

Abstract
Type 2 ryanodine receptor (RyR2) is the major calcium release channel in cardiac muscle. Phosphorylation of RyR2 by cAMP-dependent protein kinase A and by calmodulin-dependent protein kinase II modulates channel activity. Hyperphosphorylation at a single amino acid residue, Ser-2808, has been proposed to directly disrupt the binding of a 12.6 kDa FK506 binding protein (FKBP12.6) to RyR2, causing a RyR2 malfunction that triggers cardiac arrhythmias in human heart failure. To determine the structural basis of the interaction between Ser-2808 and FKBP12.6, we have employed two independent approaches to map this phosphorylation site in RyR2 by three-dimensional cryo-electron microscopy. In one approach, we inserted a green fluorescent protein (GFP) after amino acid Tyr-2801, and mapped GFP’s three-dimensional location in RyR2’s structure. In another approach, the binding site of monoclonal antibody 34C was mapped in the three-dimensional structure of skeletal muscle RyR1. The epitope of antibody 34C has been mapped to amino-acid residues 2,756 through 2,803 of the RyR1 sequence, corresponding to residues 2,722 through 2,769 of the RyR2 sequence. These locations of GFP insertion and antibody binding are adjacent to one another in domain 6 of the cytoplasmic clamp region. Importantly, the three-dimensional location of the Ser-2808 phosphorylation site is 105-120Å distance from the FKBP12.6 binding site mapped previously, indicating that Ser-2808 is unlikely to be directly involved in the binding of FKBP12.6 to RyR2, as had been proposed previously.
Sudden cardiac death (SCD) is a sudden, unexpected death caused by loss of heart function. SCD is the leading cause of natural death in the United States, producing about 330,000 deaths each year (1). SCD occurs when the electrical system in the heart malfunctions, leading to fatal arrhythmias. Most cases of SCD can be explained by cardiovascular abnormalities identifiable at autopsy, but some SCD is associated with structurally normal hearts (2). In some such cases, abnormal calcium release through a dysfunctional cardiac muscle ryanodine receptor (type 2 RyR, or RyR2) has been implicated, and molecular genetic analysis has demonstrated that mutations in the RyR2 gene can be linked to about 14 percent of these structurally normal heart SCD cases (2).
RyR2 functions as a calcium release channel in the heart, where it plays a crucial role in excitation-contraction coupling. To date, more than 60 naturally occurring mutations in RyR2 have been linked to two genetic forms of ventricular arrhythmias: catecholaminergic polymorphic ventricular tachycardia (CPVT) and arrhythmogenic right ventricular dysplasia type 2 (ARVD2; Ref. 2-6). These mutations are largely clustered in three regions of RyR2’s amino-acid sequence: region 1 is near the amino terminus, from residues 77 to 466; region 2 is near the middle of the sequence, spanning residues 1,724 through 2,958; and region 3 includes both cytoplasmic and membrane associated regions of the sequence and comprises residues 3,778 through 4,959. Intriguingly, the same three regions are homologous to the three malignant hyperthermia and central core disease mutation regions in skeletal isoform RyR (type 1 RyR, or RyR1; Ref. 7), suggesting that the three regions represent structural domains critical for the regulation of both RyR1 and RyR2, and suggesting also that the molecular pathologies underlying these RyR-linked skeletal and cardiac muscle diseases involve similar mechanisms.
The causal relationships among RyR2 mutations, the abnormal Ca2+ release function, and physiological manifestation is under investigation. Some mechanisms underlying RyR2 channel dysfunction in SCD have been proposed, including channel instability due to destabilization of the RyR2-FKBP12.6 interaction (8, 9) or domain-domain interaction (10, 11); reduced threshold for store-overload-induced Ca2+ release (12); altered channel sensitivity to cytoplasmic calcium (13); and reduced Mg2+ inhibition (14). FKBP is an isomerase that is widely expressed and involved in many cell functions. Two isoforms, FKBP12 and FKBP12.6, bind tightly to RyR1 and RyR2, respectively, and they are considered to be integral subunits of RyR (15, 16). FKBP12 and FKBP12.6 have been shown to stabilize the closed state of the RyR1 and RyR2 Ca2+ channels, thereby preventing inappropriate spontaneous channel openings (17, 18). Marx and coworkers have shown that protein kinase A phosphorylation of RyR2 dissociated FKBP12.6 and affected the channel open probability, and that hyperphosphorylation of RyR2 at a single amino acid residue Ser-2809 (in rabbit RyR2 sequence, corresponding to Ser-2808 in mouse) leads to defective channel function in failing heart (8). Subsequently, Wehrens and coworkers have shown that disease-causing mutations in RyR2 (three CPVT mutations: S2246L, R2474S, and R4497C) reduced the binding affinity of FKBP12.6 for RyR2, and increased the channel leakage that can trigger fatal cardiac arrhythmias under physical and emotional stresses (9).
However, the mechanisms proposed by Marx, Wehrens and colleagues are not consistent with other laboratories’ findings. Li et al. did not find any affect of PKA phosphorylation of the RyR2 on calcium sparks in mouse ventricular myocytes (19). Jiang et al. did not observe dissociation of FKBP12.6 from RyR2 in cardiac microsomal membranes treated with PKA (20). Stange et al. made site-directed substitutions of RyR2 at Ser-2809 (with Asp or Ala), and showed that mutant RyR2s neither abolished FKBP12.6 binding nor substantially changed channel functional properties (21). Xiao et al. found that FKBP12.6 can bind to both the Ser-2808 (mouse) phosphorylated and non-phosphorylated forms of RyR2, and that a Ser2808Asp phosphomimetic mutant retained the ability to bind FKBP12.6. Furthermore, complete phosphorylation at Ser-2808 by exogenous PKA did not disrupt the FKBP12.6-RyR2 complex (22). George et al. have demonstrated that CPVT RyR2 mutations augmented Ca2+ release, yet the RyR2-FKBP12.6 complex remained intact (23).
