

Abstract
A full understanding of the proteome will require ligands to all of the proteins encoded by genomes. While antibodies represent the principle affinity reagents used to bind proteins, their limitations have created a need for new ligands to large numbers of proteins. Here we propose a general concept to obtain protein affinity reagents that avoids animal immunization and iterative selection steps. Central to this process is the idea that small peptide libraries contain sequences that will bind to independent regions on a protein surface, and that these ligands can be combined on synthetic scaffolds to create high affinity bivalent reagents. To demonstrate the feasibility of this approach, an array of 4,000 unique 12-mer peptides was screened to identify sequences that bind to non-overlapping sites on the yeast regulatory protein Gal80. Individual peptide ligands were screened at different distances using a novel DNA linking strategy to identify the optimal peptide pair and peptide pair separation distance required to transform two weaker ligands into a single high affinity protein capture reagent. A synthetic antibody or synbody was created with 5 nM affinity to Gal80 that functions in conventional ELISA and pull-down assays. We validated our synthetic antibody approach by creating a second synbody to human transferrin. In both cases, we observed an increase in binding affinity of ∼1000-fold (ΔΔG = ∼4.1 kcal/mol) between the individual peptides and final bivalent synbody construct.
Introduction
In the post-genomic era, there exists a tremendous need for protein affinity reagents that can be used to explore the complexity and function of the proteome1-3. Although traditional antibodies are commonly used for this purpose, only a limited number of human proteins have antibodies that are available for use in standard cellular and molecular biology assays4,5. This observation is not always evident, as a disproportionate number of antibodies have been raised to a relatively small number of targets1. Antibodies are further limited by their slow production time, high cost, and poor stability. These problems have prompted researchers to develop synthetic affinity reagents that function with antibody-like properties, but avoid many of the problems associated with traditional animal immunization and hybridoma technologies6. Artificial antibodies currently being developed for this purpose include immunoglobulin domains (scFv, Fab, and Fv), a wide range of alternative protein scaffolds, nucleic acid aptamers, and some small molecule ligands7-14. While these protein affinity reagents are often easier to construct and engineer than traditional antibodies, the process of their discovery remains labor intensive and often requires iterative rounds of in vitro selection and amplification. Thus, new methods are needed to chemically synthesize protein affinity reagents on scales that are amenable to high throughput production15.
The main barrier to the development of synthetic antibodies has been the absence of effective methods for generating protein affinity reagents with high affinity to their target proteins. Most small molecule ligands isolated from combinatorial libraries have binding dissociation constants (Kd) in the micromolar range, while typical commercial monoclonal antibodies bind their targets with low nanomolar affinity. One solution to this problem is to create multivalent binding agents by combining two or more moderate affinity (1-10 μM) ligands on a synthetic tether or polymer16. Transitioning this approach to a general discovery platform requires developing methods to rapidly identify synthetic ligands to protein targets, and a simple and robust system to link these ligands into multivalent affinity reagents17.
Here we sought to develop a scalable method for generating high quality synthetic antibodies that we call synbodies. In contrast to many other synthetic antibody strategies, the central concept developed here (Figure 1) is that i) small libraries of short, presumably unstructured, polypeptides would have sufficient diversity to contain members that bind to independent sites on a protein target; ii) these peptides would be flexible enough to allow them to be linked into bivalent reagents; iii) engineerable materials such as DNA could be used to spatially constrain two or more peptides at different distances and orientations; and iv) the construct would result in a synbody with higher affinity than the individual peptide ligands alone. To test this concept, we identified peptides from a microarray that bind to independent sites on the yeast regulatory protein Gal80. We then used a novel DNA linking strategy that we refer to as combinatorial examination of ligands and linkers (CELL) to determine the optimal distance and strand orientation required to transform two weaker affinity ligands into a single high affinity reagent. The synbody produced by this process has an affinity of 5 nM, and detects Gal80 in ELISA and pull-down assays. We validated our approach by generating a second synbody to human transferrin, a common blood plasma protein. In both cases, we observed an increase in the binding affinity of ∼1000-fold between the individual peptides and final synbody construct.
Figure 1.
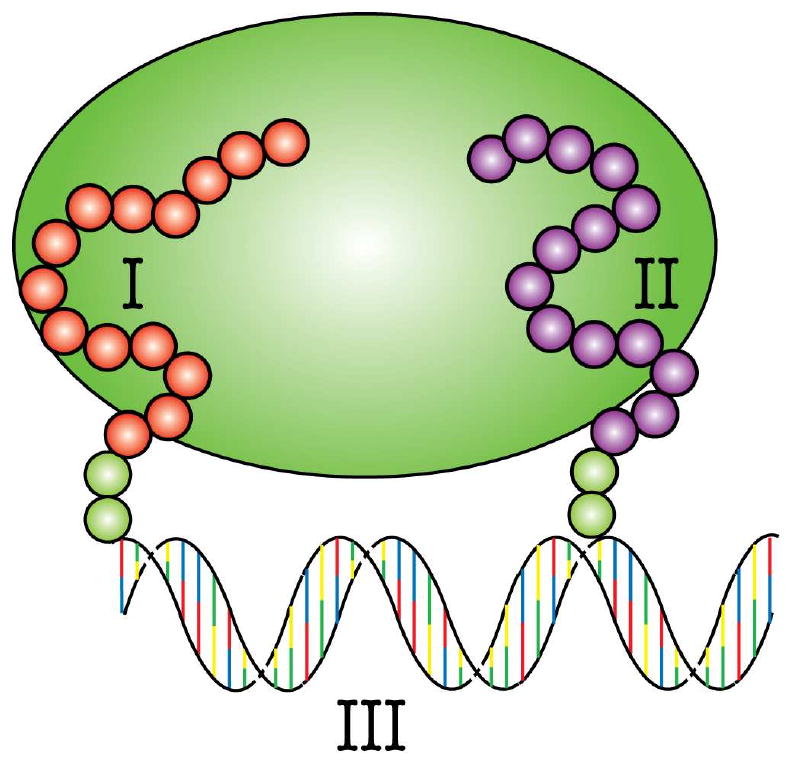
Illustration of the synbody concept. Synbodies are created by linking two peptides (I, II) that bind the target independently. In this case the linker (III) is double-stranded DNA.
Experimental Section
Protein Expression and Purification
The Gal80 protein was expressed and purified as previously described18. In brief, a pET28a plasmid containing the Gal80 insert was expressed in E. coli BL21 cells at 17 °C for 16 hrs. Soluble protein was purified by Ni-NTA affinity chromatography, separated from the column by thrombin proteolysis, and purified a second time on a heparin affinity column. The resulting Gal80 protein was concentrated and analyzed by SDS-PAGE and MALDI-TOF mass spectrometry.
