

Abstract
The etiology of immunologically mediated chronic renal allograft failure is unclear. One cause is thought to be alloantibodies. Previously in Cynomolgus monkeys, we observed a relationship among donor-specific alloantibodies (DSA), C4d staining, allograft glomerulopathy, allograft arteriopathy and progressive renal failure. To define the natural history of chronic antibody-mediated rejection and its effect on renal allograft survival, we now extend this report to include 417 specimens from 143 Cynomolgus monkeys with renal allografts. A subset of animals with long-term renal allografts made DSA (48%), were C4d positive (29%), developed transplant glomerulopathy (TG) (22%) and chronic allograft arteriopathy (CAA) (19%). These four features were highly correlated and associated with statistically significant shortened allograft survival. Acute cellular rejection, either Banff type 1 or 2, did not correlate with alloantibodies, C4d deposition or TG. However, endarteritis (Banff type 2) correlated with later CAA. Sequential analysis identified four progressive stages of chronic antibody-mediated rejection: (1) DSA, (2) deposition of C4d, (3) TG and (4) rising creatinine/renal failure. These new findings provide strong evidence that chronic antibody-mediated rejection develops without enduring stable accommodation, progresses through four defined clinical pathological stages and shortens renal allograft survival.
Keywords: Allograft, antibody, chronic, kidney, monkey, rejection
Introduction
Cellular or antibody-mediated rejections and nonrejection-mediated renal injuries from drugs, recurrent or de novo disease, vascular or viral diseases all contribute to late renal allograft loss. Distinguishing between rejection or non-rejection is not easy either clinically or pathologically. One important putative etiology of late renal allograft failure is donor-specific alloantibodies (DSA), identified in tissue by peritubular capillary staining of C4d (1,2), which correlates well with the presence of donor-reactive major histocompatibility complex (MHC) serum alloantibodies (3–15).
A substantial fraction of patients with chronic rejection have circulating antibodies and deposition of C4d (6,12,16–20), which is associated with the later development of transplant glomerulopathy (TG) (12,16,20). However, C4d is occasionally found in human renal allografts with normal function (21–24), and in one series preceded TG (20), which led us to postulate that C4d may predict later chronic antibody-mediated rejection (25,26).
To test this hypothesis, we have evaluated long-term renal allografts in Cynomolgus monkeys (Macaca fascicularis) with mixed chimerism protocols (27–33). Many of these kidney allografts survive long term without rejection, but some later develop chronic rejection with alloantibodies, TG and transplant vasculopathy (29), thereby providing a unique opportunity to study the clinical–pathological parameters involved in the development of chronic allograft rejection without the effects of exogenous immunosuppression. Previously in 17 animals we studied the association of alloantibodies, C4d deposition, TG and arteriopathy (33), but too few animals were available to test statistically the effect of alloantibodies or graft pathology on actual renal allograft survival or to test the hypothesis of four stages in the natural history of chronic alloantibody-mediated rejection. With new findings in 143 animals, we define the natural history of chronic alloantibody-mediated rejection in four stages (alloantibody, C4d deposition, TG, rising creatinine/renal failure), which markedly shortens allograft survival, and occurs without apparent enduring accommodation. In addition, ancillary findings include an association of the development of arteriopathy with prior TG, endarteritis, C4d/alloantibodies and glomerulitis and the development of interstitial fibrosis with prior TG, glomerulitis and C4d/alloantibodies.
Methods
Animals
The purpose of this study was to identify the relationship among alloantibodies, C4d, allograft pathology and late graft failure, rather than the frequency of these events in specific treatment protocols (27–33). Therefore, we evaluated all animals treated with a variety of mixed chimerism protocols from 1993 to 2007, not on chronic immunosuppression, and with renal allograft surviving more than 50 days (n = 143). The endpoint was death from any cause, including infection, spontaneous death, renal failure or euthanasia to terminate the experiment in animals with normal renal allograft function. Recipient and donor Cynomolgus monkey pairs (3–8 kg Charles River Primates, Wilmington, MA) were selected for ABO compatibility but mismatched for Cynomolgus leukocyte (CyLA) MHC antigens (27,28). All surgical procedures and postoperative care of animals were carried out in accordance with National Institute of Health guidelines and were approved by the Massachusetts General Hospital Subcommittee on Animal Research.
Regimens
The standard preparative regimen included nonlethal total body irradiation (TBI) (1.5 Gy) on day −6 and −5 relative to transplantation, local thymic irradiation (TI) (7 Gy) on day −1, i.v. ATG (ATGAM, Pharmacia and Upjohn Co., Kalamazoo, MI.) (50 mg/kg/day) on days −2, −1 and 0, and i.v. donor bone marrow transplantation (DBMT) on day 0, infused at 0.4 to 4 ×108/kg. Monkeys underwent heterotopic renal transplantation and splenectomy on day 0 and bilateral native nephrectomies under ketamine hydrochloride/diazepam anesthesia, supplemented by halothane (27). Cyclosporin (CyA, Novartis, Basel, Switzerland) was given i.m. beginning on day 1, tapered from an initial dose of 15 mg/kg/day to maintain therapeutic serum levels (>300 ng/mL), and discontinued on day 28 posttransplant, after which the serum CyA levels become undetectable by days 60 to 70. In the anti-CD40L protocol, transplantation was followed by a short course of anti-CD154 monoclonal antibody (5c8, Immerge Biotherapeutics, 20 mg/kg × 2), usually without splenectomy (34). Controls included selective elimination of major components of the protocols. Modifications of the standard protocol included substitution of peripheral blood stem cells for bone marrow cells, addition of other antibodies (e.g. antibodies to CD28, CD30, Rituxan, CTLA4Ig) or other drugs (rapamycin, fludarabine). Data from many of these animals have been published previously (27–33). The frequency of alloantibodies and positive C4d staining was indistinguishable among groups, in part because some groups contained fewer than five animals, so that data from all 143 animals were pooled for pathological review and survival analysis. Nevertheless, differences may have been missed due to sample size.
Detection of alloantibodies
Antidonor-specific alloantibodies were detected by flow cytometric analysis on sera simultaneously sampled for creatinines (29,32,33). The positive threshold was two standard deviations above pretransplant sera. Overall, 175 serum samples from 54 animals were available for correlation with pathology specimens. Sera from nine Cynomolgus animals were previous tested to verify cross-reactivity to purified human class I and class II alloantigens (32).
Pathology studies
Four hundred seventeen samples studied included 269 biopsies for elevated creatinines (incident biopsies), or stable function (protocol biopsies), 5 nephrectomies and 143 autopsies. After euthanasia, complete autopsies were performed, and sections were stained with hematoxylin and eosin and periodic acid Schiff (PAS) stains and scored using Banff criteria (35,36). TG was defined as >25% glomerular capillaries with glomerular basement membrane (GBM) duplication in the most involved glomerulus in PAS stains (Banff cg2). Chronic allograft arteriopathy (CAA) was scored only on autopsy and nephrectomy specimens due a paucity of arteries on biopsy. Electron microscopy and C4d staining was performed as previously described (33). C4d staining was considered positive if most of the capillaries in 6 or more 10 high-power fields showed circumferential C4d staining of the peritubular capillaries.
Statistical analysis
Data were analyzed using ANOVA and post-hoc corrections, Fisher exact test, Kaplan–Meier and Life Table statistics (SPSS Software, version 10, Chicago, IL).
