

Abstract
Purpose
To investigate the genotoxic effects of Lutein (LBP) & β-carotene breakdown products (β-apo-8-carotenal, BA8C) and preventive role of GSH in human retinal pigment epithelial cells (ARPE-19).
Methods
LBP- and BA8C-induced DNA damage in human retinal pigment epithelial cells (ARPE-19) was determined by Comet assay. The DNA damage was quantified by the image analysis system using Comet Score™ software. ARPE-19 cell viability was determined by CellTiter 96 AQueous one solution cell proliferation assay kit. Intracellular GSH levels were measured by Ellman’s reagent.
Results
Incubation of serum-starved ARPE-19 cells with LBP & BA8C caused significant DNA damage in a dose- and time-dependent manner. The DNA damage and cell death incurred by LBP & BA8C were significantly prevented by N-acetylcysteine (NAC) but not by α-Tocopherol + Ascorbic acid (T + AA). Furthermore, BSO-induced GSH depletion in ARPE-19 cells caused a significant elevation in LBP- & BA8C-induced DNA damage, whereas increased GSH levels in ARPE-19 cells prevented it.
Conclusions
Our results suggest that breakdown products of dietary carotenoids could be genotoxic in ARPE-19 cells. LBP-induced genotoxic effects could worsen oxidative stress. The intracellular GSH pool in ARPE-19 cells might play a critical role in carotenoid breakdown products-induced genotoxicity.
Keywords: Carotenoids, carotenoid breakdown products, lutein breakdown products, Antioxidants, DNA damage, Age-related Macular Degeneration
Introduction
Age-related macular degeneration (AMD) is considered to have a multifactorial pathogenesis1–4. The molecular events that mediate pathogenic mechanisms in AMD are not clearly defined and preventive strategies have yet to be established. However, dietary antioxidants have been considered as potential agents for AMD intervention1. In recent years a number of investigators have suggested that carotenoids such as lutein and zeaxanthin could delay or protect against AMD by acting as an antioxidant or by elevating macular pigment levels and reducing exposure to phototoxic short wavelength light5–8.
Like a number of other antioxidants, carotenoids are also reported to be pro-oxidants under various physiological conditions such as high oxygen tension and imbalanced intracellular redox status9. Under such conditions, carotenoids undergo oxidation and generate a variety of oxidized products, as demonstrated in vitro and in vivo10–12. Many carotenoid cleavage products characterized either in vitro or in vivo, are identified as aldehydes including retinaldehyde and apocarotenaldehyde, and mixtures of various long- and short-chained products. Besides aldehydes, such mixtures also contain epoxides, ionones, and other unidentified products13. The existence as well as identification of lutein, zeaxanthin and other carotenoid breakdown products (CBP) in human and primate ocular tissues has been reported12, 14–16. In case of lutein supplementation, the levels of lutein and its breakdown products (LBP) have been demonstrated to increase in human serum17 as well as in plasma and ocular tissues of monkey18. After short-term (6 months) lutein supplementation the physiological concentration of lutein and LBP in human serum is in nM range6, 17, 19. However, the physiological concentration of carotenoids and CBP could reach µM range in case of long-term carotenoid supplementation. For example, ATBC20 and CARET21 studies have reported µM concentration of β-carotene and its breakdown products after 3 years of carotenoid supplementation. However, the effects of long-term and high dose xanthophylls (lutein & zeaxanthin) supplementation on the concentration of their breakdown products in human serum and ocular tissues are not known. The ongoing AREDS-2 study is intended to investigate the effects of long-term (5 years) and high dose xanthophyll supplementation on AMD progression. Such studies may provide data on physiological concentrations of xanthophylls and their breakdown products.
The physiological role of various CBP is not known. However, various studies have reported cyto- & genotoxic effects of CBP at physiological (nM) or abnormally higher (µM) concentrations13, 22–29. Lutein & zeaxanthin supplementation as an intervention therapy for AMD, has been widely advocated with little regard for the toxic effects of lutein breakdown products (LBP). Recently, we have demonstrated that LBP could cause cell death in human retinal pigment epithelial cells (ARPE-19)22. In the present study, we have investigated the genotoxic effects of LBP as well as β-apo-8-carotenal (BA8C, β-carotene-derived aldehyde) in ARPE-19 cells. The role of intracellular GSH in the protection against lipid aldehydes-induced cyto- & genotoxicity is well established30–32. Such protection is mediated by efficient conjugation between GSH and lipid aldehydes such as 4-HNE30, 33. However, the role of GSH in protection against CBP-induced toxicity is obscure. A preliminary study has suggested that GSH can conjugate with CBP34. We observed that CBP formed a thioether linkage with the sulfhydryl moiety of GSH34. In the present study, we have shown that increase in the cellular GSH significantly prevented CBP-induced genotoxicity in ARPE-19 cells. However, α-Tocopherol + Ascorbic acid could not prevent CBP-induced genotoxicity. These results suggest that the protection rendered by GSH could be due to its conjugation with CBP followed by its possible extracellular removal. Contrary, α-Tocopherol + Ascorbic acid may not conjugate with CBP and therefore could not prevent CBP-induced genotoxicity.
Materials and Methods
Materials
Human retinal pigment epithelial cells (ARPE-19) were purchased from American Type Culture Collection (ATCC). Phosphate-buffered saline (PBS), penicillin/streptomycin (P/S) and fetal bovine serum (FBS) were purchased from GIBCO Inc. (Grand Island, NY). Glutathione-ethyl ester (GS-Et), Buthionine-[S,R]-sulfoximine (BSO), Trypsin/EDTA, N-acetylcysteine (NAC), β-apo-8-carotenal (BA8C) and Lutein were purchased from Sigma, USA. DL-all-rac-α-Tocopherol was obtained from Fluka, USA. Ascorbic acid was purchased from Mallinckrodt, USA. CellTiter96® AQueous one solution cell proliferation assay kit was purchased from Promega. USA. The Comet Assay™ kit was purchased from Trevigen, Inc. (Gaithersburg, MD). DNA purification Kit was purchased from Qiagen Inc. USA. Ellman’s Reagent was purchased from Pierce (Rockford, IL). All other chemicals were obtained from Sigma, USA.
Preparation of LBP
The LBP were prepared as described previously22. The use of sodium hypochlorite (NaOCl) to oxidize carotenoids is based on the fact that hypochlorite is a product of myeloperoxidase, hydrogen peroxide and Cl− in activated phagocytic cells35,36. Since retinal pigment epithelial cells are phagocytic cells, we used NaOCl as a model system for investigating the possible harmful effects of carotenoid oxidation products. Briefly, 5 mg of lutein dissolved in 5 ml dichloromethane and 5 ml methanol were oxidized with 80 mM NaOCl in 1.25 ml water at room temperature for 15 min. The concentration of NaOCl used was similar to that reported by Lee et al37 in activated macrophages. The LBP was extracted by a modification of the Bligh and Dyer method38, 39. To each one ml of oxidized carotenoid solution, 5 ml of dichloromethane were added, followed by vortexing for 1 min; 3.75 ml water was added and vortexing was continued for another minute. The samples were centrifuged for 2 min at 1000 × g and the lower organic phase from each sample was collected. The extraction was repeated and the organic phase from both extractions was pooled. The solvent was evaporated under Argon to approximately 0.1 ml which was diluted with sterile phosphate-buffered saline (PBS) without calcium and magnesium (pH 7.4; Cellgro) to make LBP stock solution. The excess organic phase was evaporated again. The LBP solution was ultra-filtered by centrifugation using Centricon Tube (Millipore Corp. Bedford, MA, USA) for 1 h at 10,000 × g at 4 °C. The concentration of LBP in the ultrafiltrate was determined by measuring the optical density at 220 nm on UV-2101 PC recording spectrophotometer (Shimadzu, Columbia, MD)13. The characterization of NaOCl-oxidized carotenoid products revealed the presence of apo-carotenals, epoxides, ionones and various unidentified carbonyls10. The LBP present in human macula were also characterized and showed existence of two prominent products, 3-hydroxy-β-ionone and 3-hydroxy-14’-apocarotenal12. The LBP stock solution was stored at −20° C in dark. Vehicle without Lutein was prepared by the same method.
