

Abstract
Nitrones have the general chemical formula X-CH=NO-Y. They were first used to trap free radicals in chemical systems and then subsequently in biochemical systems. More recently several nitrones including PBN (α-phenyl-tert-butylnitrone) have been shown to have potent biological activity in many experimental animal models. Many diseases of aging including stroke, cancer development, Parkinson’s disease and Alzheimer’s disease are known to have enhanced levels of free radicals and oxidative stress. Some derivatives of PBN are significantly more potent than PBN and have undergone extensive commercial development in stroke. Recent research has shown that PBN-related nitrones also have anti-cancer activity in several experimental cancer models and have potential as therapeutics in some cancers. Also in recent observations nitrones have been shown to act synergistically in combination with antioxidants in the prevention of acute acoustic noise induced hearing loss. The mechanistic basis of the potent biological activity of PBN-related nitrones is not known. Even though PBN-related nitrones do decrease oxidative stress and oxidative damage, their potent biological anti-inflammatory activity and their ability to alter cellular signaling processes can not readily be explained by conventional notions of free radical trapping biochemistry. This review is focused on our observations and others where the use of selected nitrones as novel therapeutics have been evaluated in experimental models in the context of free radical biochemical and cellular processes considered important in pathologic conditions and age-related diseases.
Keywords: Nitrones, Stroke, Glioma, Hepatocellular Carcinoma, Hearing Loss, Acoustical Trauma
Introduction
Scientific advancements regarding our understanding of free radicals in biological systems, their importance in disease processes and now the possibility of using novel therapeutics that act by influencing free radical processes has changed dramatically over the last several years. The possibility that free radicals actually may exist in biological systems was only taken seriously after radiation chemists had shown: A) that oxygen free radicals were formed by ionizing radiation and B) that ionizing radiation also causes cancer. These facts were established over 50 years ago and many discoveries since then have occurred which have made this area an important field of research. Important findings include the following demonstrations: A) that biological systems actively produce reactive oxygen species (ROS) and reactive nitric oxide species (RNS), B) that biological systems have enzymatic systems specifically to degrade these reactive species, C) that specific oxidation products are formed when biological molecules react with ROS and RNS, D) that cellular stress-related processes are communicated using ROS and RNS as signaling agents and E) that ROS and RNS are agents that play an important role in many normal processes as well as pathologic disease progression.
Following this over arching massive growth in our understanding of ROS and RNS in biological systems, an important research area in novel therapeutics has arisen. Namely that potential novel therapeutics have been discovered which act by altering the course of disease progression by acting upon ROS and RNS mediated processes. We have spent considerable effort in exploring this area mostly focused on the use of nitrones in age-related diseases. In this effort much has been learned but much more is yet to be discovered. This review is presented as an aid to enhance future effort in the pursuit of goals along this path of research.
Nitrones – From Spin Traps in Free Radical Chemistry to Experimental Animals
The nitrone chemical structure in its simplest form can be represented as X-CH=NO-Y. Nitrones began to be used in analytical chemistry applications in the late 1960s. Nitrones will react with and “trap” and stabilize free radical intermediates (Figure 1). Many chemical reactions have free radical intermediates. Since many of these intermediates exist for only a brief time they are therefore very difficult to characterize and study. Chemists showed in 1967 that some free radicals will react with nitrones to produce nitroxide free radicals [1]. The nitrone trapping of a free radical intermediate is represented in the following simple reaction where the free radical intermediate (R•) is trapped by a nitrone: X-CH=NO-Y+R• → X-CHR-NO•-Y. Janzen and Blackburn advanced the field significantly and utilized nitrones to trap free radicals and also coined the term “spin trap” for trapping free radical intermediates [2]. The reaction of the free radical species with a nitrone yields a product termed the spin-adduct (Figure 1). The nitroxyl free radical spin-adduct is usually much more stable than the free radical therefore making it possible in principle to characterize the original free radical trapped using electron paramagnetic resonance methods [3]. Shortly after beginning the use of nitrones to trap free radicals in analytical chemistry, scientists then began to use nitrones to trap free radicals in biochemical systems with reports first appearing in 1975 [4] and 1976 [5].
Figure 1.

Chemical structures of nitrones, their reaction to trap free radicals and form spin adducts and the chemical structure of PBN.