Gal80 Peptide Microarrays
Peptide microarrays were used to identify lead peptides for the Gal80 synbodies. Four custom peptide microarrays were synthesized by LC Sciences (Houston, TX) that each contained 3919 unique 12-mer peptides, synthesized from C- to N-terminus on an amino propylsilane slide spaced by a Ahx-βAla-βAla linker19. Individual sequences were chosen from eight natural amino acids (Gly, Thr, Glu, Lys, Ser, Trp, Leu, Arg) using a random sequence generator. Gal80, transferrin, and α1-antitrypsin proteins were individually labeled with Cy3 and Cy5 fluorescent dyes and applied to the arrays in four combinations: (i) α1-antitrypsin-Cy3 + transferrin-Cy5; (ii) α1-antitrypsin-Cy5 + transferrin-Cy3; (iii) Gal80-Cy5 + Gal80-Cy3 (blocked with Gal4 activation domain (AD) peptide); and (iv) Gal80-Cy3 + Gal80-Cy5 (blocked with Gal4 AD peptide). Gal80 was analyzed in the absence and presence of the synthetic Gal4 AD peptide ligand (MDQTAYNAFGITTGMFNTTTMD DVYNYLFDDEDT) by preincubating the protein with the ligand (1.5 μM) prior to microarray analysis20. In each case, the array surface was blocked with 1% BSA, 0.5% non-fat milk, and PBS-Tween (0.05% Tween-20 in PBS buffer, pH 7.4), washed three times with PBS-Tween, and incubated with the labeled protein (1 μM) in blocking buffer overnight at 4 °C. Fluorescent images of the array were acquired by scanning the array at Cy3 and Cy5 wavelengths (570 and 670 nm, respectively) using an Axon GenePix 400B Microarray Scanner (Molecular Devices).
Transferrin Peptide Microarrays
Transferrin peptide ligands were identified from a 10,000-feature microarray of 20-mer peptides. The microarray was generated in-house as described previously21. In brief, random peptides were designed to contain any amino acid with the exception of cysteine, which was added to the C-terminus of each sequence. The peptides were synthesized by Alta Biosceinces Ltd (Birmingham, UK) at >70% purity and diluted in PBS with 25% DMS to 1 mg/ml. The peptides were spotted onto a sulfo-SMCC activated polylysine slide, quenched with mercaptohexanol and stored under argon at 4 °C. The peptide microarray was blocked with 1% BSA and E. coli lysate. The slide was washed 3× with TBST buffer (tris-saline buffer with 0.05% tween) followed by 3 washes with water. Human serum transferrin protein (Sigma) was labeled with Alexa-555 and E. coli lysate was labeled with Alexa-647. Alexa-555 labeled transferfin (1.0 μM) and Alexa-647 labeled E. coli lysate were incubated with the microarry for 3 h at 24 °C. The slide was washed 3× with TBST buffer followed by 3 water washes. The slides were scanned for fluorescence at 565 and 665 nm, respectively (Figure S1, Supporting Information (SI)).
Gal80 Peptide Selection
Relative peptide binding values were calculated as the average of the Cy3 and Cy5 fluorescence intensity per peptide spot divided by the background fluorescence intensity of the array. Fluorescent binding values were obtained for all 3919 peptides against Gal80, transferrin and α1-antitrypsin. Gal80 binding peptides were identified as the subset of sequences that showed a 20-fold preference for Gal80 over two common blood proteins (transferrin and α1-antitrypsin). Gal80 binding peptides whose fluorescence intensity dropped by 4-fold or more in the blocking assay with the Gal4 AD peptide were classified as ligands overlapping the Gal80 AD binding site. These sequences were labeled AD1-4 as they mimicked the binding of Gal4 AD peptide. Sequences whose fluorescence intensity was not altered by the presence of the synthetic ligand were labeled BP 1-6, as these peptides recognized nonexclusionary regions on the protein surface. Table S1 lists the fluorescent intensity values of BP1-6 and AD1-4.
Transferrin Peptide Selection
Fluorescent binding values were determined for all 10,000 peptides present on the microarray for human transferrin. Alexa-647 labeled E. coli lysate was used to identify peptides with non-specific binding affinity. The ratio of fluorescent intensities of transferrin to E. coli lysate (λ= 565 to 665) was calculated for each peptide spot. Ten peptides were identified that had >5-fold more fluorescence for transferrin over E. coli lysate (Table S2, SI).
Peptide Characterization
Peptides BP1-6 and AD1-4 were synthesized with a Gly-Ser-Cys sequence at their C-terminus, purified by HPLC, and verified by MALDI-TOF mass spectrometry. The set of ten peptides were assayed in parallel for Gal80 binding by surface plasmon resonance using a Biacore FlexChip instrument. The surface was treated with 11-amino-1-undecanethiol hydrochloride (MUAM, Dojindo), and peptides were coupled to the array using the bifunctional coupling reagent sulfosuccinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC). Peptides were spotted at concentrations of 0.05, 0.1, 0.5, and 1.0 μM in PBS-Tween using the Spotarray 72 microarray (Perkin Elmer) with split-type pins (Telechem, SPM7 pins). Following spotting, the array was incubated in a humidity chamber for 1 h and allowed to air dry. The SPR chip was blocked for 3 cycles of 8 min each with 0.7 mg/mL BSA in PBS-Tween and 1 M mercaptohexanol. The matrix (20 × 18 immobilized peptides) was assayed for Gal80 binding by flowing the protein (1 μM) over the chip for 12 min with a flow rate of 1 mL/min and recording the protein dissociation for 10 min. Following each dissociation cycle, the surface was regenerated with a short pulse of 10 mM NaOH in 150 mM NaCl. All data was corrected by reference subtraction and analyzed using the Biacore software package.
Peptide Mapping to Gal80
The binding regions for BP1 and AD1 were determined by protein-protein interface mapping22. In brief, Gal80 was separately incubated with BP1 and AD1 at stoichiometric concentrations for 1 h in PBS buffer (pH 7.0). A negative control containing Gal80 without the BP1 or AD1 was also performed. Following incubation, a 1:1 mixture of bifunctional crosslinking reagent (BS2G-d0 and BS2G-d4, Fisher Scientific) was added to all samples and the mixture was allowed to stand for an additional 45 min. Unreacted crosslinker and peptides were removed from the reaction mixture using a 10 kDa spin-filter with an ammonium bicarbonate (pH 8.5) buffer exchange. The crosslinked samples were digested overnight at 37 °C with trypsin. Digested fragments were separated from undigested protein with a 10 kDa spin-filter and the flow-through was evaporated to dryness. Dry samples were dissolved in a minimal volume of 0.1% aqueous TFA, desalted using C-18 ZipTip (Millipore, Billerica MA) and analyzed by MALDI-TOF mass spectrometry (Figure S2A, SI). An additional analysis using Sulfo-SBED (Thermo Fisher Scientific) photoreactive crosslinker was required for the BP1 peptide due to significant formation of an intramolecular cyclic peptide product with the BS2G-d0:BS2G-d4 crosslinker. Analysis of BP1 was performed as described above with the exception that the samples were irradiated at 365 nm for 8 hours, and digest fragments were separated from undigested protein using a 10 kDa spin-filter, and the flow-through was incubated with monomeric avidin beads. The avidin beads were washed extensively and captured fragments were eluted with 50% aqueous acetonitrile containing 0.1% TFA. The eluent was evaporated and redissolved in a small volume of 50% aqueous acetonitrile containing 0.1% TFA and analyzed by MALDI-TOF mass spectrometry (Figure S2, SI).