Preparation of BA8C
The BA8C was dissolved in dichloromethane and methanol (1:1). The solution was concentrated by evaporation under Argon followed by dilution with ice-cold 50 mM sodium phosphate Buffer (pH 7.4) and the remaining solvent was evaporated again. The solution was ultra-filtered by centrifugation using Centricon Tube (Millipore Corp. Bedford, MA, USA) for 1 h at 10,000 × g at 4 °C. The concentration of BA8C in the ultrafiltrate was determined by measuring the optical density at 220 nm on UV-2101 PC recording spectrophotometer (Shimadzu, Columbia, MD)13. The stock solution was stored at −20° C in dark. For some experiments vehicle was also prepared by the same method without BA8C.
Cell culture and treatment
The ARPE-19 cells were grown to confluency in DMEM/F-12 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37° C in a humidified atmosphere of 5% CO2. Sub-confluent cells were growth-arrested in 0.1% FBS medium. The cells were subcultured using trypsin/EDTA solution with a split ratio of 1:4. The LBP or BA8C treatment was done in serum-free medium to avoid binding with serum proteins. The pretreatment of cells with NAC (1 mM) or α-Tocopherol + Ascorbic acid (T+AA) (1 mM + 100 µM) was for 1 and 24 h respectively. Control cells were incubated with vehicle, PBS. In all the experiments, the concentration of PBS was not greater than 0.2%.
The delivery of α-Tocopherol in ARPE-19 cells was carried out as described below. The stock solution of 500mM α-Tocopherol was prepared in 100 % ethanol and stored at −80° C in dark. To prepare the working stock solution, 1 ml stock solution was diluted with 1 ml heat-inactivated fetal calf serum and 3 ml of culture media without serum. Serum increases the solubility of α-Tocopherol in hydrophilic solutions. The working stock solution was further diluted 100X with culture media (without serum but containing 100µM Ascorbic acid) to get 1 mM final concentration of α-Tocopherol. The final concentration of ethanol and fetal calf serum in the media was approximately 0.2 % each.
Treatment of the cells
ARPE-19 cells (5 × 105 cells) were treated with various concentrations of LBP or BA8C or treated for varied time periods as described in the figure legends. For negative control, the cells were treated with vehicle. After the treatment the cells were detached from the well by brief trypsinization. The released cells were subsequently washed twice with ice-cold PBS and the cell viability was determined by trypan blue exclusion method. Also an aliquot was used for comet assay.
Determination of DNA damage (Comet assay)
The comet assay was carried out as per suppliers manual. Briefly, an aliquot from cell suspension was mixed with 1% low melting point (LMP) Agarose in 1:10 ratio. Immediately, 75 µl of the cell suspension were dispersed onto Comet Slides specially treated to enhance the adherence of low melting point Agarose. The slides were kept at 4° C in dark for 30 min to allow Agarose solidification. Subsequently, the slides were immersed in cold lysis buffer (10 mM Tris–HCl, 100 mM EDTA (pH 10), 2.5 M NaCl, 1% sodium lauryl sarcosinate, 1% Triton X-100) for 1 hr. The slides were then washed twice with 1× Tris-buffered EDTA solution (TBE), placed in a horizontal electrophoresis chamber and covered with TBE buffer. Electrophoresis was carried out at the rate of 1.0 V/cm for 20 min. The slides were removed from the electrophoresis chamber, washed in deionized water for 5 min and dipped in 70% alcohol for 5 min. Subsequently, the slides were air-dried, stained with SYBR-Green (1 µl/ml; 30 µl per slide) and mounted. The comet pictures were taken using Nikon Eclipse 800 epifluorescence microscope. Photographs were taken using a Roper Scientific CoolSNAP Fx monochrome cooled CCD 12-bit digital camera. At least 40 non-overlapping comets were observed per slide and each experiment was repeated at least three times. All the steps of Comet assay were conducted under red light. In order to detect the combination of DNA single strand breaks, double strand breaks, and alkali-labile sites in the DNA of ARPE-19 cell nuclei, we performed alkaline electrophoresis using TBE buffer.
Scoring
For the analysis of DNA in comet tails, the slides were examined at 250X magnification on Nixon fluorescent microscope (Nikon Eclipse 800 epifluorescence microscope). Fourty randomly selected non-overlapping cells were scored for DNA damage using the Comet Score ™ version 1.5 analysis software (TriTek Corp.). A variety of objective measurements such as total intensity (DNA content), percent DNA in tail, tail length (measured from the leading edge of the comet head) and tail moment were made. We used % DNA in tail for statistical analysis.
Cell viability assay
The ARPE-19 cells were plated (5000 cells/well) in a 96-well plate. After 24 h, the cells were serum-starved in 0.1% FBS medium for 24 h. The cells were then washed with PBS and treated with BA8C in media without serum. Cells incubated without vehicle served as control. Cell viability was determined by CellTiter 96 AQueous one solution cell proliferation assay kit. CellTiter reagent was added to each well, incubated for 3 h, and the absorbance was recorded at 490 nm using a 96-well plate spectra count (Packard, USA).
DNA laddering assay
The characteristic DNA ladder pattern for apoptosis or necrosis was determined by DNA laddering assay. ARPE-19 cells (5 × 106) were exposed to BA8C for 15 h without or with NAC pretreatment. The cells were detached by brief trypsinisation, gently washed with ice-cold PBS, and DNA was extracted using DNeasy Kit (Qiagen Inc.) according to manufacturer’s protocol. RNA was digested during DNA purification procedure. The extracted DNA was loaded onto 1.6% Agarose gel. After electrophoresis and staining with Ethidium bromide, DNA fragments were visualized and image was captured in an automatic gel documentation system (Omega System, Ultra-Lum Inc.).
Determination of Intracellular GSH using Ellman’s Reagent
ARPE-19 cells (1 × 106 cells) were treated with various concentrations of 10, 25, and 50 µM LBP for 6 h or with 100 µM BSO for 24 h or with 1 mM GS-Et for 1 h. After treatment, the cells were washed twice with ice cold PBS and scraped in 1 ml ice cold PBS. The PBS was removed after centrifugation at 1000 g for 5 min. at 4° C. The cells were homogenized in 0.3 ml of Reaction Buffer (0.1 M sodium phosphate, pH 8.0, containing 1 mM EDTA). Again the cells were centrifuged at 8000 g for 20 min. at 4° C. The resultant supernatant was used for the determination of intracellular GSH. The assay mixture was prepared by taking 100 µl standard or experimental supernatant into test tubes containing 20 µl of Ellman’s Reagent solution (4 mg Ellman’s Reagent in 1 ml of Reaction Buffer) and 1 ml of Reaction Buffer. The assay mixture was mixed gently and incubated for 15 min at room temperature. The absorbance was measured at 412 nm. A standard curve was also prepared.
Statistical Analysis
All the data represent the mean ± SD. Statistical significance of difference between untreated control and treated groups was analyzed by Student's t-test as well as ANOVA. Differences were considered statistically significant at P < 0.01.
Results
Dose- and time-dependent effect of LBP & BA8C on DNA damage in ARPE-19 cells
We examined the genotoxic effects of LBP on ARPE-19 cells. The results shown in Fig.1 suggest that LBP caused a significant DNA damage in a dose-dependent manner. The minimum concentration of LBP, 10 µM, elicited a significant (7.4 %, P<0.05) DNA damage which increased to approximately 25 % (P<0.001) with 25 µM LBP (Fig. 1 A & B). The control cells and cells treated with vehicle showed no-significant DNA damage. In order to quantify the extent of LBP-induced DNA breaks in ARPE-19 cells, the comet tail intensity was scored. As shown in Fig. 1 B, 10, 25 & 50 µM LBP caused 1.8-, 3.8- & 5.7-fold increase in % DNA in tail as compared to control cells, respectively. We also examined if a single species such as BA8C could also cause genotoxicity in retinal cells. As shown in Fig. 1, like LBP, BA8C, 10 µM, also elicited a significant 12 % (P<0.05) DNA damage which increased to approximately 30.6 % (P<0.001) with 20 µM BA8C (Fig. 1 A & B). As shown in Fig. 1 B, 10, 20 & 40 µM BA8C caused 3.5-, 7.5- & 12-fold increase in % DNA in tail as compared to control cells, respectively (Fig. 1 A & B).
