistance to treatment still remain poorly understood, and the deep research for this special tumor has been hampered by its rarity and insufficient supply of the tumoral samples 12. Indeed, only a very small number of bona fide NK leukemia-lymphoma cell lines have been described in detail 13–16 (for review, see 17).
STATs (Signal transducers and activators of transcription) are transcription factors usually activated in response to cytokines and growth factors 18. Physiologic ligand-dependent activation of STAT regulatory cascades is usually associated with modulation of cell growth and differentiation. Accordingly, a constitutive activation of STAT proteins may result in malignant transformation 19,20. In particular, a large variety of neoplastic primary cells as well as tumor-derived cell lines from patients harbor constitutively activated STAT proteins, especially STAT3 21. The oncogenic role of STAT3 was particularly well documented in anaplastic large cell lymphoma, a lymphoma also derived from cytotoxic cells 22,23,24. STAT3 activation was shown to provide a growth advantage to cells, but also confers resistance to conventional chemotherapies that rely on apoptotic machinery to eliminate tumor cells 25.
In this work, we developed a continuous cell line from a patient with fatal disseminated nasal-type NK cell lymphoma. The poor prognosis of this malignancy and its usual chemoresistance suggested us that the transcription factor STAT3 may have a role in the oncogenic process. We could show that STAT3 was involved in survival and proliferation of malignant cells. Moreover, biopsy samples obtained from 8 patients with NK cell lymphoma also harbored constitutive STAT3 activation. Altogether, our findings provide strong evidence that STAT3 plays a major role in the oncogenic mechanisms of this disease, and may represent a promising therapeutical target.
PATIENT, MATERIALS AND METHODS
Patient
A 48 year-old caucasian female patient with no past medical history was referred to our Department for recent fever with cytopenias and a 6 months lasting nasal obstruction resembling sinusitis. A computerized tomography revealed a marked mucosal thickening with a filling of both maxillar sinuses and lytic bone destruction. A right maxillary sinus biopsy was consistent with the diagnosis of nasal-type NK/T cell lymphoma according to the WHO classification. Hemoglobin level was 9.6 g/dL, white blood cell count was 19.1×109/L including 17% circulating lymphoma cells and platelets 17×109/L. Marrow aspirate showed evidence of massive erythrophagocytosis in association with hyperferritinemia and hypertriglyceridemia (35000 ng/mL and 7,2 mmol/L, respectively), consistent with the diagnosis of hemophagocytic syndrome. T cell receptor loci were in germline configuration and EBV genome was found in neoplastic cells, resulting in a final diagnosis of nasal-type NK cell lymphoma. Besides blood and marrow infiltration by malignant cells, lymphoma staging revealed liver, pancreas, gut and lung involvement. Quantification of circulating EBV DNA as assessed by LMP1 DNA amplification disclosed a high viral load (5×106 copies/mL). A chemotherapy including aracytine, etoposide and novantrone was performed with transient and partial efficacy. A salvage chemotherapy associating vincristine, methotrexate and L-asparaginase was undergone with no efficacy, and the patient died at day 10 of this treatment (and three months after the initial diagnosis) in a context of multiorgan failure due to Candida glabrata septicemia and disseminated Aspergillus fumigatus infection.
Samples
Parafin-embedded blocks of the tumour biopsies of the nasal lesion obtained at diagnosis and of the intestine at relapse were collected for histological review, immunohistochemical analysis and EBV studies. For assessment of phospho-STAT3 expression in nasal NK/T cell lymphoma, tumour samples from eight other patients with nasal-type NK/T cell lymphoma were retrieved from the file of the Departement of Pathology (Hôpital Henri Mondor, Créteil). Smooth muscle cells as well as endothelial cells were used as internal positive controls while two cases of ALK+ anaplastic large cell lymphoma were used as external positive controls. For the construction of paraffin-embedded cell block, cells were at first resuspended in 100% ethanol, then transferred onto a filter paper for automated processing of a paraffin block. Phospho-STAT3 expression was also assessed in YT, an additional NK-cell line, as well as in 2 cases of hepatosplenic T-cell lymphoma and in DERL-2 and DERL-7 cell lines, derived from a patient with an hepatosplenic T-cell lymphoma.
Normal NK cells were obtained from peripheral blood mononuclear cells (PBMC) from 6 normal individuals. NK cells were sorted by negative selection using anti-CD3 and anti-CD19 antibodies (Pharmingen, San Diego, CA, USA). CD3- and CD19-negative cells were purified using a FACSVantage cytometer (Beckton-Dickinson). The final purity of normal CD56+ cells was > 95%.
Informed consent was obtained from the patient and normal individuals. The study was approved by the institutional review board at Hôpital Saint-Antoine (Paris).
Generation of MEC04 cell line
The cell line presented here was established according to previously published guidelines 26. Peripheral blood mononuclear cells were isolated from the patient by density centrifugation of buffy coat through Ficoll-Hypaque (Nycomed Pharma AS, Oslo, Norway). Cells were cultured in RPMI 1640 supplemented with 2 mM L-glutamine and 10% heat-inactivated human serum, in the presence of recombinant interleukin-2 (rIL-2) (Laboratoires Chiron, Amsterdam) 100 U/L.
In some experiments, rIL-2 starved malignant cells were incubated with AG490, PD98056 (at concentrations ranging from 5 to 50 μM) or with a cell permeable STAT3 phosphorylated blocking peptide used at a final concentration of 2 mM (Calbiochem, La Jolla, CA, USA). This latter contains a membrane translocating sequence, a set of hydrophobic amini-acids for membrane permeability (sequence: PY*LKTK-AAVLLPVLLAAP) (Turkson et al., 2001). Vehicle was 0.1% dimethyl sulfoxide (DMSO) for AG490 or PD98056. For STAT3 blocking peptide, a peptide with the same amino-acid sequence, but not phosphorylated, was used as control (PYLKTK-AAVLLPVLLAAP) (Mimotopes, Clayton Victoria, Australia) at the same concentration. The percentage of living cells was evaluated using the trypan blue dye exclusion assay.
Animals
Six-week old male mice (NOD/SCID) were injected with MEC04 cells (10×106/mouse) intraperitoneally after a 3.5 Gy irradiation, and their outcome was compared to normal counterparts. Ten mice were examined weekly until all injected animals died. Histopathological analysis was performed in one diseased mouse. Malignant cells were stained with antibodies directed against granzyme B, Tia1, CD7 (Novocastra Reagents, Leica Microsystems GmbH) (1:80), and by in situ hybridization (ISH) with fluorescein-conjugated EBV (EBER 1 and 2) oligonucleotides (DakoCytomation, Glostrup, Denmark) complementary to nuclear RNA portions of the EBER 1 and 2 genes.
Cell staining and fluorescence-activated cell sorting (FACS) analysis
Rat monoclonal antibodies that recognize human hematopoietic cell-surface antigens were used to characterize phenotypically MEC04 cells, and were all purchased from Pharmingen (San Diego, CA, USA). We used fluoro-isothiocyanate (FITC)- and phycoerythrin (PE)-conjugated antibodies directed to surface CD2, CD3 (sCD3), intracytoplasmic CD3 epsilon chain (iCD3ε), CD4, CD7, CD8, CD16, TCRαβ and TCRγδ, CDCD19, CD56, granzyme B, CD40L, CD95 (Fas) and CD95L (Fas-ligand), CD57, and CD152 (CTLA-4) antigens. The following NK-cell antigens were also tested: killer immunoglobulin-like receptors (KIR) family (p58a, b, c, and p70), lectins family (NKG2A), and NCR family (NKP30, 44, and 46). Cells were incubated for 20 min at 4°C with each antibody. After washing, they were suspended in PBS 5% FCS supplemented with 7-amino-actinomycin D (7-AAD, Sigma) to exclude dead cells. Cell-surface labeling was analyzed by flow cytometry on a FACScan (Becton-Dickinson, San Jose, CA, USA). Data were acquired from 104 cells in list mode and analyzed with CellQuest software (Becton-Dickinson).
NK-cell cytolytic assay
MEC04 and NK92 NK-cell lines cytotoxicity was evaluated against the MHC class I-deficient human erythroleukemia K562 cell line in a standard 4-hour 51Cr release assay, as previously described 27.