The binding sites for FKBP12 and FKBP12.6 have been mapped in the three-dimensional structures of RyR1 and RyR2, respectively (24, 25). However, no information regarding the phosphorylation site Ser-2808 in the RyR’s three-dimensional architecture has been reported. We believe that a direct structural approach to localize the Ser-2808 in the three-dimensional structure of RyR is relevant to testing the proposed FKBP12.6 dissociation mechanism. Recently, we have mapped two mutation hotspots in the three-dimensional structure of RyR2 by green fluorescent protein (GFP) labeling at specific amino-acid residues, combined with cryo-electron microscopy (cryo-EM) and single-particle image processing methods (26, 27), these studies provided some structural insights into the mechanism of RyR2 mutations. In the present study, we employed the same structural approach to determine the Ser-2808 phosphorylation site in the three-dimensional structure of RyR, thereby directly testing the mechanism of PKA phosphorylation-induced FKBP12.6 dissociation that was proposed by Marks and colleagues. We have used two independent approaches to identify the three-dimensional location of the phosphorylation site. In the first approach, we inserted GFP after amino acid Tyr-2801, and mapped its three-dimensional location in RyR2’s structure. In the second approach, the binding site of monoclonal antibody 34C (mAb34C) was mapped in the three-dimensional structure of RyR1. The epitope of the 34C antibody has previously been determined to include amino-acid residues 2,756 through 2,803 of RyR1, corresponding to residues 2,722 through 2,769 of RyR2 (28). We found that the mapped GFP insertion site and the antibody binding site are adjacent to one another in domain 6 of the cytoplasmic clamp region. However, neither site is close to the FKBP12.6 binding site mapped previously.
EXPERIMENTAL PROCEDURES
Construction of RyR2A4860G and RyR2Y2801-GFP
The A4860G mutation was introduced into the mouse RyR2 as described previously (29). To generate the RyR2Y2801-GFP construct, the cDNA encoding the GFP, which was flanked by a glycine-rich linker and an AscI endonuclease restriction site, was obtained by PCR as described previously (30). An AscI site was introduced into RyR2A4860G after Tyr-2801 by overlap-extension PCR. The “outer” primers are: forward, 5’-ACCATCTATGATGCAACACCTA-3’; and reverse, 5’-CTTGGCTGTCAGTGTGTCATAA-3’. The “inner” primers are: forward, 5’-ATGGCCCTTTATGGGCGCGCCCGACGAATT TCTCAGACA-3’; reverse, 5’-AGAAATTCGTCGGGCGCGCCCATAAAGGG CCATGCTGTC-3’. The AscI fragment containing the cDNA encoding GFP and the linkers was then subcloned into the full-length RyR2A4860G after Tyr-2801 to yield RyR2Y2801-GFP, containing the A4860G mutation.
Cell culture and cDNA transfection
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium as described previously (31). HEK293 cells grown in 100-mm tissue culture dishes for 18-20 hours after subculture were transfected with 12 μg RyRA4860G or RyRY2801-GFP cDNAs using Ca2+ phosphate precipitation.
Expression and purification of RyR2A4860G and RyR2Y2801-GFP
HEK293 cells grown for 24-26 hours after transfection were washed three times with PBS plus 2.5 mM EDTA and were harvested in the same solution by centrifugation for 8 min at 700 × g in an IEC Centra-CL2 centrifuge. The cell pellets were solubilized in a lysis buffer containing 25 mM Na-PIPES (pH 7.2), 140 mM NaCl, 5 mM EGTA, 2.5 mM dithiothreitol, 1% CHAPS, 0.5% egg lecithin, and a protease inhibitor mixture (2 mM benzamidine, 4 μg/ml leupeptin, 2 μg/ml pepstatin A, 4 μg/ml aprotinin, and 0.75 mM phenylmethylsulfonyl fluoride) and were incubated on ice for one hour. Insoluble material was removed by centrifugation at 16,000 × g twice in a microcentrifuge at 4°C for 30 min, and the cell lysates were collected. Sucrose (400 mM final concentration) and NaCl (400 mM final concentration) were added to the cell lysates. Glutathione-Sepharose beads (100 μl) containing 400 μg of bound glutathione S-transferase (GST)-FKBP12.6 fusion protein were added to the cell lysates and the mixture was incubated at 4°C with rotation for 2 days. The beads were collected by centrifugation and washed four times with lysis buffer containing 0.5% CHAPS and 0.25% egg lecithin. The RyR2 proteins were eluted from the beads by incubation with 100 mM glutathione in 25 mM Na-PIPES (pH 7.2), 400 mM NaCl, 400 mM sucrose, 1 mM EGTA, 0.2 mM CaCl2, 2.5 mM dithiothreitol, 0.4% CHAPS, 0.16% egg lecithin, 200 mM Tris/HEPES (pH 7.4), and protease inhibitor mixture for 15 min. The eluate was aliquoted, frozen in liquid nitrogen, and stored at -90°C.
Ca2+ release measurement and [3H]ryanodine binding assay
The concentration of free cytosolic Ca2+ in the transfected HEK293 cells was measured with the fluorescence Ca2+ indicator dye fluo-3 as described previously (31). Equilibrium [3H]ryanodine binding to cell lysates was carried out as described previously (32).
Western blotting and fluorescence imaging
The purified RyR2A4860G and RyR2Y2801-GFP proteins were subjected to 6% SDS-PAGE and were detected by Western blotting using anti-RyR antibody (1:1000) and anti-GFP antibody (1:1000) (Affinity BioReagents, Golden, CO) as described previously (30). For detection of the green fluorescence of RyR2Y2801-GFP, HEK293 cells were grown on glass coverslips placed in a 100-mm tissue culture dish and transfected with RyR2Y2801-GFP as described above. 24 hours after transfection, the coverslips were washed three times with PBS, mounted on a glass slide, and visualized with a Leica DMRB fluorescent microscope using a 40 x objective.
MAb34C Fab purification and activity test
The mAb34C hybridoma cell line was injected into mice to produce ascites fluid. MAb34C IgG was purified from 1.2 ml of ascites fluid by affinity chromatography on immobilized Protein A using Pierce ImmunoPure IgG Purification Kit as described by the manufacturer (Perbio Science, Rockford, Illinois). Fab fragments were obtained from purified IgG by papain digestion using Pierce ImmunoPure Fab Preparation Kit. To test the binding activity of Fab, a precipitation experiment was performed: 40 μg GST-FKBP12 with glutathione-Sepharose beads, and 12 μg of purified 34c Fab with or without 20 μg RyR1, were incubated separately for 30 minutes at room temperature, and the two complexes were then mixed together and incubated for a further 30 minutes. The whole protein complexes were eluted and then analyzed by SDS-PAGE.
Purification of RyR1 and preparation of RyR1+mAb34c Fab complexes
RyR1 was isolated from terminal cisternae vesicles from rabbit skeletal muscle as previously described (33). The RyR1+mAb34c Fab complexes were formed by addition of mAb34c Fab fragments to purified RyR1 with a molar ratio 8:1 (8 Fab fragments per RyR monomer), and incubated for 30 minutes.