Results
Development of chronic rejection
The purpose of this study was to analyze the natural history of chronic rejection in all animals with renal allografts surviving 50 days and not to determine relative outcomes among protocols so that data are pooled for analysis. TG was found in 31 of 143 animals (22%, and 17% of all samples). First detected at 82–818 days with the median first appearance of 161 days for all samples, GBM duplication was usually extensive, affecting on average 89 ± 14% of the glomerular capillaries (60–100%). Most of the samples with TG also had mesangial hypercellularity (> 1 mm) (74%) and/or mononuclear cell accumulation (>1 g) (68%) in greater than 50% of glomeruli (transplant glomerulitis). In contrast, of those with less than 50% GBM duplication, 6% had mesangial hypercellularity and 17% had glomerulitis. Thus, GBM duplication was highly correlated with diffuse mesangial expansion and glomerulitis (both p < 0.001). An example of the histological progression of TG and C4d in animal 4498 with chronic antibody-mediated rejection is illustrated in Figure 1. This animal had a normal-looking renal biopsy on day 106 (Figure 1A), lacked C4d staining (Figure 1F) and had a creatinine of 1.2. Alloantibodies were first detected on day 162 and remained positive. The biopsy on day 182 showed a mild glomerulitis without TG (Figure 1B), without C4d staining (Figure 1G), and with a creatinine of 1.4. On day 225, TG was present (Figure 1C) with positive C4d staining (Figure 1H), and a slightly elevated creatinine of 1.8. TG worsened on a follow-up biopsy on day 352 (Figure 1D), and autopsy on day 371 (Figure 1E), with persistent C4d staining (Figures 1I and J) and rising creatines of 2.4 and 4.8, respectively. Previously electron microscopy confirmed prominent basement membrane duplication in five cases and identified no immune complexes (33). Seven additional autopsied kidneys from four animals with alloantibodies and C4d, and three without alloantibodies and C4d were analyzed ultrastructurally. In the three cases without alloantibodies or C4d, basement membrane duplication and excess peritubular laminations were not identified, data not shown. In the four cases with alloantibodies and C4d, the glomerular basement membrane showed frequent and marked duplication with isolated glomerular basement membrane laminations (Figure 2A, and B), plus peritubular capillary laminations, up to seven layers (Figure 2C). These findings are similar to those found in the ultrastructural analysis of human TG (37).
Figure 1. Natural history of chronic alloantibody-mediated rejection in animal no. 4498.
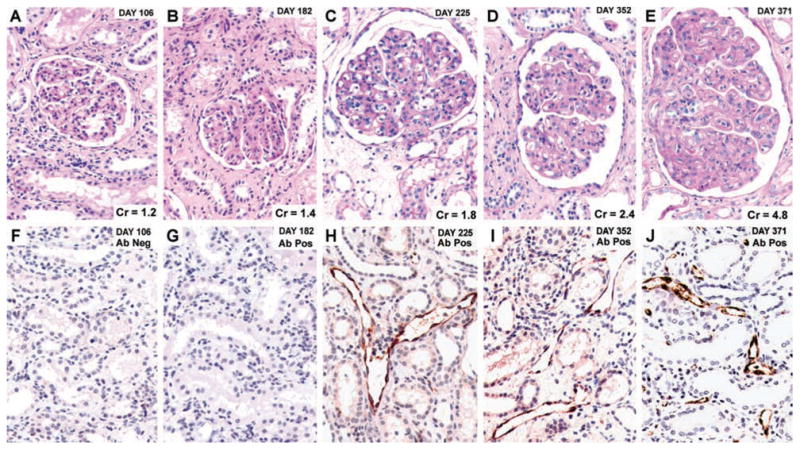
Protocol biopsies are on days 106 (Cr = 1.2), 182 (Cr = 1.4), 225 (Cr = 1.8) and 352 (Cr. 2.4) and autopsy on day 371 (Cr = 4.8). Hematoxylin-eosin stained images (A–E) and C4d immunohistochemistry and hematoxylin counterstained images (F–J). Alloantibody was detected on day 162, and C4d staining is positive and TG is present on days 225, 352 and 371. Original magnification, 400×.
Figure 2. Digital images from the electron microscopy of TG and peritubular capillary laminations.
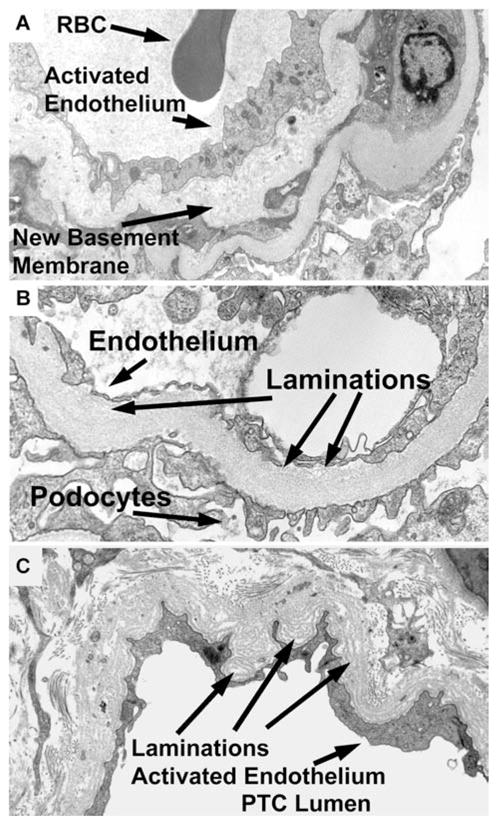
(A) Glomerular loop with new basement membrane (arrow), that is duplication, activated endothelium (arrow) and lumen with a red blood cell (RBC, arrow). Original magnification 7100×. (B) Glomerular loop with thickened basement membrane laminations (arrow), and activated endothelium (arrow). Podocytes for reference (arrow). Original magnification, 15 000×. (C) Peritubular capillary with laminations (arrow). and activated endothelium (arrow). Peritubular capillary lumen for reference (PTC lumen). Original magnification, 9100×.
C4d deposition
Samples from 29% of the animals showed prominent C4d deposition in peritubular capillaries in at least one sample (41/143), and 31% of all samples were C4d positive (79/251) (33) (Figure 1). The first time of appearance of PTC C4d deposition was 65–770 days after transplant (median of 117 days). Twenty-two animals with C4d-positive biopsies remained positive on repeat biopsies (1–5 subsequent biopsies); one was negative at autopsy 10 days later and 18 did not have a subsequent biopsy. Of the 11 animals with focal C4d (less than 50% of fields) four became positive on subsequent biopsies (22, 65, 91 and 111 days later), two became negative (4 and 42 days later), one remained focal and four had no follow-up sample.
Negative controls including 9 native kidney controls, 6 biopsies of allografts with polyomavirus interstitial nephritis and 7 allograft biopsies with posttransplant acute tubular injury (ATN) all showed no peritubular capillary C4d staining (data not shown).
Experimental protocols and antibody to donor T cells and B cells
Overall, alloantibodies were detected in 48% of the animals tested (26/54). The antibodies were first detected 53–839 days after transplantation (median of 106 days) (29,31–33).
Correlation between C4d and alloantibodies
For individual animals, alloantibodies correlated with C4d deposition in the allografts of 54 animals tested. Nineteen of 26 animals with alloantibody (73%) had positive C4d, whereas only 3 of 28 without alloantibody (10%) had positive C4d. This relationship is statistically significant, p < 0.001. Similarly among concurrent samples, alloantibodies and C4d strongly associated (p < 0.001, n = 177) (Table 1).