ELISA
Quantification of interferon-γ, IL-10, vascular-endothelial growth factor (VEGF), transforming growth factor (TGF)β and tumor necrosis factor (TNF)α and β in culture supernatant was performed using an enzyme immunoassay kit (R&D System) after overnight rIL-2 deprivation. Supernatants of normal human NK cells grown in the same conditions were used as control.
Quantification of EBV DNA and detection of LMP-1 variant
Quantification of EBV-DNA was performed as well from MEC04 cell line established from PBMC of the patient as from culture supernatant using quantitative real time PCR assay as previously described 28. Furthermore, LMP-1 gene variant was detected on the basis of overlapping PCR assay amplifying LMP-1 sequence. The assay used the designed primers as follows: forward: 5′ atg aca tgg taa tgc cta gaa gta aag 3′; reverse: 5′aca att gat gga aga ggt tga aaa 3′. PCR products were analyzed by electrophoresis in a 3% agarose gel containing 2 μg/ml ethidium bromide. A 231 bp length from B95-8 EBV strain is considered as wild-type LMP-1 gene after sequencing, whereas a deleted fragment is of lower length (i.e., 201 bp).
Immunohistochemistry, immunocytochemistry, and confocal microscopy
Immunohistochemistry and immunocytochemistry were performed as previously described 29, by using an anti-Y705 STAT3 antibody (Biolabs, Cell Signaling technology) (dilution 1:50, antigen retrieval with 1x citrate buffer pH 6.0, incubation overnight at 4°C). The detection of EBV infected cells was performed using either immunohistochemistry with a monoclonal antibody to LMP-1 (CS1-4, DakoCytomation) or ISH with fluorescein-conjugated EBV (EBER1 and 2) oligonucleotides (DakoCytomation, Glostrup, Denmark) complementary to nuclear RNA portions of the EBER 1 and 2 genes. The ISH procedure was performed on deparaffinized tissue sections, as previously reported 3.
For confocal study, cells were cytospun on slides (5.104 cells/slide) during 2 minutes at 180g, fixed with cold methanol for 10 minutes and paraformaldehyde 4% for 20 minutes at room temperature, and then permeabilized with 0.2% Triton X-100 for 5 minutes at room temperature. The slips were washed 3 times with PBS 1X, and cells were blocked with 10% horse serum, bovine serum albumin 1%, NaN3 0.02% in PBS 1x for 45 minutes at room temperature. Cells were incubated overnight with polyclonal anti-STAT3 primary antibody (1:100) (Biolabs, Cell Signaling technology) in PBS 1X and bovine serum albumin at 4°C. After 3 washes, cells were incubated with second antibody labeled with PE (1:200) for 1 hour. Mounting was performed using Vectashield medium (Vector Laboratories, Burlingame, CA) including DAPI (4′, 6-diamino-2-phenyindole) fluorochrome for nuclei staining. Slides were examined using a laser confocal microscope (Leica Microsystems, Heidelberg GmbH).
Western blotting and antibodies
Western-blotting experiments were performed as previously described (Coppo et al., 2003). Antibodies were purchased from Biolabs (anti-phospho-Y705 STAT3, anti-phospho-Ser727 STAT3, anti-STAT3, anti-phospho-Erk1/2, and anti-Erk1/2 antibodies), Santa Cruz Biotechnology (anti-phospho-Y1007/Y1008 JAK2 antibody), Upstate Biotechnology (anti-phospho-Y694 STAT5, and anti-STAT5 antibodies) and DakoCytomation (anti-LMP-1 antibodies, CS1-4). They were diluted 1:1000 (anti-STAT3, anti-STAT5, anti-Erk1/2 and anti-LMP-1 antibodies) or 1:500 (anti-phospho-Y705 STAT3, anti-phospho-Ser727 STAT3, anti-phospho-JAK2, and anti-phospho-Erk1/2 antibodies).
Purification of TAT proteins
Purification of TAT proteins was performed as previously described (Harir et al., 2007). Briefly, BL21 (DE3) pLysS bacteria (Stratagene, Amsterdam, The Netherland) expressing the TAT fusion proteins were grown in LB broth medium containing 50 μg/ml ampicillin and 34 μg/ml chloramphenicol at 37°C. TAT-STAT3β, TAT-STAT3 Y705F, and TAT-wild-type STAT5 protein expression was induced by addition of 1 mM IPTG at 37°C during 4 hours. Cells were harvested and sonicated in buffer A (8 M urea, 20 mM HEPES (pH = 8.0), and 500 mM NaCl). Lysates were clarified by centrifugation at 15 600g for 4 minutes at 4°C. The supernatants containing the recombinant TAT fusion proteins were equilibrated in 12 mM imidazole and then applied to Ni-Nta agarose columns (Qiagen, Courtaboeuf, France). Columns were washed with 6 bed volumes of buffer B (4 M urea, 20 mM HEPES (pH = 8.0), 500 mM NaCl, and 12 mM imidazole). After washing, bound proteins were eluted with buffer B containing 250 mM imidazole. Eluates were dialyzed against PBS or 0.9% NaCl with 4 buffer changes overnight at 4°C. The purity and concentration of TAT fusion proteins were assessed by Coomassie blue-stained SDS-polyacrylamide gel electrophoresis (PAGE) using standard concentrations of BSA.
In vitro proliferation studies of cells transduced with TAT proteins
MEC04 cells were incubated as triplicates in flat-bottom 24-well plates at a density of 2 × 104 cells/well in RPMI 1640 medium with 10% FCS and 100 U/L rIL-2 and supplemented with the different TAT fusion proteins at the indicated concentration for 24 or 48 hours. The percentage of living cells was evaluated using the trypan blue dye exclusion assay.
Quantitative RT-PCR analysis of Bcl-XL and c-myc mRNA transcripts
Total RNA was purified from MEC04 cell line transduced or not with TAT-STAT3β and TAT-STAT3 Y705F by using Trizol reagent (Invitrogen, Carlsbad, CA), followed by treatment with DNAse I (Invitrogen) to discard any contaminating DNA. First strand cDNA was synthesized from 2 μg of total RNA using the Superscript II reverse transcriptase (Invitrogen) with 200 ng of random hexamers. Real time PCR was performed using an ABI PRISM 7700 Sequence Detection System (PE Applied Biosystems, Foster City, CA). C-myc, Bcl-XL and glyceraldehyde-3′-phosphate deshydrogenase (GAPDH) transcripts copy numbers were detected by the first PCR cycle during real-time analysis in which the initiation of exponential cDNA template amplification exceeds background by 10-fold (the threshold cycle (CT)). Primer sequences are listed in supplemental data (Table S1). PCR were performed in duplicate using the Taqman® master mix (PE Applied Biosystems) with 10 ng of cDNA. Calibration curves were established to verify that PCR efficiencies were similar for target genes and GAPDH. The relative quantification of target gene products of MEC04 cell line transduced or not with TAT-STAT3β and TAT-STAT3 Y705F was calculated by the 2−ΔΔct method 30. Genes were considered to be up-regulated when 2−ΔΔct was > 1, and down-regulated if < 1.
Statistical analysis
For analysis of cell proliferation and mortality, values represent the mean ± SD for at least 3 separate experiments. The significance of differences between experimental variables was determined by the use of the student t test, and P less than 0.05 was considered as significant.
RESULTS
1. Morphological and phenotypic analysis of primary cells
The original lymphoma biopsies at presentation (nasal) and at relapse (stomach) showed a pleomorphic proliferation of medium to large-sized atypical lymphoid cells with numerous apoptotic figures (Figure 1A), associated with an angiocentric growth pattern, angioinvasive features and areas of necrosis. Malignant cells had a CD3ε+(weak), CD5−, CD2+, CD7+, CD4−/CD8− and CD56+ phenotype (Figure 1B) with an activated cytotoxic (Tia1+) profile and expression of LMP-1 (not shown). In situ hybridization with EBER 1 and 2 probes showed that most tumor cells were positive for EBV (Figure 1C). These results were consistent with a diagnosis of nasal-type NK/T cell lymphoma.
Figure 1. Histopathology.