After the original early reports in the mid 1970s demonstrated the potential for the use of nitrone spin traps to detect and characterize free radical intermediates in subcellular biological systems, increasing research was conducted using nitrones in biochemical research for the next 10–20 years. In this regard excellent research came from several major laboratories including, Mason’s laboratory [6–10], Piette’s laboratory [11–14], Kalyanaraman’s laboratory [15–18], our laboratory [19–24], as well as many others. McCay’s laboratory from the very early days began to focus more on the use of nitrones to trap free radicals in experimental animals. For example, trapped radicals were demonstrated in rats administered CCl4 [25,26], experimental animals given ethanol [27], as well as during exposure to gamma radiation [28]. Bolli, Janzen and McCay studied the free radicals produced in “stunned” hearts of rats [29–33] and McCay, Janzen and Bray made seminal observations on the pharmacokinetics and metabolism of the commonly used spin-trap PBN i.e. α-phenyl-tert-butyl nitrone [33–35].
Early Experiments Testing Nitrones as Pharmaceutical Agents
It was in the context of the increasing use of nitrones to trap free radicals in experimental animals undergoing oxidative stress that the extrapolation to the experimental testing of nitrones as pharmaceutical agents began. We and others have reviewed the early phases of the use of nitrones as pharmaceutical agents previously [36–39]. The first early reports of the use of nitrones as potential pharmaceutical agents were those reported by Noveli in 1985 and 1986 [40–42] demonstrating that administered PBN protected rats from the trauma and death brought on by subjecting them to confinement in a rotating drum as well as the shock trauma brought on by LPS injection. The protective activity of PBN was replicated and extended by more rigorous experiments of septic shock using LPS by McKechnie et al [43] Hamburger and McCay [44] and Pogrebniak et al [45]. PBN was found to be protective in these models only if administered prior to the LPS challenge. It was soon after these observations (Dec, 1988) that Floyd and Carney made the first observations demonstrating that PBN had neuroprotective activity in the Mongolian gerbil stroke model [46–48]. This discovery arose from follow-up experiments where we had demonstrated using salicylate to trap hydroxyl free radicals that ischemia/reperfused gerbil brains produced reactive oxygen species [49]. PBN was then utilized in an attempt to trap putative secondary free radicals induced by hydroxyl free radical reactions with biological molecules. This approach failed because the secondary free radicals that may have been trapped to form the nitroxyl PBN spin adducts were rendered paramagnetically silent due to their reduction to the hydroxylamine products. Despite the failure to realize our experimental goals, serendipitous success was soon realized since the follow-up experiments showed that PBN was protective against stroke-induced brain injury even if it was administered up to 1 hr after the stroked brain was reperfused [48]. These results were soon reproduced [50] and extended [51,52].
Observations Extending the Biological Activity of PBN
Following these early observations other laboratories as well as our own extended the neuroprotective activity of PBN to models involving excitotoxicity induced neuronal damage [53], toxin induced brain seizures [54,55], and bacterial meningitis caused by group B streptococci [56]. Hillered’s group also extensively studied the effects of traumatic brain injury in animal models and demonstrated that PBN and the 2-sulfonyl-PBN derivative i.e. S-PBN have protective roles [57–62]. In addition to brain, PBN was shown to be protective in hearing loss in rats caused by the combined exposure to carbon monoxide and loud noise [63]. PBN was also shown to be effective in preventing streptozotocin-induced diabetes [64], thalidomide-induced birth defects [65], ischemia/reperfusion-induced acute renal failure [66] and in light-induced damage to eyes affecting vision [67–69]. Several studies have also suggested that PBN increased life-span in normal [70] and senescence-accelerated mice [71] and in rats [72] and also prevented age-related decline in behavioral parameters in rats [72] as well as gerbils [47,73]. Pertinent to the anti-aging effect of PBN, in rigorously controlled and replicated studies at three different experimental sites using a genetic model that simulates outbred wild mice, it was demonstrated that the PBN metabolite 4-hydroxy-PBN administered in the diet from a young age did not cause an increase in life span [74,75].
Wide-Spread Biological Activity of PBN – What General Mechanism is Involved?
The studies noted above as well as those not cited in this review clearly demonstrate that PBN-related nitrones have potent biological activity. In fact, it can be generalized that these compounds have shown activity in the majority of experimental models of either pathologic conditions or age-related diseases where they have been tested. New observations continue to be made. For example, recent observations include the demonstrations that: A) PBN ameliorates hippocampal injury and improved learning and memory in juvenile rats following neonatal exposure to LPS [76], B) PBN decreased the incidence of external malformations in a mouse model of LPS-induced teratogenesis [77] and C) PBN decreased the damage to striatal neurons and loss in motor skills in neonatal rats exposed to hypoxia-ischemia [78].