Table 1.
| Concurrent pathology |
|||||
|---|---|---|---|---|---|
| Pathology on biopsy | Banff arterial inflammation >v0 | Presence of alloantibodies | TG> cg1 | Positive C4d PTC | Banff acute> suspicious |
| TG> cg1 | 0.600 | <0.001 | <0.001 | 0.012 | |
| Fibrosis ≥50% | 0.820 | 0.001 | <0.001 | <0.001 | 0.77 |
| Banff acute > suspicious | <0.001 | 0.110 | 0.100 | 0.060 | |
| Banff arterial inflammation >v0 | 0.970 | 0.910 | 0.990 | <0.001 | |
| Positive C4d PTC | 0.150 | <0.001 | <0.001 | 0.15 | |
| Presence of alloantibodies | 0.970 | <0.001 | <0.001 | 0.22 | |
| Banff infiltrate >i1 | 0.230 | 0.280 | 0.380 | 0.670 | <0.001 |
| Banff glomerular inflammation >g1 | 0.001 | 0.001 | <0.001 | 0.001 | 0.002 |
| N > 150 | |||||
| Later pathology |
|||||
|---|---|---|---|---|---|
| Pathology on biopsy | Banff acute > suspicious | Positive C4d PTC | Chronic arteriopathy | TG > cg1 | Fibrosis ≥50% |
| TG > cg1 | >0.05 | <0.001 | <0.001 | <0.001 | |
| Fibrosis ≥50% | >0.05 | 0.050 | 0.150 | 0.040 | |
| Banff acute > suspicious | 0.110 | 0.630 | 0.120 | 0.67 | |
| Banff arterial inflammation >v0 | <0.001 | 0.600 | <0.001 | 0.730 | 0.74 |
| Positive C4d PTC | >0.05 | <0.001 | <0.001 | <0.001 | |
| Presence of alloantibodies | 0.150 | <0.001 | <0.001 | <0.001 | <0.001 |
| Banff infiltrate >i1 | < 0.001 | 0.720 | 0.680 | 0.360 | 0.36 |
| Banff glomerular inflammation >g1 | >0.05 | <0.001 | <0.001 | <0.001 | 0.001 |
| N > 150; for CAA (autopsies) >47 | |||||
Alloantibodies and C4d are risk factors for TG
Alloantibodies are a risk factor for the development of TG (Table 1). Fourteen of 26 animals with alloantibody developed TG (54%), whereas only 1 of 28 animals (4%) without antibodies developed TG (p < 0.001). C4d is also a risk factor for the development of TG. Among the 54 animals with known alloantibody status, 14 of 26 animals positive for C4d (54%) developed TG, whereas only 1 of 28 (4%) without C4d developed TG (p < 0.001). Using all animals, including 89 with an unknown antibody status, 9 of 15 (60%) with C4d developed TG, whereas only 7 of 74 (9%) without C4d developed TG (p < 0.001). In 93 animals with adequate numbers of biopsies to determine if C4d proceeded the development of TG, in 19 animals C4d preceded TG, whereas in only 3 animals TG was identified before C4d. Both C4d and TG were occurring concurrently in 71 animals. Among concurrent samples alloantibodies, C4d and TG were strongly associated (p < 0.001, n = 264). The presence of either alloantibodies or C4d staining correlated with the later development of TG if not present concurrently (p < 0.001).
Correlation of alloantibodies, C4d and CAA
CAA was found in 27 of 143 (19%) of the autopsy and nephrectomy samples (Table 1). Using only autopsies to assure adequate sampling of arteries, alloantibodies before autopsy were a risk factor for CAA (p < 0.001). Overall, 13 of 26 animals with alloantibodies had CAA at autopsy (50%), whereas only one animal of 24 without alloantibodies had CAA at autopsy (4%). Positive C4d staining before autopsy predicted CAA. Using 110 animals surviving 62 days or longer because the earliest detection of CAA was 62 days, of the 39 with C4d peritubular capillary staining, 20 had CAA (51%), but of 71 without C4d, only 8 had CAA (11%, p = 0.001). TG before autopsy and arteriopathy at autopsy were strongly associated. Using 96 animals surviving 82 days or longer because the earliest detection of TG was 82 days, of the 30 animals with TG, 18 also had CAA (60%), whereas of 66 without TG, only 7 had CAA (11%; p < 0.001) (Table 1).
Additional pathological correlations
Fibrosis of greater than 50% of the cortex was correlated with peritubular C4d deposition, TG and alloantibodies on concurrent specimens, (p < 0.001) but not with acute cellular rejection (Banff types 1, 2 or suspicious) (Table 1). Concurrent or prior episodes of acute cellular rejection were not associated with the presence of alloantibodies, C4d or TG, but TG on biopsy was associated with later development of CAA, and fibrosis. Endarteritis (Banff acute cellular rejection, type 2) was a risk factor for CAA (p < 0.001) but not for C4d or TG (all p > 0.5) because 12 of 15 animals (80%) with a type 2 rejection had CAA, whereas only 8 of 60 without a type 2 rejection had CAA (13%). C4d staining of peritubular or glomerular capillaries on biopsy predicted the later development of CAA, TG and fibrosis (p < .001) (Table 1).
Sequential stages of chronic alloantibody-mediated rejection
If alloantibodies, C4d deposition, TG and renal failure are causally related, a temporal sequence should exist (Figure 3). To test this hypothesis in 18 autopsied animals with allografts surviving 200 days or longer and with adequate interval sampling, the geometric mean of the first day of appearance of alloantibodies, C4d, TG and rising creatinine was 158, 210, 252 and 367 days with respective mean creatinines of 1.4, 1.9, 2.2 and 6.0 (Figure 3, top) with diagrammatic representation of the stages of chronic alloantibody-mediated rejection (Figure 3, bottom). First to appear was circulating alloantibodies (42% of cases), C4d (18% of cases) or a combination of C4d and alloantibodies (37% of cases). Only rarely (3%) was the first manifestation TG without C4d or alloantibodies.
Figure 3. Sequential appearance of alloantibody, C4d deposition, TG and rising creatinine in 18 animals.
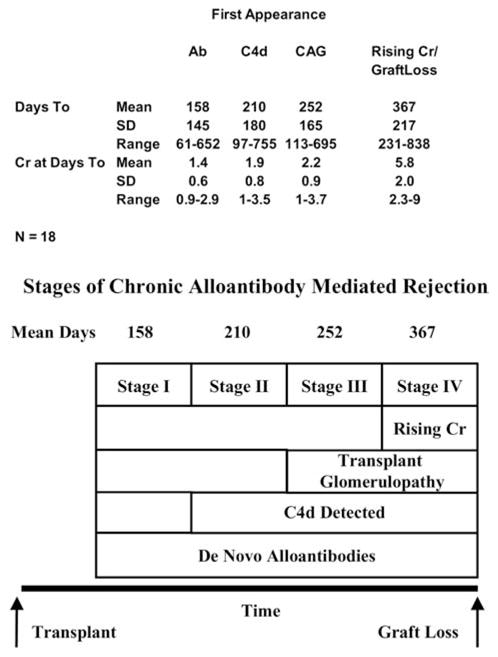
Stages of chronic alloantibody-mediated rejection. (Top) The first appearance of alloantibody, C4d, transplant glomerulopathy and rising creatinines in 18 animals. (Bottom) Proposed four stages of chronic alloantibody-mediated rejection.
Over time alloantibodies, C4d, TG generally became congruent (Figure 4) supporting our hypothesis that these are three closely related events. In 18 animals with alloantibodies, 9 (50%) were C4d positive at the time alloantibodies was detected or within 34 days. Eight more became C4d positive 35–225 days later so that 94% of antibody-positive animals were or became C4d positive (Figure 4). In 18 animals with alloantibodies, 5 were TG positive (28%) at the time alloantibodies was detected or within 28 days. Ten more became TG positive 37–543 days later so that 89% of alloantibodies positive animals were or became TG positive.
Figure 4.
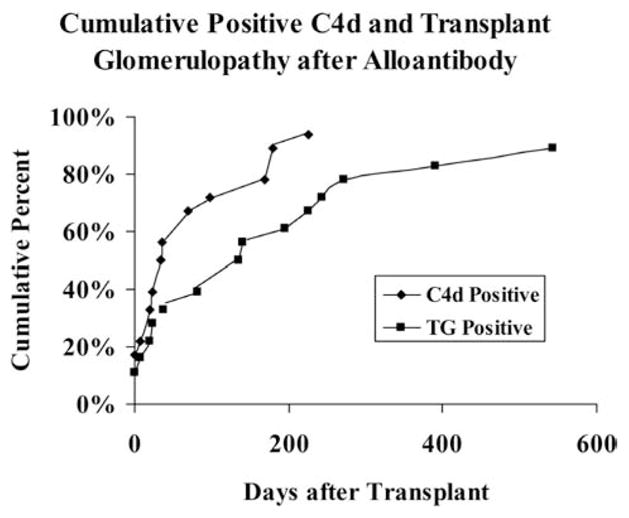
Cumulative increasing frequencies of positive transplant glomerulopathy and C4d staining.