Peptide Mapping to Transferrin
Transferrin peptides, TRF23 and TRF26, were immobilized in separate spin-column vials (ThermoFisher Scientific) on 25 μL of UltraLink Iodoacetyl Resin (ThermoFisher Scientific) via the C-terminal cysteine using the manufacturer's recommended protocol. A third spin-column vial containing UltraLink Resin quenched with β-mercaptoethanol was prepared as a negative control. 10 μM transferrin prepared in 1× PBS buffer was incubated with the prepared resin for 60 min., after which 0.5% (v:v) formaldehyde was added to the samples for 15 min. Formaldehyde crosslinking was quenched with the addition of 100 mM Tris pH 8.5. The resin was washed 3 times with 100 mM Tris pH 8.5, then 3 times with 10 mM glycine pH 2.5, 3 times with nanopure H2O and finally 3 times with 100 mM Tris pH 8.5. 300 nM proteomics grade Trypsin (Sigma-Aldrich) was prepared in 100 mM Tris pH 8.5 and incubated with the samples for 4 hours at 37 °C. After trypsin digestion, the resin was washed 3 times with 100 mM Tris pH 8.5, then 3 times with 10 mM glycine pH 2.5 and finally 3 times with nanopure H2O then dried by centrifugation in the spin-column vials. Approximately 20 μL of nanopure H2O was added to the bottom of each spin-column vial, below (not in contact with) the resin bed and the vial was partially closed with a screw-cap thereby creating a humid environment inside the vial. Formaldehyde crosslinks were reversed with heat by placing the vials in an oven at 70 °C overnight. Following crosslink reversal, 25 μL of 75% nanopure H2O 25% acetonitrile was added to the resin to dissolve free transferrin digested peptide fragments. This solution was spun to the bottom of the spin-column vial in a centrifuge then evaporated in a vacuum centrifuge leaving a faint white residue at the bottom. This residue was redissolved with 2 μL 1:1 Acetonitrile:H2O containing 0.1% trifluoroacetic acid and saturated α-cyano-4-hydroxycinammic acid matrix, then spotted on a MALDI-MS target plate and analyzed with MALDI-MS (Table S3, SI).
Synbody Construction
The peptides were conjugated to synthetic DNA (Table S4 and S5, SI) using standard amine coupling chemistry23 (Scheme 1, SI). The amine modified DNA oligonucleotide (Keck Facility, Yale University) was treated with 66 μL of succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) (1 mg/mL) in acetonitrile with 200 μL of DNA (20 nmol) in 0.1 M K2HPO4 buffer, pH 7.2. The mixture was incubated for 30 min at 24°C, ethanol precipitated, re-suspended in 200 μL of K2HPO4 buffer (0.1 M, pH 7.2), and incubated with 200 μL of peptide (100 nmol) for 3 h at 24°C. Conjugated DNA-peptide molecules were purified by denaturing PAGE, re-suspended in H2O, and quantified by UV absorbance. The template strand was conjugated to a second DNA molecule containing a 3′-biotin or thiol moiety by UV cross-linking. This was achieved by annealing the template-peptide conjugate (2 nmol) with a complementary DNA strand containing a 5′-psoralen and 3′-biotin or 3′-thiol (4 nmol) in cross-linking buffer (100 mM KCl, 1 mM spermidine, 200 mM Hepes, and 1 mM EDTA, pH 8.0), and irradiating the mixture with ultra violet light (366 nm) for 15 min. Cross-linked DNA-peptide fusions were again purified by denaturing PAGE, re-suspended in H2O, and quantified by UV absorbance. The disulfide bond on the cross-linked DNA was reduced prior to use by incubating the molecule in 10 mM tris(2-carboxyethyl) phosphine hydrochloride (TCEP) for 30 min at 24°C. Bivalent DNA-peptide fusion molecules were assembled by annealing the strands in 50 mM NaCl.
Gal80 Synbody Distance Screen
Bivalent DNA-peptide conjugates were printed onto an activated Flexchip surface using the same protocol described above for peptide printing. Following the immobilization step, the chip surface was blocked for 3 cycles of 8 min each with 0.7 mg/mL BSA in PBS-tween and 1 M mercaptohexanol. Two concentrations of Gal80 protein (1 μM and 100 nM) were prepared in running buffer and tested for binding to the 20 × 18 matrix of immobilized synthetic antibody constructs. Each protein solution was flowed across the chip surface for 12 min at 1 ml/min and protein dissociation was recorded for another 10 min under the same flow rate. At the end of each dissociation cycle, the surface was regenerated with a short pulse of 10 mM NaOH in 150 mM NaCl. All data were corrected by reference subtraction and analyzed using the Biacore software package.
Transferrin Synbody Distance Screen
Transferrin synbody constructs were assayed for relative binding using a Biacore T100 surface plasmon resonance instrument. Transferrin was immobilized on a CM5 chip using standard NHS-amine chemistry and each synbody construct (1 μM) was passed over the transferrin chip in PBS-tween buffer with a flow rate of 30 μL/min. The binding response of each transferrin synbody construct was measured, with all sensograms double reference subtracted from buffer injections and the reference cell.
Affinity Determination by SPR
Binding affinities for the synbodies and individual components were determined using a Biacore T100 surface plasmon resonance instrument. Gal80 protein was immobilized on a CM5 chip using standard amine coupling chemistry, which resulted in 18,500 response units immobilized to the chip giving an Rmax of 289 for the peptides and 4800 for the bivalent DNA-peptide conjugates. Binding assays conducted at lower immobilization levels (6,000 RU) produced similar Kd values (1.2 nM for the synbody). Transferrin protein was immobilized on a separate CM5 chip using standard amine coupling chemistry, yielding 6,962 response units immobilized. Individual binding assays were performed at multiple concentrations in standard PBS-tween buffer with a flow rate of 30 μL/min. Each assay consisted of a 100 sec contact time followed by a 300 sec dissociation time. All sensograms were double referenced using buffer injections and the reference cell to subtract nonspecific background binding. Solution binding affinity values were determined using the affinity fits in the Biacore software package using a 1:1 binding model and represent the average of at least two independent trials. Example affinity plots for Gal80 SC-13 and peptides are shown in Figure S4, SI.
Fluorescent Anisotropy
Fluorescein-labeled synbodies, constructed from a fluorescein-labeled oligonucleotide, were diluted into 1× HBS buffer containing 5 mM MgCl2 to a final concentration that was 10-fold below the expected Kd. This solution was incubated with increasing concentrations of protein that spanned the Kd values. The complex was incubated in a 96-well black plate in a total volume of 100 μL at room temp for 1 hr. The plate was excited at 490 nm with vertically polarized light (Ivv), with vertical and horizontal components (Ivh) detected at 525 nm (SpectroMax plate reader, Spectro). Fluorescent anisotropy values (r) were calculated by the SpectroMax software using the equation r = (Ivv-GIvh)/((Iv+2GIvh). A G-value of 1.2 was used and the data was subtracted from a buffer-only control (rf) to give Δr values (Δr = r-rf). Experiments were performed in triplicate and the final Kd was determined using GraphPad Prism to fit a hyperbolic curve to the data (Figure 5B, 6D).
Figure 5.
