

Summary
We consider the problem of comparing cumulative incidence functions of non-mortality events in the presence of informative coarsening and the competing risk of death. We extend frequentist-based hypothesis tests previously developed for non-informative coarsening and propose a novel Bayesian method based on comparing a posterior parameter transformation to its expected distribution under the null hypothesis of equal cumulative incidence functions. Both methods use estimates derived by extending previously published estimation procedures to accommodate censoring by death. The data structure and analysis goal are exemplified by the AIDS Link to the Intravenous Experience (ALIVE) study, where researchers are interested in comparing incidence of human immunodeficiency virus seroconversion by risk behavior categories. Coarsening in the forms of interval and right censoring and censoring by death in ALIVE are thought to be informative, thus we perform a sensitivity analysis by incorporating elicited expert information about the relationship between seroconversion and censoring into the model.
Keywords: Bayesian Analysis, Frequentist Analysis, Hypothesis Test, Interval Censoring, Markov Chain Monte Carlo, Sensitivity Analysis
1. Introduction
In prospective studies examining incidence of non-mortality outcomes, event times are often assessed at regular pre-scheduled appointments. These event times can be interval censored when study participants miss visits and return after a hiatus having experienced the event of interest. Censoring also occurs when participants never return or die before returning after missing visits. Such data are usually analyzed by assuming non-informative censoring, a special case of coarsening at random (CAR) [1–3]. However, the censoring process may be related to the event process. That is, the coarsening mechanism may be coarsened not at random (CNAR). Unfortunately, observed data are not sufficient to estimate the relationship between these processes. Therefore, analyzing coarsened data involves making unverifiable assumptions about the relationship between coarsening and event incidence.
There have been several recent methodologic developments to address coarsening not at random. Bayesian and frequentist methodologies were developed for interval-censored data to estimate cumulative incidence for (potentially) CNAR data by incorporating elicited expert information into the model [4]. A tool for quantifying local sensitivity to informative coarsening in the context of censoring has been proposed [5]. A test for dependent censoring has been developed using auxiliary visit-compliance information outside of the censoring interval [?]. Assuming a proportional hazards model, inference methods based on the collection of all attainable hazards ratios have been proposed [7]. The book by Sun [8] provides a compendium of analytical methods to handle noninformative interval censoring and a brief treatment of informative interval censoring. However, none of the aforementioned developments includes hypothesis assessment methods for informatively interval-censored data without imposing a proportional hazards model.
In this paper, we aim to develop formal hypothesis assessment procedures for comparing cumulative incidence functions using estimates derived by extending previously published methods [4] to accommodate censoring by death. For frequentist inference, we extend the logrank [9] and a two-sided version of the integrated weighted difference (IWD) [10] tests to allow informative censoring. We also generalize IWD to more than two groups. For Bayesian inference, we propose parameter transformations of posterior event-time probabilities, motivated by the logrank and IWD tests.
We apply our methods to AIDS Link to the Intravenous Experience (ALIVE), an ongoing prospective observational study of risk factors for human immunodeficiency virus (HIV) infection among injection drug users (IDUs) in Baltimore, Maryland [11–13]. In this study, HIV serostatus, a proxy for HIV infection status, was determined by subsequent regularly scheduled laboratory blood tests. For those who attended every visit on schedule, time to seroconversion (operationalized as years from enrollment here) is known, resulting in discrete event-time data. However, ALIVE participants often missed visits or attended visits off schedule, sometimes resulting in interval-censored seroconversion times only known within a range of years. Also, some seropositive participants never tested positive during the study due to loss to follow up, administrative censoring, or death. We compare ten-year seroconversion incidence (1988-1998) between those who self-reported sharing needles for injecting drugs during the six months prior to enrollment and those who did not. To perform a sensitivity analysis, we use information about the relationship between incidence and censoring elicited from ALIVE investigators and an AIDS epidemiologist. After estimating cumulative incidence functions, we assess the null hypothesis of equal HIV incidence between needle sharers and non-sharers.
2. Data Structure, Coarsening Models, and Inference
2.1. Data structure
Let T = t denote seroconversion during year t, where E = {t : t = 1, …, M + 1} is the support of T. M denotes last year of follow-up from enrollment, and T is arbitrarily set to be M + 1 for individuals who did not seroconvert during follow-up. Observed data for an individual may be a set of years from E, [L, R] = {t ∈ E : L ≤ t ≤ R}, where [L, R] is a coarsening of T because T ∈ [L, R]. For example, if a participant tests negative for HIV at a visit in year 2, returns for a visit in year 5 and tests positive, then serconversion could have occurred as early as year 2 (after the year 2 visit) and as late as year 5 (before the year 5 visit). Therefore L = 2, R = 5, and T ∈ [2, 5]. However, if the participant never returned after the year 2 visit, then seroconversion could have occurred anytime during or after year 2 within the study period, after the end of the study, or never. Therefore, L = 2, R = M + 1, and T ∈ [2, M + 1]. If seroconversion is known to occur in year t, t=1, …, M, then L = R = t. For example, if the participant would have returned later in year 2 and tested positive, then L = 2, R = 2, and thus T = 2. In general, if seroconversion did not occur during follow-up, then L = R = M + 1, and if knowledge about T is incomplete, then L < R. Those with L < R = M + 1 are right-censored drop-outs, and those with L < R < M + 1 are interval-censored returners [4].
We generalize the above ideas to accommodate the competing risk of death encountered in ALIVE. Those with first missed visit in year l who die in year r (r > l) either seroconverted in [L = l, R = r] or died seronegative. For those censored by death, [L = l, R = r] has an altered interpretation: R = r denotes death at year r, and possible event times are {l, …, r, M + 1}. Therefore, T = M + 1 denotes not seroconverting while at risk. For example, if a participant who tested negative during year 2 and missed subsequent visits died with unknown serostatus during year 5, then seroconversion either did not occur (T = M + 1) or occurred between years 2 and 5, inclusive. Therefore T ∈ {2, …, 5, M + 1}. Let Δ = δ, δ ∈ {0, 1}, indicate whether R is year of death. Thus, the aforementioned participant would have L = 2, R = 5, and Δ = 1. In general, if R < M + 1 and serostatus is unknown at year R due to death, then Δ = 1, otherwise Δ = 0.
Let G denote number, and 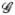 denote the set, of groups to compare. We assume that, for those in group g, g ∈ , we observe ng i.i.d. copies of the data. Pg(·) refers to probabilities for those in group g. Where necessary, the subscript i will denote subject-specific data.
denote the set, of groups to compare. We assume that, for those in group g, g ∈ , we observe ng i.i.d. copies of the data. Pg(·) refers to probabilities for those in group g. Where necessary, the subscript i will denote subject-specific data.