Fig. 1.
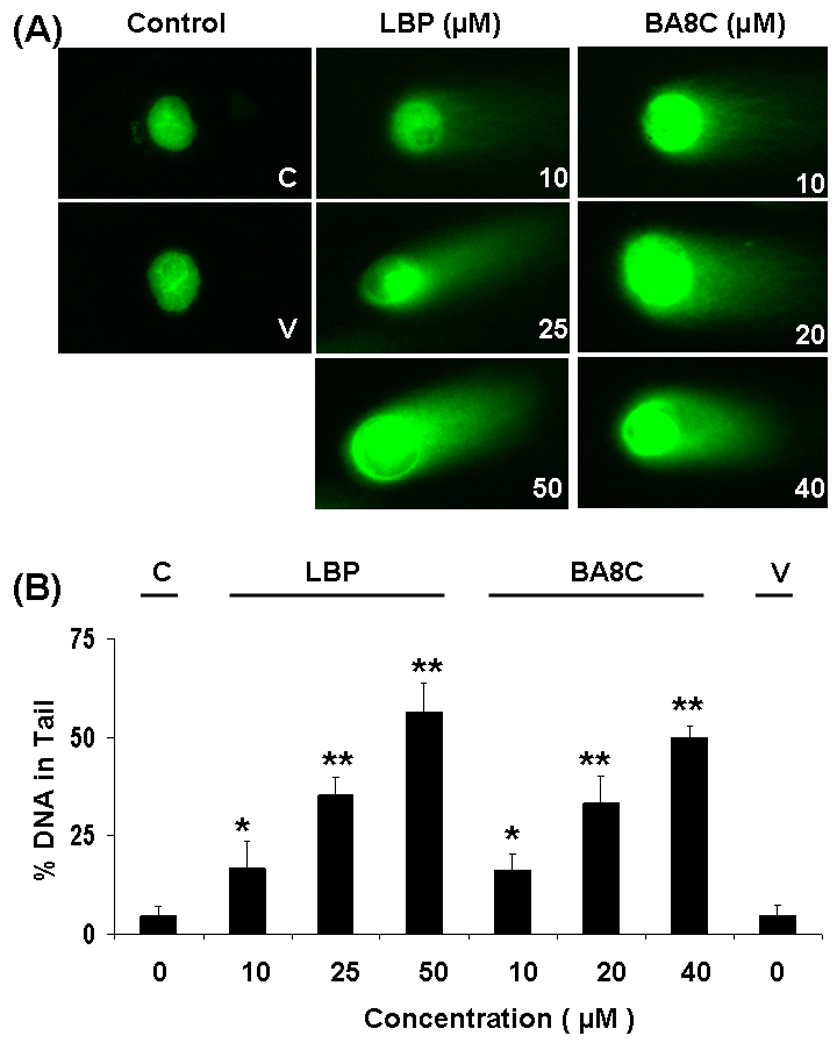
Dose-dependent genotoxic effects induced by LBP & BA8C in ARPE-19 cells. (A) ARPE-19 cells were incubated with 10, 25 & 50 µM LBP and 10, 20 & 40 µM BA8C and vehicle for 9 h and single cell gel electrophoresis was performed to determine the DNA damage. The figure shows the representative comet tails indicative of DNA damage. (B) The bars indicate %DNA in tail (mean ± SD) from three independent experiments. Mean was calculated from three to four parallel slides, 40 comets were evaluated per slide (*P < 0.05, **P < 0.001 vs. control).
We also examined time-dependent effects of LBP and BA8C on DNA damage in ARPE-19 cells. As shown in Fig. 2, with 50 µM LBP the comet tails were visible as early as 3 h after LBP treatment but the extended incubation time caused highly significant (P<0.001) (12 fold) damage as measured by tail length which was highly significant. Moreover, with 40 µM BA8C, the comet tails were also visible as early as 3 h after BA8C treatment but the extended incubation time elicited 9.4 fold (P<0.001) DNA damage (Fig. 2 A & B).
Fig. 2.
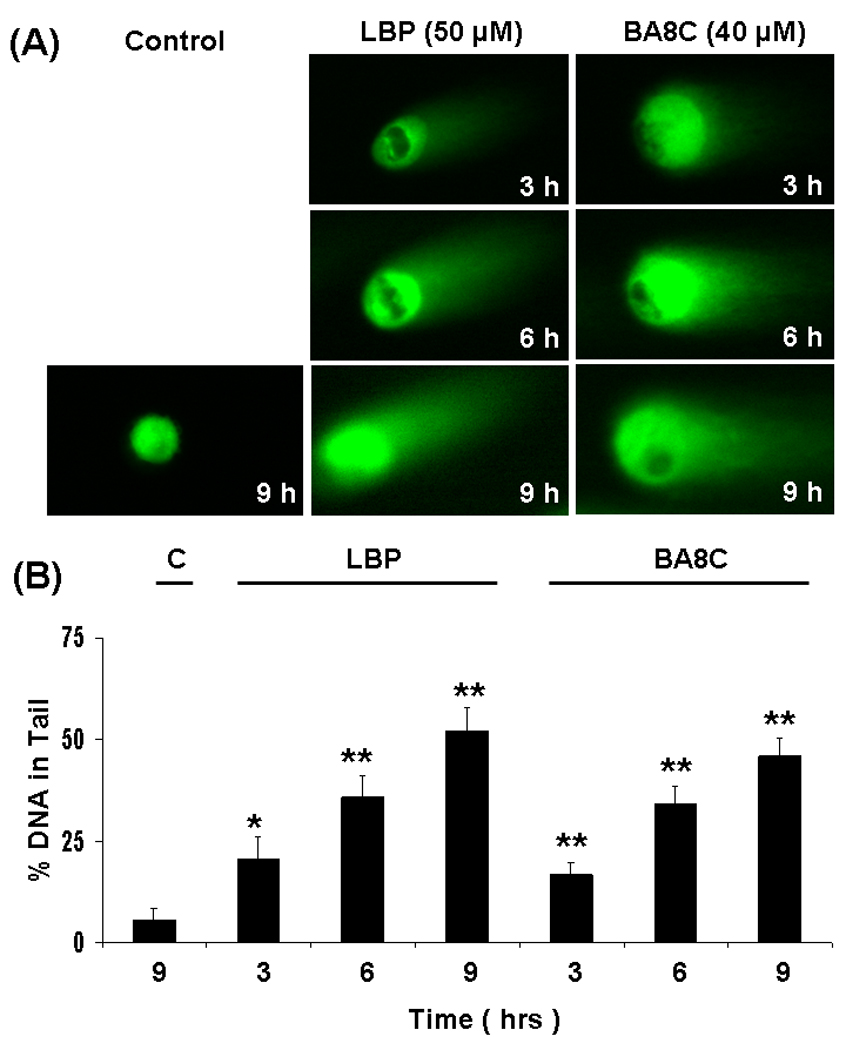
Time-dependent genotoxic effects induced by LBP & BA8C in ARPE-19 cells. (A) ARPE-19 cells were incubated with 50 µM LBP and 40 µM BA8C for 3, 6 and 9 h and single cell gel electrophoresis was performed to assess the DNA damage. The figure shows the representative comet tails indicative of DNA damage. (B) The bar diagram shows tail intensity (%DNA in tail) (mean ± SD) from three independent experiments. The results are given as mean ± SD. The mean in each case was calculated from three to four parallel slides, 40 comets were evaluated per slide (*P < 0.05, **P < 0.001 vs. control).