Survival
To test the relationship between survival times and presence of alloantibodies, survival times were analyzed in two ways. In the first method animals were separated into three groups, (Figure 5): (1) acute cellular rejections, (2) other (nonrejections, mostly obstruction) and (3) long-term survivors, which were partitioned to test if the presence or absence of alloantibodies, positive C4d staining, TG or CAA were risk factors for survival of long-term animals. In this analysis, survival time was defined as death from any cause or the survival interval at the time of a normal-looking biopsy if the animal was alive. Of these long-term survivors, the presence of alloantibodies, C4d, TG or CAA reduced the respective mean and median survival time by at least 58% (Table 2 and Figure 5). In the second method Kaplan–Meirer analysis was performed censoring animals that died from nonrejection causes (mostly obstruction). This analysis showed that some animals without alloantibody (or C4d, TG or CAA) might survive indefinitely when nonrejection causes are censored from the data (Table 2 and Figure 5) and that the presence of alloantibody (or C4d TG, or CAA) markedly shortened the survival times (Table 2, p < 0.01).
Figure 5. Survival curves of 143 animals.
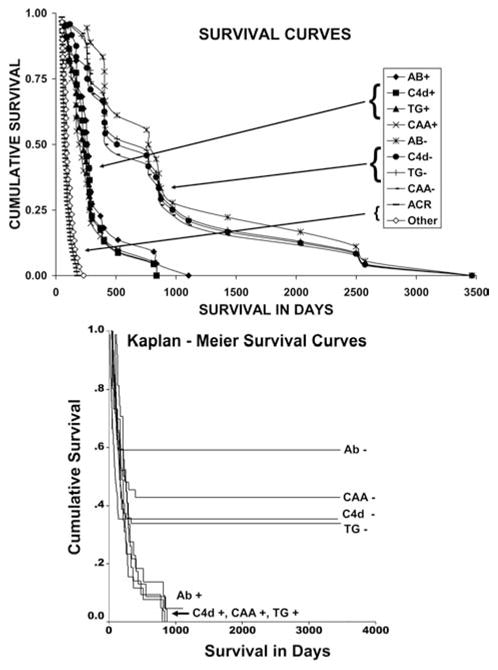
(Top) Survival interval until death from any cause including termination of the experiment. Number of animals: Alloantibody negative (18), alloantibody positive (22), C4d negative (24), C4d positive (22), TG positive and negative (23), chronic arteriopathy positive (26) and negative (20), ACR (64) and other (33). ACR = acute cellular rejection (mostly ACR2, mean and median survival, 87 or 76 days). Other is death from nonrejection: obstruction (most cases) or infection, BK nephritis (1), or posttransplant lymphoproliferative disease (1) (mean or median survival, 105 or 90 days). P < 0.001 for respective positive and negative groups. (Bottom) Kaplan–Meier survival curves of 143 animals with data censored for nonrejection. Number of animals: Alloantibody negative (15), C4d negative (52), TG negative (57), CAA negative (37), alloantibody positive (25), C4d positive (25), TG positive (26) and CAA positive (26). P < 0.01 for respective positive and negative groups.
Table 2.
| Survival times (days) |
||||||
|---|---|---|---|---|---|---|
| N | Mean | SD | Median | SD | p-Value | |
| Antibody negative | 18 | 1056 | 222 | 770 | 79 | |
| Antibody positive | 22 | 348 | 56 | 274 | 18 | p <0 .001 |
| C4d negative | 24 | 950 | 185 | 755 | 282 | |
| C4d positive | 22 | 303 | 43 | 256 | 33 | p <0 .001 |
| TG negative | 23 | 964 | 186 | 755 | 282 | |
| TG positive | 23 | 287 | 42 | 230 | 35 | p <0.001 |
| CAA negative | 20 | 912 | 174 | 508 | 287 | |
| CAA positive | 26 | 286 | 49 | 215 | 38 | p <0.001 |
| Kaplan–Meier survival times (days) |
||||||
|---|---|---|---|---|---|---|
| N | Mean | SD | Median | SD | p-Value | |
| Antibody negative | 15 | 2086 | 277 | |||
| Antibody positive | 25 | 313 | 54 | 252 | 31 | p <0.01 |
| C4d negative | 52 | 1288 | 187 | |||
| C4d positive | 25 | 282 | 42 | 230 | 30 | p <0 .01 |
| TG negative | 57 | 1252 | 184 | |||
| TG positive | 26 | 267 | 41 | 195 | 36 | p <0.01 |
| CAA negative | 37 | 1559 | 220 | |||
| CAA positive | 26 | 237 | 40 | 174 | 6 | p <0.01 |
p-value refers to either medians or means.
p-value refers to means.
Discussion
This study confirms that nonhuman primates develop chronic alloantibody-mediated renal allograft rejection, very similar to humans (12,16,20), without confounding nonrejection etiologies of late renal allograft failure often present in humans. Major new findings document markedly shortened allograft survival caused by alloantibodies, consistent with the excess renal allograft loss in humans associated with alloantibodies (38–40). The clinical pathological progression strongly supports the four proposed stages of chronic alloantibody-mediated rejection (25,26). Additionally, the overall progressive nature of antibody-mediated chronic rejection and renal dysfunction observed in this study fails to support the hypothesis that persistent MHC alloantibody and C4d deposition merely identify a state of quiescent accommodation without ongoing or eventual pathological injury and allograft dysfunction (41). In addition, ancillary findings include an association of the development of arteriopathy with prior TG, endarteritis, C4d/alloantibodies and glomerulitis, and the development of interstitial fibrosis with prior TG, glomerulitis and C4d/alloantibodies.
The absence of maintenance immunosuppression in this model makes extrapolation to human data difficult because most humans generally receive continuous treatment. In this study, the average interval between alloantibodies and renal failure was 0.6 years compared in humans of 2.7 years for class I and 3.9 years for class II alloantibodies (40). This variation between monkeys and humans may reflect treatment in humans or biological variation in the disease itself. Using a variety of tolerance protocols in Cynomolgus monkeys, we have pooled data for statistical and survival analysis. The tightness of the observed correlations, the temporal sequence and the ultimate congruence of alloantibodies with these pathological findings provide strong evidence that they are closely associated events, if not causally related, and support the etiological relevance of alloantibodies over minor variations in treatments and make this pattern highly relevant to human renal allografted patients, who all receive variations in therapy (38–40). Other investigators using different models also emphasize the value of nonhuman primate models of chronic rejection to describe similar pathologic features, TG and CAA, although without C4d (42,43).
Although C4d and alloantibodies were strongly correlated, a fraction of discordant cases exist with C4d deposition but without detectable alloantibodies or the reverse on any given biopsy similar to occasional discordance observed in humans (3,8,44–46). Positive C4d staining without concurrently detectable serum alloantibodies (which occurs in about 10% of human biopsies) might be due to non-HLA alloantibodies (47–49) or adsorption of antibodies by the graft. This study supports the later possibility because most of the alloantibody-negative, C4d-positive animals became alloantibody positive on subsequent testing or lost the C4d staining. Other investigators have shown that eluates of renal allograft nephrectomies removed for chronic rejection contain alloantibodies in 71% of cases but only 31% of these patients had detectable alloantibodies in the circulation (50). Why C4d staining occasionally lags the presence of serum alloantibodies (Figures 2–4) is unclear but may involve complement regulatory proteins or accommodation (51–53), lack of complement fixing antibodies (54) or just insufficient antibody to activate complement. Our serial studies also showed that most examples of discordance between C4d and alloantibodies subsequently became congruent on follow-up, that is, increasing frequency of C4d or TG over time (Figure 4). However, congruence was not absolute, and a few animals with alloantibodies never acquired C4d or TG. Some human studies have reported that a minority of TG cases were associated with alloantibodies and positive C4d (55–57), whereas Sis et al. identified that in 54 allograft kidneys with TG, 70% had alloantibodies and 36% had positive C4d (58). Nevertheless, 27% of cases could not be associated with either alloantibodies or positive C4d (58). Some of these human cases might be attributable to calcineurin inhibitor-induced thrombotic microangiopathy. Similarly, in our study of 31 animals with TG, 8 lacked C4d or alloantibody (26%). With increased sampling (Figure 4) 11% of animals with TG had no antecedent alloantibody or C4d, suggesting that sampling error and selection bias may increase the frequency of alloantibody-associated TG. Other human studies provide support that the strongest predictor of TG is C4d deposition (12,18,20,59). Also supporting the relationship between alloantibodies and C4d is the inability in this study to correlate acute cellular rejection and TG. The slightly lower frequency of human alloantibody-associated TG compared to monkeys is unclear but could be related to continuous immunosuppression especially calcineurin inhibitors, which are known to cause thrombotic microangiopathy (TMA)-associated TG. Twelve animals in this study were identified without both positive C4d positive peritubular capillary (PTC) staining, glomerulitis (Table 1) (60) or concurrent TG. Seven of these 12 animals later developed TG also suggesting that all cases of TG cannot be attributed solely to alloantibody/C4d or calcineurin inhibitor TMA. Variations in antibody titer, complement regulation, endothelial gene expression, accommodation or antibody/complement-independent mechanisms may all contribute to the heterogeneity of TG development.