2.2. Coarsening Models
2.2.1. Coarsening at random
Given L = l, R = r, and Δ = δ, let A(l, r, δ) denote possible values of T induced by censoring, where A(l, r, 0) = [l, r] and A(l, r, 1) = {[l, r], M + 1}. Within group g, CAR means
| (1) |
for all [l, r] ∈ E* = {[l, r] : l ≤ r, l, r ∈ E} and δ ∈ {0, 1}. Using Bayes' rule with 1, it can be shown that CAR also means
for all t ∈ A(l,r,δ) [3].
In words, CAR means that among those in group g, the coarsening process provides no information about the seroconversion process beyond knowing that the true year of seroconversion resides in the set of years induced by censoring. As a result, estimated event-time probabilities for censored individuals only depend on estimated probabilities for years in that set.
2.2.2. CNAR models
The coarsening (in this case, censoring) mechanism cannot be identified from observed data [3], therefore we consider a class of CNAR models indexed by a (possibly group-specific) censoring bias function that allows elicited expert information to determine whether event probabilities for coarsened event times should be made stochastically larger at later times (seroconversions tend to occur late in the censoring set) or earlier times (seroconversions tend to occur early in the censoring set) relative to CAR [4]. We ‘exponentially tilt’ [14] the model assuming CAR for each group g, g ∈ :
| (2) |
where cg(l, r, δ; qg) = Σs∈A(l,r,δ) Pg(T = s | T ∈ A(l, r, δ)) exp{qg(s, l, r, δ)}, and qg(t, l, r, δ) is a specified censoring bias function of (t, l, r, δ) for those in group g. If qg(·) does not depend on t, no tilting is performed, and CAR is assumed for group g. Information about death is only utilized to define possible seroconversion times and in qg(·) to allow estimation of the seroconversion process without requiring estimation of the death process.
Using Bayes' rule, equation (2) can be represented as a selection model:
| (3) |
for t, t′ ∈ A(l, r, δ). From (3), qg(t, l, r, δ) is the group-g difference in log probability of having censoring set A(l,r,δ) comparing those with T = t to those with T equal to some reference value, tref, such that qg(tref, l, r, δ) = 0.
2.2.3. Low-dimensional parameterization of qg(·)
We temporarily return to ALIVE to describe a proposed censoring bias function. To facilitate a sensitivity analysis, we parameterize a censoring bias function by a small set of unidentified censoring bias parameters to capture key features of ALIVE. The function parameters differentiate between those who are interval censored, right-censored alive, and censored by death. We allow the censoring mechanism to differ between non-sharers (g = n) and needle sharers (g = s). Let ϕ = {ϕg: g = n, s} denote group-specific censoring bias parameters. The proposed censoring bias function is
| (4) |
where ϕg = {ϕg1, ϕg2, ϕg3}, and I(·) denotes the indicator function. From equation (3), exp{ϕg1} is the probability ratio of having L = 1, R = 5, and Δ = 0 comparing those with T = 5 to those with T = 1, among those in group g, g = n, s. The factor 9/4 is used to attain this interpretation for elicitation. Similarly, exp{ϕg2} is the needle sharing-specific probability ratio of having L = 1, R = M + 1, and Δ = 0, comparing those with T = M + 1 to those with T = 1. Lastly exp{ϕg3} is the needle sharing-specific probability ratio of having L = 1, R = M + 1, and Δ = 1, comparing those with T = M + 1 to those with T = 1. Note that ϕg1, ϕg2, and ϕg3 refer to returning, dropping out alive, and being censored by death, respectively, for those in group g.
From (2), we see that when exp{ϕg1} > 1 (< 1), returners are assumed to be more (less) likely to seroconvert late than seroconvert early. When exp{ϕg2} > 1 (< 1), drop-outs who remain alive are assumed more (less) likely to seroconvert late or never than seroconvert early. Lastly, When exp{ϕg3} > 1 (< 1), drop-outs who die with unknown serostatus are assumed more (less) likely to seroconvert late or never than seroconvert early.
The form of Equation 4 was chosen to capture key features of ALIVE, such as the presence of both interval-censoring and drop-outs as well as the competing risk of death. Equation 4 also allows us to explore sensitivity to different assumptions about needle sharers and non-sharers because the purpose of the analysis is to compare HIV incidence between these two groups. Lastly, we specified Equation 4 to be a scientifically interpretable low-dimensional function to facilitate elicitation from subject matter experts. Thus, for specifying a censoring bias function for other applications, one should consider study design features, scientific hypotheses, interpretability of parameters, and ease of elicitation.
2.3. Inference
2.3.1. Frequentist inference
Let pgj be the event probability during year j among those in group g. Frequentist estimation of pg = pg1, …, pg(M+1) is performed via the expectation-maximization algorithm [15] by replacing coarsened event times with their expected values given L, R, and Δ [4]. See Appendix I for details.
Probability estimates are used to estimate Fg(·), the cumulative incidence function for group g,
. Once estimates of p = {pg, g ∈ }, denoted p̂, and standard errors are obtained, statistics can be derived for testing the null hypothesis H0 : Fg(·) = Fg′(·), g = g′ ∀g, g′ ∈ using the delta method, including logrank (LR) and IWD tests.
Let LR = {LRg, g ∈ }t be a vector of length with gth component
, where dgj = ngp̂gj is the estimated number of seroconverts in group g during year j,
is the estimated number at risk in group g during year j, dj = Σg∈Gdgj, and nj = Σg∈Gngj. Thus LRg takes the form of the logrank test numerator. The variance of LR, ΣLR, is a × matrix estimated by Σ̂LR (see Appendix I). The test statistic is
, where
denotes generalized inverse of Σ̂LR. Under the null hypothesis,
has a χ2 distribution with G − 1 degrees of freedom. The generalized inverse is needed owing to the loss of one degree of freedom from estimating pj by dj/nj.