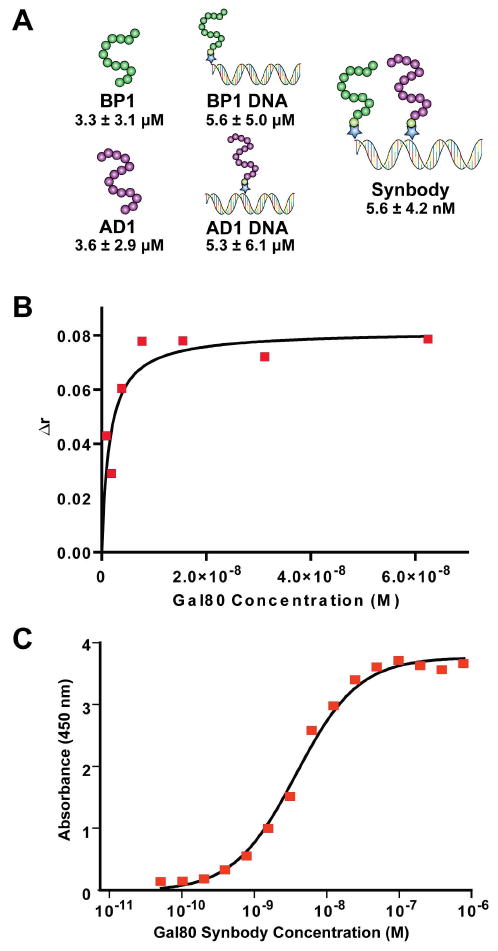
Characterization of the Gal80 synbody. (A) Dissociation constants for BP1 and AD1 were measured as individual peptides, individual peptides on the DNA scaffold, and as a bivalent synbody. The bivalent synbody improves the affinity of the peptides by 1000-fold to produce a synthetic antibody with a Kd of 5.6 nM. (B) Solution phase binding affinity of SC-13 was determined by fluorescence anisotropy. Gal80 protein was titrated against fluorescein-labeled synbody and fluorescent anisotropy was measured by exciting at 480 nm and emitting at 525 nm. The average of three experiments resulted in a Kd for SC-13 of 3.0 ± 1.3 nM. (C) The dissociation constant of the synbody was validated by ELISA and was determined to be 3.9 ± 0.3 nM.
Figure 6.
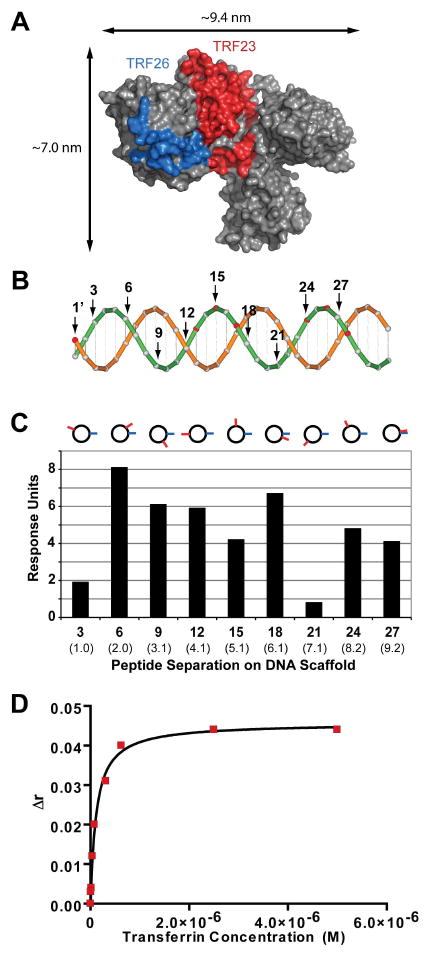
Generation of a transferrin synbody. (A) The transferrin peptides, TRF23 and TRF26, were mapped using PyMOL to the X-ray crystal of transferrin (PDB ID: 2HAV) by protein crosslinking experiments shown in red and blue, respectively. (B) A cartoon of the dsDNA scaffold used to spatially separate TRF23 and TRF26 peptides. TRF23 was conjugated to the 1′ base pair position on the template strand (orange) and TRF26 was conjugated to one of every third base pair position on the complementary strand (green). (C) The peptide distance assay was used to screen TRF23 and TRF26 at nine different base pair positions on the DNA scaffold. The synbody with the highest relative response to transferrin occurred when TRF26 was at the 1′ position and TRF23 was at the 6 base pair position termed TRF SC-6. Approximate linear distances in nanometers are given in parenthesis and the cartoon above the graph indicates the peptide positions when looking down the dsDNA helix. (D) The average of three fluorescence anisotropy experiments of TRF SC-6 resulted in a Kd of 86.5 ± 18.6 nM.
Affinity Determination by ELISA
Enzyme linked immunosorbant assays (ELISA) were conducted by incubating 200 ng of Gal80 protein in 0.1 M sodium bicarbonate, pH 9.8, in a nontreated Maxisorb NUNC 96-well plate overnight at 4°C in a humidifier. The solution was removed and replaced with 100 μL of blocking buffer (2% BSA in PBS, pH 7.4), which was incubated for 1 h at 37°C in the humidifier. The solution was removed and the plate was washed three times with PBS-tween and tapped dry. The biontylated Gal80 synbody, dsDNA, and peptides were added to the plate at varying concentrations in PBS-tween. The ligands were incubated with the plate for 1 h at 37°C. The ligand solution was removed and the plate was washed three times with PBS-tween. Horseradish peroxidase conjugated streptavidin was diluted 1:1000 in 0.1% BSA in PBS-tween, and 50 μL was added to each well and incubated for 1 h at 37°C. The strepavidin solution was removed and the plate was washed three times with buffer PBS-tween. 50 μL of TMB (3,3′, 5,5′-tetramethylbenzidine) was added to each well and the solution was incubated for 15 min at 24°C. 50 μL of 0.5 M HCl was added to stop the reaction and the plate was scanned immediately using a SpectroMax plate reader. The transferrin synbody was assayed for binding affinity in the same manner with the exception that the transferrin synbody was labeled with fluorescein, which enabled direct fluorescent measurements to be made by scanning the plate for fluorescence at 525 nm. These assays were conducted in triplicate, the data were then normalized by subtracting all fluorescent values from the no protein control, plotted, and fit using GraphPad Prism.
Pull-down Assay
Biotinylated Gal80 synbody was immobilized onto prewashed streptavidin coated magnetic beads (DynaBeads, Invitrogen) by incubating 150 pmoles synbody with 30 μl bead slurry at room temperature for 30 min. Gal80 protein (80 pmoles) and E. coli lysate (10 μL of A280-27.8) were incubated with the synbody beads for 30 min at 4 °C. The beads were washed three times with 30 μl of 1× HBS buffer supplemented with 5 mM MgCl2. Gal80 was eluted by incubating the synbody beads with 20 μl protein loading buffer (50 mM Tris, pH 6.8, 10% sodium dodecyl sulfate, 1% β-mercaptoethanol, 50% glycerol, 0.05% bromophenol blue). 0.1 μl ladder (Novex sharp, Invitrogen), 0.5 μl pure protein (1.6 μM), 0.1 μl spiked lysate, and 5 μl elution were run on a 4-12% SDS-PAGE gel (NuPAGE, Invitrogen) for 45 min at 200V and imaged using SilverXpress silver staining kit (Invitrogen). Beads without the synbody were used as a control to evaluate non-specific binding to the resin. A second control was conducted using E. coli lysate not expressing Gal80 to assess the potential for the Gal80 synbody to bind other proteins present in E. coli lysate (Figure S6, SI).