The pathogenesis of CAA is unclear but is likely related to T cell, alloantibody or natural killer (NK) cell-mediated mechanisms (26). This study does not distinguish among these potential mechanisms but identified alloantibody/C4d and/or ACR2 (endarteritis) as risk factors for CAA. Of 26 animals with CAA, two lacked precursor alloantibodies/C4d or endothelialitis, 7 had only precursor endothelialitis, 10 only had precursor alloantibodies/C4d and 7 had both alloantibodies/C4d and endothelialitis. These data support the hypothesis that the pathological injury from either cellular or antibody-mediated rejection contributes to CAA, consistent with other observations (43,61). Alternatively, alloantibodies may not mediate CAA per se but are just a marker of sensitization for the cellular immunity that actually causes CAA (endothelialitis) but was missed due to sampling error because endothelialitis is more prominent in arcuate-sized renal arteries (62,63). Consistent with this hypothesis is that in autopsies CAA was observed more frequently with Banff type 2 rejections. Possibly, alloantibody and complement activation can recruit or activate T cells or dendritic cells (64) or B cells acting as antigen-presenting cells could promote acute cellular rejection (65,66). NK cells by ligation of Fc receptors and alloantibody could mediate allograft vasculopathy by interferon activation (67,68). CAA was predicted by positive C4d staining and alloantibodies. Overall, 68% of the monkeys that had CAA showed C4d-positive peritubular staining, comparable to 61% reported in humans (6).
How alloantibodies and complement activation promote glomerulopathy or possibly arteriopathy are unknown. Antibodies to class I MHC antigens can stimulate endothelial and smooth muscle proliferation and expression of fibroblast growth factor (FGF) receptors (69). Terminal complement components (C5b-9) trigger the production of FGF and platelet-derived growth factor (PDGF) by endothelial cells (70). Thus, antibodies and activated complement might cause chronic antibody-mediated rejection by inducing gene products that promote endothelial activation with subsequent basement membrane duplication and arterial smooth muscle proliferation (71,72).
To establish accommodation in some models requires acute complement depletion (73), especially the absence of activation of the terminal complement cascade (74,75), the up-regulation of complement regulatory components (DAF and CD59) (51,76) or the induction of anti-apoptotic proteins (Bcl-2, Bcl-xl, HO-1) or other cytoprotective proteins (nitric oxide, indolamine 2,3 dioxygenase) (52,77,78). Low alloantibody titer or sub-saturating quantities of alloantibody may preferentially induce anti-apoptotic gene expression and accommodation (79,80). More likely chronic antibody-mediated rejection may represent partially accommodated acute antibody-mediated rejection (26), in which accommodation only mitigates the mechanisms of acute injury but does not prevent gene expression of proteins that eventually causes capillary, interstitial or vascular remodeling, the pathological hallmarks of injury associated with clinical dysfunction. Our studies suggest that in the absence of immunosuppression anti-MHC alloantibodies may not be associated with stable accommodation in contrast to carbohydrate antigen/ABO incompatibility, which may lead to more stable accommodation (81–84).
Alloantibodies and chronic rejection are significant problems in tolerance protocols (42,85,86). This study confirms the difficulty of inducing tolerance in some types of T helper cells and in B cells, stressing the importance of the indirect pathway in allograft rejection (87–89) and are especially relevant to those clinical protocols designed to induce tolerance, T regulatory cells or to reduce or eliminate immunosuppression because the activation requirements of Th1, Th2, T regulatory cells or B cells may be different (90). Because complement activation is a T-cell costimulatory pathway, persistent complement activation in an allograft may increase the risk for the development of acute cellular rejection (64,65). Our results also emphasize the importance of timely and serial serological assays for alloantibodies and C4d staining because among various assays presently available only these predicted the critically important but clinically silent progressive intragraft pathological changes that can ultimately lead to graft loss from chronic antibody-mediated rejection (14,32,91).
In summary, the natural history of chronic antibody rejection in this model takes place in the absence of antigen-independent etiologies of chronic allograft nephropathy, occurs in four progressive stages, shortens renal allograft survival, is likely mediated by alloantibodies and develops without enduring stable accommodation.
Acknowledgments
The authors thank Patricia Della Pelle and Nicole Brousaides for superb technical assistance. This work was supported in part by a grant from the National Institutes of Health (HL18646).
References
- 1.Feucht HE, Felber E, Gokel MJ, et al. Vascular deposition of complement-split products in kidney allografts with cell-mediated rejection. Clin Exp Immunol. 1991;86:464–470. doi: 10.1111/j.1365-2249.1991.tb02954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feucht HE, Schneeberger H, Hillebrand G, et al. Capillary deposition of C4d complement fragment and early renal graft loss. Kidney Int. 1993;43:1333–1338. doi: 10.1038/ki.1993.187. [DOI] [PubMed] [Google Scholar]
- 3.Collins AB, Schneeberger EE, Pascual MA, et al. Complement activation in acute humoral renal allograft rejection: Diagnostic significance of C4d deposits in peritubular capillaries. J Am Soc Nephrol. 1999;10:2208–2214. doi: 10.1681/ASN.V10102208. [DOI] [PubMed] [Google Scholar]
- 4.Mauiyyedi S, Colvin RB. Humoral rejection in kidney transplantation: New concepts in diagnosis and treatment. Curr Opin Nephrol Hypertens. 2002;11:609–618. doi: 10.1097/00041552-200211000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Mauiyyedi S, Crespo M, Collins AB, et al. Acute humoral rejection in kidney transplantation: II. Morphology, immunopathology, and pathologic classification. J Am Soc Nephrol. 2002;13:779–787. doi: 10.1681/ASN.V133779. [DOI] [PubMed] [Google Scholar]
- 6.Mauiyyedi S, Pelle PD, Saidman S, et al. Chronic humoral rejection: Identification of antibody-mediated chronic renal allograft rejection by C4d deposits in peritubular capillaries. J Am Soc Nephrol. 2001;12:574–582. doi: 10.1681/ASN.V123574. [DOI] [PubMed] [Google Scholar]
- 7.Crespo M, Pascual M, Tolkoff-Rubin N, et al. Acute humoral rejection in renal allograft recipients: I. Incidence, serology and clinical characteristics. Transplantation. 2001;71:652–658. doi: 10.1097/00007890-200103150-00013. [DOI] [PubMed] [Google Scholar]
- 8.Bohmig GA, Exner M, Habicht A, et al. Capillary C4d deposition in kidney allografts: A specific marker of alloantibody-dependent graft injury. J Am Soc Nephrol. 2002;13:1091–1099. doi: 10.1681/ASN.V1341091. [DOI] [PubMed] [Google Scholar]