Cryo-electron microscopy and image processing
EM grids were prepared for cryo-EM by an FEI Vitrobot computer-controlled freeze-plunging instrument (FEI Company, Hillsboro, Oregon). Micrographs were recorded using low-dose protocols on an FEI Tecnai F20 field emission gun transmission electron microscope operated at 200 kV, equipped with an Oxford CT3500 cryo-transfer holder (Gatan, Inc., Warrendale, Pennsylvania). The temperature of the grids was maintained at around -170°C. The defocus of the micrographs ranged between 1.5 and 4.5 μm, at a magnification of 50,760x (± 2%). To obtain more side-view information of the RyR molecules, we tilted the grids between 0° and 50° during data collection. Each exposure corresponded to an electron dose of ~10 e-/Å2. Micrographs were checked for drift, astigmatism, and presence of Thon rings by optical diffraction. Selected electron micrographs were digitized on a Zeiss/Imaging scanner (Z/I Imaging Corporation, Huntsville, Alabama) with a step size of 14 μm.
Images were processed using the SPIDER/WEB software package (34). For two-dimensional multi-reference classification analysis, each particle image was assigned to one group of a set of different orientations after the first round of projection matching, and two-dimensional averages for each group were computed for both the control RyR1 and the RyR1+mAb34C Fab complex. A set of multi-reference images was generated by mixing the control RyR1 and RyR1+mAb34C Fab complex two-dimensional averages (with 0 to 4 Fab fragments occupied the binding sites). The particles from the RyR1+mAb34C Fab complex dataset were subjected to a supervised classification employing the multi-reference image templates, and particles were assigned to a class based on the highest similarity (judged by the cross-correlation coefficient, Ref. 35). Any particles without 4 Fab fragments bound were then eliminated from the dataset in the following image processing.
Three-dimensional reconstructions were obtained from images of particles lying in all available orientations, through a projection matching procedure (36). The final three-dimensional reconstructions of RyR2A4860G, RyR2Y2801-GFP, control RyR1, and the RyR1+mAB34C Fab complex were computed from 7,591, 6,086, 16,043 and 16,782 particle images, respectively, ensuring that the particle orientations covered the entire angular space, and that each view would be represented by almost equal numbers of particles. Four-fold symmetry was enforced in all three-dimensional reconstructions. The final resolutions were estimated to be 27Å by the Fourier shell correlation with a cut-off of 0.5 (37) for both RyR2A4860G and RyR2Y2801-GFP, and 25Å for both control RyR1 and the RyR1+mAB34C Fab complex.
Results
Construction, expression, functional characterization, and purification of RyR2A4860G and RyR2Y2801-GFP
We have previously shown that mutation A4860G, located in the pore inner helix of RyR2, displays a reduced basal channel activity and a reduced sensitivity to activation by caffeine (29). These observations suggest that the A4860G mutation stabilizes the closed state of the channel. We reasoned that such a mutant channel would be superior to the wild-type channel for structural studies. Indeed, we found that the A4860G mutant protein can be readily purified and visualized by cryo-EM as intact, square-shaped particles, and there is no structural difference between the wild-type and A4860G RyR2s. Accordingly, the A4860G mutant RyR2 (RyR2A4860G) was subsequently used as the control for our structural studies. To determine the location of the phosphorylation site, Ser-2808, in the three-dimensional structure of RyR2, GFP was inserted after Tyr-2801 in the RyR2A4860G (Figure 1A). The resulting GFP-RyR2 fusion protein, RyR2Y2801-GFP, was expressed in HEK293 cells. As shown in Figure 1B, HEK293 cells expressing RyR2Y2801-GFP were readily detected by their green fluorescence, indicating proper folding of the inserted GFP. To determine whether RyR2Y2801-GFP functions as a Ca2+ release channel, we measured intracellular Ca2+ release, using the fluorescent Ca2+ indicator dye fluo-3 acetoxymethyl ester (fluo-3 AM) in HEK293 cells transfected with RyR2A4860G or RyR2Y2801-GFP. As shown in Figure 1C, HEK293 cells transfected with RyR2Y2801-GFP cDNA displayed multiple Ca2+ release events in response to repeated stimulation by caffeine, a well known RyR activator. Interestingly, the addition of ryanodine led to a diminished response to multiple caffeine stimulations (Figure 1D). This diminution most likely resulted from modulation of the channel by ryanodine, which is known to bind only to the open state of the channel, and to dramatically increase the channel open probability. As a consequence of binding, the ryanodine-modified channel no longer responded to further caffeine stimulation (30). These results suggest that RyR2Y2801-GFP is sensitive to caffeine and ryanodine modification. Importantly, the Ca2+ release responses of RyR2Y2801-GFP to caffeine and ryanodine were similar to those observed for the control RyR2A4860G. Furthermore, RyR2Y2801-GFP and RyR2A4860G showed similar Ca2+ dependence of [3H]ryanodine binding (Figure 1E). The EC50 for Ca2+ of RyR2A4860G and RyR2Y2801-GFP are 0.32 ± 0.01 μM and 0.31± 0.02 μM (mean ± SEM, n = 6), respectively. These data suggest that the RyR2Y2801-GFP protein retains proper folding and channel function.
Figure 1. CDNA construction and biochemical characterizations of RyR2A4860G and RyR2Y2801-GFP.