Results
Ligand Discovery by Peptide Microarray
We used custom peptide microarrays to identify peptides that bound to independent sites on the surface of the yeast regulatory protein Gal80. Microarrays were synthesized with ∼4,000 unique 12-mer polypeptide sequences. The peptide sequence at each position in the array was generated randomly, selecting one of the eight amino acids, Gly, Thr, Gln, Lys, Ser, Trp, Leu, and Arg, for each residue in the sequence. Based on experience with peptide phage library selections24, all peptides were designed to contain at least one tryptophan residue in their sequence. Gal80 protein labeled with a fluorescent tag was incubated in the presence and absence of the synthetic Gal4 activation domain peptide (Gal4 AD, residues 847-881)20, which binds the Gal80 repressor site with high affinity, and both samples were deposited onto separate peptide microarrays. Analysis was performed to distinguish peptides that bind the Gal80 repressor site from peptides that bind elsewhere on the protein surface (Figure 2A-B). The two array images revealed that 1,644 peptides bind the Gal80 protein with fluorescence intensity greater than 2- fold above the background fluorescence of the microarray. Of these peptides, 957 sequences were blocked by the Gal4 AD peptide, suggesting that this group of peptides binds the repressor site of Gal80. Peptides blocked by Gal4 AD were termed activation domain (AD) peptides, while peptides that were not blocked by the AD peptide were termed Gal80 binding peptides (BP).
Figure 2.
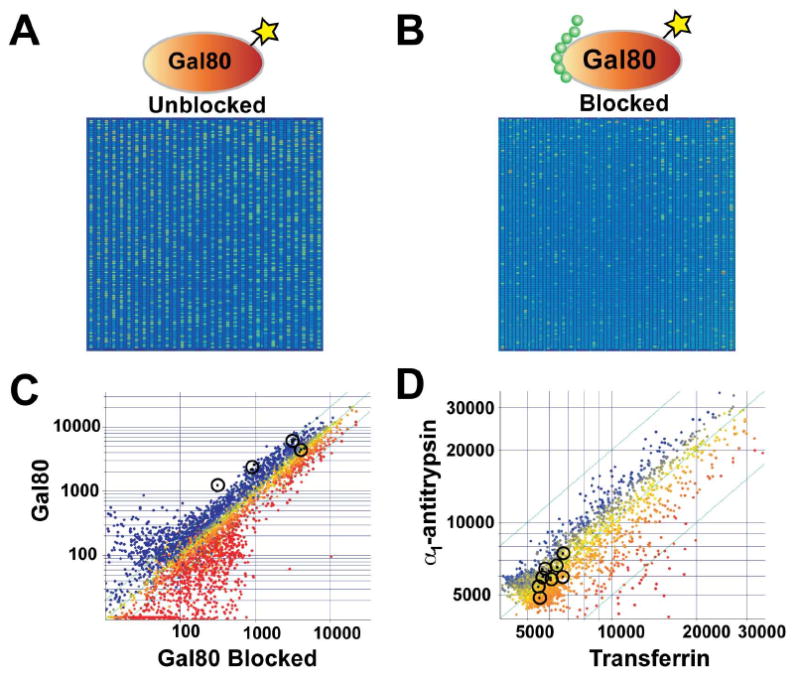
Protein ligand discovery using peptide microarrays. Gal80 binding peptides were identified from a peptide microarray containing ∼4,000 unique features. Fluorescently labeled Gal80 protein was incubated with the peptide microarray in the absence (A) and presence (B) of a known ligand to the Gal80 repressor site. Higher overall fluorescence observed when Gal80 was incubated in the absence of the synthetic ligand indicates that many peptides on the microarray bind the repressor site. (C-D), scatter plot analysis (non-normalized) was used to identify peptides with high affinity to non-overlapping sites on Gal80 and low affinity to α1-antitrypsin and transferrin. Black circles indicate peptide sequences that were identified with a high fluorescent ratio of Gal80 to α1-antitrypsin and transferrin.
To distinguish peptides with specific affinity to Gal80 from peptides that non-specifically bind to any protein surface, a second set of microarray assays were performed using the human serum proteins, α1-antitrypsin and transferrin, to represent two common blood proteins. Array analysis revealed that 28% and 52% of the selected peptides showed significant binding to α1-antitrypsin and transferrin, respectively, demonstrating that this collection of Gal80 binding peptides had high non-specific binding. The remaining peptides exhibited varying degrees of affinity for Gal80, and a ranking of the fluorescent intensity for Gal80 relative to α1-antitrypsin and transferrin allowed a subset of Gal80 binding peptides to be identified (Figure 2C-D) that showed high fluorescence for Gal80 and low fluorescence for α1-antitrypsin and transferrin (Table S1, SI). In this collection, four peptides were identified that bound the Gal80 repressor site (peptides AD1-4), and six peptides that recognized a different region or regions on the Gal80 protein surface (peptides BP1-6). Each of the ten candidate peptides was synthesized, and assayed on a Biacore surface plasmon resonance (SPR) Flexchip for affinity to recombinant Gal80 protein. The peptides BP1 and AD1 were found to have the highest relative binding to Gal80 (Figure 3A).
Figure 3.
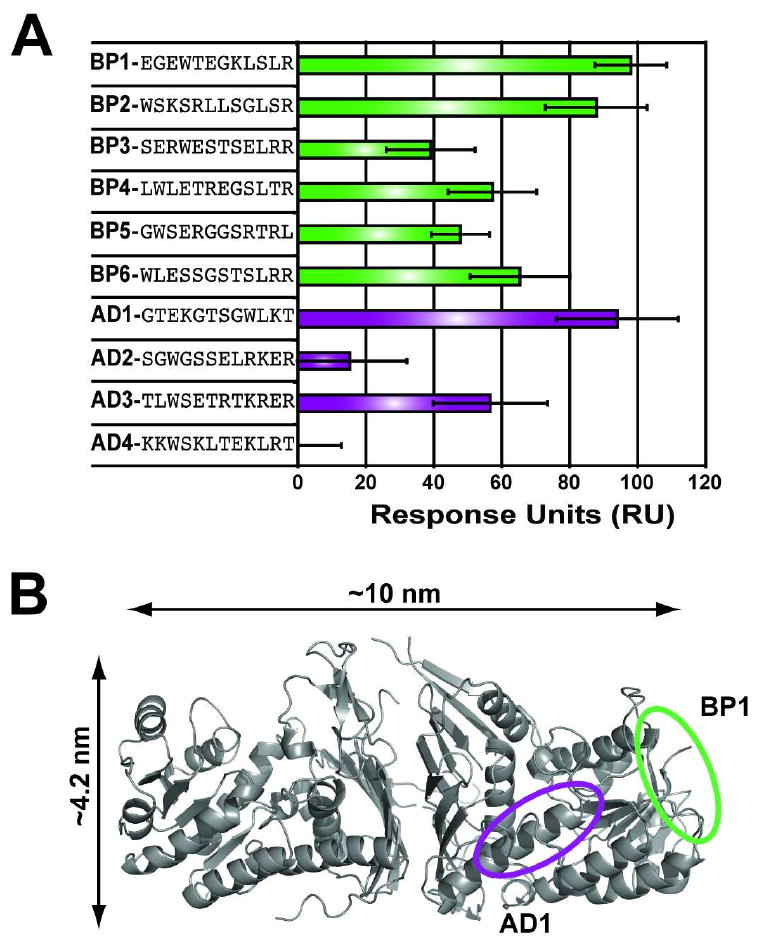
Analysis of the Gal80 binding peptides. (A) The relative binding affinity of each peptide was determined by SPR. BP1 and AD1 were identified as the two peptides that bound Gal80 with the highest response. (B) Separate, non-overlapping binding sites were validated by mapping BP1 and AD1 to the surface of the Gal80 protein. Gal80 regions cross-linked to BP1 and AD1 are circled on the crystal structure (PDB ID: 3BTV) in green and purple, respectively.