The IWD test was originally proposed to perform a one-sided hypothesis for G = 2 [10]. We generalize it to a two-sided test. The two-sample test with weight w(·), estimated by ŵ(·), has numerator
, where
. Let
denote the variance of IWD, estimated by
(Appendix I). The test statistic, Zobs = IWD/σ̂IWD, can be compared to a standard normal distribution [10]. When G ≥ 2, we modify the test by comparing F̂g(j), g∈ , to the estimated overall cumulative function: F̂(j) = Σg∈GngF̂g(j)/n. Let IWD = {IWDg, g ∈ }t be a vector of length G with gth component
. The variance of IWD is a G × G matrix, ΣIWD, estimated by Σ̂IWD (Appendix I). The test statistic,
, is distributed χ2 with G − 1 degrees of freedom under the null hypothesis due to estimation of F̂(j)
2.3.2. Bayesian inference
We specify a Dirichlet prior density for pg to obtain posterior cumulative incidence. Conjugate analysis cannot be performed, therefore we use a Markov Chain Monte Carlo (MCMC) algorithm described in Appendix II [4]. Simulated p's are then transformed into an interpretable one-dimensional quantity summarizing the difference between G cumulative incidence functions.
One proposed quantity is motivated by the logrank test. Let Nsim denote the Markov chain length. Let p(s) be the vector of simulated p at iteration s, and let LR(p(s)) denote the posterior logrank transformation, a vector of length G with gth component , where , and . Let ΣLR(p(s)) be a G × G matrix motivated by the variance of the logrank test numerator when the null hypothesis is true (Appendix II). The transformation involves calculating . The median of this posterior parameter transformation under the null hypothesis is the median of a χ2 distribution with G − 1 degrees of freedom, approximately μG = G − 1 − 2/3 + 4/[27(G − 1)] − 8/[729(G − 1)2]. The vector of logrank parameter transformations is denoted by . Let ω denote the observed data, where ωigj is the indicator that tj ∈ [Lig, Rig], i.e., that time tj is a possible event time for person i in group g. A posterior tail probability summarizing the degree of overlap between the observed distribution and expected distribtution under the null can be calculated by . For a visual representation, the can be plotted with a kernel, the distribution of the logrank test statistic when all G event-time distributions are equal. When G = 2, a transformation can be calculated by ZLR(p(s)) = LRg(p(s))/σLR(p(s)), where σLR(p(s)) is the standard deviation of LRg(p(s)). Let ZLR(p) = {ZLR(p(1)), ⋯, ZLR(p(Nsim))}; ZLR(p) can be plotted with a standard normal kernel, and the tail probability can be calculated as 2 [min{P (ZLR(p) ≥ 0 | ω), P (ZLR(p) ≤ 0 | ω)}].
The second quantity considered is motivated by the IWD test. Let IWD(p(s)) denote the posterior IWD transformation, a vector of length G with gth component , where , and w(s)(j) is a weight function. Let ΣIWD(p(s)) be a G × G matrix motivated by the variance of the IWD test numerator (Appendix II). The parameter transformation is . When G = 2, inference is performed like that for ZLR(p) using ZIWD(p) = {ZIWD(p(1)), ⋯, ZIWD(p(Nsim))}, where ZIWD(p(s)) = IWD2(p(s))/σIWD(p(s)), and σIWD(p(s)) is the same form as the standard deviation of IWD2(p(s)).
These posterior tail probabilities are interpreted differently from frequentist p-values. Instead of calculating the tail probability of a test statistic under H0 at the observed value, we calculate the posterior tail probability of a parameter transformation at its expected value when H0 is true. This tail probability summarizes the degree of overlap between the observed posterior parameter transformation distribution and that expected under H0, where tail probabilities of 1 and 0 denote perfect and no overlap, respectively. Comparing the posterior distribution to some reference posterior (in this case, the expected posterior under H0) to assess Bayesian hypotheses is thought to be more appropriate than inference by Bayes' factors for continuous parameters, because calculating Bayes' factors requires hypotheses to have strictly positive prior probabilities [16]. The reference posterior approach has the additional benefit of being simpler to perform than calculation Bayes' factors because it does not require running the MCMC multiple times.
3. Simulation Study
Simulations were performed for two-sample and G-sample logrank and IWD tests with G = 3 and = {1, 2, 3}, allowing left, interval, and right censoring, but no competing risks. Event times were simulated from a multinomial distribution using the continuation ratio logistic model with M = 4. Let ρij = P(Ti = j | Ti ≥ j, Zi) for j = 0, …, M + 1. The continuation ratio model for the three-sample simulation is logit
, j = 0, …, M, where β = {β1, β2} and Zi = {Zi1, Zi2}, where Z1 = I(g = 2) and Z2 = I(g = 3). Replacing
with β1Z1i results in the continuation ratio model used for the two-sample simulation study. For the three-group simulation study, groups two and three are assumed to have the same distribution (e.g., a control group and two exchangeable treatments), β1 = β2 = β. For each group, censoring intervals were simulated given T. The function q(·) and the distribution of T for group g do not identify P(Li = l, Ri = r | Zi, Ti) = Pg(Li = l, Ri = r | Ti). The number of free parameters in this distribution for each group equals (M + 2)(M + 1)/2, the number of intervals minus the number of event times. These parameters (interval probabilities) were fixed at values satisfying the constraints Pg(T = t) > Pg(T = t, L = l, R = r), for g ∈ . The strict inequality allows positive probability for each combination of l and r including t. The remaining M + 2 interval probabilities were identified from the constraints Σl≤rPg(L = l, R = r) = 1 and Σ{l,r}: l≤t≤rPg(T = t | L = l, R = r)Pg(L = l, R = r) = Pg(T = t).
True event times, T, were drawn given g, with β ∈ {0, 0.75} and θ = {−0.65, −0.55, −0.45, −0.15, −0.05}. Let ϕ = {ϕg: g ∈ } be the vector of group-specific censoring bias parameters for the censoring bias function qg(t, l, r) = ϕg(t−l)/(r−l). The true censoring bias parameters were combinations of {−log(2), 0, log(2)}. In this study, ϕ2 = ϕ3.
The empirical sizes of the tests were estimated assuming β = 0. Empirical power was estimated for the alternative hypothesis H1 : Fg(·) ≠ Fg′ (·) when β = 0.75, where g = 1 and g′ = 2 for the two-sample test and where g ≠ g′ for some g, g′ ∈ for the G-sample test. We chose ng = 100, 200, 500 and performed 1000 simulations for each specification. Simulations were also performed on uncensored data. Values of the true parameters were chosen to produce between 86% and 97% censoring (i.e., P(L ≠ R)), depending on ϕ and β. For censored data, two weight functions were used for the IWD test, ŵ(j) = 1, and ŵ(j) = w*(j), where
, and K̂g(j) is the sample proportion of individuals in group g with known serostatus in year j [10]. When data are uncensored, w*(j) simplifies to 1.