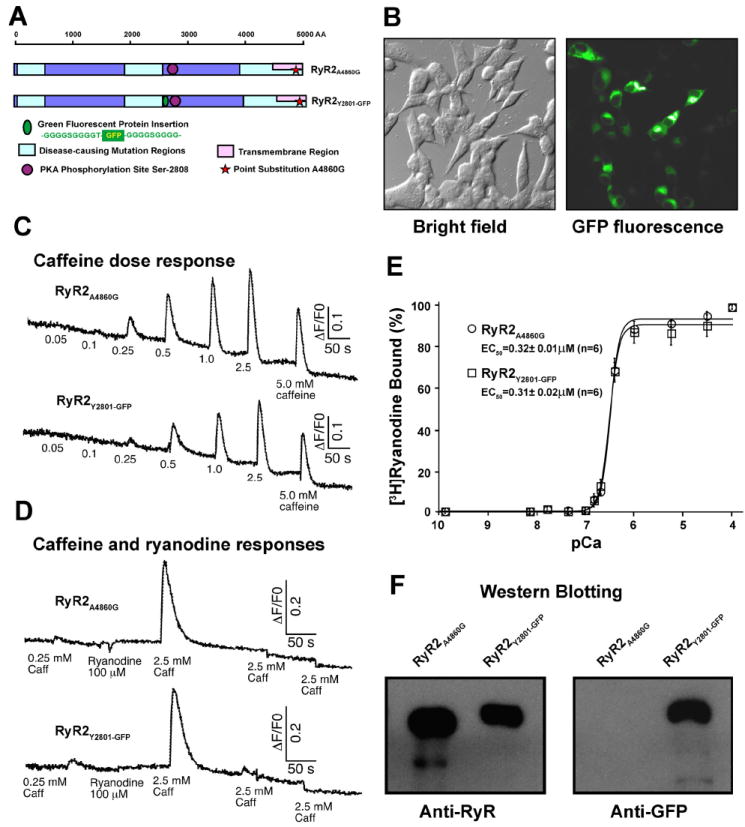
(A) Schematic illustration of cDNA construction of RyR2A4860G and RyR2Y2801-GFP. The blue strip represents the RyR’s ~5,000 amino acid sequence. Mutations in RyR2 sequence implicated in sudden cardiac death are largely clustered in three regions (light blue boxes). The carboxy-terminal region (~500 amino acids) composes the trans-membrane domain (pink box). Ser-2808, the PKA phosphorylation site is indicated (purple circle), as is the point substitution of Ala4860Gly (red star). The sequence for GFP flanked by two glycine rich spacers was inserted after Tyr-2801 (green oval). (B) Bright field (left) and fluorescence microscopy (right) images of HEK293 cells expressing RyR2Y2801-GFP. (C-D) Measurement of Ca2+ release activity of RyR2A4860G and RyR2Y2801-GFP following the addition of the various concentrations of RyR2 activator, caffeine (C), and in the presence of ryanodine (D). (E) [3H]ryanodine binding to the cell lysates of RyR2A4860G and RyR2Y2801-GFP at various Ca2+ concentrations. (F) Western blot of purified RyR2A4860G and RyR2Y2801-GFP.
The RyR2A4860G and RyR2Y2801-GFP proteins were purified from cell lysates by affinity chromatography, with GST-FKBP12.6 used as the affinity ligand (30). The purified protein was analyzed by SDS-PAGE and western blotting. A single high molecular mass band, which migrated at a slightly slower rate than the RyR2A4860G band did (as expected, due to the additional mass of GFP), was detected in the purified sample of RyR2Y2801-GFP. This band was recognized by both the anti-RyR and anti-GFP antibodies. On the other hand, the purified RyR2A4860G protein was recognized only by the anti-RyR antibody (Figure 1F).
Three-dimensional localization of Y2801-GFP
The RyR2A4860G and RyR2Y2801-GFP samples were diluted to a suitable concentration, applied to carbon-coated EM grids, blotted and rapidly frozen, and imaged by electron microscopy. Figure 2A and Figure 2B shows typical electron micrographs in which individual RyR2A4860G and RyR2Y2801-GFP channels are visible, most of these channels show standard RyR morphology (38).
Figure 2. Cryo-electron microscopy and three-dimensional reconstructions of RyR2A4860G and RyR2Y2801-GFP.

A portion of a cryo-EM micrograph is shown for purified RyR2A4860G (A) and for RyR2Y2801-GFP (B) with protein particles embedded in a thin layer of vitreous ice. The tetrameric structure of RyR2 is well preserved. Several individual particles are marked with white circles. Scale bar, 500Å. (C-E) Surface representations of the three-dimensional reconstructions of RyR2A4860G (C, shown in blue) and RyR2Y2801-GFP (D, shown in green). The three-dimensional reconstructions are depicted in three views: left, top view showing the cytoplasmic surface that in situ would face the transverse tubule membrane; middle, side view; right, bottom view showing the surface that would face the sarcoplasmic reticulum lumen. The numerals 1-10 on the cytoplasmic assembly indicate the distinguishable domains, according to our earlier nomenclature (38, 39). Difference map (E, RyR2Y2801-GFP – RyR2A4860G) shown in green and superimposed on the three-dimensional reconstruction of the RyR2A4860G (in blue).
Image processing was performed using the SPIDER/WEB software package (34). Three-dimensional reconstructions were obtained by a projection matching procedure (36). Figure 2C and Figure 2D show surface representations of the three-dimensional reconstructions of RyR2A4860G and RyR2Y2801-GFP in three principal orientations. For both the reconstructed structure consists of two major components: a large cytoplasmic assembly composed of at least 10 distinct domains (labeled by numerals 1-10; ref. 38, 39), and a smaller transmembrane assembly (labeled “TA”). For both of the reconstructions, the final resolution was estimated to be 27 Å using the Fourier shell correlation criterion with 0.5 as the cutoff value (37). Overall, the two three-dimensional reconstructions are very similar, but there are still some differences. The most noticeable difference was found in the region between domains 5 and 6; specifically, the volume of the bridge linking domains 5 and 6 of RyR2Y2801-GFP appears to be larger than the corresponding bridge volume in RyR2A4860G (highlighted in red circles). To more precisely delineate the differences, we generated a three-dimensional difference map by subtracting the volume of RyR2A4860G from that of RyR2Y2801-GFP. The difference regions were displayed in green and superimposed on the three-dimensional reconstruction of RyR2A4860G (Figure 2E). This difference map clearly indicates one significant difference, located in each bridge connecting domains 5 and 6 in the cytoplasmic assembly. GFP was inserted into each RyR2 monomer and, since RyR2 is a homo-tetramer composed of four identical monomers, the difference is expected to repeat four times in the three-dimensional difference map.
We are confident that the extra density in the map represents the excess mass contributed by the GFP insertion in RyR2Y2801-GFP, because it is the only significant difference that is seen when the three-dimensional difference map is displayed with the same density threshold as we used to image the RyR2Y2801-GFP and RyR2A4860G reconstructions, no additional filtering or masking was need. Furthermore, the calculated volume of one of the four repeated difference in Figure 2E corresponds to a molecular mass of 28 kDa, assuming a protein density of 1.37g/cm3; this value agrees well with the molecular mass of GFP (38). Other minor differences, only visible at a much lower density threshold, are not consistent with the inserted GFP in terms of either volume or density. In summary, Tyr-2801, as indicated by the inserted GFP, is located in the bridge between domains 5 and 6, in each of the corner (clamp) regions of RyR2.