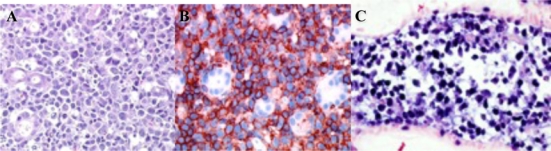
(A) Primary lesions show invasion of tumor cells to small blood vessels (Hematoxylin-eosin). (B) Tumor cells expressed CD56 (red staining). (C) In situ hybridization with EBER 1 and 2 probes was positive in primary malignant cells (blue staining). Original magnification, × 400. Images were captured with a Zeiss Axioscop2 microscope (Oberkochen, Germany) and Neofluar 100×/0.1 NA optical lenses (Zeiss). Photographs were taken with a DP70 Olympus camera (Tokyo, Japan). Image acquisition was performed with Olympus DP Controller 2002, and images were processed with Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
2. MEC04 NK cell line characteristics
Peripheral blood mononuclear cells obtained from the patient on diagnosis were cultured in the presence of rIL-2 in January, 2004. A continuous cell line, named MEC04, could be maintained for more than 3 years. This cell line is available upon request from the corresponding author. Cells remained IL-2-dependent and were mycoplasm free. Their doubling time was 20 hours. Cells had large nuclei with abundant basophilic cytoplasm containing scarce azurophilic granules (Figure 2A). T cell receptor (TCR)γ and immunoglobulin H (Framework region [Fr]1, Fr2 and Fr3]) loci were in the germinal conformation. Cytogenetical analysis disclosed a near tetraploid pattern with a complex formula with numerous numerical and structural chromosome changes as follows: 50–82, −Xx4[13],−3[12], add(3)(p?24)[3], −4[12], −5[12], del(6)(q12)[2], +del(6)(q12)[11], add(6)(q26) [11], del(7)(p1?2p1?5)[9], add(7)(p1?2)[7], −7[4], −8, −8[12],−9[3], −9,−9[10], −10, −10[13], −11[13], add(11)(p11)[4], +add(11)(p11)[8], add(11)(q22)[10], +add(11)(q22)[2], −12[9], −13[11], −14[3], −15[4], −16,[3], −17[4], −17, −17[8], add(18)(p11)x2[13], +20[3], +mar1[12], +1-7mars[11] cp[13]. As some structural abnormalities are present in duplicate, they occurred before endoreduplication, probably as some numerical one, while some others obviously occurred as secondary events (Figure 2B). Twelve abnormalities were systematically observed in 11 to 13 of the analyzed cells, which provides evidence for cell clonality. This finding also rules out a possible cross-contamination with another established cell line. The cell line was immunophenotyped 3 times within 2 years. Table 1 shows the last detailed immunophenotypical features of MEC04 cells performed 2 years after the cell line establishment. Immunophenotyping of MEC04 cells was similar to this of primary leukemic cells, with the exception for CD94, which expression was lost. This peculiar phenotypic profile strongly suggested that MEC04 originates from the CD56+bright NK cell population with regulatory functions. These cells normally comprise about 10% of circulating NK, and have the capacity to produce variable levels of cytokines such as interferon-γ, IL-10, VEGF, TGFβ, IL-13 and granulocyte-monocyte growth factor (GM-CSF). They also have a low natural cytotoxicity and express adhesion molecules such as CD62L (L-selectin) 31. To confirm this hypothesis, we tested the cytotoxic properties of MEC04 cells against K562 cells. We found that MEC04 cells were poorly cytotoxic against K562 cells when compared to the control NK92 cell line (Figure 2C). Moreover, MEC04 cells spontaneously released very high amounts of IL-10, and to a lesser extent interferon-γ and VEGF in the culture supernatant as compared to normal NK cells (Table 2). By contrast, there was no significant secretion of TNFα, TNFβ, or TGFβ (not shown). Taken together, these results provide evidence that MEC04 cells correspond to the CD56+bright NK cell subset 31.
Figure 2. Characterization of MEC04 NK cell line.
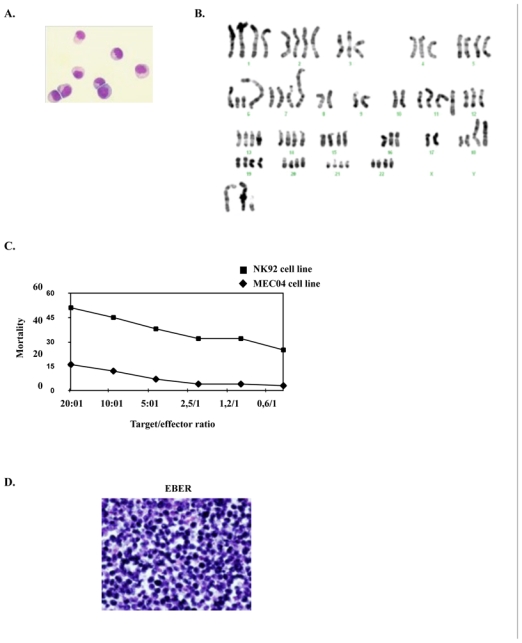
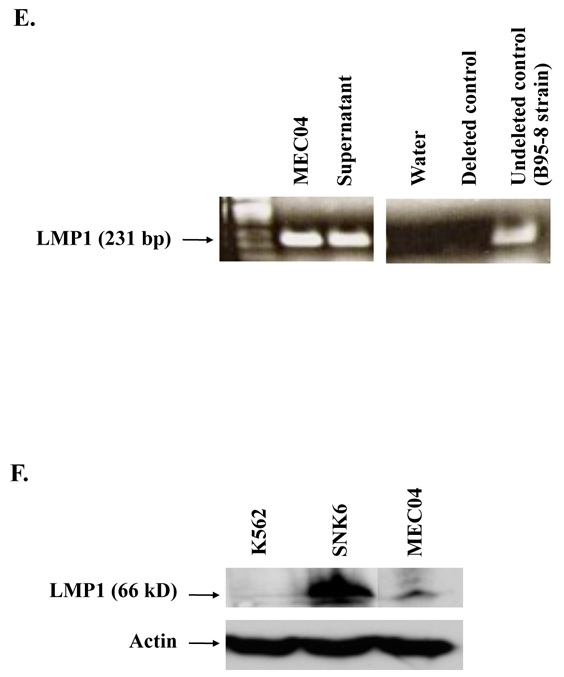
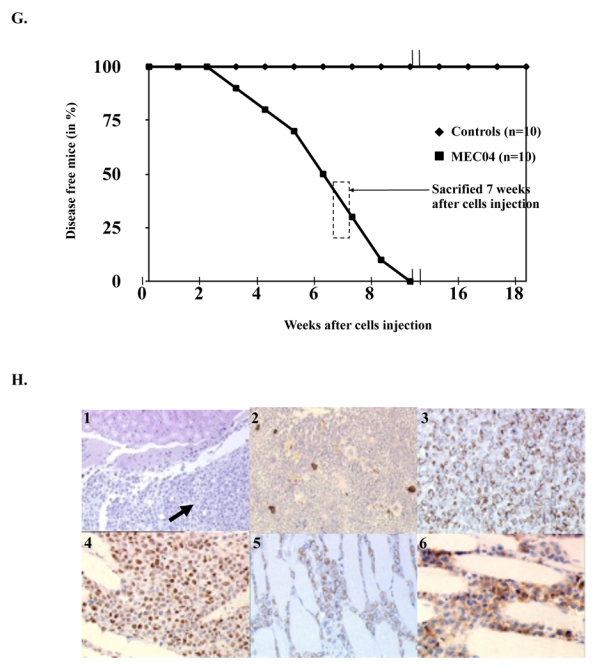
(A) Morphology of established MEC04 cell line (May-Grünwald Giemsa staining, original magnification, × 500). (B) A representative cytogenetical analysis of MEC04 cells. (C) Cytotoxicity of MEC04 CD56+bright NK cell line (◆) compared with CD56+dim cell line NK92 (■) against K562 target cells. Cytotoxicity for both NK cell lines was measured by 51Cr release assay against K562 target cells. (D) In situ hybridization of MEC04 cells with EBER1 and 2 probes showed that cells were associated with EBV (blue staining). (E) Evidence of lmp-1 DNA by PCR in MEC04 cells and culture supernatant. Lmp1 was amplified by quantitative PCR, electrotransferred in a 3% agarose gel. A single band corresponding to wild-type lmp-1 fragment (231 pb) was found in MEC04 cells and culture supernatant. (F) MEC04 cells were analyzed for the expression of LMP-1 protein by western-blotting. K562 is an erythroleukemic cell line used as a negative control, whereas SNK6 is a NK cell line expressing LMP-1 protein and used as a positive control. (G) Kaplan-Meier plot of NOD/SCID mice that were injected intraperitoneally with MEC04 cells (n = 10) (■) versus control NOD/SCID mice (◆) (n = 10). Mice that received MEC04 cells injection died within 3 to 9 weeks after peritoneal injection. The experiment was performed 3 times; a representative experiment is shown. One diseased mouse was killed and analyzed 7 weeks after cells injection (dashed box). (H) Typical tissue infiltration of mice that received MEC04 cells injection and killed 7 weeks after injection. Kidneys (infiltration of malignant mononuclear cells (black arrow)) (1), bone marrow (2), and peritoneum (3 to 6) were massively infiltrated. MEC04 cells were stained with antibodies against granzyme B (2, 5), Tia1 (3), EBER (4), and CD7 (6), brown staining. Original magnification, × 200 (panels 1, 3–6), × 100 (panel 2), Zeiss Axioscope.