The wide-spread activity suggests that there is a universal mechanism that can explain the mechanistic-basis of the general activity of PBN. If this is true it has not been elucidated thus far. Since PBN was brought to the attention of scientists because of its ability to trap free radicals it is rational to assume that its biological action is due to its spin-trapping activity. This can not be completely ruled out, however the conventional notions of mass action solution chemistry spin-trapping is highly unlikely to explain the biological action of PBN and related compounds. This is because the general reaction rate of PBN with most free radicals is quite slow, i.e. usually about 105–107M−1sec−1 [3]. This dictates, as has been demonstrated in solution chemistry trapping studies, that PBN then must be present at very high concentrations (i.e. 10–50 mM) in order for PBN to trap a significant fraction of the free radicals being produced. As we have argued previously [36,79,80], PBN is rarely higher than 10–50 µM and almost never more than 0.5 mM in the target tissue of systems where it has been shown to be active. Based on a solution chemistry based perspective, the low target tissue PBN levels strongly suggest that PBN would be capable of spin-trapping only a very small fraction of free radical species produced which strongly argues against this explanation to account for its biological action. If it however is taken into consideration that the interior of cells has physical properties of a gel rather than bulk water and that PBN may have specific strong affinities for specific proteins or other biological molecules then it is possible that PBN may spin-trap critical free radicals on the surface of these macromolecules which may be of importance in the biological processes investigated. PBN has been shown to trap protein thiyl free radicals [81] but it has not been proven that these or other protein radicals are of critical importance in biological processes where PBN has been shown to have a significant effect. In the case of the spin trap DMPO (5, 5-dimethyl-1-pyrroline N-oxide) new methodology has shown that many proteins retain a DMPO molecule attached covalently as revealed by mass spectroscopy [82–86] and by immunological methods [87–89]. Modifications of this new methodology may become available to help address the questions of the importance of PBN reacting with free radicals on biological macromolecules.
PBN-Possible Mechanisms Other than Spin-Trapping
Mechanisms other than spin-trapping have been investigated to account for the biological activity of PBN and related compounds. In general PBN has been shown to have some activity and therefore influence many biological processes. As discussed earlier various notions other than spin-trapping to explain the mechanistic action of PBN have been considered. These include A) its antioxidant properties, B) its action on important membrane enzymes including ion transport proteins and C) its action as an anti-inflammatory agent. With regard to PBN as an antioxidant, at least two studies have demonstrated that it is a very poor antioxidant in simple systems [90,91]. In biological lipid peroxidation systems it was shown that PBN at ~5 mM was an effective antioxidant but the antioxidant BHT or γ-tocopherol was as active at about 5 µM or less, i.e. they were about thousand-fold more effective than PBN [90]. Since PBN solubilizes into lipid bilayers it is not surprising that it may influence enzymes especially if the concentration is in the high range. In this regard PBN has been shown to block calcium channels that are important in pulmonary artery relaxation [92]. The ED50 for PBN was shown to be 1.93 mM in this regard. PBN also has been shown to inhibit acetylcholinesterase activity with a Ki of 0.58 mM [93]. It was shown that PBN has no influence on muscarinic or glutamate receptors though [94,95]. Many studies in our laboratory and Kotake’s laboratory have demonstrated that PBN has general anti-inflammatory activity. The anti-inflammatory activity of PBN is manifested as the curtailment of inflammatory cytokine expression and the accompanying expression of genes associated with the action of proinflammatory cytokines. Examples of gene induction products which are in general suppressed by PBN include inducible nitric oxide synthase (iNOS) and COX-2. The induction of these genes have been shown to be curtailed by PBN administration in a wide range of biological models and systems [37,64,96–103]. The resultant curtailment of the production of enhanced nitric oxide (NO) and inflammatory prostaglandins is expected to have significant influence on biological parameters important in pathologic conditions and age-related diseases as has been noted in several neurodegenerative models [55,96,99] and in cancer development [80,104,105].