- 9.Haas M, Ratner LE, Montgomery RA. C4d staining of perioperative renal transplant biopsies. Transplantation. 2002;74:711–717. doi: 10.1097/00007890-200209150-00021. [DOI] [PubMed] [Google Scholar]
- 10.Nickeleit V, Zeiler M, Gudat F, Thiel G, Mihatsch MJ. Detection of the complement degradation product C4d in renal allografts: Diagnostic and therapeutic implications. J Am Soc Nephrol. 2002;13:242–251. doi: 10.1681/ASN.V131242. [DOI] [PubMed] [Google Scholar]
- 11.Koo DD, Roberts IS, Quiroga I, et al. C4d deposition in early renal allograft protocol biopsies. Transplantation. 2004;78:398–403. doi: 10.1097/01.tp.0000128328.68106.54. [DOI] [PubMed] [Google Scholar]
- 12.Sijpkens YW, Joosten SA, Wong MC, et al. Immunologic risk factors and glomerular C4d deposits in chronic transplant glomerulopathy. Kidney Int. 2004;65:2409–2418. doi: 10.1111/j.1523-1755.2004.00662.x. [DOI] [PubMed] [Google Scholar]
- 13.Herman J, Lerut E, Van Damme-Lombaerts R, Emonds MP, Van Damme B. Capillary deposition of complement C4d and C3d in pediatric renal allograft biopsies. Transplantation. 2005;79:1435–1440. doi: 10.1097/01.tp.0000158420.26623.0f. [DOI] [PubMed] [Google Scholar]
- 14.Scornik JC, Guerra G, Schold JD, Srinivas TR, Dragun D, Meier-Kriesche HU. Value of posttransplant antibody tests in the evaluation of patients with renal graft dysfunction. Am J Transplant. 2007;7:1808–1814. doi: 10.1111/j.1600-6143.2007.01855.x. [DOI] [PubMed] [Google Scholar]
- 15.Worthington JE, McEwen A, McWilliam LJ, Picton ML, Martin S. Association between C4d staining in renal transplant biopsies, production of donor-specific HLA antibodies, and graft outcome. Transplantation. 2007;83:398–403. doi: 10.1097/01.tp.0000251430.11723.b6. [DOI] [PubMed] [Google Scholar]
- 16.Horita S, Nitta K, Kawashima M, et al. C4d deposition in the glomeruli and peritubular capillaries associated with transplant glomerulopathy. Clin Transplant. 2003;17:325–330. doi: 10.1034/j.1399-0012.2003.t01-1-00014.x. [DOI] [PubMed] [Google Scholar]
- 17.Cardarelli F, Pascual M, Tolkoff-Rubin N, et al. Prevalence and significance of anti-HLA and donor-specific antibodies long-term after renal transplantation. Transpl Int. 2005;18:532–540. doi: 10.1111/j.1432-2277.2005.00085.x. [DOI] [PubMed] [Google Scholar]
- 18.Vongwiwatana A, Gourishankar S, Campbell PM, Solez K, Halloran PF. Peritubular capillary changes and C4d deposits are associated with transplant glomerulopathy but not IgA nephropathy. Am J Transplant. 2004;4:124–129. doi: 10.1046/j.1600-6143.2003.00294.x. [DOI] [PubMed] [Google Scholar]
- 19.Sijpkens YW, Doxiadis II, van Kemenade FJ, et al. Chronic rejection with or without transplant vasculopathy. Clin Transplant. 2003;17:163–170. doi: 10.1034/j.1399-0012.2003.00039.x. [DOI] [PubMed] [Google Scholar]
- 20.Regele H, Bohmig GA, Habicht A, et al. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: A contribution of humoral immunity to chronic allograft rejection. J Am Soc Nephrol. 2002;13:2371–2380. doi: 10.1097/01.asn.0000025780.03790.0f. [DOI] [PubMed] [Google Scholar]
- 21.Mengel M, Bogers J, Bosmans JL, et al. Incidence of C4d stain in protocol biopsies from renal allografts: Results from a multicenter trial. Am J Transplant. 2005;5:1050–1056. doi: 10.1111/j.1600-6143.2005.00788.x. [DOI] [PubMed] [Google Scholar]
- 22.Haas M, Montgomery RA, Segev DL, et al. Subclinical acute antibody-mediated rejection in positive crossmatch renal allografts. Am J Transplant. 2007;7:576–585. doi: 10.1111/j.1600-6143.2006.01657.x. [DOI] [PubMed] [Google Scholar]
- 23.Mengel M, Bogers JP, Bosmans JL, Seron D, Gwinner W, Haller H. Incidence of C4d staining and morphology of acute humoral rejection in protocol biopsies of renal allografts: A multicenter study. J Am Soc Nephrol. 2003;14:11A. [Google Scholar]
- 24.Nickerson P, Gibson IW, Karpinski M, et al. C4d deposition in protocol biopsies from flow cytometry crossmatch positive or negative renal transplant recipients. J Am Soc Nephrol. 2002;13:567A. [Google Scholar]
- 25.Takemoto SK, Zeevi A, Feng S, et al. A national conference to assess antibody mediated rejection in solid organ transplantation. A J Transplant. 2004;4:1033–1041. doi: 10.1111/j.1600-6143.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 26.Colvin RB, Smith RN. Antibody-mediated organ allograft rejection. Nat Rev Immunol. 2005;5:807–817. doi: 10.1038/nri1702. [DOI] [PubMed] [Google Scholar]
- 27.Kawai T, Cosimi AB, Colvin RB, et al. Mixed allogeneic chimerism and renal allograft tolerance in cynomolgus monkeys. Transplantation. 1995;59:256–262. [PubMed] [Google Scholar]
- 28.Kimikawa M, Sachs DH, Colvin RB, Bartholomew A, Kawai T, Cosimi AB. Modifications of the conditioning regimen for achieving mixed chimerism and donor-specific tolerance in cynomolgus monkeys. Transplantation. 1997;64:709–716. doi: 10.1097/00007890-199709150-00008. [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Poncelet A, Sachs DH, et al. Long-term outcome and alloantibody production in a non-myeloablative regimen for induction of renal allograft tolerance. Transplantation. 1999;68:1767–1775. doi: 10.1097/00007890-199912150-00022. [DOI] [PubMed] [Google Scholar]
- 30.Kawai T, Wee SL, Bazin H, et al. Association of natural killer cell depletion with induction of mixed chimerism and allograft tolerance in non-human primates. Transplantation. 2000;70:368–374. doi: 10.1097/00007890-200007270-00023. [DOI] [PubMed] [Google Scholar]
- 31.Abe M, Kawai T, Futatsuyama K, et al. Postoperative production of anti-donor antibody and chronic rejection in renal transplantation. Transplantation. 1997;63:1616–1619. doi: 10.1097/00007890-199706150-00014. [DOI] [PubMed] [Google Scholar]
- 32.Boskovic S, Kawai T, Smith RN, et al. Monitoring antidonor alloantibodies as a predictive assay for renal allograft tolerance/long-term observations in nonhuman primates. Transplantation. 2006;82:819–825. doi: 10.1097/01.tp.0000234786.26511.a4. [DOI] [PubMed] [Google Scholar]
- 33.Smith RN, Kawai T, Boskovic S, et al. Chronic antibody mediated rejection of renal allografts: Pathological, serological and immunologic features in nonhuman primates. Am J Transplant. 2006;6:1790–1798. doi: 10.1111/j.1600-6143.2006.01351.x. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Abrahamian G, Sogawa H, et al. Costimulatory blockade for induction of mixed chimerism and renal allograft tolerance in nonhuman primates. Transplant Proc. 2001;33:221–222. doi: 10.1016/s0041-1345(00)01982-5. [DOI] [PubMed] [Google Scholar]
- 35.Racusen LC, Colvin RB, Solez K, et al. Antibody-mediated rejection criteria – an addition to the Banff 97 classification of renal allograft rejection. Am J Transplant. 2003;3:708–714. doi: 10.1034/j.1600-6143.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 36.Racusen LC, Solez K, Colvin RB, et al. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999;55:713–723. doi: 10.1046/j.1523-1755.1999.00299.x. [DOI] [PubMed] [Google Scholar]