Three-dimensional localization of mAb34C Fab fragment
In the second approach, we localized the binding site of a sequence specific monoclonal antibody, mAb34C, on skeletal isoform RyR1 (Figure3A). MAb34c is an antibody that is commercially available and is widely used to detect RyR in biochemical assays; it recognizes all three isoforms of RyR. The epitope of mAb34C has previously been determined to involve amino-acid residues 2,756 through 2,803 of RyR1, corresponding to residues 2,722 through 2,769 of RyR2 (28), about 40 amino acid residues away from the RyR2’s PKA phosphorylation site Ser-2808. The primary sequence and three-dimensional structure are highly conserved between RyR1 and RyR2, and the binding site of FKBP12 in RyR1 is located in the same region as is the FKBP12.6 binding site in RyR2 (24, 25). Thus, through comparison of the three-dimensional locations of the mAb34C binding site and the FKBP binding site, we will be able to assess the likelihood of interaction between FKBP and the PKA phosphorylation site.
Figure 3. Binding site, purification, and binding activity test for mAb34C.
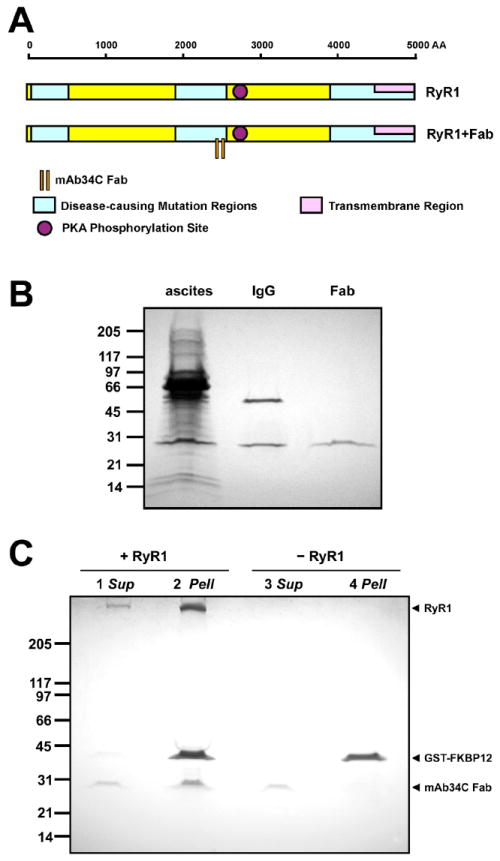
(A) Schematic illustration of the mAb34C Fab fragment binding site in RyR1’s amino-acid sequence. The epitope of mAb34C comprises amino-acid residues 2,756 through 2,803 of RyR1, corresponding to residues 2,722 through 2,769 of RyR2, thus the mAb34C binding site is close to the PKA phosphorylation site, Ser-2808 (purple circle). (B) SDS-PAGE of mAb34C ascites, IgG, and Fab fragment. IgG was purified from mouse ascites, and Fab fragments were obtained from purified IgG by papain digestion. (C) Co-precipitation of mAb34C Fab fragments and RyR1 by GST-FKBP12 pull-down. Sup, supernatant; Pell, pellet.
A mAb34C hybridoma cell line was use to produce milligram amounts of antibodies. MAb34C IgG was purified from mouse ascites, and Fab fragments were obtained from purified IgG by papain digestion. We used Fab fragments instead of IgGs in our single particle cryo-EM, because one IgG molecule has two antigen-binding sites, and could thus cross-link two RyR molecules. The purified mAb34c IgG and Fab were checked by SDS-PAGE. Figure 3B shows the result of purification. Two bands are evident in the IgG lane, one at 25kDa, and the other at 50kDa. These positions are consistent with the known molecular masses of the light chain and heavy chain of IgG, respectively. There is only one band (25kDa) in the Fab lane, corresponding to the light chain and the truncated heavy chain. No other band was observed in either lane, indicating that both the IgG and Fab fragment are pure.
The binding activity of the mAb34C Fab was tested by co-precipitation with RyR1. Fusion protein GST-FKBP12 binds specifically to RyR1, but not to mAb34C Fab. MAb34C Fab was incubated with or without RyR1 before mixing with GST-FKBP12, and it was then precipitated by glutathione-Sepharose beads. After precipitation, the supernatant and pellet were analyzed by SDS-PAGE. Figure 3C shows that in the presence of RyR1 (Lane 1 and 2), the mAb34C Fab was pulled down by GST-FKBP12 and glutathione-Sepharose beads, but it was not pulled down in the absence of RyR1 (Lane 3 and 4). This precipitation test clearly demonstrates that the purified Fab fragment of mAb34C can bind to detergent-solubilized RyR1 specifically.
The purified mAb34C Fab fragments were incubated with RyR1 for complex formation. Both the mAb34C Fab+RyR1 complexes and control RyR1 (without added Fab fragments) were imaged by cryo-EM. Figure 4A and Figure 4B show typical electron micrographs in which individual particles of control RyR1 and mAb34C Fab-RyR1 complex are visible.
Figure 4. Cryo-electron microscopy and three-dimensional reconstructions of RyR1 and RyR1+mAb34c Fab complex.

A portion of a cryo-EM micrograph is shown for control RyR1 (A) and for the RyR1+mAb34c Fab complex (B). Several individual particles are marked with white circles. Scale bar, 500Å. (C-E) Surface representations of the three-dimensional reconstructions of control RyR1 (C, shown in yellow) and RyR1+mAb34c Fab complex (D, shown in orange). Difference map (E, RyR1+mAb34c Fab complex – control RyR1) shown in orange, is superimposed on the three-dimensional reconstruction of the control RyR1 (in yellow).