Nitrones as Therapeutics for Stroke
Basic Science Discoveries Leading to Commercial Development
The activity of PBN-related nitrones has been a subject of intense research and commercial development for over 20 years. As noted in the Introduction section, Floyd and Carney in 1988–1992 made the original discoveries in experimental stroke that led to most of this activity [46–48]. The fact that PBN administered up to 1 hr after an ischemia/reperfusion insult to the brain of Mongolian gerbils showed protective activity was surprising and clearly suggested possible therapeutic potential for the indication of acute ischemic stroke in humans. Centaur Pharmaceuticals beginning in 1993 aided by NIH SBIR grants helped implement much of the commercial research and development activity in this arena until 2000 when Renovis, Inc became the primary commercial company guiding this effort. Centaur partnered with Astra that later became AstraZeneca which funded all of the major development research and clinical studies from Phase I through Phase III stages from mid 1995 through 2007. The PBN derivative 2,4-disulfophenyl-N-tert-butylnitrone, referred to as NXY-059, was the candidate drug that was taken through the many stages of clinical development for the acute ischemic stroke indication.
Nitrone Stroke Drug NXY-059 Preclinical Studies
Green et al provided a most extensive review of the nitrone data (NXY-059, PBN, as well as 2-sulfophenyl-N-tert-butylnitrone) in relation to the preclinical research on NXY-059 in stroke [106]. NXY-059 is a less competitive compound for trapping free radicals in vitro systems than PBN or S-PBN [107] It should be noted however that PBN is a poor antioxidant when compared to the well known antioxidants α-tocopherol and butylated hydroxytoluene [90,91]. However as a protective agent in experimental stroke models, NXY-059 is much more effective than PBN in the amount of compound needed as well as the time window after the stroke when administration was started [106]. NXY-059 showed effectiveness when administered at a dose of 0.3 mg/kg/hr starting 1 hr after a 2 hr occlusion of the middle cerebral artery allowing regional brain reperfusion in a rat model. However PBN administered at a comparable dose rate showed no neuroprotective effect. In addition to the transient focal ischemia models in rats, NXY-059 was also shown to have significant neuroprotective activity in the permanent focal ischemia model in rat [106] as well as in the marmoset primate model [106,108,109]. In the rat middle cerebral artery permanent focal ischemia model, the degree of neuroprotection as judged by decreased infarct volume was directly proportional to the plasma free concentration of NXY-059 in the 0–120 µmol/L range [106]. In this model neuroprotection was significant even if NXY-059 was administered starting 4 hrs after middle cerebral artery ligation. Probably the most critical preclinical experiments were conducted in the marmoset model. This permanent focal middle cerebral ischemia model was studied when NXY-059 was administered either immediately after artery ligation [108] or starting at 4 hrs after artery ligation [110]. NXY-059 had significant neuroprotective activity in both studies as judged by behavior parameters as well as by prevention of brain death. Behavioral parameters obtained from both the hill and valley tests were used. In these tests the use of the lateral and contralateral arms was evaluated in trained animals at 3 and 10 weeks after artery ligation. Significant neuroprotection was afforded by NXY-059 in all the neuroprotection parameters measured. These critical experiments in addition to all the rodent studies conducted in several independent laboratories as well as the fact that NXY-059 had shown no significant toxicity in humans in several Phase I and Phase II clinical trials [106,111] were important in the evaluation conducted by AstraZeneca that led to proceeding into Phase III clinical trials.
Phase I and II Clinical Trials
Much of the background development research leading up to Phase II clinical trials has been thoroughly summarized previously [39,106,108,109,111]. NXY-059 was shown to be a very safe drug in all the clinical trials [106,111–113]. If a stroke drug alters hemostasis parameters, this may have an adverse effect. NXY-059 was studied in 30 healthy adults where it was demonstrated that it had no effect on bleeding time or platelet aggregation or adhesion [112]. It was also shown that NXY-059 has no effect on the action of recombinant tissue-type plasminogen activation on the clearance of human thrombi in vitro [113]. Studies of NXY-059 pharmacokinetics administered intravenously in healthy young and elderly humans showed that the plasma elimination half-life was short, being 2–3 hrs, renal elimination was predominant, it was not reabsorbed by the kidney and the recovery of the unchanged drug in the urine was 80–90% of the total dose irrespective of age of the subjects [111]. A follow up study confirmed that renal elimination of NXY-059 was dominate and that there was no reabsorption by the drug. It was also demonstrated that NXY-059 excretion in the kidney was due to active elimination by the organic ion transporter [114]. The drug clearance from plasma did fit a 2 compartment model indicating that it did have some penetration into tissue. Studies in rats showed that there was some penetration in brain. Studies in sham operated rats as well as those receiving a permanent middle cerebral artery occlusion demonstrated that NXY-059 did penetrate into the brain cortex region of sham operated rats (6.26 ± 1.26 nmol/g) but it penetrated even less in the stroked rats in the region of the artery ligation (3.84 ± 0.80 nmol/g) than the region of the contralateral side (6.14 ± 2.18 nmol/g). This is expected if the main supply of blood is blocked from coming into the stroked region. Another study important to the penetration of NXY-059 into stroked brain was conducted by Dehouck et al [110]. In this study the penetration of PBN, S-PBN as well as NXY-059 was examined in an in vitro model of the blood-brain barrier. As expected, the uptake of S-PBN and NXY-059 was low but PBN readily penetrated. However, importantly the penetration rate of NXY-059 increased several-fold if the blood-brain barrier was subjected to hypoxia or ischemia [110].