- 37.Wavamunno MD, O’Connell PJ, Vitalone M, et al. Transplant glomerulopathy: Ultrastructural abnormalities occur early in longitudinal analysis of protocol biopsies. Am J Transplant. 2007;7:2757–2768. doi: 10.1111/j.1600-6143.2007.01995.x. [DOI] [PubMed] [Google Scholar]
- 38.Terasaki PI. Humoral theory of transplantation. Am J Transplant. 2003;3:665–673. doi: 10.1034/j.1600-6143.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 39.Lee PC, Terasaki PI, Takemoto SK, et al. All chronic rejection failures of kidney transplants were preceded by the development of HLA antibodies. Transplantation. 2002;74:1192–1194. doi: 10.1097/00007890-200210270-00025. [DOI] [PubMed] [Google Scholar]
- 40.Worthington JE, Martin S, Al-Husseini DM, Dyer PA, Johnson RW. Posttransplantation production of donor HLA-specific antibodies as a predictor of renal transplant outcome. Transplantation. 2003;75:1034–1040. doi: 10.1097/01.TP.0000055833.65192.3B. [DOI] [PubMed] [Google Scholar]
- 41.Platt JL. C4d and the fate of organ allografts. J Am Soc Nephrol. 2002;13:2417–2419. doi: 10.1097/01.asn.0000030140.74450.0b. [DOI] [PubMed] [Google Scholar]
- 42.Torrealba JR, Fernandez LA, Kanmaz T, et al. Immunotoxin-treated rhesus monkeys: A model for renal allograft chronic rejection. Transplantation. 2003;76:524–530. doi: 10.1097/01.TP.0000075788.72614.D4. [DOI] [PubMed] [Google Scholar]
- 43.Wieczorek G, Bigaud M, Menninger K, et al. Acute and chronic vascular rejection in nonhuman primate kidney transplantation. Am J Transplant. 2006;6:1285–1296. doi: 10.1111/j.1600-6143.2006.01307.x. [DOI] [PubMed] [Google Scholar]
- 44.Feucht HE. Complement C4d in graft capillaries – the missing link in the recognition of humoral alloreactivity. Am J Transplant. 2003;3:646–652. doi: 10.1034/j.1600-6143.2003.00171.x. [DOI] [PubMed] [Google Scholar]
- 45.Abe M, Sawada T, Horita S, Toma H, Yamaguchi Y, Teraoka S. C4d deposition in peritubular capillary and alloantibody in the allografted kidney suffering severe acute rejection. Clin Transplant. 2003;17(Suppl 10):14–19. doi: 10.1034/j.1399-0012.17.s10.7.x. [DOI] [PubMed] [Google Scholar]
- 46.Kato M, Morozumi K, Takeuchi O, et al. Complement fragment C4d deposition in peritubular capillaries in acute humoral rejection after ABO blood group-incompatible human kidney transplantation. Transplantation. 2003;75:663–665. doi: 10.1097/01.TP.0000053402.87256.6B. [DOI] [PubMed] [Google Scholar]
- 47.Perrey C, Brenchley PE, Johnson RW, Martin S. An association between antibodies specific for endothelial cells and renal transplant failure. Transpl Immunol. 1998;6:101–106. doi: 10.1016/s0966-3274(98)80024-5. [DOI] [PubMed] [Google Scholar]
- 48.Martin S, Brenchley PE, Postlethwaite RJ, Johnson RW, Dyer PA. Detection of anti-epithelial cell antibodies in association with pediatric renal transplant failure using a novel microcytotoxicity assay. Tissue Antigens. 1991;37:152–155. doi: 10.1111/j.1399-0039.1991.tb01863.x. [DOI] [PubMed] [Google Scholar]
- 49.Wu GD, Jin YS, Salazar R, et al. Vascular endothelial cell apoptosis induced by anti-donor non-MHC antibodies: A possible injury pathway contributing to chronic allograft rejection. J Heart Lung Transplant. 2002;21:1174–1187. doi: 10.1016/s1053-2498(02)00457-6. [DOI] [PubMed] [Google Scholar]
- 50.Martin L, Guignier F, Mousson C, Rageot D, Justrabo E, Rifle G. Detection of donor-specific anti-HLA antibodies with flow cytometry in eluates and sera from renal transplant recipients with chronic allograft nephropathy. Transplantation. 2003;76:395–400. doi: 10.1097/01.TP.0000078895.24606.45. [DOI] [PubMed] [Google Scholar]
- 51.Cornell LD, Della Pelle P, Brousiadies N, Collins AB, Colvin RB. Endothelial response to rejection: Enhanced expression of complement regulatory proteins decay accelerating factor (DAF, CD55) and protectin (CD59) in human renal allografts. Mod Pathol. 2004;17(Suppl 1):285A. [Google Scholar]
- 52.Otterbein LE, Zuckerbraun BS, Haga M, et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat Med. 2003;9:183–190. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- 53.Soares MP, Lin Y, Sato K, Stuhlmeier KM, Bach FH. Accommodation. Immunol Today. 1999;20:434–437. doi: 10.1016/s0167-5699(99)01530-3. [DOI] [PubMed] [Google Scholar]
- 54.Sund S, Hovig T, Reisaeter AV, Scott H, Bentdal O, Mollnes TE. Complement activation in early protocol kidney graft biopsies after living-donor transplantation. Transplantation. 2003;75:1204–1213. doi: 10.1097/01.TP.0000062835.30165.2C. [DOI] [PubMed] [Google Scholar]
- 55.Akalin E, Dinavahi R, Dikman S, et al. Transplant glomerulopathy may occur in the absence of donor-specific antibody and C4d staining. Clin J Am Soc Nephrol. 2007;2:1261–1267. doi: 10.2215/CJN.02420607. [DOI] [PubMed] [Google Scholar]
- 56.Gloor JM, Sethi S, Stegall MD, et al. Transplant glomerulopathy: Subclinical incidence and association with alloantibody. Am J Transplant. 2007;7:2124–2132. doi: 10.1111/j.1600-6143.2007.01895.x. [DOI] [PubMed] [Google Scholar]
- 57.Namba Y, Oka K, Moriyama T, et al. Incidence of positive C4d deposition in long-term survival cases over 10 yr after renal transplantation. Clin Transplant. 2006;20(Suppl 15):20–25. doi: 10.1111/j.1399-0012.2006.00545.x. [DOI] [PubMed] [Google Scholar]
- 58.Sis B, Campbell PM, Mueller T, et al. Transplant glomerulopathy, late antibody-mediated rejection and the ABCD tetrad in kidney allograft biopsies for cause. Am J Transplant. 2007;7:1743–1752. doi: 10.1111/j.1600-6143.2007.01836.x. [DOI] [PubMed] [Google Scholar]
- 59.Regele H, Exner M, Watschinger B, et al. Endothelial C4d deposition is associated with inferior kidney allograft outcome independently of cellular rejection. Nephrol Dial Transplant. 2001;16:2058–2066. doi: 10.1093/ndt/16.10.2058. [DOI] [PubMed] [Google Scholar]
- 60.Magil AB, Tinckam K. Monocytes and peritubular capillary C4d deposition in acute renal allograft rejection. Kidney Int. 2003;63:1888–1893. doi: 10.1046/j.1523-1755.2003.00921.x. [DOI] [PubMed] [Google Scholar]
- 61.Kuypers DR, Chapman JR, O’Connell PJ, Allen RD, Nankivell BJ. Predictors of renal transplant histology at three months. Transplantation. 1999;67:1222–1230. doi: 10.1097/00007890-199905150-00005. [DOI] [PubMed] [Google Scholar]
- 62.Colvin RB, Cohen AH, Saiontz C, et al. Evaluation of pathologic criteria for acute renal allograft rejection: Reproducibility, sensitivity, and clinical correlation. J Am Soc Nephrol. 1997;8:1930–1941. doi: 10.1681/ASN.V8121930. [DOI] [PubMed] [Google Scholar]
- 63.Nickeleit V, Vamvakas EC, Pascual M, Poletti BJ, Colvin RB. The prognostic significance of specific arterial lesions in acute renal allograft rejection. J Am Soc Nephrol. 1998;9:1301–1308. doi: 10.1681/ASN.V971301. [DOI] [PubMed] [Google Scholar]