Mapping Peptide Binding to Gal80 protein
To determine whether BP1 and AD1 do indeed recognize separate, non-overlapping sites on the Gal80 protein surface, peptide mapping was used to determine the binding location of each peptide. Similar to traditional protein-protein interface mapping22, BP1 and AD1 were separately cross-linked to the Gal80 protein using a bifunctional crosslinking reagent. The protein-peptide complex was digested with trypsin and analyzed by MALDI-TOF mass spectrometry. The resulting mass spectra (Figure S2, SI) showed that Gal80 amino acid residues 384-420 crosslink to residues 1-4 of peptide AD1, and Gal80 amino acid residues 1-8 crosslink to residues 9-12 of peptide BP1. Mapping the peptide binding sites to the surface of the X-ray crystal structure of Gal80 (Figure 3B) demonstrated that the two peptides recognized different sites on the Gal80 surface, with AD1 binding the Gal4 AD-binding region of Gal8018.
Synbody Design
Because BP1 and AD1 could potentially bind Gal80 in many different conformations, it was unclear what distance and which type of linker would be required to transform the two peptides into a high affinity protein capture reagent. While previous approaches to this problem have relied on structural knowledge and extensive chemical synthesis25,26, we sought to develop a general strategy that could be performed on any water-soluble protein without prior structural information. We hypothesized that unstructured peptides might be less sensitive to linker restrictions as was the case in previous constructs25. For this purpose, we developed an assay that we refer to as combinatorial examination of ligands and linkers (CELL), which combines the multiplex capability of SPR with the nanoscale precision of DNA self-assembly to simultaneously search multiple peptide pairs and peptide pair distances in a single binding assay27. The molecular design (Figure 4A) with DNA as a linker relies on DNA-peptide conjugates that self-assemble into bivalent DNA-peptide fusion molecules with two peptides positioned at different distances along the DNA backbone. DNA is a logical choice for a synthetic scaffold as DNA adopts a helical structure that is predictable, rigid over short distances, and easy to engineer. Using standard amine coupling chemistry23, the template strand was conjugated to the C-terminus of either BP1 or AD1 (Scheme S1, SI). This strand was then annealed to a complementary strand that contained either the BP1 or AD1 peptide conjugated at nucleotide positions 9, 13, 15, 17, 24, 26, and 28, thereby creating a small combinatorial library of bivalent fusion molecules separated by distances of 3-9 nanometers.
Figure 4.
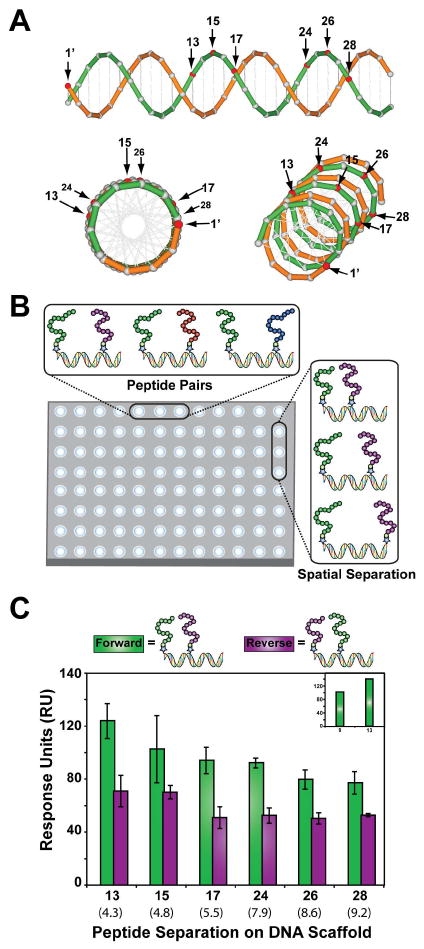
Combinatorial examination of ligands and linkers. (A) A model of the DNA backbone showing the spatial separation between individual peptide positions. Modified nucleotide positions (red) indicate the locations where peptides were conjugated to the DNA. (B) Combinatorial analysis of bivalent DNA-peptide fusion molecules was performed in a single step using a Biacore Flexchip that measured 400 independent binding interactions. (C) The combinatorial peptide pair and peptide pair distance assay was used to screen BP1 and AD1 in two orientations at six different base pair distances on the DNA scaffold. (inset) binding response of the forward synbodies at nucleotide positions 9 and 13. The synbody with the highest relative response to Gal80 was determined to be the combination with BP1 and AD1 at positions 1′ and 13, respectively. Approximate linear distances in nanometers are given in parenthesis.
SPR analysis on a Flexchip (Figure 4B) allowed all possible homo- and hetero- BP1 and AD1 peptide pairs to be analyzed in a single experiment. The set of bivalent DNA-peptide fusion molecules were immobilized on the Flexchip, and relative Gal80 binding was determined by flowing Gal80 over the surface. Bivalent complexes constructed of hetero-peptide pairs (Figure 4C) showed higher overall binding to Gal80, although the homo-peptide pairs (Figure S3, SI) did show substantial binding to Gal80, possibly because Gal80 is a dimeric protein in solution. Of the hetero-pairs tested, a clear trend emerged in which BP1 was favored on the template strand and AD1 on the complementary strand. One reason for this might be that the bivalent affinity reagent has a chiral preference due to the helicity of natural B-form DNA. The synbody with the highest binding response for Gal80 occurred at a distance of ∼4.3 nm with BP1 at position 1′ on the template strand and AD1 at position 13 on the complementary strand. We refer to this complex as synbody construct-13 (SC-13). It is interesting that SC-13, which has almost 180° separation between the two peptides shows the highest binding response to Gal80, while the remaining constructs, including SC-9 (Figure 4C insert), trend toward lower binding response units. This suggests that multiple distances will yield synbodies with enhanced binding, since the inherent flexibility of the peptide is able to conform to the protein surface.
Synbody Affinity Measurements
The dissociation constant (Kd) of SC-13 was determined by SPR and this value was compared to the affinity of the individual peptide ligands. A summary of this data is given in Figure 5A. The individual peptides, either alone or coupled to double-stranded DNA, bound Gal80 with affinity constants of ∼5 μM. No binding was observed for the DNA linker alone (Kd > 400 μM) demonstrating that the DNA linker itself does not interact directly with the target protein. When both peptides were positioned on the DNA linker at the optimal orientation and spatial separation distance, the affinity of SC-13 for Gal80 increased 1000-fold to yield an equilibrium dissociation constant of 5 nM. The affinity of SC-13 and individual peptides was validated independently using an ELISA-type assay (Figure 5C) and by fluorescence anisotropy (Figure 5B). This dramatic change in binding affinity demonstrates that synbodies can be created with affinity constants similar to traditional antibodies.
Transferrin Ligand Discovery and Peptide Mapping
The CELL process was used to generate a second synbody to the human serum protein transferrin. Transferrin ligands were identified from a custom microarray consisting of 10,000 individual 20-mer peptides. We hypothesized that this larger array would cover sufficient sequence space to identify peptides with affinity to different sites on the transferrin protein without the need for a discrete transferrin-blocking agent. In these experiments, Alexa-555 labeled transferrin was incubated with the 10,000-peptide microarray in competition with Alexa-647 labeled E. coli lysate. The bacterial lysate served as competitor to aid in the identification of peptides with high specificity to transferrin. The fluorescent ratio of Alexa-555 transferrin to Alexa-647 E. coli lysate was calculated for each peptide on the microarray. Ten peptides were identified with more than 5-fold specificity to transferrin (Table S2).