A problem inherent in this approach is that among the more than ten thousands receptor images that were selected for computation of the three-dimensional structures of mAb34C Fab+RyR1 complex, some receptors will have less than the full complement of four bound Fab molecules (RyR1 is a homo-tetramer and thus has four Fab binding sites). This situation contrasts with that for the GFP insertion approach, in which cDNA of GFP is inserted into that of RyR2, and thus all of the expressed receptors have the GFP insertion (HEK293 cells do not possess endogenous RyR). In the antibody approach, since the binding affinity between RyR1 and mAb34C Fab is estimated to be in the micromolar range, a small proportion of RyR1 subunits are expected to be free of bound Fab. A simple calculation, using an estimated binding affinity of 1μM, reveals that 98.8% of RyR1 subunits are bound to Fab under our experimental conditions. To estimate the actual percentage of RyR1 that are bound to Fab molecules, we performed a two-dimensional classification analysis (multi-references based alignment, follow by a variation statistics analysis, ref. 35) at the beginning of image processing, to assess the extent of this heterogeneity. Our result indicated that about 93% of the RyR1 particles have 4 Fab fragments bound, whereas 7% RyR1 have fewer. Most of the latter Fab-depleted RyR1 particles were then eliminated from the dataset in the following image processing. However, this two-dimensional classification is accurate for low-tilt angle data (below 30°), so we can not rule out that a small percentage of Fab-depleted RyR1 from high-tilted angles data remains in the final dataset (less than 2% of the total number of particles). Since the three-dimensional reconstruction is calculated from over 16,000 particle images, a strong density feature due to bound Fab fragment is expected, and in fact observed, at our current level of resolution (see results below).
Figure 4C and Figure 4D show surface representations of the three-dimensional reconstructions of control RyR1 and the mAb34C Fab+RyR1 complex in three views. The final resolution was estimated to be 25 Å. Overall, the two three-dimensional structures are also very similar, the differences appear mainly in domain 6, as shown in the side-views in the two panels (highlighted by red circles), where domain 6 of the mAb34C Fab+RyR1 complex appears “taller” at its outer margin. We generated a three-dimensional difference map by subtracting the volume of the control RyR1 from the volume of the mAb34C Fab+RyR1 complex. The difference regions, displayed in orange, are superimposed on the three-dimensional reconstruction of control RyR1 (Figure 4E). This difference map clearly indicates that there is one difference per RyR1 subunit, located at periphery of domain 6.
Discussion
FKBP12.6 binds tightly to cardiac muscle RyR2, and it is thought to modulate RyR2 channel gating by stabilizing the closed state of the channel, and by promoting transitions between closed and open states (i.e. eliminating sub-conductance states; Ref. 16). A highly homologous protein, FKBP12, binds to skeletal muscle RyR1 and stabilizes RyR1 in a similar manner (17). Dissociation of FKBP from RyR by drugs (e.g., FK506) or antibiotics (e.g. rapamycin) causes an increased channel open probability, higher frequency of sub-conductance states, and sarcoplasmic reticulum calcium leakage (18). The Marks group has proposed that PKA hyperphosphorylation of a unique amino acid, Ser-2809 in RyR2, dissociates FKBP12.6, and that such a dissociation affects RyR2 functioning, thereby contributing to the ventricular arrhythmias that often occur during heart failure (8). In a later paper from the same group, it was proposed that FKBP12.6 dissociation occurs through an electrostatic repulsion between the negatively charged phosphate group at Ser-2809 of RyR2 and a negatively charged residue, Asp-37, of FKBP12.6 near the hydrophobic binding pocket (9). Mutations in RyR2 that cause CPVT and AVRD2 are hypothesized to weaken the affinity of RyR2 for FKBP12.6 (9). As a result, disruption of the FKBP12.6-RyR2 interaction at Ser-2809 was proposed as a novel mechanism for cardiac muscle dysfunction in sudden cardiac death and heart failure.
However, the proposed mechanisms are not consistent with other laboratories’ findings (19-23), and our present results contribute to the growing body of evidence that disfavors the proposal that PKA hyperphosphorylation at Ser-2809 (or Ser-2808 in different species) in RyR2 induces dissociation of FKBP12.6. The FKBP12 and FKBP12.6 binding sites were previously mapped to the same region in the three-dimensional structures of RyR1 and RyR2, at or near the junction of domains 3 and 9 (red in Figure 5A; Ref. 24, 25). In the present study, we used two independent approaches localize two regions within RyR2’s amino-acid sequence that are near the phosphorylation site in the three-dimensional structure of RyR. The GFP insertion at Tyr-2801, which is seven amino acid residues upstream of Ser-2808, is located in the in the bridge between domains 5 and 6 (green in Figure 5A). The binding site for the Fab fragment derived from monoclonal antibody mAb34C, whose epitope is about 40 amino acid residues away from Ser-2808, is located in domain 6 (orange in Figure 5A). These two sites are adjacent to one another in each of the clamp structures in RyR’s cytoplasmic region, and their center to center separation is about 40Å. The three-dimensional location of the phosphorylation site Ser-2808 is also close to a previously mapped mutation site at Ser-2367, which is located in the bridge between domains 5 and 6 (pink in Figure 5A)26. Considering the GFP mapping to be the most accurate determination of the position of Ser-2808 in the three-dimensional structure of RyR, we conclude that Ser-2808 is not close to the binding site of FKBP12.6. Within the homo-tetramer structure of RyR (with its four phosphorylation sites and its four FKBP12.6 binding sites), we consider the two FKBP12.6 binding sites that are nearest to the phosphorylation site, as shown in Figure 5A, the FKBP12.6 binding site in the same clamp region is about 105Å apart (green double-ended arrow) from the Ser-2808 site, while the other one in the adjacent clamp regions is about 120Å away (red double-ended arrow).
Figure 5. The PKA phosphorylation site Serine-2808 is not close to FKBP12.6 binding site.
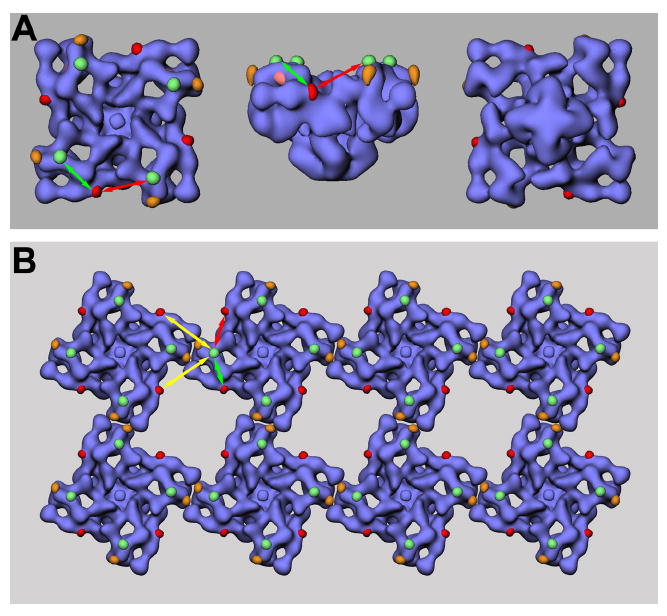
(A) A three-dimensional reconstruction of RyR2 is shown in blue, the site of the GFP insertion after Tyr-2801 is shown in green, and the binding site of mAb34C is shown in orange. The two locations are close to a previously mapped mutation site Ser-2367 (in pink, visible in middle panel). If the GFP site id considered to be a marker of Ser-2808’s location, the distance to the FKBP12.6 site on the same clamp structure is 105Å (green double-headed arrow), and the distance is 120Å to the FKBP12.6 bound to an adjacent clamp structure (red double-ended arrow). (B) Ser-2808 is not close to the FKBP12.6 binding site in the nearest-neighbor RyR. The three-dimensional mapping of Ser-2808 and the FKBP12.6 binding site are superimposed onto the structural model of two-dimensional ordered arrays of RyR. The distance between Ser-2808 and the FKBP12.6 in the nearest-neighbor RyR is over 130Å (yellow double-ended arrow).