- 64.Heeger PS, Lalli PN, Lin F, et al. Decay-accelerating factor modulates induction of T cell immunity. J Exp Med. 2005;201:1523–1530. doi: 10.1084/jem.20041967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baldwin WM, Ota H, Rodriguez ER. Complement in transplant rejection: Diagnostic and mechanistic considerations. Springer Semin Immunopathol. 2003;25:181–197. doi: 10.1007/s00281-003-0133-3. [DOI] [PubMed] [Google Scholar]
- 66.Rahimi S, Qian Z, Layton J, Fox-Talbot K, Baldwin WM, 3rd, Wasowska BA. Non-complement- and complement-activating antibodies synergize to cause rejection of cardiac allografts. Am J Transplant. 2004;4:326–334. doi: 10.1111/j.1600-6143.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- 67.Uehara S, Chase CM, Kitchens WH, et al. NK cells can trigger allograft vasculopathy: The role of hybrid resistance in solid organ allografts. J Immunol. 2005;175:3424–3430. doi: 10.4049/jimmunol.175.5.3424. [DOI] [PubMed] [Google Scholar]
- 68.Kondadasula SV, Roda JM, Parihar R, et al. Co-localization of the IL-12 receptor and Fc{gamma}RIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-{gamma} Blood. 2008;111:4173–4183. doi: 10.1182/blood-2007-01-068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bian H, Reed EF. Anti-HLA class I antibodies transduce signals in endothelial cells resulting in FGF receptor translocation, down-regulation of ICAM-1 and cell proliferation. Transplant Proc. 2001;33:311. doi: 10.1016/s0041-1345(00)02022-4. [DOI] [PubMed] [Google Scholar]
- 70.Benzaquen LR, Nicholson-Weller A, Halperin JA. Terminal complement proteins C5b-9 release basic fibroblast growth factor and platelet-derived growth factor from endothelial cells. J Exp Med. 1994;179:985–992. doi: 10.1084/jem.179.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, Reed EF. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin, and focal adhesion kinase in an actin-dependent manner. J Immunol. 2002;168:5415–5423. doi: 10.4049/jimmunol.168.11.5415. [DOI] [PubMed] [Google Scholar]
- 72.Gong KW, Jin YP, Lepin EJ, Chen HW, Lee H, Reed EF. Class I mediated signal transduction in smooth muscle cells. Hum Immunol. 2003;64(10 Suppl):S60. [Google Scholar]
- 73.Lin Y, Soares MP, Sato K, et al. Accommodated xenografts survive in the presence of anti-donor antibodies and complement that precipitate rejection of naive xenografts. J Immunol. 1999;163:2850–2857. [PubMed] [Google Scholar]
- 74.Suhr BD, Black SM, Guzman-Paz M, Matas AJ, Dalmasso AP. Inhibition of the membrane attack complex of complement for induction of accommodation in the hamster-to-rat heart transplant model. Xenotransplantation. 2007;14:572–579. doi: 10.1111/j.1399-3089.2007.00422.x. [DOI] [PubMed] [Google Scholar]
- 75.Wang H, Arp J, Liu W, et al. Inhibition of terminal complement components in presensitized transplant recipients prevents antibody-mediated rejection leading to long-term graft survival and accommodation. J Immunol. 2007;179:4451–4463. doi: 10.4049/jimmunol.179.7.4451. [DOI] [PubMed] [Google Scholar]
- 76.Ding JW, Zhou T, Ma L, et al. Expression of complement regulatory proteins in accommodated xenografts induced by anti-alpha-Gal IgG in a rat-to-mouse model. Am J Transplant. 2008;8:32–40. doi: 10.1111/j.1600-6143.2007.02016.x. [DOI] [PubMed] [Google Scholar]
- 77.Heslan JM, Renaudin K, Thebault P, Josien R, Cuturi MC, Chiffoleau E. New evidence for a role of allograft accommodation in long-term tolerance. Transplantation. 2006;82:1185–1193. doi: 10.1097/01.tp.0000236573.01428.f3. [DOI] [PubMed] [Google Scholar]
- 78.Lin Y, Soares MP, Sato K, et al. Long-term survival of hamster hearts in presensitized rats. J Immunol. 2000;164:4883–4892. doi: 10.4049/jimmunol.164.9.4883. [DOI] [PubMed] [Google Scholar]
- 79.Cecka JM, Zhang Q, Reed EF. Preformed cytotoxic antibodies in potential allograft recipients: Recent data. Hum Immunol. 2005;66:343–349. doi: 10.1016/j.humimm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 80.Narayanan K, Jendrisak MD, Phelan DL, Mohanakumar T. HLA class I antibody mediated accommodation of endothelial cells via the activation of PI3K/cAMP dependent PKA pathway. Transpl Immunol. 2006;15:187–197. doi: 10.1016/j.trim.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 81.Shimizu I, Smith NR, Zhao G, Medof E, Sykes M. Decay-accelerating factor prevents acute humoral rejection induced by low levels of anti-alphaGal natural antibodies. Transplantation. 2006;81:95–100. doi: 10.1097/01.tp.0000188176.18666.68. [DOI] [PubMed] [Google Scholar]
- 82.King KE, Warren DS, Samaniego-Picota M, Campbell-Lee S, Montgomery RA, Baldwin WM., 3rd Antibody, complement and accommodation in ABO-incompatible transplants. Curr Opin Immunol. 2004;16:545–549. doi: 10.1016/j.coi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 83.Segev DL, Simpkins CE, Warren DS, et al. ABO incompatible high-titer renal transplantation without splenectomy or anti-CD20 treatment. Am J Transplant. 2005;5:2570–2575. doi: 10.1111/j.1600-6143.2005.01031.x. [DOI] [PubMed] [Google Scholar]
- 84.Takahashi K. Recent findings in ABO-incompatible kidney transplantation: Classification and therapeutic strategy for acute antibody-mediated rejection due to ABO-blood-group-related antigens during the critical period preceding the establishment of accommodation. Clin Exp Nephrol. 2007;11:128–141. doi: 10.1007/s10157-007-0461-z. [DOI] [PubMed] [Google Scholar]
- 85.Kirk AD, Burkly LC, Batty DS, et al. Treatment with humanized monoclonal antibody against CD154 prevents acute renal allograft rejection in nonhuman primates. Nat Med. 1999;5:686–693. doi: 10.1038/9536. [DOI] [PubMed] [Google Scholar]
- 86.Kirk AD, Harlan DM, Armstrong NN, et al. CTLA4-Ig and anti-CD40 ligand prevent renal allograft rejection in primates. Proc Natl Acad Sci USA. 1997;94:8789–8794. doi: 10.1073/pnas.94.16.8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Auchincloss H, Jr, Lee R, Shea S, Markowitz JS, Grusby MJ, Glimcher LH. The role of “indirect” recognition in initiating rejection of skin grafts from major histocompatibility complex class II-deficient mice. Proc Natl Acad Sci USA. 1993;90:3373–3377. doi: 10.1073/pnas.90.8.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gould DS, Auchincloss H., Jr Direct and indirect recognition: The role of MHC antigens in graft rejection. Immunol Today. 1999;20:77–82. doi: 10.1016/s0167-5699(98)01394-2. [DOI] [PubMed] [Google Scholar]
- 89.Steele DJ, Laufer TM, Smiley ST, et al. Two levels of help for B cell alloantibody production. J Exp Med. 1996;183:699–703. doi: 10.1084/jem.183.2.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kirk AD, Baldwin WM, Cascalho MI, Chong AS, Sykes M, West LJ. American society of transplantation symposium on B cells in transplantation: Harnessing humoral immunity from rodent models to clinical practice. Am J Transplant. 2007;7:1464–1470. doi: 10.1111/j.1600-6143.2007.01815.x. [DOI] [PubMed] [Google Scholar]
- 91.Pollinger HS, Stegall MD, Gloor JM, et al. Kidney transplantation in patients with antibodies against donor HLA class II. Am J Transplant. 2007;7:857–863. doi: 10.1111/j.1600-6143.2006.01699.x. [DOI] [PubMed] [Google Scholar]