Effect of BA8C on ARPE-19 cell viability
In our previous study22 we demonstrated that LBP cause ARPE-19 cell death. Therefore, we examined whether single aldehyde species of carotenoid (i.e. BA8C) cause cell death in retinal cells. When ARPE-19 cells were incubated with BA8C for 24 h, a concentration-dependent (0 – 100 µM BA8C) decrease in cell viability was observed (Fig. 3 A & B). The LD50 for BA8C was approximately 40 µM as determined by CellTiter 96® AQueous one solution cell proliferation assay. No apparent change in cell viability was observed when the cells were treated with vehicle. The characteristic DNA ladder pattern of apoptotic and/or necrotic cells was also determined by DNA laddering assay. DNA fragments were examined as irregular bands (“smear”), indicating BA8C-induced necrosis in a dose-dependent manner in ARPE-19 cells (Fig. 3 C). However, NAC prevented BA8C-induced cell death in ARPE-19 cells. These results suggest that CBP could be genotoxic to retinal cells.
Fig. 3.
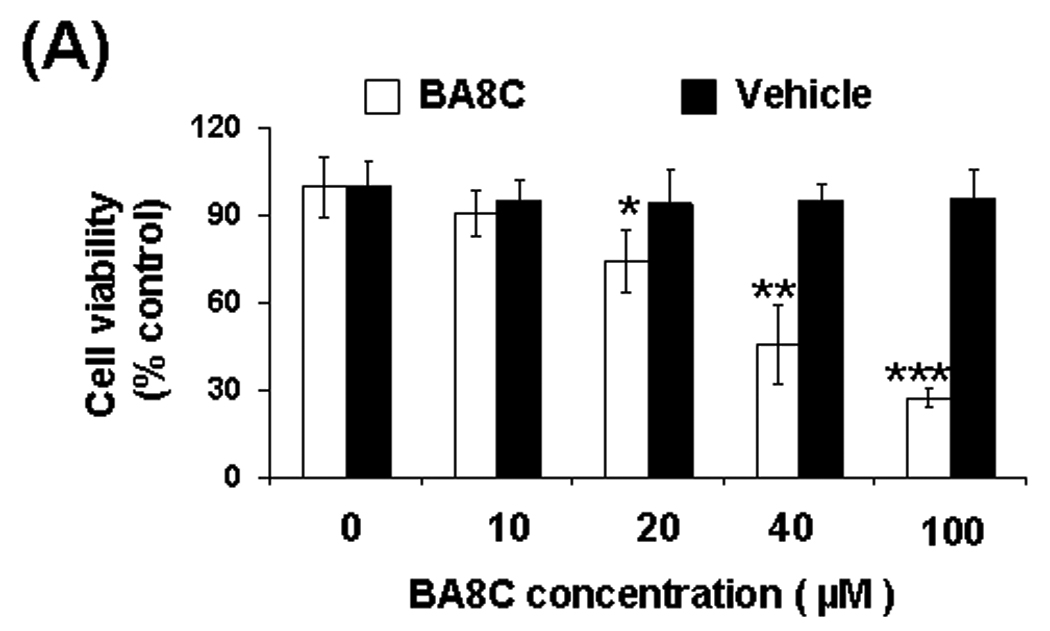
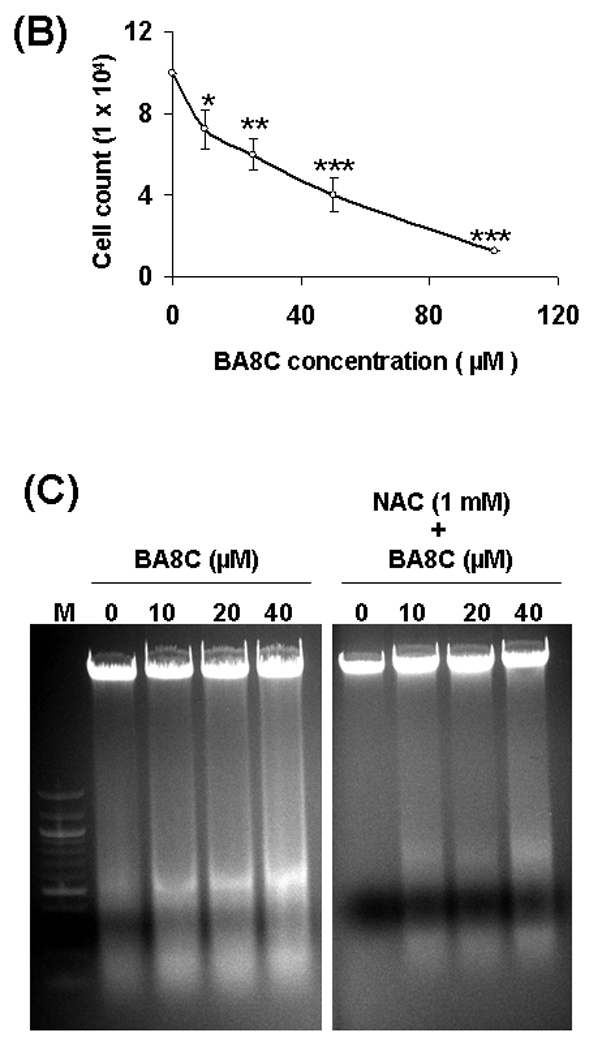
(A & B) Effect of BA8C on ARPE-19 cell viability. BA8C caused concentration-dependent decrease in ARPE-19 cell viability. Cells were exposed to various concentrations of BA8C (0–100 µM) as well as corresponding amount of vehicle and after 24 h cell viability was determined by MTT assay (A) or trypan blue exclusion method (B). Data represents the mean ± S.D. of three experiments (*P < 0.05, **P < 0.01, ***P < 0.001). (C) Effects of BA8C on DNA in ARPE-19 cells. The figure shows Ethidium bromide stained gels containing DNA from ARPE-19 cells treated with 10, 20 & 40 µM BA8C for 15 h without or with pretreatment of 1 mM NAC for 1 h. DNA fragmentation is observed as a smear from BA8C-treated cells which was prevented by NAC. Lane 1. DNA marker; Lanes 2 - 4. DNA from BA8C (0, 10, 20 & 40 µM, respectively) treated cells. Lanes 5 - 8. DNA from BA8C (0, 10, 20 & 40 µM respectively)- treated cells with pretreatment of NAC (1mM).
Effect of NAC and α-Tocopherol + Ascorbic Acid on LBP- and BA8C-induced DNA damage in ARPE-19 cells
In order to examine the effect of NAC on LBP- and BA8C-induced genotoxicity, ARPE-19 cells were incubated with 1 mM NAC for 1h before LBP or BA8C treatment. As shown in Fig. 4, NAC decreased the LBP- & BA8C-induced DNA damage in ARPE-19 cells. The scoring of comet tails in terms of % DNA in tail showed significant (P<0.001) reduction in DNA damage compared to cells treated with LBP or BA8C (Fig. 4 B). The protective and neutralizing effect of NAC could involve its conjugation with carotenoid products through thioether linkage34. This phenomenon could be confirmed by using other physiologically relevant antioxidants which may not conjugate with carotenoid products. Therefore, we examined the effect of α-Tocopherol + Ascorbic Acid on DNA damage induced by LBP in ARPE-19 cells. The incubation with α-Tocopherol + Ascorbic Acid prior to LBP or BA8C treatment could not prevent the DNA damage induced by LBP (Fig. 4). These results indicate that the difference in protective action of NAC as well as α-Tocopherol + Ascorbic Acid could be determined by their ability to conjugate with the carotenoid aldehydes.
Fig. 4.