Phase III Clinical Trials
As noted earlier, the marmoset experiments plus the safety profile of NXY-059 contributed to the decision to begin Phase III clinical trials. Phase III clinical trials were conducted as a series of two major studies. The two trials were referred to as the Stroke-Acute Ischemic NXY Treatment (SAINT) trials or SAINT I and SAINT II trials. The SAINT I trial ran from May 2003 through November 2004 at 158 hospitals in 24 countries [115]. The patients were older than 18 yrs and had a clinical diagnosis of acute stroke that commenced within 6 hrs before entry into the study. It was required that the patients had limb weakness and had a score of at least 6 on the National Institutes of Health Stroke Scale (NIHSS). Patients were randomly assigned to receive an i.v. infusion of NXY-059 or placebo within 6 hrs after the onset of stroke. The initial infusion rate was 2270 mg per hour and then reduced to 480 to 960 mg for a further 71 hours with the aim of maintaining a free NXY-059 plasma concentration of 260 µmol per liter. Treatment outcome was based on a clinical assessment which included functional measures that were dependent primarily on the modified Rankin scale (range 0–5) and the Barthel index [115]. Therefore the SAINT I trial was a randomized, double-blind, placebo-controlled trial involving 1722 patients with acute ischemic stroke and randomly assigned to receive 72-hr infusion of placebo or intravenous NXY-059 beginning within 6 hrs after the onset of stroke. In the final outcome, 1699 subjects were included in the efficacy analysis. It was shown that NXY-059 significantly improved the overall distribution of scores on the modified Rankin scale, as compared with placebo (P = 0.038) by the Cochran-Martel-Haenszel test [115]. The common odds ratio for improvement across all categories of the modified Rankin scale was 1.20 (95 percent confidence interval, 1.01 to 1.42). In contrast to the modified Rankin scale noted above, NXY-059 did not show significant improvement in neurological functioning as measured by the NIHSS test. There was no difference in mortality or adverse events between the treated and untreated group.
Results of SAINT I Clinical Trial
The first report of the success of NXY-059 in the SAINT I trial was hailed as the first experimental drug to receive this distinction amid a large number of failures for the treatment of acute ischemic stroke. After the first report there was further analysis of the data by others leading to conclusions that differed. For instance one critical review concluded that in general terms that for every 100 stroke patients with NXY-059 about 10 would benefit and none would be harmed by the treatment [116]. Another review concluded that the treatment provided little evidence for the demonstration of efficacy [117]. Actually the primary investigator’s conducted a much more thorough analysis of the data where the outcome parameters were evaluated at 7 days, 30 days and 90 days [118]. The authors concluded from this more detailed analysis that NXY-059 provided beneficial effects at 7 and 30 days after the stroke but not at 90 days. They also concluded that the SAINT I trial was underpowered to measure effects on the neurological examination. These conclusions were apparently important in causing the initiation of another Phase III clinical trial containing many more stroke patients which was referred to as the SAINT II trial.