Simulation test results are shown in Tables I and II. The first row of each sample size-specific study shows results for uncensored data. The first column shows the true ϕ that generated the censoring intervals for simulations with censoring. The second column shows the assumed ϕ for the model with censored data, either CAR or the true ϕ. Both tables show results for six tests: the IWD test with w = 1 and w = w* and the logrank test, all for G = 2 and G = 3. The empirical size results in Table I show the tests perform well with no censoring, and the performance improves as the sample size increases. When ϕ are correctly specified, or when the bias for both parameters is of equal magnitude in the same direction (e.g., true ϕ are {−log(2), −log(2)}, but CAR is assumed), the tests perform well. Empirical size differs most from nominal size when ϕ are biased in different directions (e.g., true ϕ are {−log(2), log(2)}, but CAR is assumed). No single test performs uniformly better than the others, however, the logrank test tends to be more anticonservative than the IWD test for smaller samples sizes, even with no censoring. When the data are censored, the three-group IWD test produces the most conservative (n = 500, true ϕ = {−log(2), −log(2)}) and most anticonservative (n = 200, true ϕ = {−log(2), 0}) results. Empirical power is shown in Table II. In general, the test with weight w* is more powerful than the analogous test with w = 1. With no censoring, the logrank test is more powerful than the IWD test. However, with censoring in smaller sample sizes, the IWD test tends to be more powerful. In larger sample sizes, the difference is negligible. The true underlying distribution has hazard ratio 2.12 (exp{0.75}) comparing groups 2 and 3 to group 1. When groups 2 and 3 are biased to have greater (lower) hazards relative to group 1, power is increased (decreased).
Table I.
Simulation results: empirical size of integrated weighted difference (IWD) and logrank (LR) tests with nominal size 0.05. 1000 iterations, M = 4, n1 = n2 = n3 = n., ϕ2 = ϕ3 when G = 3, no censoring or 86% censoring.
| n. | True {ϕ1, ϕ2} | Modeled ϕ | 2 groups w = 1 |
2 groups w* |
2 groups LR |
3 groups w = 1 |
3 groups w = w* |
3 groups LR |
|---|---|---|---|---|---|---|---|---|
| 100 | (no censoring) | 0.054 | 0.061 | 0.055 | 0.064 | |||
| −log(2), −log(2) | Truth | 0.043 | 0.038 | 0.048 | 0.043 | 0.042 | 0.059 | |
| CAR | 0.055 | 0.056 | 0.066 | 0.063 | 0.060 | 0.075 | ||
| −log(2), 0 | Truth | 0.050 | 0.058 | 0.048 | 0.052 | 0.052 | 0.057 | |
| CAR | 0.129 | 0.127 | 0.131 | 0.128 | 0.131 | 0.137 | ||
| −log(2), log(2) | Truth | 0.058 | 0.059 | 0.062 | 0.050 | 0.054 | 0.061 | |
| CAR | 0.311 | 0.292 | 0.297 | 0.311 | 0.306 | 0.304 | ||
| 200 | (no censoring) | 0.054 | 0.055 | 0.055 | 0.059 | |||
| −log(2), −log(2) | Truth | 0.044 | 0.044 | 0.036 | 0.049 | 0.054 | 0.051 | |
| CAR | 0.060 | 0.059 | 0.059 | 0.054 | 0.058 | 0.054 | ||
| −log(2), 0 | Truth | 0.055 | 0.052 | 0.051 | 0.072 | 0.071 | 0.066 | |
| CAR | 0.208 | 0.201 | 0.209 | 0.197 | 0.198 | 0.204 | ||
| −log(2), log(2) | Truth | 0.053 | 0.052 | 0.058 | 0.052 | 0.055 | 0.050 | |
| CAR | 0.500 | 0.490 | 0.464 | 0.528 | 0.518 | 0.511 | ||
| 500 | (no censoring) | 0.050 | 0.049 | 0.052 | 0.054 | |||
| −log(2), −log(2) | Truth | 0.054 | 0.052 | 0.053 | 0.062 | 0.062 | 0.061 | |
| CAR | 0.045 | 0.043 | 0.050 | 0.061 | 0.059 | 0.059 | ||
| −log(2), 0 | Truth | 0.054 | 0.056 | 0.056 | 0.059 | 0.060 | 0.063 | |
| CAR | 0.389 | 0.377 | 0.366 | 0.364 | 0.364 | 0.365 | ||
| −log(2), log(2) | Truth | 0.047 | 0.048 | 0.049 | 0.034 | 0.035 | 0.040 | |
| CAR | 0.891 | 0.880 | 0.863 | 0.917 | 0.909 | 0.894 |
Table II.
Simulation results: empirical power of IWD and LR tests with size 0.05. 1000 iterations, β = 0.75 (hazard ratio = 2.12), M = 4, n1 = n2 = n3 = n, ϕ2 = ϕ3 when G = 3, 97% censoring when Z = 1 and 86% censoring when Z = 0 or no censoring.