Table 1.
Immunophenotypic analysis of NK cells on diagnosis and of the cell line.
| Antigen | Leukemic cells | MEC04 |
|---|---|---|
| NK antigens | ||
| CD16 | 3 | 4 |
| CD56 | 99 (bright) | 100 (bright) |
| p58a (CD158a) | 0 | 1 |
| p58b (CD158b) | 1 | 0 |
| p58c | 1 | 0 |
| p70 | 0 | 0 |
| NKG2A (CD159a) | 100 | 0 |
| NKP30 (CD337) | 2 | 2 |
| NKP44 (CD336) | 2 | 2 |
| NKP46 (CD335) | 3 | 7 |
| T cell antigens | ||
| CD1a | 8 | 0 |
| CD2 | 99 | 99 |
| Surface CD3 | 0 | 0 |
| Intracytoplasmic CD3ε | 21 | 19 |
| CD4 | 10 | 6 |
| CD5 | 20 | 18 |
| CD7 | 84 | 92 |
| CD8 | 3 | 3 |
| TCRαβ | 0 | 0 |
| TCRγδ | 0 | 0 |
| B cell antigens | ||
| CD19 | 0 | 0 |
| Adhesion molecules | ||
| CD62L | 0 | 0 |
| Activation antigens | ||
| CD69 | 58 | 100 |
| CD45 and its isoforms | ||
| CD45RA | 60 | ND |
| CD45RO | 2 | ND |
| Others | ||
| CD152 (CTLA-4) | 0 | ND |
| CD40L | 0 | ND |
| CD95/CD95L | 0 | ND |
Data are expressed as a percentage of positive cells. The phenotypic characterization was repeated 3 times for MEC04 cell line, with reproducible results. Data are referred to one representative characterization.
Table 2.
Quantification of cytokines concentration in culture medium.
| Control | MEC04 | |||
|---|---|---|---|---|
| − IL-2 | + IL-2 | − IL-2 | + IL-2 | |
| Interferon-γ (μg/ml) | 0 (0) | 47.5 (31) | 11.8 (2.5) | 81 (11) |
| IL-10 (pg/ml) | 0 (0) | 10.6 (14) | 4287 (1897) | 17616 (2282) |
| VEGF (pg/ml) | 0 (0) | 7.6 (10.8) | 41.2 (23.4) | 59 (16) |
IL-2: interleukin-2. IL-10: interleukin-10. VEGF: vascular-endothelial growth factor.
Values are given in pg/ml (±SE) for 106 cells/ml and represent the mean of at least 3 independent experiments. Control was a NK cell population from a healthy donor.
In situ hybridization with EBERs probes disclosed EBV genome in virtually all MEC04 cells (Figure 2D). LMP-1 genome amplification by polymerase chain reaction (PCR) performed from MEC04 cells and on the culture supernatant found a unique wild-type LMP-1 band, consistent with a clonal origin of MEC04 cells (Figure 2E). No LMP-1 deleted variant was detected in MEC04 cells or in the supernatant, warranting the absence of dual variants. The virions detected in the culture supernatant were sensitive to DNAse (not shown), suggesting that MEC04 cell line released cell-free fragments of EBV DNA by cellular lysis. By western-blot analysis, we could evidence that LMP-1 protein was expressed in MEC04 cells; the expression level was mild when compared to SNK6 cells (Figure 2F).
When injected intraperitoneally (10×106) to 10 NOD/SCID mice after a 3.5 Gy irradiation, MEC04 cells resulted in death in all animals within 4 to 9 weeks (Figure 2G). In our experiments, we chose to analyze mice at 7 weeks after injection. At autopsy, mice showed a massive infiltration of various organs (kidneys, peritoneum, and bone marrow) by MEC04 cells, which were stained for granzyme B, Tia1, CD3, CD7 (Figure 2H) and CD56 (not shown). None of these organs were found to display images suggesting hemophagocytic syndrome.
3. STAT3 is constitutively phosphorylated in MEC04 cell line. Involvement of JAK2 and MAP-Kinase/Erk pathway
The resistance to therapy as observed in our patient, as well as the high amounts of VEGF released spontaneously by malignant cells 32 suggested us that STAT3 was a promising candidate protein to account for oncogenesis in nasal NK-cell lymphoma. We thus examined phospho-STAT3 expression in primary cells from a gastric biopsy. We found that STAT3 was constitutively phosphorylated in the Y705 dimerization residue and localized in the nuclei of malignant cells (Figure 3A). Confocal microscopy analysis also confirmed that STAT3 was constitutively expressed in the nuclei of MEC04 cells (Figure 3B). From these results, we assumed that constitutive STAT3 activation might be a yet unrecognized target for intervention in nasal NK-cell lymphoma. Therefore, we studied this process in more detail.
Figure 3. STAT3 localization in primary biopsy and in MEC04 cells.
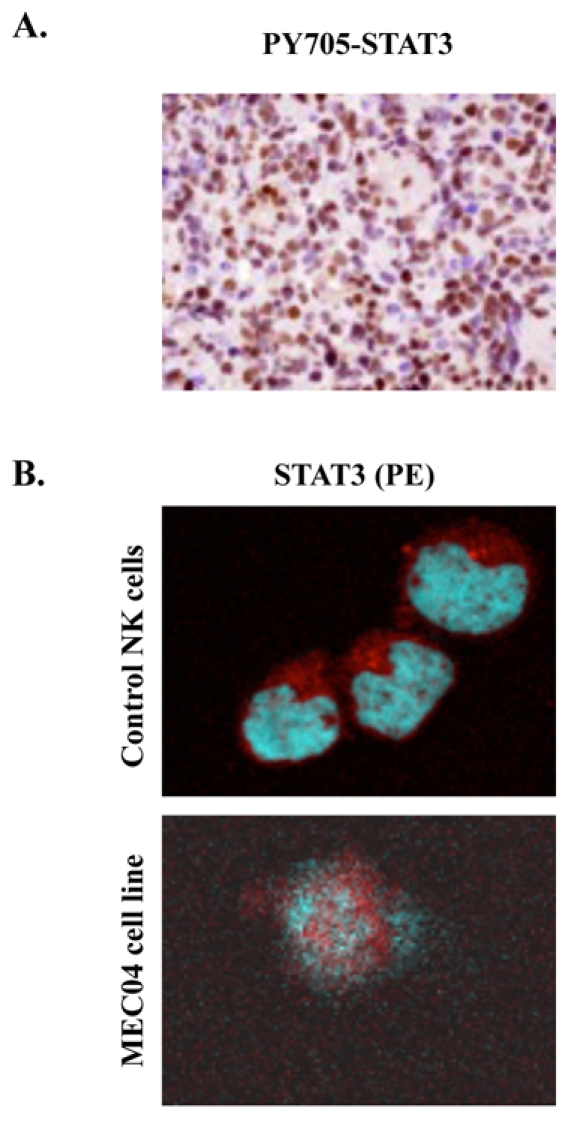
(A) Immunochemistry with anti-Y705-STAT3 shows a constitutive phosphorylation of Y705 STAT3 with a nuclear localization in primary cells from a gastric biopsy (brown staining) (original magnification, × 200, Zeiss Axioscope). (B) Evidence of STAT3 translocation in MEC04 cell line by confocal microscopic analysis. MEC04 cells were washed and were resuspended in culture milieu without rIL-2 overnight. Cells were fixed, permeabilized, and stained with DAPI (nucleus marker, blue), with anti-STAT3 (1:200), and visualized with anti-mouse PE (red)-labeled secondary antibody (1:100). Images were acquired on a Leica TCS SP2 laser scanning confocal microscope (Leica Microsystems, Heidelberg GmbH) equipped with a HCX PL Apochromat CS 63×1.40 Oil objective, using Leica Confocal Software (version 2.61). STAT3 is mainly detected in nucleus in MEC04 cells only. In contrast, control NK cells exhibit a cytoplasmic pattern of STAT3 staining.