In both skeletal and cardiac muscle, RyRs self-assemble into two-dimensional ordered arrays at the terminal cisternae membrane of the sarcoplasmic reticulum. One question arises: can PKA phosphorylation of a Ser-2808 in one RyR disrupt the binding of FKBP12.6 to a neighboring RyR in the arrays. Paolini and coworkers have defined the orientation of the two-dimensional arrays of RyR (40), and we have superimposed our three-dimensional mapping of Ser-2808 and FKBP12.6 binding site onto their structural model (Figure 5B). Even in two-dimensional arrays, the distance between the phosphorylation site Ser-2808 (which takes the place of Tyr-2801) and the closest FKBP12.6 binding site in the neighboring RyR exceeds 130Å (yellow double-ended arrow).
In additional to the direct structural mappings, we have found two lines of indirect evidences to corroborate that the phosphorylation site Ser-2808 is not close to the FKBP binding site. First, in the purification of RyR2Y2801-GFP, we used glutathione-Sepharose beads containing pre-bound GST-FKBP12.6 fusion protein as the affinity binding ligand. The successful purification indicated that the insertion of GFP near Ser-2808 did not prevent the binding of FKBP12.6 to RyR2. Second, in the co-precipitation test of mAb34C Fab fragments, we used glutathione-Sepharose beads with pre-bound GST-FKBP12 to pull down the mAb34C Fab+RyR1 complex. The positive results further indicate that the binding of FKBP12 does not interfere with the binding of mAb34C Fab fragments.
The site of the GFP insertion is only seven amino acid residues away from Ser-2808 in the primary sequence, but the rather large distance between the GFP site and the actual FKBP12.6 binding location as found by three-dimensional cryo-EM (> 100Å) is not consistent with the idea that phosphorylation site is directly involved in binding of FKBP12.6. Other recent studies have also raised doubts about the role of this phosphorylation site in the FKBP12.6-RyR2 interaction. A carboxy-terminal truncation analysis demonstrated that a region between residues 1815 and 1855, near divergent region 3, is essential for FKBP12.6 interaction (41). Consistent with this finding, we used cryo-EM to map a GFP insertion at Thr-1874 (in the middle of divergent region 3) to domain 9, a position adjacent to the FKBP12.6 binding site (41). In contrast to the FKBP dissociation mechanism (8, 9), recent studies have found that PKA phosphorylation of Ser-2808 in RyR2 does not result in dissociation of FKBP12.6, because FKBP12.6 can bind to both the Ser-2808 phosphorylated and non-phosphorylated forms of RyR2 (22). RyR2s with the mutation S2808D or S2808A at this conserved phosphorylation site also retained the ability to bind FKBP12.6 (21).
RyR2 is known to be phosphorylated at Ser-2808 by PKA, CaMKII and PKG, and at Ser-2814 by CaMKII. Phosphorylation of RyR2 at these residues modulates the activity of the channel. However, little is known about the structural basis of regulation of RyR2 by phosphorylation. Our data clearly demonstrate that the phosphorylation motif that encompasses the Ser-2808 and Ser-2814 is far away from the FKBP12.6 binding site, thus excluding the possibility that the phosphorylation of RyR2 at Ser-2808 directly disrupts the binding of FKBP12.6, causing its dissociation from the channel, as proposed previously. On the other hand, it is of interest to note that the three-dimensional location of the Ser-2808 site is close to a previous mapped Ser-2367 site located in the central disease-causing mutation hot spot (pink in Figure 5A; Ref. 26), suggesting that the effect of phosphorylation on the function of RyR2 may be similar to that of the disease-causing mutations.
There are three diseases-causing mutation hot spots in the RyR2 sequence that harbor diseases-causing mutations, region 1 is near the amino terminus of the channel, from residues 77 to 466; region 2 is near the middle of the sequence, spanning residues 1,724 through 2,958; and region 3 includes part of the cytoplasmic and membrane associated regions of the channel and comprises residues 3,778 to 4,959. Two mutation hotspots lie in cytoplasmic regions of RyR2, the amino-terminal region and the central region, are well separated in the primary sequence (> 1,200 amino acids), however according to the hypothesis proposed by Ikemoto and colleagues, these two regions occur in structural domains that physically interact with each another in the three-dimensional space, and changes in the strength of their interaction due to diseases-causing mutations can affect channel gating (10). More specifically, the interactions between the amino-terminal and central regions (domains zipping) stabilize RyR in the closed state, and reduction in the affinity of the interaction causes separation of these regions (domains unzipping) and thereby causing RyR to open more frequently. In support of this hypothesis, we have mapped a mutation in the amino-terminal region (Ser-437) into a region between domain 5 and 9 (27), and a mutation in the central region (Ser-2367) to a site between domain 5 and 6 (26). These findings suggest that the amino-terminal mutations are located in domain 5, while the central mutations are in domain 6, and that there are domain-domain interactions between them to stabilize the RyR channel (27). Since the three-dimensional location of the Ser-2808 PKA phosphorylation site is located near to the domain-domain boundary, it is possible that PKA hyperphosphorylation at Ser-2808 may alter the domain-domain interactions between the N-terminal and the central regions of RyR2, and thus the gating properties of the channel.
In summary, evidence is continuing to mount against the proposal that PKA hyperphosphorylation at Ser-2808 induces dissociation of FKBP12.6 from RyR2 via a direct steric repulsion between the negatively charged phosphate group at Ser-2808 and the negatively charged residue Asp-37 of FKBP12.6. If the FKBP12.6-RyR2 interaction were weakened by PKA hyperphosphorylation, this weakening action would have to occur via an indirect mechanism, such as an allosteric effect operating over a distance of tens of Angstroms (i.e. a long-range conformational change), or alternating an effect induced by one or more RyR regulatory factors, including small molecules like Ca2+ and Mg2+, and large proteins like calmodulin, PKA, CaMKII, protein phosphatase 1, protein phosphatase 2a, and DHPR.