From the list of ten transferrin-binding peptides, peptides 23 (TRF23) and 26 (TRF26) were found to bind different sites on the transferrin surface. Each peptide was immobilized on beads, crosslinked to transferrin using formaldehyde, and digested by trypsin. The formaldehyde crosslinked fragments were reversed with heat and analyzed by MALDI-TOF mass spectrometry. The mass spectra data (Table S3, SI) showed that TRF23 was crosslinked to transferrin amino acid residues 415-433, 582-599, and 664-679 and TRF26 was crosslinked to transferrin amino acid residues 435-447 and 448-470. The peptide binding sites were mapped to the surface of the X-ray crystal structure of transferrin (Figure 6A), which revealed that TRF23 and TRF26 bind different non-overlapping sites on the transferrin protein.
Transferrin Synbody Construction and Screen
Transferrin synbody constructs were generated using TRF23 and TRF26 conjugated to different nucleotide positions along the dsDNA scaffold. For the transferrin synbodies, we spatially separated the two peptides at every third position along the DNA scaffold (base pair position 3, 6, 9, 12, 15, 18, 21, 24, and 27) as shown in Figure 6B. The small library of transferrin synbodies spanned a distance of ∼1.0-9.2 nanometers. The synbodies were screened against transferrin for relative binding response using a Biacore T100 SPR instrument (Figure 6C). The synbody with the highest binding response for transferrin occurred at base pair 6 with a distance of ∼2.0 nm and is referred to as transferrin synbody construct-6 (TRF SC-6).
Transferrin Synbody Affinity Measurements
The transferrin peptides and TRF SC-6 were assayed for affinity to transferrin using SPR. Transferrin was immobilized on the SPR chip surface and each ligand was passed over the surface while response units were measured. TRF23 and TRF26 had moderate affinity for transferrin with apparent Kd values of 17.4 μM and 120 μM, respectively. We noticed that the dissociation constants for the transferrin ligands were higher than what was previously observed for the Gal80 ligands. This difference could be due to a larger entropic penalty associated with the longer peptides used in the transferrin study (12-mer vs 20-mer peptides). TRF SC-6 resulted in a Kd of 117 nM by SPR, which is a ∼1,000-fold improvement in binding affinity over the weaker affinity peptide sequence. We validated the binding affinity of TRF SC-6 by fluorescence anisotropy, which produced a Kd of 86. 5 nM (Figure 6D), and demonstrated that TRF SC-6 functions in an ELISA like assay with a dissociation constant of 68.4 nM (Figure S5, SI).
Specificity Measurements
A standard pull-down experiment was performed to evaluate the specificity of Gal80 SC-13. This synbody was chosen because it exhibited 10-fold higher affinity for its desired target than TRF SC-6. The synbody was modified with biotin, immobilized on streptavidin coated magnetic beads, and the resin was incubated with total soluble E. coli lysate that contained 3% recombinant Gal80 protein. The beads were washed thoroughly with buffer and the protein that remained bound to the resin was eluted with SDS and analyzed by denaturing gel electrophoresis. A silver-stained image of the resulting gel (Figure 7) indicated that the Gal80 synbody successfully enriched Gal80 protein from a crude mixture of E. coli proteins. Close inspection of the gel revealed the presence of four low intensity bands at 18, 26, 40, and 70 kDa, respectively. A control experiment performed with E. coli lysate devoid of Gal80 failed to reproduce these bands, which suggests that these four proteins are due to interactions with Gal80 and not the SC-13 synbody (Figure S6, SI).Comparison of lanes 4 and 5 reveal that three additional bands observed in the elution lane were due to non-specific binding to the streptavidin coated magnetic beads. These data demonstrate that the Gal80 synbody functions as a strong protein capture reagent capable of discriminating Gal80 from many other proteins present in a complex biological mixture.
Figure 7.
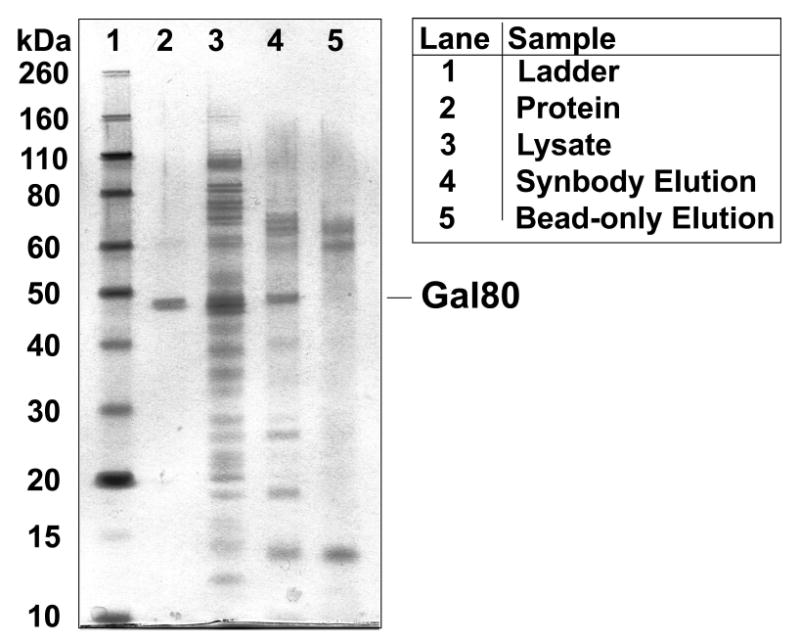
Specificity assay of Gal80 synbody. A standard pull-down assay was used to evaluate the specificity of the Gal80 synbody. Immobilized SC-13 was able to pull Gal80 protein out of a complex mixture of E. coli lysate.
Discussion
In this work we describe a new approach to creating synthetic protein affinity reagents called synbodies. We first demonstrated that two peptides that bind different sites on the Gal80 protein could be isolated from a relatively small peptide library of ∼4,000 unique 12-mers displayed on a microarray surface. The independent binding of the peptides was supported by a crosslinking assay. A novel DNA linking strategy was designed to spatially separate the two peptides at different distances and orientations. One peptide was linked to DNA at a constant position, while the second peptide was linked to different positions on a complementary strand. Annealing the two strands created 24 unique bivalent synbodies, which were simultaneously assayed in a single SPR experiment. The synbody with the highest binding response (SC-13) was further characterized and resulted in a ∼1,000 fold improvement in affinity over the individual peptides. The affinity of the best synbody (5 nM) is comparable to a typical antibody affinity and functions in conventional ELISA and pull-down assays. A second set of experiments was performed on the human blood plasma protein transferrin, and these assays resulted in a similar improvement in binding affinity over the individual peptides. Together, these experiments show how a synthetic protein affinity reagent can be created without resorting to animal immunization methods or iterative rounds of in vitro selection and amplification.
During the course of our experiments, it was discovered that a large portion (∼40%) of peptide sequences displayed on the microarray exhibited significant affinity to the target protein. This outcome stands in contrast to most directed evolution experiments where selections often yield only one or a limited number of solutions, most of which bind the dominant epitope of a given target protein12. One interpretation of this result is that polypeptide ligands are relatively common in protein sequence space, and therefore large combinatorial libraries are not necessary to find small sequences with relatively simple functions. Indeed, it was discovered that a simple screen could be used to identify ligands that recognized their cognate target with low micromolar binding affinity.