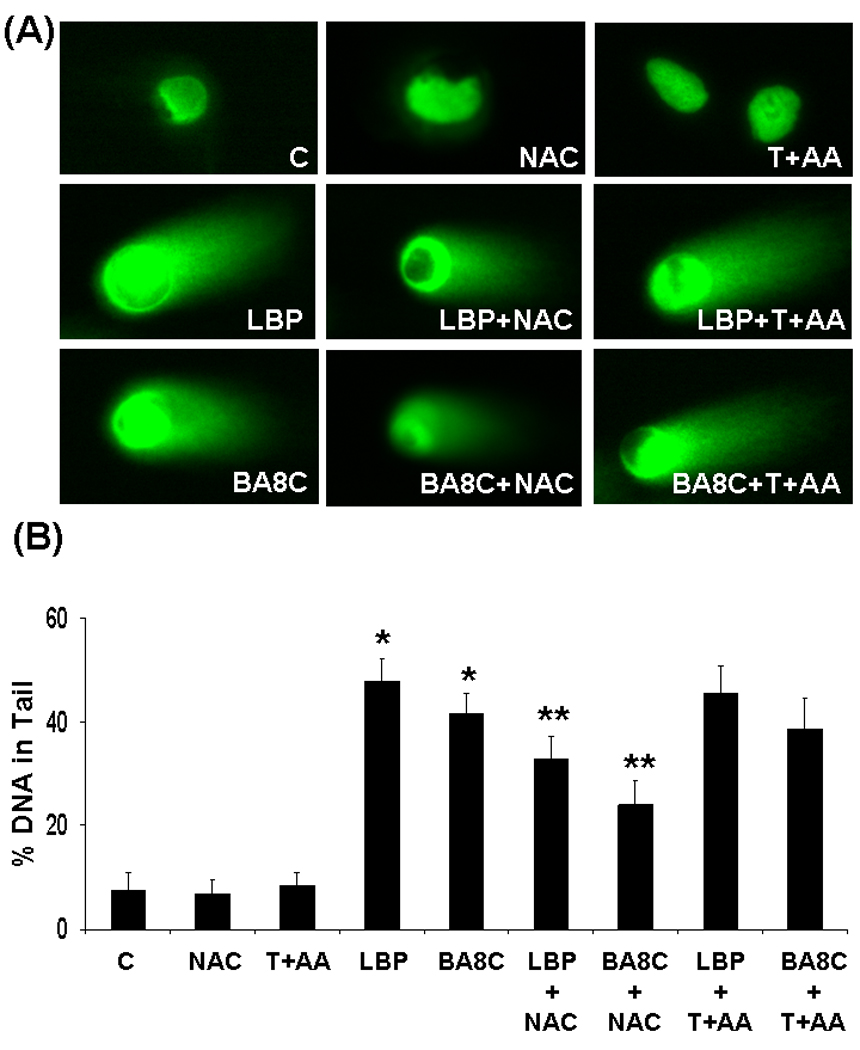
Effect of NAC and α-Tocopherol + Ascorbic Acid (T+AA) on genotoxic effects induced by LBP & BA8C in ARPE-19 cells. (A) ARPE-19 cells pretreated with NAC (1 mM) or T+AA (1 mM + 100 µM) for 1 or 24 h respectively and incubated with 50 µM LBP or 40 µM BA8C for 9 h and single cell gel electrophoresis was done to assess the DNA damage. The figure shows representative comet tails from each group indicative of DNA damage. (B) The bar diagram shows tail intensity (%DNA in tail) (mean ± SD) from three independent experiments. The results are given as mean ± SD. The mean in each case was calculated from three to four parallel slides, 40 comets were evaluated per slide (*P < 0.001 vs. control; **P < 0.001 vs. LBP or BA8C).
Role of cellular GSH in LBP- & BA8C-induced DNA damage in ARPE-19 cells
It is demonstrated that GSH regulates 4-hydroxynonenal-induced genotoxicity in cancer cells30. Therefore, we examined if cellular GSH could also regulate LBP- & BA8C-induced genotoxicity in retinal cells. The effects of LBP on intracellular GSH levels in ARPE-19 cells were examined first. As shown in Fig. 5 A, LBP caused significant decline in GSH pool in retinal cells after 6 h of treatment. The decline in GSH was ~22% (73 nmol/mg protein, p<0.05), ~30% (65 nmol/mg protein, p<0.05), and ~61% (36 nmol/mg protein, p<0.001) by 10, 25, and 50 µM LBP, respectively. The effects of CDA on DNA of retinal cells with increased or decreased GSH levels were determined by treating the cells with GS-Et or BSO, respectively. The GSH levels (94 nmol/mg protein) in ARPE-19 cells were depleted (~77%) by pretreatment with 100 µM BSO for 24 h (Fig. 5 B). The GSH levels in ARPE-19 cells were elevated significantly (~23%, p<0.01, without BSO treatment and ~213%, p<0.001, with BSO treatment) after 1 h incubation with 1mM GS-Et (Fig. 5 C). When the intracellular GSH was depleted by BSO, there was a significant increase in DNA damage by LBP as well as BA8C. However, the DNA damage decreased significantly when the intracellular GSH was elevated. As shown in Fig. 5D, LBP- as well as BA8C-induced DNA damage was significantly (P<0.001 and P<0.01, respectively) enhanced due to pretreatment of BSO as compared to BSO untreated cells. These results demonstrate that GSH could play a pivotal role in the protection of ARPE-19 cells against CBP-induced genotoxicity. The comet scores in terms of % DNA in tail increased in cells pretreated with BSO prior to LBP or BA8C treatment compared to cells treated with LBP or BA8C only. Moreover, GS-Et-pretreatment significantly (P<0.01 and P<0.001 respectively) prevented LBP- and BA8C-induced DNA damage in cells treated without or with BSO (Fig. 5 D). The results demonstrate that adequate cellular GSH levels could protect against LBP- and BA8C-induced DNA damage in ARPE-19 cells.
Fig. 5.
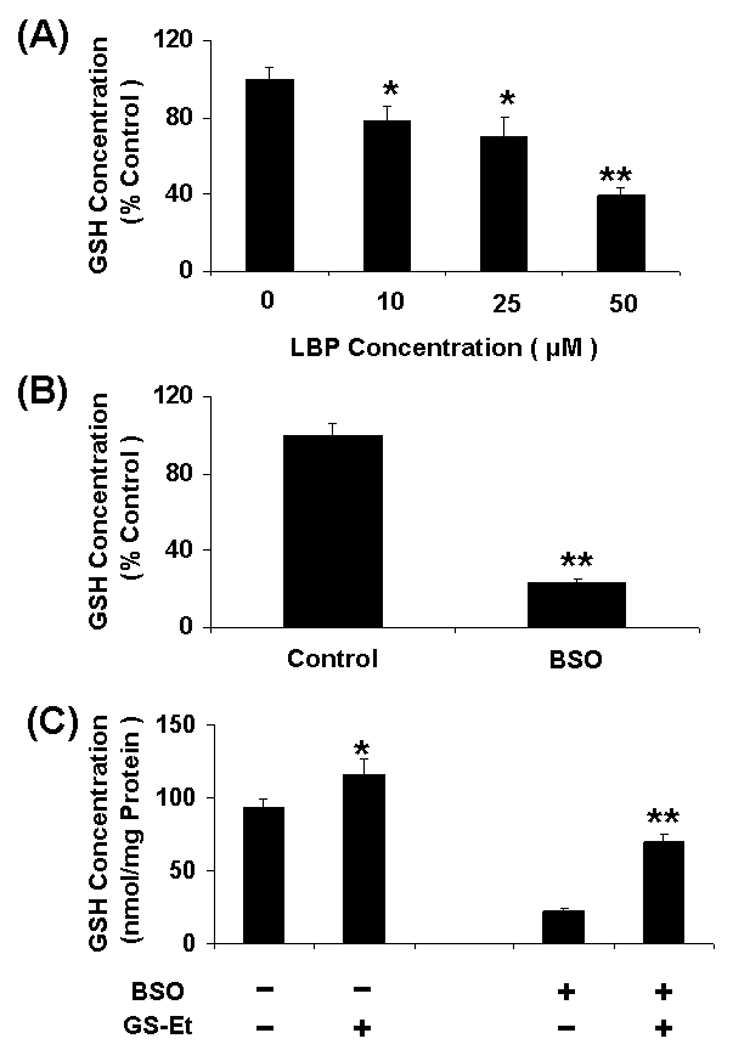
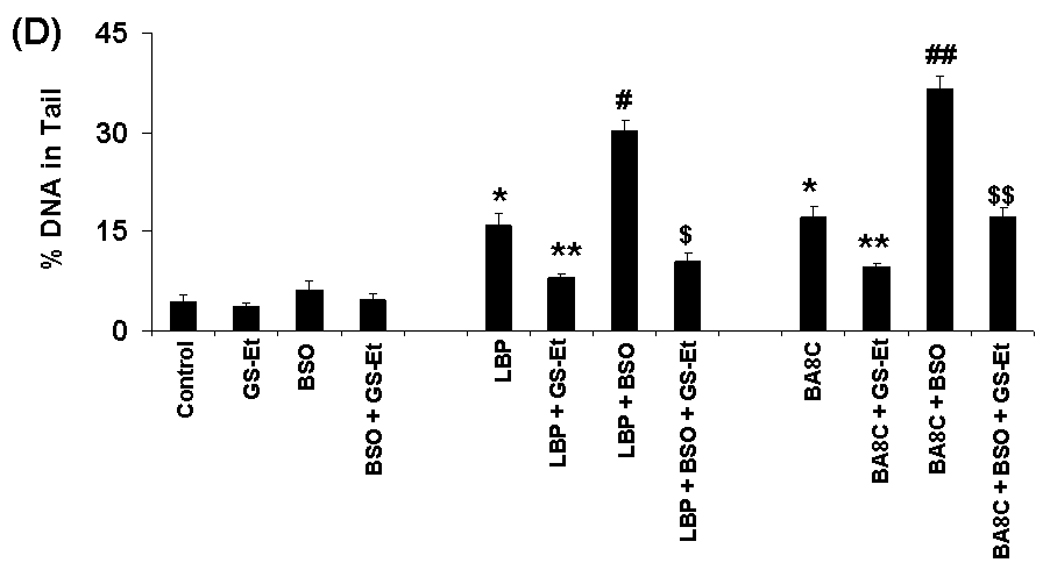
Role of GSH in CBP-induced genotoxicity. (A & B) Depletion of GSH levels induced by LBP or BSO in ARPE-19 cells. The GSH levels were determined in the cytosolic fraction of the cells treated with 10, 25 & 50 µM LBP or 100 µM BSO for 6 h or 24 h, respectively. Data represents the mean ± S.D. of three experiments (*P < 0.05, **P < 0.001). (C) Elevation of GSH levels after glutathione-ethyl-ester (GS-Et) treatment in ARPE-19 cells. The GSH levels were determined in the cytosolic fraction of the cells treated without or with 100 µM BSO for 24 h, followed by incubation with 1mM GS-Et for 1 h. Data represents the mean ± S.D. of three experiments (*P < 0.01 vs control, **P < 0.001 vs BSO control). (D) Effect of GSH depletion and/or supplementation on LBP- & BA8C-induced genotoxicity in ARPE-19 cells. For GSH depletion the cells were pretreated with 100 µM BSO for 20 h. For GSH supplementation the cells were pretreated with 1mM GS-Et for 1 h. After pretreatment the cells were treated with 10 µM LBP or 10 µM BA8C for 9 h and single cell gel electrophoresis was done to assess the DNA damage. The bar diagram shows tail intensity (%DNA in tail) (mean ± SD) from three independent experiments. The results are given as mean ± SD. The mean in each case was calculated from three to four parallel slides, 40 comets were evaluated per slide (*P < 0.01 vs. control; **P < 0.01 vs. LBP or BA8C only; #P < 0.001 vs. LBP only, ¤P < 0.01 vs. LBP + BSO, ##P < 0.01 vs. BA8C only, ¤¤P < 0.001 vs. BA8C + BSO).
Discussion
Lutein and zeaxanthin are widely used as prophylactic agents in AMD5–8. They can undergo oxidation and form various short and long chain cleavage products. The effects of CBP have largely been ignored. This study demonstrates that the CBP of dietary carotenoids can be genotoxic. This may have implications for dietary supplementation with carotenoids in AMD.