Acknowledgments
This work was supported by the American Heart Association grant 0430076N to Z.L., by National Institutes of Health grant AR40615 to T.W., and by research grants from the Canadian Institutes of Health Research and the Heart and Stroke Foundation of Alberta to S.R.W.C. We thank the Wadsworth Center’s Molecular Genetics, Immunology, and Electron Microscopy Core Facilities, and the Resource for Visualization of Biological Complexity (NIH Biotechnological Resource Grant RR01219).
References
- 1.American Heart Association. Heart Disease and Stroke Statistics —2007 Update. Dallas, Tex: [Google Scholar]
- 2.Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Mayo Clin Proc. 2004;79:1380–1384. doi: 10.4065/79.11.1380. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 4.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K. Circulation. 2001;103:485–490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FES, Vignati G, Benatar A, DeLogu A. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 6.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Circulation. 2004;110:2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 7.McCarty TV, Quane KA, Lynch PJ. Hum Mutat. 2000;15:410–417. doi: 10.1002/(SICI)1098-1004(200005)15:5<410::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 8.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 9.Wehrens XHT, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song L-S, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 10.Ikemoto N, Yamamoto T. Front Biosci. 2002;7:d671–683. doi: 10.2741/A803. [DOI] [PubMed] [Google Scholar]
- 11.George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA. Circ Res. 2006;98:88–97. doi: 10.1161/01.RES.0000199296.70534.7c. [DOI] [PubMed] [Google Scholar]
- 12.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SRW. Proc Natl Acad Sci USA. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas NL, Lai FA, George CH. Biochem Biophys Res Commun. 2005;331:231–238. doi: 10.1016/j.bbrc.2005.02.194. [DOI] [PubMed] [Google Scholar]
- 14.Lehnart SE, Wehrens XHT, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Circulation. 2004;109:3208–3214. doi: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 15.Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P, Marks AR. J Biol Chem. 1992;267:9474–9477. [PubMed] [Google Scholar]
- 16.Jekakumar LH, Ballester L, Cheng DS, McIntyre JO, Chang P, Olivey HE, Rollins-Smith L, Barnett JV, Murray K, Xin H-B, Fleischer S. Biochem Biophys Res Commun. 2001;281:979–986. doi: 10.1006/bbrc.2001.4444. [DOI] [PubMed] [Google Scholar]
- 17.Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasová E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 18.Kaftan E, Marks AR, Ehrlich BE. Circ Res. 1996;78:990–997. doi: 10.1161/01.res.78.6.990. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Kranias EG, Mignery GA, Bers DM. Circ Res. 2002;90:309–316. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 20.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Circ Res. 2002;91:1015–1022. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 21.Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. J Biol Chem. 2003;278:51693–51702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- 22.Xiao B, Sutherland C, Walsh MP, Chen SRW. Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- 23.George CH, Higgs GV, Lai FA. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 24.Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin H-B, Fleischer S. J Biol Chem. 1997;272:32463–32471. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- 25.Sharma MR, Jeyakumar LH, Fleischer S, Wagenknecht T. Biophys J. 2006;90:164–172. doi: 10.1529/biophysj.105.063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Wang R, Zhang J, Chen SRW, Wagenknecht T. J Biol Chem. 2005;280:37941–37947. doi: 10.1074/jbc.M505714200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R, Chen W, Cai S, Zhang J, Bolstad J, Wagenknecht T, Liu Z, Chen SRW. J Biol Chem. 2007 doi: 10.1074/jbc.M700660200. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong J, Oyamada H, Demaurex N, Grinstein S, McCarthy TV, MacLennan DH. J Biol Chem. 1997;272:26332–26339. doi: 10.1074/jbc.272.42.26332. [DOI] [PubMed] [Google Scholar]
- 29.Wang R, Bolstad J, Kong H, Zhang L, Brown C, Chen SRW. J Biol Chem. 2004;279:3635–3642. doi: 10.1074/jbc.M311367200. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Zhang J, Li P, Chen SRW, Wagenknecht T. J Biol Chem. 2002;277:46712–46719. doi: 10.1074/jbc.M208124200. [DOI] [PubMed] [Google Scholar]
- 31.Chen SRW, Li X, Ebisawa K, Zhang L. J Biol Chem. 1997;272:24234–24246. doi: 10.1074/jbc.272.39.24234. [DOI] [PubMed] [Google Scholar]
- 32.Du GG, Imredy JP, MacLennan DH. J Biol Chem. 1998;273:33259–33266. doi: 10.1074/jbc.273.50.33259. [DOI] [PubMed] [Google Scholar]
- 33.Inui M, Saito A, Fleischer S. J Biol Chem. 1987;262:1740–1747. [PubMed] [Google Scholar]
- 34.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 35.Frank J. In: Three-dimensional Electron Microscopy of Macromolecular Assemblies. Frank J, editor. Oxford University Press; New York, NY: 2006. pp. 145–192. [Google Scholar]
- 36.Liu Z, Zhang J, Sharma M, Li P, Chen SRW, Wagenknecht T. Proc Natl Acad Sci USA. 2001;98:6104–6109. doi: 10.1073/pnas.111382798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malhotra A, Penczek P, Agrawal RK, Gabashvili IS, Grassucci RA, Junemann R, Burkhardt N, Nierhaus KH, Frank J. J Mol Biol. 1998;280:103–116. doi: 10.1006/jmbi.1998.1859. [DOI] [PubMed] [Google Scholar]
- 38.Radermacher M, Rao V, Grassucci R, Frank J, Timerman AP, Fleischer S, Wagenknecht T. J Cell Biol. 1994;127:411–423. doi: 10.1083/jcb.127.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samso M, Wagenknecht T, Allen PD. Nature Struct Mol Biol. 2005;12:539–544. doi: 10.1038/nsmb938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paolini C, Protasi F, Franzini-Armstrong C. J Mol Biol. 2004;342:145–153. doi: 10.1016/j.jmb.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Liu Z, Masumiya H, Wang R, Jiang D, Li F, Wagenknecht T, Chen SRW. J Biol Chem. 2003;278:14211–14218. doi: 10.1074/jbc.M213164200. [DOI] [PubMed] [Google Scholar]