| n. | True {ϕ1, ϕ2} | Modeled ϕ | 2 groups w = 1 |
2 groups w* |
2 groups LR |
3 groups w = 1 |
3 groups w* |
3 groups LR |
|---|---|---|---|---|---|---|---|---|
| 100 | (no censoring) | 0.966 | 0.972 | 0.976 | 0.982 | |||
| −log(2), −log(2) | Truth | 0.860 | 0.860 | 0.850 | 0.909 | 0.900 | 0.907 | |
| CAR | 0.638 | 0.668 | 0.601 | 0.697 | 0.723 | 0.684 | ||
| −log(2), 0 | Truth | 0.810 | 0.837 | 0.785 | 0.867 | 0.884 | 0.865 | |
| CAR | 0.929 | 0.939 | 0.912 | 0.970 | 0.976 | 0.963 | ||
| −log(2), log(2) | Truth | 0.756 | 0.784 | 0.750 | 0.820 | 0.841 | 0.823 | |
| CAR | 1.000 | 1.000 | 0.999 | 1.000 | 1.000 | 1.000 | ||
| 200 | (no censoring) | 0.999 | 0.999 | 1.000 | 1.000 | |||
| −log(2), −log(2) | Truth | 0.993 | 0.989 | 0.991 | 0.998 | 0.998 | 0.999 | |
| CAR | 0.916 | 0.931 | 0.893 | 0.953 | 0.968 | 0.941 | ||
| −log(2), 0 | Truth | 0.973 | 0.979 | 0.971 | 0.992 | 0.994 | 0.988 | |
| CAR | 0.996 | 0.997 | 0.996 | 0.999 | 0.999 | 0.997 | ||
| −log(2), log(2) | Truth | 0.961 | 0.973 | 0.959 | 0.982 | 0.991 | 0.985 | |
| CAR | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | ||
| 500 | (no censoring) | 1.000 | 1.000 | 1.000 | 1.000 | |||
| −log(2), −log(2) | Truth | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | |
| CAR | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | ||
| −log(2), 0 | Truth | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | |
| CAR | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | ||
| −log(2), log(2) | Truth | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | |
| CAR | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
We repeated the simulation study with n=500 and 1000 iterations by including a competing risk. In this case, the number of free parameters is (M + 2)2 because there are twice as many intervals with a competing risk than without a competing risk i.e., specify probabilities for intervals L = l, R = r, Δ = 0 and L = l, R = r, Δ = 1 rather than only L = l, R = r. Interval probabilities were fixed at values satisfying the constraints Pg(T = t) > Pg(T = t, L = l, R = r, Δ = δ), for g ∈ . Analogous to the case with no competing risk, the strict inequality allows positive probability for each combination of l, r, δ including t. Similarly, the remaining M + 2 interval probabilities were identified from the constraints Σδ∈{0,1}Σl≤rPg(L = l, R = r, Δ = δ) = 1 and Σδ∈{0,1} Σ{l,r}:t∈A(l,r,δ) Pg(T = t | L = l, R = r, Δ = δ)Pg(L = l, R = r, Δ = δ) = Pg(T = t). The empirical type I error from this simulation ranged from 0.048 (ϕ = {−log(2), 0}, IWD test with w = w* and G = 2) to 0.062 (ϕ = {−log(2), −log(2)}, IWD test with w = w* and G = 2; ϕ = {−log(2), 0}, IWD test with w = 1 and logrank test, both with G = 3). The empirical type I errors were, on average, closer to the nominal type I error than those in Table I obtained without the competing risk. Trends in empirical power with the competing risk were similar to those without the competing risk in Table II (not shown). The empirical distribution of the test statistic is graphically compared with the chi-square distribution in Figure 1. Regardless of ϕ, number of groups, and presence of a competing risk, Figure 1 shows that the distributions of all three test statistics (ID, IWD, and LR) approximate a chi-square distribution well.
Figure 1.

Simulation study results, Ng = 500, Nsim = 1000. Empirical distribution of integrated weighted difference (ID, w = 1; IWD, (w = w*) and logrank (LR) tests compared to a chi-square distribution (two groups, df=1; three groups, df=2), with and without a competing risk.
4. Example Data Analysis: ALIVE
We apply our proposed inference methods to ALIVE to compare ten-year cumulative incidence functions of seroconversion between those who self-reported needle sharing at enrollment and those who did not. Among the 2,205 ALIVE participants with complete needle-sharing information, 1,527 reported sharing needles, and 678 did not. Among those reporting sharing needles, 190, 1,135, 144, and 58 participants were censored by death, right-censored by drop-out or end of study, interval censored, and exactly observed, respectively. The respective numbers among those reporting not sharing needles were 73, 522, 54, and 29. Among needle sharers, 242 (15.8%) total died within ten years of enrollment. Similarly, 100 (14.7%) total non-sharers died during the study. The relationship between needle-sharing and seroconversion may be driven by differential death rates between groups. However, logrank test results (p = 0.52) do not support this hypothesis. Death as a primary endpoint was not addressed in the sensitivity analysis.
4.1. Elicitation and sensitivity analysis
To elicit values of ϕ in equation (4), two ALIVE investigators were seperately interviewed to obtain ranges of the parameters and hyperparameters found in Table III (columns 4-5, and 6-7, respectively). Prior distributions of p and ϕ were elicited from an external AIDS epidemiologist and an ALIVE investigator, respectively. The AIDS epidemiologist was interviewed regarding the expected seroconversion time distribution and the weight of expert opinion relative to ALIVE data. An expert unaffiliated with ALIVE was consulted to obtain opinion prior to ALIVE. The elicited weight of prior opinion was 10 percent of final results (ALIVE data weighted 90 percent). Prior information about seroconversion probabilities was not specific to needle-sharing status to reflect the “null” hypothesis of equal cumulative incidence functions. Elicited ranges of exp{ϕ} were centered and scaled parameters ranged 0 to 1 with assumed beta distributions. Plots of beta distributions were used to elicit the beta hyperparameters from an ALIVE investigator. To accommodate correlations between the parameters, we consider the normal approximation to the beta distribution. Using scatterplots of pairwise correlations between parameters to elicit covariances, the ALIVE investigator's prior variance-covariance matrix involved positive correlations for several combinations of needle-sharing group and censoring type:
Table III.
Elicited range of exp (ϕ) and hyperparameters for beta distributions used in Bayesian analyses.
| (1) | (2) | (3) | (4) | (5) | (6) | (7) |
|---|---|---|---|---|---|---|
| Needle-sharing | Censoring | ϕ | Range of exp (ϕ) | Shape | Scale | |
| Yes | Interval-censored | ϕs1 | 1.75−1 | 2.75 | 2.00 | 7.75 |
| Dropped out | ϕs2 | 1.50 | 3.00 | 5.25 | 2.75 | |
| Dead | ϕs3 | 2.00 | 2.50 | 2.00 | 1.00 | |
| No | Interval-censored | ϕn1 | 1.15−1 | 2.50 | 2.00 | 9.50 |
| Dropped out | ϕn2 | 1.75 | 2.50 | 3.75 | 2.00 | |
| Dead | ϕn3 | 2.00 | 2.50 | 2.00 | 1.00 | |
Additional details about the elicitation process and rationale for ranges of parameters and their hyperparameters can be found elsewhere [17].
4.2. Frequentist results
Frequentist inference was performed using estimates assuming CAR and combinations of minimum and maximum elicited values of ϕ [4]. For each combination, needle-sharing specific seroconversion probabilities were estimated, and logrank and IWD tests were performed with weights w(j) = 1 and w*(j).
Table IV shows estimated needle-sharing specific one-, five-, and ten-year cumulative incidence and 95 percent confidence intervals (using the complementary log-log transformation), and p-values for three values of ϕ: ϕ = 0 (CAR assumed), {max(ϕn), min(ϕs)}, and {min(ϕn), max(ϕs)}. When CAR is assumed, estimated probabilities are similar across groups, corroborated by large p-values for IWD and logrank tests. The minimum p-values were produced when needle sharers are assumed to seroconvert stochastically early in their observed sets (minimum ϕs) and non-sharers are assumed to seroconvert stochastically late in their observed sets (maximum ϕn) according to elicited ranges for ϕ. Estimated cumulative incidence is lower for non-sharers than for needle-sharers under this assumption. Similarly, when the opposite assumption is made (minimum ϕn, maximum ϕs), estimated cumulative incidence was higher for non-sharers than for needle sharers. Test results were most sensitive to values of ϕs2 (not shown), because needle-sharing drop-outs alive at the end of year ten is the largest needle-sharing group-by-censoring type category in ALIVE, and experts expressed the most uncertainty about them.