Since STAT3 is usually activated by JAK proteins, we first attempted to inhibit STAT3 by using AG490, a chemical compound that inhibits JAK proteins, especially JAK2. For this, we exposed MEC04 cells 24 and 48 hours to the JAK2 inhibitor AG490 in the absence of rIL-2, and the number of living cells was estimated by the trypan blue exclusion method. We found that AG490 led to a significant dose-dependent growth inhibition (Figure 4A). By Western-blot analysis, JAK2 inhibition resulted in a decrease in the level of phospho-Y705 STAT3 in a dose-dependent manner. A dose-dependent decrease in phospho-Y1007/Y1008 JAK2 was also observed. Inhibition of JAK2 by AG490 also led to a dose-dependent inhibition of phospho-Ser727 STAT3 (Figure 4B). To explore further the mechanisms involved in STAT3 phosphorylation, we asked whether MAP-Kinase/Erk1/2 pathway could be involved in Ser727 STAT3 phosphorylation. For this purpose, we exposed MEC04 cells 24 hours to PD98056, a specific MEK1 inhibitor, the immediate upstream protein involved in Erk1/2 activation. We found that inhibition of MEK1 by PD98056 led to a decrease of Ser727 phosphorylation in a dose-dependent manner (Figure 4C). Comparable results were obtained by using UO126, a MEK1/2 inhibitor (not shown). These results suggest that in this model, JAK2 is dysregulated and leads to cell growth advantage by constitutive phosphorylation of Y705 STAT3, but also Ser727 through MAP-Kinase/Erk pathway.
Figure 4. Mechanisms involved in constitutive STAT3 phosphorylation.
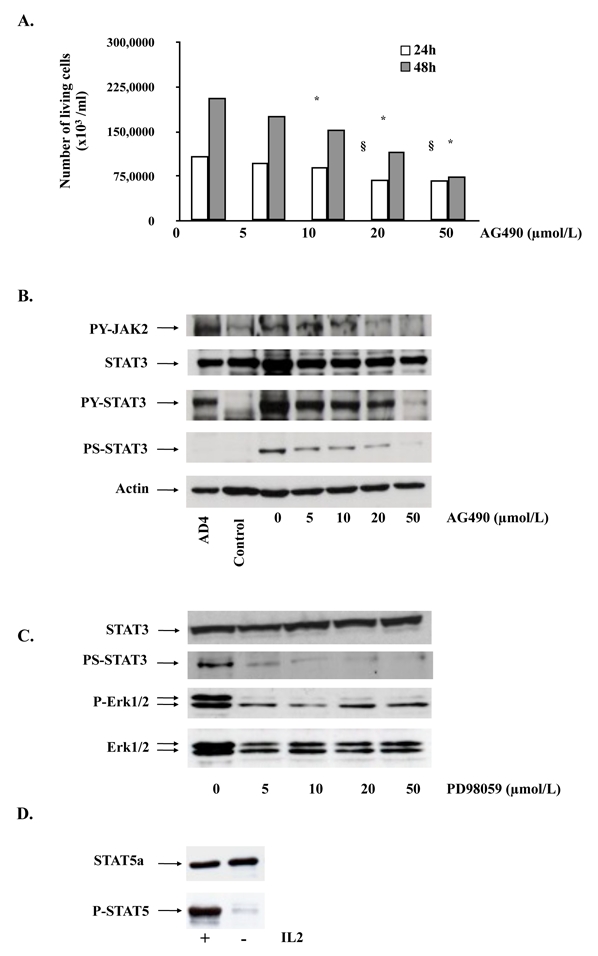
(A) MEC04 cells were exposed to increasing concentrations of AG490, and the cells were counted at 24 and 48 hours after treatment using the trypan blue dye assay. Results are mean of 3 independent experiments. The statistical significance was calculated by the Student t test. *, p<0.01; §, p<0.05. (B) MEC04 cells and control NK cells were cultured without rIL-2 for 24 hours and collected. MEC04 cells were exposed to AG490 or (C) PD98056, and collected 24 hours after treatment. Cell lysates were analyzed by western-blotting with the indicated antibodies. AD4 are murine embryonic stem (ES) cells expressing BCR-ABL oncogene and stimulated with murine leukemia inhibitory factor (LIF), which were used as a positive control of STAT3 phosphorylation. (D) MEC04 cells were cultured with or without rIL-2 overnight and collected. Cell lysates were analyzed by western-blotting for Y694-STAT5 phosphorylation.
We also attempted to assess whether STAT5, another STAT protein involved in oncogenesis would be involved in this model. For this purpose, MEC04 cells were analyzed for STAT5 phosphorylation by Western-blot analysis after rIL-2 deprivation overnight. We found that constitutive STAT5 phosphorylation was only barely detectable in MEC04 cells (Figure 4D).
4. Selective inhibition of constitutive activation of STAT3 induces growth inhibition and cell death of MEC04 cell line
To assess more accurately the oncogenic role of STAT3 in our cell line, we first selectively inhibited this molecule by overexpressing two distinct mutated forms of STAT3. The first one, STAT3β, is an isoform of STAT3 which was reported to act as a dominant-negative form of this latter 33. The second one bears a single point mutation on the tyrosine residue involved in the dimerization and the nuclear translocation of STAT3, which was changed to phenylalanine (Y705F) (Figure 5A). STAT3Y705F has been previously shown to block the activation of endogenous STAT3 in various cell types, possibly by titrating out receptor docking sites 34. As we failed to inhibit STAT3 with STAT3Y705F by using retroviral infections with MIGR-MSCV vector, we generated recombinant TAT-STAT3β and TAT-STAT3Y705F fusion proteins (Figure 5A). The use of TAT fusion proteins was reported to be an efficient approach to transduce either cell lines or primary cells 35. TAT-STAT3β and TAT-STAT3Y705F proteins were purified and quantified as described in “Patients, materials, and methods” and incubated (100 nM) with MEC04 cells for 24 and 48 hours in the absence of rIL-2. The number of living cells and cell mortality was estimated (Figure 5B). Since STAT5 was not constitutively phosphorylated in our cells, recombinant TAT-STAT5 fusion protein was used as control. Transduction of TAT-STAT3β and STAT3Y705F proteins resulted in 50% growth inhibition of MEC04 cell line (Figure 5B). By contrast, transduction of wild-type STAT5 (100 nM) did not affect growth and survival of MEC04 cells. Western-blot analysis with anti-phospho-Y705 STAT3 antibody showed a significant decrease in Y705 STAT3 phosphorylation 48 hours after transduction of TAT-STAT3β and STAT3Y705F proteins (Figure 5C). Cells were efficiently transduced as shown by western-blot analysis using anti-HA and anti-STAT3 antibodies (Figure 5C). We also verified by quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) that inhibition of endogenous STAT3 by STAT3β and STAT3Y705F resulted in the inhibition of STAT3 target genes such as Bcl-XL and c-myc (Figure 5D).
Figure 5. Specific inhibition of STAT3 activation.