The physiological concentration of lutein and LBP in human serum has been reported to be in nM range after short term lutein supplementation for 6 months6, 17,19, but their actual concentration in human serum and ocular tissues after long-term lutein supplementation is not known. The ongoing AREDS-2 study (5 years) may provide data on physiological concentration of xanthophylls (lutein and zeaxanthin) and their breakdown products after long-term xanthophylls supplementation. Nevertheless, the ATBC20 and CARET21 studies have reported µM concentration of β-carotene and its breakdown products (For example, average Vit. A concentration - 2.2 µM) after 3 years of carotenoid supplementation. Moreover, van Helden et al23 demonstrated that β-carotene breakdown products could enhance inflammation-induced oxidative DNA damage in lung epithelial cells at physiologically relevant concentrations. Based on this fact, in the present study, we used µM concentration of LBP and BA8C to investigate their effect on DNA damage in ARPE-19 cells. The concentrations of breakdown products though high based on short term lutein supplementation studies but may be in physiological range based upon long term use of xanthophylls.
The DNA damage and genetic alterations are major risk factors in various oxidative stress-related diseases including AMD40, 41. The results of present study demonstrate that LBP as well as BA8C contribute to DNA damage in a concentration- and time-dependent manner (Fig. 1 & Fig. 2). To assess the quantity and extent of DNA damage in ARPE-19 cells by CBP, a highly sensitive comet assay method was used to demonstrate the severity of genotoxicity. We used LD50 dose for LBP as well as BA8C to study genotoxic effects as reported by us earlier in ARPE-19 cells22. The genotoxic effects of β-carotene (20 µM) and BA8C (2, 5, and 20 µM) have been demonstrated in A549 cells29. Moreover, BA8C is also demonstrated to form 1,N2 –etheno-2’-deoxyguanosine adduct which could lead to DNA strand breaks and mutations42. These results are concordant with our findings which show significant DNA damage in ARPE-19 cells treated with similar concentrations of LBP and BA8C. The mechanism of DNA damage could be either direct through apoptosis22, or indirect through damaging plasma membrane and inducing necrosis as observed in this study. Thus, genotoxic effects of LBP at concentrations used in this study could potentially cause or exacerbate retinal pathology.
The extent of geno- & cytotoxicity induced by various agents is dependent on cellular GSH levels.30, 43. It is well known that under oxidative stress, GSH is oxidized to GSSG and transported out44, 45 which eventually results in significantly lower cellular GSH levels. The increased carotenoid oxidation and CBP formation coupled with decreased availability of GSH could exacerbate the genotoxicity of CBP. Since GSH is known to be the first line of defense in various tissues against oxidative stress, we investigated the effects of increased as well as decreased intracellular GSH levels in CBP-induced genotoxicity in retinal cells. When the cells with basal cytosolic GSH levels were treated with CBP, there was a dose-dependent increase in the DNA damage (Fig. 1) which corresponded to decline in basal GSH levels in cytosol (Fig. 5 A). These results indicate that the extent of CBP-induced DNA damage in ARPE-19 cells is associated with cytosolic GSH levels. The CBP, being hydrophobic, could interact with hydrophilic molecules such as GSH which can be translocated to nucleus where it could cause increased DNA damage and thereby decreased intracellular GSH levels. The conjugate of GSH and CBP formed by thioether linkage could also be readily translocated out of the cells thereby causing decreased intracellular levels of GSH as well as CBP. This would be the protective role of GSH against the genotoxic effects of CBP. This hypothesis is supported by our observation that BSO treatment, which decreased the GSH levels, significantly increased DNA damage (Fig. 5 D). However, it also suggests that CBP translocation to nucleus, under depleted GSH conditions, could also be facilitated by other mechanisms. Although, the precise mechanism for the transportation of CBP from cytosol to nucleus is not known, β-carotene and its breakdown products have been demonstrated to accumulate in crude nuclei of NCL-H69 cells46. Furthermore, the GSH supplementation significantly prevented the CBP-induced DNA damage in the ARPE-19 cells (Fig. 5 D). Our results thus indicate that increasing GSH levels in the cells could attenuate CBP-induced DNA damage.