Step two of our synthetic antibody process involved developing a general approach that could be used to transform any two peptide ligands that showed affinity to separate, non-overlapping sites into a single high affinity protein capture reagent. This challenge required designing a strategy that could be broadly applied to a wide range of protein targets. While earlier work on the structure-activity-relationship of protein ligands almost always required some structural knowledge of the target protein 13,25, we sought to create a strategy that functioned independent of any protein information. Our solution to this problem was to use DNA to systematically explore different peptide pairs and peptide pair separation distances in a single binding assay. DNA is an ideal building block material for this purpose as it allows for small subtle differences in the length to be explored in a systematic fashion. Coupling a combinatorial library of bivalent DNA-peptide fusion molecules to the surface of an SPR Flexchip, and screening the different complexes for binding made it possible to rapidly search different peptide pairs and peptide pair distances for optimal binding to the target protein. Through this process, it was discovered that two modest affinity ligands could be transformed into a single high affinity protein-binding reagent. Characterization of the resulting molecule demonstrated that purely chemical methods enabled a synthetic antibody to be created that functioned as an effective antibody mimic.
One interesting phenomenon to come from this study was the observation that two non-competing ligands optimally spaced on a synthetic scaffold improved the binding affinity of the individual peptides by 1000-fold. In a perfect cooperative binding event, where both ligands recognize two independent sites, one might expect the binding affinity of a bivalent affinity reagent to be at or near the product of the affinities of the two peptides16. In the current study, for example, that would have produced a synbody with an affinity for Gal80 of ∼25 pM, which is 200-fold better than the affinity we observed in our binding assays. Determining whether the binding constants of bivalent affinity reagents scale linearly with the affinity of their individual ligands, and how the chemical composition of the linker impacts the net increase in binding affinity remain two very interesting questions. If for example, the affinity constant of bivalent synbodies do indeed scale linearly with the affinity of the individual ligands, then improving the quality, and possibly orientation of the ligands should lead to the creation of synbodies that are able to more closely approximate the cooperativity of a perfect bivalent binding event.
In conclusion, we describe a novel strategy that could be use to develop synthetic antibodies from available chemical building blocks without resorting to protein design, in vitro selection, or animal immunization. The simplicity of this technique suggests that this technology should be amenable to automation, which would make it possible to rapidly generate synbodies to larger numbers of protein targets. These molecules could then be used to investigate the complexity and function of the human proteome—a task currently limited by the availability of high quality antibodies.
Supplementary Material
Acknowledgments
We would like to thank L. Joshua-Tor and P.R. Kumar for providing the Gal80 plasmid and the initial protein, A. Maganty for expressing Gal80, M. Hahn and L. Howell for peptide synthesis and purification, and P. Hunter for writing the sequence generator program. We would also like to thank P. Stafford and Z. Zhao for helpful discussions. B.W. and M.G. were supported in part by an IGERT Fellowship from the NSF. This work was supported in part by start-up funds to S.J. and by grants from the National Institutes of Health (R21 CA126622-01) and the Science Foundation of Arizona (CAA 0265-08) to J.C.
Footnotes
Supporting information includes the following: (1) transferrin ligand discovery using peptide microarrays, (2) mass spectra of the Gal80-peptide mapping assay; (3) binding response for the homo-peptide pair Gal80 synbodies; (4) example SPR affinity plots for Gal80 SC-13, BP1, and AD1; (5) TRF SC-6 affinity determination by ELISA; (6) control pull-down gel; (7) scheme of peptide-DNA oligonucleotide conjugation; (8) table of binding responses from the Gal80 peptides; (9) table of binding responses from the TRF peptides; (10) mass spectra data of the TRF-peptide mapping assay; (11) table of DNA oligonucleotides used to make the Gal80 synbodies; (12) table of DNA oligonucleotides used to make the TRF synbodies; and (13) the complete author list for reference number 6. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Blow N. Nature. 2007;447:741–744. doi: 10.1038/447741a. [DOI] [PubMed] [Google Scholar]
- 2.Hober S, Uhlen M. Curr Opin Biotech. 2008;19:30–35. doi: 10.1016/j.copbio.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Uhlen M. Mol Cell Proteomics. 2007;6:1455–1456. [PubMed] [Google Scholar]
- 4.Chambers RS. Curr Opin Chem Biol. 2005;9:46–50. doi: 10.1016/j.cbpa.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 5.Zichi D, Eaton B, Singer B, Gold L. Curr Opin Chem Biol. 2008;12:78–85. doi: 10.1016/j.cbpa.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Taussig MJ, et al. Nat Methods. 2007;4:13–17. [Google Scholar]
- 7.Hoogenboom HR. Nat Biotechnol. 2005;23:1105–1116. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 8.Hudson PJ, Souriau C. Nat Med. 2003;9:129–134. doi: 10.1038/nm0103-129. [DOI] [PubMed] [Google Scholar]
- 9.Hey T, Fiedler E, Rudolph R, Fiedler M. Trends Biotechnol. 2005;23:514–522. doi: 10.1016/j.tibtech.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Bradbury ARM, Marks JD. J Immunol Methods. 2004;290:29–49. doi: 10.1016/j.jim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Hosse RJ, Rothe A, Power BE. Prot Sci. 2006;15:14–27. doi: 10.1110/ps.051817606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson DS, Szostak JW. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- 13.Erlanson DA, Wells JA, Braisted AC. Annu Rev Biophys Biomol Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- 14.Williams BAR, Lin L, Lindsay SM, Chaput JC. J Am Chem Soc. 2009;131:6330–6331. doi: 10.1021/ja900916p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kodadek T, Reddy MM, Olivios HJ, Bachhawat-Sikder K, Alluri PG. Acc Chem Res. 2004;37:711–718. doi: 10.1021/ar030145l. [DOI] [PubMed] [Google Scholar]
- 16.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Reddy MM, Bachhawat-Sikder K, Kodadek T. Chem Biol. 2004;11:1127–1137. doi: 10.1016/j.chembiol.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Kumar PR, Yu Y, Sternglanz R, Johnston SA, Joshua-Tor L. Science. 2008;319:1090–1092. doi: 10.1126/science.1151903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pelloise JP, Zhou X, Srivannavit O, Zhou T, Gulari E, Gao X. Nat Biotechnol. 2002;20:922–926. doi: 10.1038/nbt723. [DOI] [PubMed] [Google Scholar]
- 20.Johnston SA, Salmeron JM, Dincher SS. Cell. 1987;50:143–146. doi: 10.1016/0092-8674(87)90671-4. [DOI] [PubMed] [Google Scholar]
- 21.Boltz K, Gonzalez-Moa MJ, Stafford P, Johnston SA, Svarovsky SA. Analyst. 2009;134:650–652. doi: 10.1039/b823156g. [DOI] [PubMed] [Google Scholar]
- 22.Sinz A, Kalkhof S, Ihling C. J Am Soc Mass Spectr. 2005;16:1921–1931. doi: 10.1016/j.jasms.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 23.Harrison JG, Balasubramanian S. Nuc Acids Res. 1998;26:3136–3145. doi: 10.1093/nar/26.13.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barry MA, Dower WJ, Johnston SA. Nat Med. 1996;2:229–305. doi: 10.1038/nm0396-299. [DOI] [PubMed] [Google Scholar]
- 25.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 26.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 27.Homola J, Vaisocherova H, Dostalek J, Piliarik M. Methods. 2005;37:26–36. doi: 10.1016/j.ymeth.2005.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.