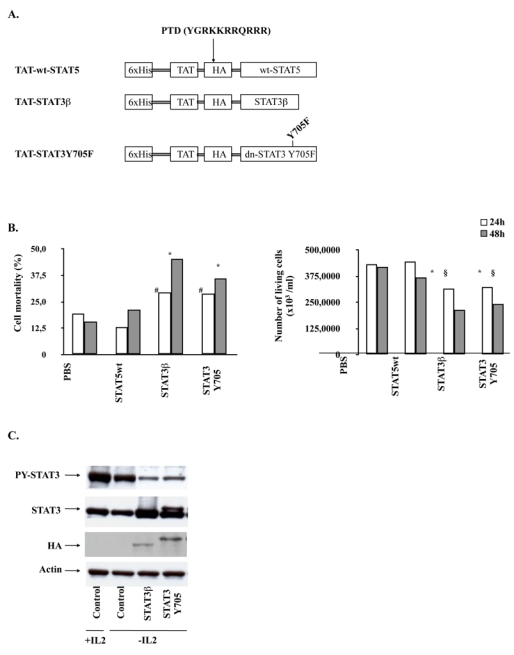
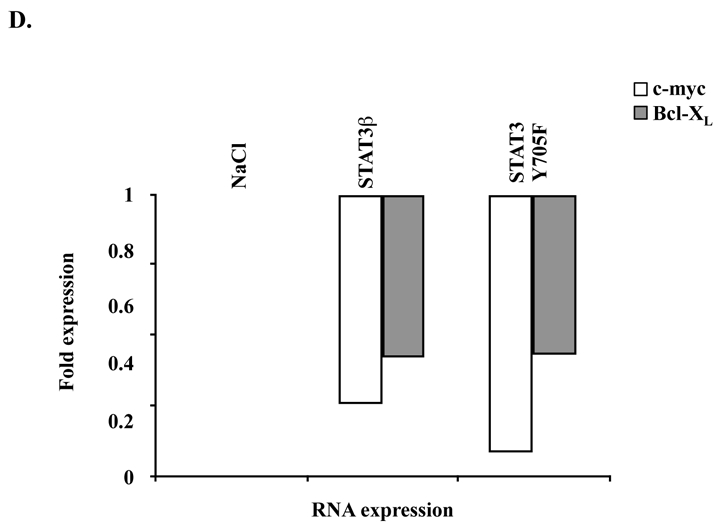
(A) Schematic representation of TAT-STAT3 and TAT-STAT5 constructs. The different cDNAs were introduced into the bacterial expression pTAT-HA. Resulting recombinant STAT3 (STAT3β or STAT3Y705F) and STAT5 proteins were fused in their N-terminal part to a 6 × His-Tag followed by the protein transduction domain (PTD) of the TAT protein and a HA Tag sequence. (B) MEC04 cells were transduced or not with 100 nM of the different TAT-STAT3 and TAT-STAT5 proteins during 48 hours. The mortality and the number of living cells was determined using the trypan blue dye exclusion assay. Results are the mean ± SD of 3 independent experiments. The statistical significance was calculated by the Student t test. #, <0.02; *, p<0.01; §, p<0.05 (C) After extensive washes, lysates from transduced MEC04 cells were prepared and analyzed by western-blotting with the indicated antibodies. (D) RNA was isolated from MEC04 cells 48 hours after transduction with TAT-STAT3β or TAT-STAT3Y705F recombinant proteins. Expression of c-myc and Bcl-XL mRNAs were evaluated by quantitative RT-PCR.
As an independent confirmation of the role of STAT3 in MEC04 cells oncogenesis, we also incubated these cells with a cell-permeable peptide that has been shown to block the effects of tyrosine-phosphorylated STAT3 36 by selectively preventing its binding to DNA 36. Incubation of MEC04 cells with 2 mM of STAT3 inhibitor peptide for 24 to 48 hours without rIL-2 resulted in a reduction of the cell number by almost 50% with a comparable increase in cell mortality (Supplemental data, figure S1A). By western-blot analysis, we showed that exposure of MEC04 cells to the STAT3 inhibitor peptide for 48 hours resulted in a decrease in Y705 STAT3 phosphorylation (Supplemental data, figure S1B).
5. Constitutive Y705 STAT3 phosphorylation in NK cell lines and in tumor cells from patients with nasal-type NK/T cell lymphomas
In order to determine whether the potential oncogenic role of STAT3 observed in MEC04 cells can be extended to patients with nasal-type NK/T cell lymphoma, we analyzed the expression of the phosphorylated form of STAT3 in YT (another NK cell line) and in 9 other patients with associated nasal NK/T cell lymphoma using an anti-phospho-Y705 STAT3 antibody. All these cases were positive for LMP and/or EBER (not shown). In 8/9 patients with interpretable results, the nuclei of the malignant cells were stained, providing evidence for a constitutive phosphorylation of Y705 STAT3 in these tumors (Figure 6 and data not shown). The percentage of positive cells were variable from 5% to more than 50%, and the staining intensity was heterogeneous, from weak to strong nuclear immunoreactivity (Table 3). STAT3 phosphorylation was also observed in YT cell line (Figure 6). By contrast, no Y705 phosphorylation was evidenced in 2 hepatosplenic T-cell lymphoma cell lines (DERL-2 and DERL-7), neither in 2 biopsies from patients with hepatosplenic T-cell lymphoma (data not shown).
Figure 6. STAT3 phosphorylation in biopsies of patients with nasal-type NK cell lymphoma and in cell lines.
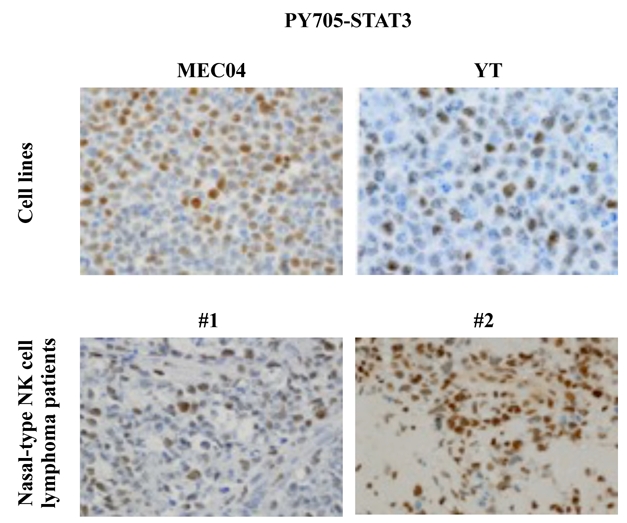
Immunochemistry shows a constitutive phosphorylation of Y705 STAT3 and a nuclear localization of this latter on MEC04 and YT cells and on neoplastic cells of 2 cases of nasal-type NK-cell lymphomas (brown staining) (original magnification, × 200, Zeiss Axioscope).\
Table 3.
Results of PY705-STAT3 immunostaining on blocks of the tumour biopsies.
| Sample # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| % of positive cells | < 5% | > 50% | 5–50% | 5–50% | 5–50% | 5–50% | > 50% | 5–50% | > 50% |
| Staining | W | M/S | W/M | W | W | W | S | W/S | W/M |
W: weak; M: moderate; S: strong
DISCUSSION
We report herein a new human NK cell line termed MEC04, derived from a nasal-type NK/T-cell lymphoma. Besides clinical presentation, arguments for the NK cell origin of our cell line is the NK-cell phenotype (i.e., negativity for typical B and T cell-lineage markers and positivity for CD56 and NKG2A markers), the germline configuration of T cell receptor (TCR) and immunoglobulin loci and the association of MEC04 cell line with EBV. Of note, this latter feature along with negativity for CD4 marker allowed us to rule out the diagnosis of plasmacytoid dendritic cell leukemia, initially considered as a blastic NK-cell lymphoma, and now considered as the malignant counterpart of normal plasmacytoid dendritic cells 37. Human NK cells are viewed as divided into two subsets based on the level of expression of CD56 at their surface, i.e., CD56bright and CD56dim, each with distinct properties 31. Different results presented here are in agreement with the fact that MEC04 cells originally correspond to the CD56bright NK cell subset. First, their immunophenotype was typical of CD56bright NK cells, i.e., a bright positivity for CD56 and no expression of CD16 neither of classical NK receptors, apart from NKG2A, which was found strongly positive in all cells on diagnosis. Second, MEC04 cells were poorly cytotoxic, and third, they released spontaneously very high amounts of interferon-γ and IL-10, which are cytokines reported to be classically released by CD56+bright NK cells. It is interesting to note that the massive cytokines secretion may at least in part account for the clinical picture of the patient. Indeed, the high interferon-γ production may result in hemophagocytic syndrome 38, whereas IL-10 may account for the wasting and the profoundly altered performans status. Of note, though MEC04 cells induced a fatal disease in mice, they could not reproduce a hemophagocytic syndrome in mice. This observation may be explained by the fact that interferon-γ has species-specific biological activities 39.