During oxidative stress GSH readily and rapidly conjugates with lipid aldehydes, such as HNE, catalysed by Glutathione-S-transferase (GST)47,48. The GST-mediated conjugation of HNE with GSH is a major detoxification pathway against lipid aldehyde-induced DNA damage30. Thus GST could be the body's major defense against lipid aldehyde-induced protein alteration as well as DNA damage through conjugation of aldehydes. Reduction of lipid aldehydes to corresponding alcohols by Aldose reductase (AR) and transporting lipid aldehydes out of cells could be the major defense of cells30. Glutathione conjugates with CBP could also be the major defense of cells against products of carotenoid oxidation. In case of carotenoid products, we have observed efficient conjugation of CBP with GSH or NAC34. However, the role of GST in expediting conjugation and protection of cells against CBP is not known. Moreover, the role of AR in lipid aldehydes-induced DNA damage as well as cytotoxicity is well established30, 33, 49. Further studies are required to investigate the possible role of AR and other reductases in the protection against CBP-induced genotoxicity or cytotoxicity.
In summary, the results of this study suggest that (1) LBP could contribute to dose- and time-dependent increase of DNA damage in ARPE-19 cells, (2) short term dietary supplementation with lutein, which results in only nM serum levels, may be quite safe since µM levels are required to cause damage and, (3) the modulation of cellular GSH levels could ameliorate LBP-induced genotoxic effects in retinal cells. Randomized long-term studies such as AREDS-2 would greatly help in understanding the beneficial versus harmful effects of lutein and zeaxanthin commonly used in the prevention and treatment of AMD.
Acknowledgment
This study was supported by the National Institutes of Health (NIH) Grants GM71036 (to K.V.R.) DK36118 (to S.K.S.) and Wilkins AMD Fund & Research to Prevent Blindness Grant (to F.J.G.M.v K).
Footnotes
Declaration of Interest:
The authors report no conflicts of interest. The authors alone are responsible for content and writing of the paper.
References
- 1.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Opthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 2.Holz FG, Pauleikhoff D, Klein R, Bird AC. Pathogenesis of lesions in late age-related macular disease. Am. J. Ophthalmol. 2004;137:504–510. doi: 10.1016/j.ajo.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 3.Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s Aß-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA. 2002;99:11830–11835. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004;122:598–614. doi: 10.1001/archopht.122.4.598. [DOI] [PubMed] [Google Scholar]
- 5.Delcourt C, Carrière I, Delage M, Barberger-Gateau P, Schalch W. The POLA Study Group. Plasma Lutein and Zeaxanthin and Other Carotenoids as Modifiable Risk Factors for Age-Related Maculopathy and Cataract: The POLA Study. Invest. Ophthalmol. Vis. Sci. 2006;47:2329–2335. doi: 10.1167/iovs.05-1235. [DOI] [PubMed] [Google Scholar]
- 6.Johnson EJ, Chung HY, Caldarella SM, Snodderly DM. The influence of supplemental lutein and docosahexaenoic acid on serum, lipoproteins, and macular pigmentation. Am. J. Clin. Nutr. 2008;87:1521–1529. doi: 10.1093/ajcn/87.5.1521. [DOI] [PubMed] [Google Scholar]
- 7.Leung IY, Sandstrom MM, Zucker CL, Neuringer M, Max Snodderly D. Nutritional manipulation of primate retinas. IV. Effects of n--3 fatty acids, lutein, and zeaxanthin on S-cones and rods in the foveal region. Exp. Eye Res. 2005;81:513–529. doi: 10.1016/j.exer.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Neuringer M, Sandstrom MM, Johnson EJ, Snodderly DM. Nutritional manipulation of primate retinas, I: effects of lutein or zeaxanthin supplements on serum and macular pigment in xanthophyll-free rhesus monkeys. Invest. Ophthalmol. Vis. Sci. 2004;45:3234–3243. doi: 10.1167/iovs.02-1243. [DOI] [PubMed] [Google Scholar]
- 9.Palozza P, Serini S, Di Nicuolo F, Piccioni E, Calviello G. Prooxidant effects of β-carotene in cultured cells. Mol. Aspects Med. 2003;24:353–362. doi: 10.1016/s0098-2997(03)00031-1. [DOI] [PubMed] [Google Scholar]
- 10.Handelman GJ, van Kuijk FJGM, Chatterjee A, Krinsky NI. Characterization of products formed during the autoxidation of β-carotene. Free Radic. Biol. Med. 1991;10:427–437. doi: 10.1016/0891-5849(91)90051-4. [DOI] [PubMed] [Google Scholar]
- 11.Hurst JS, Contreras JE, Siems WG, van Kuijk FJGM. Oxidation of carotenoids by heat and tobacco smoke. Biofactors. 2004;20:23–35. doi: 10.1002/biof.5520200103. [DOI] [PubMed] [Google Scholar]
- 12.Prasain JK, Moore R, Hurst JS, Barnes S, van Kuijk FJGM. Electrospray tandem mass spectrometric analysis of zeaxanthin and its oxidation products. J. Mass Spectrom. 2005;40:916–923. doi: 10.1002/jms.868. [DOI] [PubMed] [Google Scholar]
- 13.Hurst JS, Saini MK, Jin G, Awasthi YC, van Kuijk FGJM. Toxicity of oxidized β-carotene to cultured human cells. Exp. Eye Res. 2005;81:239–243. doi: 10.1016/j.exer.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Bernstein PS, Khachik F, Carvalho LS, Muir GJ, Zhao DY, Katz NB. Identification and quantitation of carotenoids and their metabolites in the tissues of the human eye. Exp. Eye. Res. 2001;72:215–223. doi: 10.1006/exer.2000.0954. [DOI] [PubMed] [Google Scholar]
- 15.Bhosale P, Bernstein PS. Quantitative measurement of 3'-oxolutein from human retina by normal-phase high-performance liquid chromatography coupled to atmospheric pressure chemical ionization mass spectrometry. Anal. Biochem. 2005;345:296–301. doi: 10.1016/j.ab.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 16.Khachik F, Bernstein PS, Garland DL. Identification of lutein and zeaxanthin oxidation products in human and monkey retinas. Invest. Ophthalmol. Vis. Sci. 1997;38:1802–1811. [PubMed] [Google Scholar]
- 17.Khachik F, de Moura FF, Chew EY, Douglass LW, Ferris FL, 3rd, Kim J, Thompson DJ. The Effect of Lutein and Zeaxanthin Supplementation on Metabolites of These Carotenoids in the Serum of Persons Aged 60 or Older. Invest. Ophthalmol. Vis. Sci. 2006a;47:5234–5242. doi: 10.1167/iovs.06-0504. [DOI] [PubMed] [Google Scholar]
- 18.Khachik F, London E, de Moura FF, Johnson M, Steidl S, Detolla L, Shipley S, Sanchez R, Chen XQ, Flaws J, Lutty G, McLeod S, Fowler B. Chronic Ingestion of (3R, 3’R, 6’R)-Lutein and (3R, 3'R)-Zeaxanthin in the Female Rhesus Macaque. Invest. Ophthalmol. Vis. Sci. 2006b;47:5476–5486. doi: 10.1167/iovs.06-0194. [DOI] [PubMed] [Google Scholar]
- 19.Huang LL, Coleman HR, Kim J, de Monasterio F, Wong WT, Schleicher RL, Ferris FL, 3rd, Chew EY. Oral supplementation of lutein/zeaxanthin and omega-3 long chain polyunsaturated fatty acids in persons aged 60 years or older, with or without AMD. Invest Ophthalmol Vis Sci. 2008;49:3864–3869. doi: 10.1167/iovs.07-1420. [DOI] [PubMed] [Google Scholar]
- 20.Albanes D, Heinonen OP, Taylor PR, Virtamo J, Edwards BK, et al. Alpha-Tocopherol and beta-carotene supplements and lung cancer incidence in the alpha-tocopherol, beta-carotene cancer prevention study: effects of base-line characteristics and study compliance. J Natl Cancer Inst. 1996;88:1560–1570. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 21.Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88:1550–1559. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]