Table IV.
ALIVE frequentist results. Estimated cumulative incidence (F̂) and 95% confidence intervals (CI) at years 1, 5, and 10; logrank and integrated weighted difference (IWD) p-values across censoring assumptions.
| IWD |
||||||||
|---|---|---|---|---|---|---|---|---|
| Non Sharers |
Needle Sharers |
Logrank | w=1 | w = w* | ||||
| Assumption | Year | F̂ | 95% CI | F̂ | 95% CI | p-value | p-value | p-value |
| CAR | 1 | 0.07 | 0.05, 0.10 | 0.05 | 0.04, 0.07 | 0.68 | 0.86 | 0.87 |
| 5 | 0.16 | 0.13, 0.20 | 0.18 | 0.15, 0.20 | ||||
| 10 | 0.23 | 0.19, 0.28 | 0.24 | 0.22, 0.28 | ||||
| Max ϕn, Min ϕs | 1 | 0.05 | 0.03, 0.07 | 0.05 | 0.03, 0.06 | 0.11 | 0.17 | 0.18 |
| 5 | 0.11 | 0.09, 0.14 | 0.14 | 0.12, 0.17 | ||||
| 10 | 0.17 | 0.14, 0.21 | 0.21 | 0.18, 0.24 | ||||
| Min ϕn, Max ϕs | 1 | 0.06 | 0.04, 0.08 | 0.03 | 0.02, 0.05 | 0.68 | 0.53 | 0.52 |
| 5 | 0.12 | 0.10, 0.16 | 0.12 | 0.10, 0.14 | ||||
| 10 | 0.19 | 0.15, 0.23 | 0.18 | 0.16, 0.20 | ||||
4.3. Bayesian results
Prior beliefs about seroconversion probabilities and censoring bias parameters were converted into Dirichlet and multivariate normal hyperparameters, respectively. First, exp{ϕ} were centered and scaled to have range [0, 1] and marginal beta distributions, inducing means and variances used to make normal approximations to beta densities. Elicited correlation coefficients induced an approximate multivariate normal joint distribution for centered and scaled exp{ϕ}, and the logit transformation was used for simulating from a multivariate normal distribution.
Bayesian analysis was first performed for fixed ϕ. We examined results assuming CAR and the same two extreme specifications used in frequentist analysis, {max(ϕn), min(ϕs)} and {min(ϕn), max(ϕs)}. MCMC was run for 500 burn-in and 5000 additional iterations. A previously published diagnostic scheme was used for all Bayesian analyses [18]. Needle-sharing specific mean posterior one-, five-, and ten-year cumulative incidence, 95 precent credible intervals, and parameter transfomation tail probabilities are shown in Table V. The IWD parameter transformation was only calculated for w = 1 because, given imputed complete data, w* = 1. Mean posterior cumulative incidence was higher than analogous results from frequentist analyses due to shrinkage to the prior, which suggested more accelerated seroconversion than estimates from data alone. Also, credible intervals are slightly narrower than analogous confidence intervals, especially for the ten-year cumulative incidence, due to additional information from the prior and many drop outs. The tail probabilities were similar to analogous frequentist p-values.
Table V.
ALIVE Bayesian results. Mean posterior cumulative incidence (F̂) and 95% credible intervals (CI) at years 1, 5, and 10; logrank (LR) and integrated weighted difference (IWD) with w = 1 tail probabilities (tail prob.) across censoring assumptions.
| Assumption | Year | Non Sharers | Needle Sharers | LR | IWD | ||
|---|---|---|---|---|---|---|---|
| F̂ | 95% CI | F̂ | 95% CI | tail prob. | tail prob. | ||
| CAR | 1 | 0.07 | 0.05, 0.10 | 0.06 | 0.04, 0.07 | 0.74 | 0.84 |
| 5 | 0.18 | 0.15, 0.22 | 0.19 | 0.16, 0.22 | |||
| 10 | 0.26 | 0.22, 0.30 | 0.26 | 0.24, 0.29 | |||
| Max ϕn, Min ϕs | 1 | 0.05 | 0.03, 0.07 | 0.05 | 0.06, 0.04 | 0.12 | 0.13 |
| 5 | 0.13 | 0.11, 0.16 | 0.16 | 0.14, 0.18 | |||
| 10 | 0.20 | 0.16, 0.23 | 0.23 | 0.20, 0.26 | |||
| Min ϕn, Max ϕs | 1 | 0.06 | 0.04, 0.08 | 0.04 | 0.03, 0.05 | 0.63 | 0.47 |
| 5 | 0.14 | 0.12, 0.16 | 0.14 | 0.12, 0.16 | |||
| 10 | 0.21 | 0.18, 0.25 | 0.20 | 0.18, 0.22 | |||
| Random ϕ | 1 | 0.05 | 0.04, 0.07 | 0.04 | 0.03, 0.06 | 0.74 | 0.82 |
| 5 | 0.14 | 0.11, 0.17 | 0.14 | 0.12, 0.16 | |||
| 10 | 0.20 | 0.17, 0.24 | 0.21 | 0.18, 0.23 | |||
Fully Bayesian analysis was then performed, averaging over the posterior distribution of ϕ. MCMC was burned in for 1000 iterations and run for 10000 more. Metropolis-Hastings [19] acceptance was 64 and 86 percent for needle sharers and non-sharers, respectively. Prior and posterior densities for exp {ϕ} were approximately equal, reflecting no information about these parameters in the data (not shown). Prior and posterior densities for one-, five-, and ten-year cumulative incidence are reported in Figure 2. Posterior densities are tighter than priors, due to small weight given to elicited information relative to the data. Cumulative incidences are between those obtained using extreme elicited censoring bias parameter values, and tail probabilities were similar to those assuming CAR (Table V). Figure 3 shows that the LR and IWD posterior parameter transformations differ little from a standard normal kernel, suggesting seroconversion cumulative incidence functions do not differ across needle-sharing status. Posterior mean cumulative incidence and tail probabilities in Table V corroborate this conclusion.