From the study of our cell line, we bring herein evidence for the role of STAT3 in nasal-type NK/T cell lymphoma oncogenesis. We first found using the MEC04 immortalized cells derived from a patient with a fatal nasal NK-cell lymphoma at a leukemic stage that STAT3 was constitutively translocated in the nuclei of malignant cells. In addition, STAT3 was constitutively phosphorylated on Y705 and Ser727 residues despite rIL-2 starvation. We showed that the oncogenic role of STAT3 was specific since its inhibition by two distinct STAT3 proteins with dominant-negative effect resulted in a significant decrease in survival and proliferation of malignant cells. Similar results were obtained with STAT3 inhibitory peptides. On the opposite, STAT5 had apparently no role in the oncogenesis in our model since rIL-2 starvation led to a complete inhibition of its phosphorylation. Of importance, we showed by immunochemistry a constitutively phosphorylated Y705 STAT3 in neoplastic cells of most primary nasal NK/T-cell lymphoma samples tested, as well as in YT, another NK cell line. These results indicate that the constitutive activation of STAT3 is widely involved in the pathogenesis of nasal NK/T cell lymphoma.
STAT3 is known to play a crucial role in regulating cell growth, survival, motility and cell transformation by regulating a large spectrum of proteins such as myeloid cell leukemia sequence 1 (Mcl-1), survivin, cyclin D1, Bcl-XL and Myc 25. One of the potential mechanisms implicated in STAT3 oncogenesis might be the over-expression of Bcl-XL, one of the most important survival factors expressed in constitutively activated STAT3-associated cancers 19. Indeed, promoters of Bcl-XL, as well as myc harbor multiple STAT3-binding sites, named acute-phase response elements or APRE sequences. In our model, we found that inhibition of activated STAT3 resulted in the down-regulation of Bcl-XL and c-myc expression, which suggests a role for these 2 target genes in resistance to apoptosis, cell cycling, and treatment refractoriness in nasal NK/T cell lymphomas.
In malignant cells, STAT3 may be activated through different signaling pathways 40,41. By the use of inhibitors, we found here that JAK2 protein is involved in the constitutive phosphorylation of both Y705 and Ser727 STAT3 residues, and that Ser727 STAT3 phosphorylation by JAK2 also involved MAP-Kinase/Erk1/2 pathway. In the light of these findings, JAK2 may be suggested as an additional promising candidate for targeted therapies, though these results need further confirmation with a more specific inhibition of JAK2. The mechanisms leading to a dysregulation of JAK2/STAT3 pathway in our model remain to be defined. Interestingly, these former may involve STAT3 itself, since STAT3 was reported to induce the expression of DNMT1, a DNA-methyl transferase involved in the methylation of tumor suppressor genes such as SHIP-1, which is involved in the dephosphorylation of activated JAK and STAT3 proteins 42,43. Activated STAT3 was also reported to have the capacity to amplify the transcription of its own gene, by binding specific consensus sequences within the promoter of this latter 44,45. The high amounts of cytokine release by MEC04 cells may be an additional mechanism by which JAK2/STAT3 pathway is dysregulated in our model. Indeed, several tumor-associated factors including IL-10 and VEGF are activators of STAT3 46,47,48, and may therefore be involved in sustained STAT3 activation through an autocrine and paracrine loop. Overall, these findings provide interesting evidences to suggest that dysregulated STAT3 may subsequently amplify and exacerbate its own oncogenic activity.
MEC04 cells were found to release wild-type LMP1 genome in culture supernatants as well as in vivo, consistent with a prior infection of the original NK cell by EBV, a characteristic feature of nasal-type NK/T-cell lymphomas 2. PCR analysis from MEC04 cells revealed a single wild-type LMP1 genome band, which shows that our cell line is clonal. The relationship between EBV infection and sustained activation of STAT3 is questionable. It might be hypothesized that a physical interaction between the cytoplasmic signaling domain of LMP-1 and JAK2, as reported for JAK3 49 could play a role. We attempted to co-immunoprecipitate LMP-1 and JAK2 in MEC04 cells, but we failed to provide evidence for such association (data not shown). However, intermediate proteins which may interact between LMP-1 and JAK2 such as TRAFs or TRADD may also be involved 50. Specific inhibition of LMP-1 would be of interest to assess the question whether LMP-1 is oncogenic in our system.
In conclusion, our work supports the first evidence that STAT3 has a major and selective role in the pathophysiology of nasal-type NK cell malignancies. The properties of MEC04 cells as well as the pattern of cytokines they secrete may explain number of clinical features of nasal-type NK cell lymphomas. Finally, STAT3 may account for the high resistance of malignant cells to chemotherapy, and specific therapies aimed at targeting STAT3 should be highly efficient in this disease. Further studies are now required to assess the possible involvement of additional signalling pathways, which may act in concert with STAT3 in the transforming process. The cell line we developed, as well as our model developed from mouse injected with MEC04 cells, may represent an interesting model to study the efficiency of pharmacological compounds in vitro and in vivo, and help to clarify the oncogenic role of EBV in such diseases.
Supplementary Material
Acknowledgments
We are indebted to Véronique Debuysscher, Aline Regnier (Inserm E0351, Faculté de Médecine d’Amiens, France), and Sandrine Malot (Service d’Hématologie et de Thérapie Cellulaire, Hôpital Saint-Antoine, Paris, France) for technical assistance, Peter Moller for providing the protocol for the construction of the paraffin-embedded cell block. We also thank Christian Schmitt (Inserm U841, équipe 2, Créteil, France) for providing DERL-2, DERL-7 and SNK6 cell lines and for invaluable comments.
Supported in part by a Grant from the GIS-Institut des Maladies Rares (GIS MR0428).
Footnotes
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu).