- 22.Kalariya NM, Ramana KV, Srivastava SK, van Kuijk FJGM. Carotenoid derived aldehydes-induced oxidative stress causes apoptotic cell death in human retinal pigment epithelial cells. Exp. Eye Res. 2008;86:70–80. doi: 10.1016/j.exer.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Helden YG, Keijer J, Knaapen AM, Heil SG, Briedé JJ, van Schooten FJ, Godschalk RW. Beta-carotene metabolites enhance inflammation-induced oxidative DNA damage in lung epithelial cells. Free Radic Biol Med. 2009;46:299–304. doi: 10.1016/j.freeradbiomed.2008.10.038. [DOI] [PubMed] [Google Scholar]
- 24.Alija AJ, Bresgen N, Sommerburg O, Langhans CD, Siems W, Eckl PM. Beta-carotene breakdown products enhance genotoxic effects of oxidative stress in primary rat hepatocytes. Carcinogenesis. 2006;27:1128–1133. doi: 10.1093/carcin/bgi342. [DOI] [PubMed] [Google Scholar]
- 25.Nara E, Hayashi H, Kotake M, Miyashita K, Nagao A. Acyclic carotenoids and their oxidation mixtures inhibit the growth of HL-60 human promyelocytic leukemia cells. Nutr. Cancer. 2001;39:273–283. doi: 10.1207/S15327914nc392_18. [DOI] [PubMed] [Google Scholar]
- 26.Salerno C, Capuozzo E, Crifò C, Siems W. alpha-Tocopherol increases caspase-3 up-regulation and apoptosis by beta-carotene cleavage products in human neutrophils. Biochim. Biophys. Acta. 2007;1772:1052–1056. doi: 10.1016/j.bbadis.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 27.Salgo M, Cueto R, Winston G, Pryor W. β-carotene and its oxidation products have different effects on microsome mediated binding of benzo[a]pyrene to DNA. Free Radic. Biol. Med. 1999;26:162–173. doi: 10.1016/s0891-5849(98)00172-5. [DOI] [PubMed] [Google Scholar]
- 28.Siems W, Sommerburg O, Schild L, Augustin W, Langhans CD, Wiswedel I. β-Carotene cleavage products induce oxidative stress in vitro by impairing mitochondrial respiration. FASEB J. 2002;16:1289–1291. doi: 10.1096/fj.01-0765fje. [DOI] [PubMed] [Google Scholar]
- 29.Yeh SL, Wu SH. Effects of quercetin on beta-apo-8'-carotenal-induced DNA damage and cytochrome P1A2 expression in A549 cells. Chem. Biol. Interact. 2006;163:199–206. doi: 10.1016/j.cbi.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Yadav UCS, Ramana KV, Awasthi YC, Srivastava SK. Glutathione level regulates HNE-induced genotoxicity in human erythroleukemia cells. Toxicol. Appl. Pharmacol. 2008;227:257–264. doi: 10.1016/j.taap.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakajima A, Yamada K, Zou LB, Yan Y, Mizuno M, Nabeshima T. Interleukin-6 protects PC12 cells from 4-hydroxynonenal-induced cytotoxicity by increasing intracellular glutathione levels. Free Radic. Biol. Med. 2002;32:1324–1332. doi: 10.1016/s0891-5849(02)00845-6. [DOI] [PubMed] [Google Scholar]
- 32.Cao Z, Hardej D, Trombetta LD, Li Y. The role of chemically induced glutathione and glutathione S-transferase in protecting against 4-hydroxy-2-nonenalmediated cytotoxicity in vascular smooth muscle cells. Cardiovasc. Toxicol. 2003;3:165–177. doi: 10.1385/ct:3:2:165. [DOI] [PubMed] [Google Scholar]
- 33.Ramana KV, Bhatnagar A, Srivastava S, Yadav UC, Awasthi S, Awasthi YC, Srivastava SK. Mitogenic responses of vascular smooth muscle cells to lipid peroxidation-derived aldehyde 4-hydroxy-trans-2-nonenal (HNE): role of aldose reductase-catalyzed reduction of the HNE-glutathione conjugates in regulating cell growth. J. Biol. Chem. 2006;281:17652–17660. doi: 10.1074/jbc.M600270200. [DOI] [PubMed] [Google Scholar]
- 34.Kalariya NM, Ramana KV, Srivastava SK, van Kuijk FJGM. Association for Research in Vision & Ophthalmology (ARVO) Fort Lauderdale, FL: 2008. Post-translational protein modification by Lutein derived aldehydes. Abstract # 4968. [Google Scholar]
- 35.Krasowska A, Konat GW. Vulnerability of brain tissue to inflammatory oxidant, hypochlorous acid. Brain Res. 2004;997:176–184. doi: 10.1016/j.brainres.2003.09.080. [DOI] [PubMed] [Google Scholar]
- 36.Whiteman M, Rose P, Siau JL, Cheung NS, Tan GS, Halliwell B, Armstrong JS. Hypochlorous acid-mediated mitochondrial dysfunction and apoptosis in human hepatoma HepG2 and human fetal liver cells: role of mitochondrial permeability transition. Free Radic Biol Med. 2005;38:1571–1584. doi: 10.1016/j.freeradbiomed.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 37.Lee CS, Jang YY, Song JS, Song JH, Han ES. Ambroxol inhibits peroxynitrite-induced damage of alpha1-antiproteinase and free radical production in activated phagocytic cells. Pharmacol Toxicol. 2002;91:140–149. doi: 10.1034/j.1600-0773.2002.910309.x. [DOI] [PubMed] [Google Scholar]
- 38.Bligh EG, Dyer WJ. A rapid method for total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 39.van Kuijk FJ, Thomas DW, Stephens RJ, Dratz EA. Gas chromatography-mass spectrometry method for determination of phospholipid peroxides; I. Transesterification to form methyl esters. J. Free Radic. Biol. Med. 1985;1:215–225. doi: 10.1016/0748-5514(85)90121-7. [DOI] [PubMed] [Google Scholar]
- 40.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003;76:397–403. doi: 10.1016/s0014-4835(03)00023-x. [DOI] [PubMed] [Google Scholar]
- 42.Marques SA, Loureiro AP, Gomes OF, Garcia CC, Di Mascio P, Medeiros MH. Induction of 1,N(2)-etheno-2'-deoxyguanosine in DNA exposed to beta-carotene oxidation products. FEBS Lett. 2004;560:125–130. doi: 10.1016/S0014-5793(04)00084-5. [DOI] [PubMed] [Google Scholar]
- 43.Ebert MN, Beyer-Sehlmeyer G, Liegibel UM, Kautenburger T, Becker TW, Pool-Zobel BL. Butyrate-induces glutathione S-transferase in human colon cells and protects from genetic damage by 4-hydroxynonenal. Nutr. Cancer. 2001;4:156–164. doi: 10.1080/01635581.2001.9680627. [DOI] [PubMed] [Google Scholar]
- 44.Meister A, Anderson ME, Hwang O. Intracellular cysteine and glutathione delivery systems. J. Am. Coll. Nutr. 1986;5:137–151. doi: 10.1080/07315724.1986.10720121. [DOI] [PubMed] [Google Scholar]
- 45.Srivastava SK, Beutler E. The transport of oxidized glutathione from the erythrocytes of various species in the presence of chromate. Biochem. J. 1969;114:833–837. doi: 10.1042/bj1140833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prakash P, Jackson CL, Gerber LE. Subcellular accumulation of beta-carotene and retinoids in growth-inhibited NCI-H69 small cell lung cancer cells. Nutr Cancer. 1999;34:76–82. doi: 10.1207/S15327914NC340111. [DOI] [PubMed] [Google Scholar]
- 47.Alin P, Danielson UH, Mannervik B. 4-Hydroxyalk-2-enals are substrates for glutathione transferase. FEBS Lett. 1985;179:267–270. doi: 10.1016/0014-5793(85)80532-9. [DOI] [PubMed] [Google Scholar]
- 48.Zimniak P, Singhal SS, Srivastava SK, Awasthi S, Sharma R, Hayden JB, Awasthi YC. Estimation of genomic complexity, heterologous expression, and enzymatic characterization of mouse glutathione S-transferase mGSTA4-4 (GST 5.7) J. Biol. Chem. 1994;269:992–1000. [PubMed] [Google Scholar]
- 49.Srivastava S, Dixit BL, Cai J, Sharma S, Hurst HE, Bhatnagar A, Srivastava SK. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: role of aldose reductase. Free Radic. Biol. Med. 2004;29:642–651. doi: 10.1016/s0891-5849(00)00351-8. [DOI] [PubMed] [Google Scholar]