Figure 2.

ALIVE Bayesian results. Posterior (solid line, needle sharers; dashed line, non-sharers) and prior (dotted line) densities of one-, five-, and ten-year cumulative incidence.
Figure 3.

ALIVE Bayesian Inference. Posterior density for ZLR(p) (solid line) and ZIWD(p) (dashed line) parameter transformations with standard normal kernel (dotted line). Mean posterior (solid line, needle sharers; dashed line, non-sharers) and prior (dotted line) cumulative incidence.
5. Discussion
We developed two approaches for comparing cumulative incidence functions for informatively coarsened event-time data using estimates derived from previously published methods [4] and applied them to ALIVE. We accounted for the competing risk of death in the analysis, extended two test statistics to accommodate informatively coarsened data, and proposed a novel approach for Bayesian hypothesis assessment that is interpretable and easily calculated after performing a single MCMC run. We also extended the IWD test to be two sided and to accommodate more than two groups. We showed that these tests perform well when the censoring mechanism is correctly specified. The simulation study was performed using a regression model appropriate for discrete event-time data. Future research includes estimation and variable selection using this model in the presence of informative coarsening.
CAR-based analyses of ALIVE data suggest baseline needle sharing is not associated with time to seroconversion. These conclusions are robust to elicited assumptions about the visit-compliance process. CAR-based and random-ϕ posterior cumulative incidence varied little. Had our sensitivity analysis results produced conflicting conclusions, the test results could be displayed using contour plots like in [20] to allow readers to see conclusions as a function of ϕ. Needle sharing status poses a short term risk for HIV and may change over time, thus future research involves developing methods to accommodate time-varying risk factors.
The proposed inference methods are beneficial in that they facilitate a sensitivity analysis across assumptions about the censoring mechanism using results from a previously published estimation method. The methods are generalizable to any scientific application where the goal is to compare cumulative incidence across groups in the presence of informative censoring. However, the form of the censoring bias function should differ as appropriate, and interpretations of the censoring bias parameters would be context specific. The approaches proposed here are preferable to other ad hoc sensitivity analyses involving single imputation of interval endpoints or interval midpoint imputation, because these approaches underestimate standard errors [21]. Also, interval endpoint imputation involves extreme assumptions that may not be scientifically plausible. In particular, imputing L and R for T is equivalent to specifying ϕ = −∞ and ϕ = ∞, respectively.
Despite the benefits, these methods have limitations. In particular, results may be sensitive to distributional assumptions. For example, the correlation structure of Dirichlet priors does not take advantage of time ordering of visits. Also, the proposed methods are limited to small numbers of groups. Lastly, utilizing these methods requires a potentially challenging elicitation process and difficult choice of censoring bias function.
Despite these challenges, assumptions must be made to analyze coarsened data, because the coarsening process is not identifiable from observed data [22], thus statistical methods that can incorporate ranges of assumptions are needed. Our methods fulfill this need by allowing investigators to explicitly communicate their assumptions and perform a sensitivity analysis to explore the impact of these assumptions on analysis results, facilitating more honest reporting of results from scientific studies.
Acknowledgments
This research was supported by National Institute of Aging grant T32 AG00247, National Institute of Health grants 1-R29-GM48704-04, 5R01A132475, R01CA74112, 1-R01-MH56639-01A1, and 1-R01-DA10184-01A2, and National Institute on Drug Abuse grant DA 04334. The authors thank Drs. Tom Louis and Mike Daniels, and Samuel Friedman for helpful discussions.
Contract/grant sponsor: National Institute of Aging; contract/grant number: T32-AG00247
Contract/grant sponsor: National Institute of Health; contract/grant number: 1-R29-GM48704-04, 5R01A132475, R01CA74112, 1-R01-MH56639-01A1, and 1- R01-DA10184-01A2
6. Appendix I: Frequentist Methods
6.1. Estimation
The complete-data likelihood is , where Iigj is the event indicator at time j for person i in group g. Initial estimates of pg are used to evaluate the expected complete-data log likelihood, given observed data (E-step). The E-step at iteration s is
where λg are Lagrange multipliers, and
Q(p; p(s−1)) is maximized (M-step) to obtain updated estimates of pg. The M-step results in a reweighted version of Turnbull's self-consistency equation [23] for each group g, g ∈ :
Estimated standard errors for probabilities in each group can be calculated using Louis's [24] method for estimating the observed information matrix.
To derive the observed information matrix for group g, Ipg, let denote the complete-data information matrix, and let denote the missing-data information matrix, where . Using Louis's method, , where log[Li(pg)] is the complete-data log likelihood and ωig is the observed information for individual i in group g. Also, where , the complete-data score equation for individual i in group g. Then, is a (M + 1) × (M +1) diagonal matrix with jth diagonal element . Next, is a (M + 1) × (M + 1) matrix with j, k element . The variance-covariance matrix for p̂g is then , estimated by plugging p̂g into and .
6.2. Test Statistics
To calculate Σ̂IWD, let be the estimated variance of , where
| (5) |
and is the estimated covariance between p̂gj′ and p̂gk′, the j′, k′ entry of , the inverse estimated observed information matrix for group g.
Let f̂g = ng/n. Then, the g,h (g h ∈ ) entry of Σ̂IWD equals
| (6) |
To calculate Σ̂LR, we define to be the block-diagonal G(M + 1) × G(M + 1) inverse estimated observed information matrix. Then, using the multivariate delta method,
where is a G × G(M + 1) matrix with g, (h − 1)(M + 1) + j′ entry, i.e., , equal to
7. Appendix II: Bayesian Methods
7.1. Estimation
The Bayesian algorithm is a G-group version of that previously described [4]. Let Ig be complete data and ωg be observed data (e.g., L, R, and Δ) for all individuals in group g.
For the Dirichlet prior, let Bg = {bg1, ⋯, bg(M+1)} be a base measure defined on E for those in group g, the prior mean of pg. A precision parameter, α*, describes concentration of the distribution around Bg, where elements of Bg sum to 1. Let αgj = α*bgj, for j = 1, …, M +1. The Dirichlet density is given by:
where pg1, ⋯, pg(M+1) ≥ 0; ; the αg = {αg1,…, αg(M+1)} are all positive; and αgj's are interpreted as ‘prior counts’ of seroconverts during year j in group g. Data are incomplete, thus we use the Gibbs sampler [25] with data augmentation [26] and a Metropolis-Hastings step [19].