References
- 1.Jaffe ES HN, Stein H, Vardiman JW. Pathology and genetics, tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press; 2001. World Health Organization classification of tumours. [Google Scholar]
- 2.Jaffe ES. Pathobiology of peripheral T-cell lymphomas. Hematology Am Soc Hematol Educ Program. 2006:317–322. doi: 10.1182/asheducation-2006.1.317. [DOI] [PubMed] [Google Scholar]
- 3.Kanavaros P, Lescs MC, Briere J, Divine M, Galateau F, Joab I, et al. Nasal T-cell lymphoma: a clinicopathologic entity associated with peculiar phenotype and with Epstein-Barr virus. Blood. 1993;81:2688–2695. [PubMed] [Google Scholar]
- 4.Kanavaros P, Briere J, Emile JF, Gaulard P. Epstein-Barr virus in T and natural killer (NK) cell non-Hodgkin’s lymphomas. Leukemia. 1996;10 (Suppl 2):s84–87. [PubMed] [Google Scholar]
- 5.Bossard C, Belhadj K, Reyes F, Martin-Garcia N, Berger F, Kummer JA, et al. Expression of the granzyme B inhibitor PI9 predicts outcome in nasal NK/T-cell lymphoma: results of a Western series of 48 patients treated with first-line polychemotherapy within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2007;109:2183–2189. doi: 10.1182/blood-2006-07-033142. [DOI] [PubMed] [Google Scholar]
- 6.Quintanilla-Martinez L, Kremer M, Keller G, Nathrath M, Gamboa-Dominguez A, Meneses A, et al. p53 Mutations in nasal natural killer/T-cell lymphoma from Mexico: association with large cell morphology and advanced disease. Am J Pathol. 2001;159:2095–2105. doi: 10.1016/S0002-9440(10)63061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takakuwa T, Dong Z, Nakatsuka S, Kojya S, Harabuchi Y, Yang WI, et al. Frequent mutations of Fas gene in nasal NK/T cell lymphoma. Oncogene. 2002;21:4702–4705. doi: 10.1038/sj.onc.1205571. [DOI] [PubMed] [Google Scholar]
- 8.Takahara M, Kishibe K, Bandoh N, Nonaka S, Harabuchi Y. P53, N- and K-Ras, and beta-catenin gene mutations and prognostic factors in nasal NK/T-cell lymphoma from Hokkaido, Japan. Hum Pathol. 2004;35:86–95. doi: 10.1016/j.humpath.2003.08.025. [DOI] [PubMed] [Google Scholar]
- 9.Jeon YK, Kim H, Park SO, Choi HY, Kim YA, Park SS, et al. Resistance to Fas-mediated apoptosis is restored by cycloheximide through the downregulation of cellular FLIPL in NK/T-cell lymphoma. Lab Invest. 2005;85:874–884. doi: 10.1038/labinvest.3700291. [DOI] [PubMed] [Google Scholar]
- 10.Aozasa K, Takakuwa T, Hongyo T, Yang WI. Nasal NK/T-cell lymphoma: epidemiology and pathogenesis. Int J Hematol. 2008;87:110–117. doi: 10.1007/s12185-008-0021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi M, Kita K, Miwa H, Nishii K, Oka K, Ohno T, et al. Frequent expression of P-glycoprotein/MDR1 by nasal T-cell lymphoma cells. Cancer. 1995;76:2351–2356. doi: 10.1002/1097-0142(19951201)76:11<2351::aid-cncr2820761125>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 12.Zhao S, Tang QL, He MX, Yang F, Wang H, Zhang WY, et al. A novel nude mice model of human extranodal nasal type NK/T-cell lymphoma. Leukemia. 2008;22:170–178. doi: 10.1038/sj.leu.2404945. [DOI] [PubMed] [Google Scholar]
- 13.Tsuchiyama J, Yoshino T, Mori M, Kondoh E, Oka T, Akagi T, et al. Characterization of a novel human natural killer–cell line (NK-YS) established from natural killer cell lymphoma/leukemia associated with Epstein-Barr virus infection. Blood. 1998;92:1374–1383. [PubMed] [Google Scholar]
- 14.Kagami Y, Nakamura S, Suzuki R, Iida S, Yatabe Y, Okada Y, et al. Establishment of an IL-2-dependent cell line derived from ‘nasal-type’ NK/T-cell lymphoma of CD2+, sCD3-, CD3epsilon+, CD56+ phenotype and associated with the Epstein-Barr virus. Br J Haematol. 1998;103:669–677. doi: 10.1046/j.1365-2141.1998.01029.x. [DOI] [PubMed] [Google Scholar]
- 15.Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000;14:922–930. doi: 10.1038/sj.leu.2401769. [DOI] [PubMed] [Google Scholar]
- 16.Nagata H, Konno A, Kimura N, Zhang Y, Kimura M, Demachi A, et al. Characterization of novel natural killer (NK)-cell and gammadelta T-cell lines established from primary lesions of nasal T/NK-cell lymphomas associated with the Epstein-Barr virus. Blood. 2001;97:708–713. doi: 10.1182/blood.v97.3.708. [DOI] [PubMed] [Google Scholar]
- 17.Drexler HG, Matsuo Y. Malignant hematopoietic cell lines: in vitro models for the study of natural killer cell leukemia-lymphoma. Leukemia. 2000;14:777–782. doi: 10.1038/sj.leu.2401778. [DOI] [PubMed] [Google Scholar]
- 18.Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood. 2003;101:2940–2954. doi: 10.1182/blood-2002-04-1204. [DOI] [PubMed] [Google Scholar]
- 19.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 20.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 21.Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–1148. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21:1038–1047. doi: 10.1038/sj.onc.1205152. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol. 2002;168:466–474. doi: 10.4049/jimmunol.168.1.466. [DOI] [PubMed] [Google Scholar]
- 24.Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11:623–629. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 25.Aggarwal BB, Sethi G, Ahn KS, Sandur SK, Pandey MK, Kunnumakkara AB, et al. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann N Y Acad Sci. 2006;1091:151–169. doi: 10.1196/annals.1378.063. [DOI] [PubMed] [Google Scholar]
- 26.Drexler HG, Matsuo Y. Guidelines for the characterization and publication of human malignant hematopoietic cell lines. Leukemia. 1999;13:835–842. doi: 10.1038/sj.leu.2401428. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen S, Dhedin N, Vernant JP, Kuentz M, Al Jijakli A, Rouas-Freiss N, et al. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood. 2005;105:4135–4142. doi: 10.1182/blood-2004-10-4113. [DOI] [PubMed] [Google Scholar]
- 28.Clave E, Rocha V, Talvensaari K, Busson M, Douay C, Appert ML, et al. Prognostic value of pretransplantation host thymic function in HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2005;105:2608–2613. doi: 10.1182/blood-2004-04-1667. [DOI] [PubMed] [Google Scholar]
- 29.Chen CL, Hsieh FC, Lieblein JC, Brown J, Chan C, Wallace JA, et al. Stat3 activation in human endometrial and cervical cancers. Br J Cancer. 2007;96:591–599. doi: 10.1038/sj.bjc.6603597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 32.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 33.Yoo JY, Huso DL, Nathans D, Desiderio S. Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell. 2002;108:331–344. doi: 10.1016/s0092-8674(02)00636-0. [DOI] [PubMed] [Google Scholar]
- 34.Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harir N, Pecquet C, Kerenyi M, Sonneck K, Kovacic B, Nyga R, et al. Constitutive activation of Stat5 promotes its cytoplasmic localization and association with PI3-kinase in myeloid leukemias. Blood. 2007;109:1678–1686. doi: 10.1182/blood-2006-01-029918. [DOI] [PubMed] [Google Scholar]
- 36.Turkson J, Ryan D, Kim JS, Zhang Y, Chen Z, Haura E, et al. Phosphotyrosyl peptides block Stat3-mediated DNA binding activity, gene regulation, and cell transformation. J Biol Chem. 2001;276:45443–45455. doi: 10.1074/jbc.M107527200. [DOI] [PubMed] [Google Scholar]
- 37.Garnache-Ottou F, Feuillard J, Saas P. Plasmacytoid dendritic cell leukaemia/lymphoma: towards a well defined entity? Br J Haematol. 2007;136:539–548. doi: 10.1111/j.1365-2141.2006.06458.x. [DOI] [PubMed] [Google Scholar]
- 38.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104:735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 39.Hemmi S, Bohni R, Stark G, Di Marco F, Aguet M. A novel member of the interferon receptor family complements functionality of the murine interferon gamma receptor in human cells. Cell. 1994;76:803–810. doi: 10.1016/0092-8674(94)90355-7. [DOI] [PubMed] [Google Scholar]
- 40.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Blenis J, Li HC, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–1994. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–1064. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narimatsu M, Maeda H, Itoh S, Atsumi T, Ohtani T, Nishida K, et al. Tissue-specific autoregulation of the stat3 gene and its role in interleukin-6-induced survival signals in T cells. Mol Cell Biol. 2001;21:6615–6625. doi: 10.1128/MCB.21.19.6615-6625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–947. [PubMed] [Google Scholar]
- 46.Boulland ML, Meignin V, Leroy-Viard K, Copie-Bergman C, Brière J, Touitou R, et al. Human interleukin-10 expression in T/natural killer-cell lymphomas: association with anaplastic large cell lymphomas and nasal natural killer-cell lymphomas. Am J Pathol. 1998;153:1229–1237. doi: 10.1016/S0002-9440(10)65667-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kasprzycka M, Marzec M, Liu X, Zhang Q, Wasik MA. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc Natl Acad Sci U S A. 2006;103:9964–9969. doi: 10.1073/pnas.0603507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 49.Gires O, Kohlhuber F, Kilger E, Baumann M, Kieser A, Kaiser C, et al. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. Embo J. 1999;18:3064–3073. doi: 10.1093/emboj/18.11.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eliopoulos AG, Young LS. LMP1 structure and signal transduction. Semin Cancer Biol. 2001;11:435–444. doi: 10.1006/scbi.2001.0410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.