First, starting values are chosen for censoring bias parameters, ϕ(0), and event-time probabilities, p(0). The Gibbs sampler involves three simulation steps per iteration. For iteration s = 1, …, Nsim simulate:
from ,
from ,
ϕ(s) from ,
where p(·) denotes the density.
The vector of imputed event indicators, , for person i in group g are simulated from a truncated multinomial distribution,
The are aggregated into group-by-time frequencies. Let denote the simulated event count during interval j among those in group g at iteration S, and . Conditional on , pg is independent of ϕ and ωg. Therefore, can be simulated in Step 2 from , a Dirichlet distribution with hyperparameters . Metropolis-Hastings is used in Step 3 to simulate ϕ. Let I(s) denote the iteration s vector of simulated event indicators and ω denote observed data across all groups. The candidate, ϕ*, is simulated from the jumping distribution at iteration s, Js(ϕ* | ϕ(s−1)), and is accepted with probability min(1,rMH), where
In this application, Js(ϕ* | ϕ(s−1)) was a multivariate normal distribution.
7.2. Posterior Parameter Transformations
To calculate the logrank parameter transformation, let
For the IWD parameter transformation, elements of ΣIWD(p(s)) take the same form as Equation (6), with /nj replacing in Equation (5).
Footnotes
Prepared using simauth.cls
References
- 1.Heitjan DF, Rubin DB. Ignorability and coarse data. The Annals of Statistics. 1991;19:2244–2253. [Google Scholar]
- 2.Heitjan DF. Ignorability and coarse data: Some biomedical examples. Biometrics. 1993;49:1099–1109. [PubMed] [Google Scholar]
- 3.Gill RD, van der Laan MJ, Robins JM. Coarsening at random: characterizations, conjectures and counter-examples. In: Lin DY, Fleming TR, editors. State of the Art in Survival Analysis. Springer; New York: 1997. pp. 255–294. [Google Scholar]
- 4.Shardell M, Scharfstein DO, Bozzette SA. Survival curve estimation for informatively coarsened discrete event-time data. Statistics in Medicine. 2007;26:2184–202. doi: 10.1002/sim.2697. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Heitjan DF. A simple local sensitivity analysis tool for nonignorable coarsening: application to dependent censoring. Biometrics. 2006;62:1260–1268. doi: 10.1111/j.1541-0420.2006.00580.x. [DOI] [PubMed] [Google Scholar]
- 6.Betensky RA, Finkelstein DM. Testing for dependence between failure time and visit compliance with interval-censored data. Biometrics. 2002;58:58–63. doi: 10.1111/j.0006-341x.2002.00058.x. [DOI] [PubMed] [Google Scholar]
- 7.Park Y, Tian L, Wei LJ. One- and two-sample nonparametric inference procedures in the presence of a mixture of independent and dependent censoring. Biostatistics. 2006;7:252–67. doi: 10.1093/biostatistics/kxj005. [DOI] [PubMed] [Google Scholar]
- 8.Sun J. Statistical analysis of interval-censored failure time data. Springer; New York: 2006. [Google Scholar]
- 9.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consieration. Cancer Chemother Rep. 1966;50:163–70. [PubMed] [Google Scholar]
- 10.Petroni GR, Wolfe RA. A two-sample test for stochastic ordering with interval-censored data. Biometrics. 1994;50:77–87. [PubMed] [Google Scholar]
- 11.Vlahov D, Anthony JC, Munoz A, et al. The ALIVE study, a longitudinal study of HIV-1 infection in intravenous drug users: description of methods and characteristics of participants. NIDA Res Monogr. 1991;109:75–100. [PubMed] [Google Scholar]
- 12.Strathdee SA, Galai N, Safaiean M, et al. Sex differences in risk factors for hiv seroconversion among injection drug users: a 10-year perspective. Arch Intern Med. 2001;161:1281–8. doi: 10.1001/archinte.161.10.1281. [DOI] [PubMed] [Google Scholar]
- 13.Nelson KE, Galai N, Safaiean M, et al. Temporal trends in the incidence of human immunodeficiency virus infection and risk behavior among injection drug users in Baltimore, Maryland, 1988-1998. Am J Epidemiol. 2002;56:641–53. doi: 10.1093/aje/kwf086. [DOI] [PubMed] [Google Scholar]
- 14.Barndorff-Nielsen OE, Cox DR. Asymptotic techniques for use in statistics. Chapman & Hall; London: 1989. [Google Scholar]
- 15.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. (B).Journal of the Royal Statistical Society. 1977;39:1–22. [Google Scholar]
- 16.Bernardo JM, Rueda R. Bayesian hypothesis testing: A reference approach. International Statistical Review. 2002;70:351–72. [Google Scholar]
- 17.Shardell M, Scharfstein DO, Vlahov D, Galai N. Inference for survival curves with informatively coarsened discrete event-time data: application to ALIVE. Johns Hopkins University, Dept of Biostatistics Working Papers Working Paper 150. 2007 http://www.bepress.com/jhubiostat/paper150.
- 18.Cowles KP, Carlin BP. Markov chain Monte Carlo convergence diagnostics: A comparative review. Journal of the American Statistical Association. 1996;91:883–904. [Google Scholar]
- 19.Hastings WK. Monte Carlo sampling methods using Markov chains and their applications. Biometrika. 1970;57:97–109. [Google Scholar]
- 20.Scharfstein D, Robins JM, Eddings W, Rotnitzky A. Inference in randomized studies with informative censoring and discrete time-to-event endpoints. Biometrics. 2001;57:404–413. doi: 10.1111/j.0006-341x.2001.00404.x. [DOI] [PubMed] [Google Scholar]
- 21.Rubin DB. Multiple Imputation for Nonresponse in Surveys. Wiley; New York: 1987. [Google Scholar]
- 22.Kadane JB. Subjective Bayesian analysis for surveys with missing data. The Statistician. 1993;42:415–426. [Google Scholar]
- 23.Turnbull BW. The empirical distribution function with arbitrarily grouped, censored and truncated data. (B).Journal of the Royal Statistical Society. 1976;38:290–295. [Google Scholar]
- 24.Louis TA. Finding the observed information matrix when using the EM algorithm. (B).Journal of the Royal Statistical Society. 1982;44:226–233. [Google Scholar]
- 25.Geman S, Geman D. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Transactions in Pattern Analysis and Machine Intelligence. 1984;6:721–741. doi: 10.1109/tpami.1984.4767596. [DOI] [PubMed] [Google Scholar]
- 26.Tanner MA, Wong WH. The calculation of posterior distributions by data augmentation. Journal of the American Statistical Association. 1987;82:528–540. [Google Scholar]