Results of SAINT II Clinical Trial and Pooled Summary
The SAINT II trial was conducted during the period of May 2003 through June 2006 and enrolled 3306 patients having the same criteria as in SAINT I trial except the study involved 362 centers from 31 countries [119]. The primary endpoint was the distribution of disability scores on the modified Rankin scale at 90 days. In addition scores on neurologic and activities of daily living scales were secondary endpoints. The efficacy analysis was based on 3195 patients. The results showed that the distribution on the modified Rankin scale did not differ between the NXY-059 treatment group and the placebo group [119]. In addition there was no evidence of efficacy for any of the secondary endpoints either. Following the publication of the SAINT II results a pooled analysis of the SAINT I and SAINT II trials was then published [120] which concluded that NXY-059 was not effective in the combined trials. The details provided of the pooled trials shows that these trials included 498 centers from 38 countries and that the design and content of the 2 SAINT studies were developed by a steering committee comprising stroke experts from Europe, North America and Australia. The steering committee had complete access to the data and was responsible for writing the manuscript [120]. These results were clearly disappointing to those in the stroke treatment community who had expected a positive outcome based on the SAINT I trials. It should be noted that the trials, both preclinical and clinical, had been developed in close accordance with the guidelines proposed by an academic-industry roundtable group (STAIR) [121–123] and therefore the failure of NXY-059 was considered a serious blow to the neuroprotection concept that guided stroke scientists for some time [124].
Critiques of Clinical Trials
There have been many reviews and critiques of the steps in the commercial development of NXY-059 [125] as well as questioning the neuroprotection concept that has guided stroke scientists for some time. Since NXY-059 is a compound that has been grouped into the loosely defined category of antioxidants, it is important for the future of potential therapeutics that may be placed in this category to learn from the important points revealed by the lessons to be gleaned from the NXY-059 development history. It is important to list deficiencies in the preclinical trials that scientists have pointed out. Savitz has pointed out that the preclinical data have several short comings including a lack of strenuous testing that reproducibly showed robust protection in extended time windows in clinically relevant stroke models at several different academic research laboratories [125]. He also states that the clinical trials of NXY-059 were inadequately designed, in part, because of inappropriate treatment windows and inclusion of diverse stroke patients. Others seem to agree with this assessment [122,126]. Another important failure pointed out by Feuerstein et al was that more attention was needed to characterize the nature of the drug candidate-target interaction and its relationship to pharmacodynamic treatment end points [123].
Lessons Learned Pertinent to Antioxidant Therapeutics
An insightful review focused specifically on free radical scavengers and stroke was provided by Wang and Shuaib [127]. They noted that in addition to NXY-059 only 3 other therapeutic antioxidant medications had progressed into clinical trials for stroke; these include Ebselen, Tirilazad and Edavarone. Both Ebselen and Tirilazad were tested in stroked patients but their development has been terminated and therefore only Edavarone has succeeded in being used for stroke but only in Japan. They conclude that antioxidants as potential therapeutics is still valid as a goal, but that certain key elements need to be addressed in future development. First, there is a need to more thoroughly understand mechanistically how neurons die especially in human cerebral ischemia. Second, ischemic cell death is the result of complex noxious processes including oxidative stress, excitotoxicity, functional failure of ionic pumps, inflammatory reactions and activation of apoptotic death pathways and that termination of one of these cascades may not be sufficient to reduce the brain damage that occurs. Thirdly, therefore a combination therapy with different compounds targeting different pathways may offer better chance than single medications. Two other elements were suggested, these included the concept that DNA regulatory elements in the promoter regions of genes that were upregulated by ischemic tolerance of cells may be a valid target and finally the need to test compounds in patients that had been more carefully selected using newer technologies such as MRI screening. These are excellent suggestions and are clearly issues that need to be considered in future drug development for stroke and probably other pathologic conditions where exacerbated oxidative stress is considered an etiological agent.
Anti-Cancer Activity of PBN-Related Nitrones
Early Studies on Hepatocellular Carcinoma
We discovered that PBN has anti-cancer activity in the well known dietary choline deficiency rat liver cancer model [128–130]. Our research on the influence of PBN on hepatocarcinogenesis induced by a choline-deficiency diet started in 1997. Development of hepatocellular carcinoma in many animal models begins as preneoplastic lesions that appear as “small islands” of “altered cells” in the liver. In the case of the choline deficiency model as well as in several other models there are hundreds to thousands of preneoplastic nodules (islands) that develop in the liver. With time and sustained oncogenic pressure a few adenomas arise from some of these islands and also with time carcinomas may arise from the adenomas or from the preneoplastic lesions per se. The choline-deficiency model was tested because reactive oxygen species and lipid peroxidation had been shown to be very important primary agents in this model by several laboratories [131–135]. Since PBN had been shown to suppress oxidative damage in several biological systems with very little toxicity, it seemed a reasonable choice. The first experiments were conducted on the Wistar rat model and involved three levels of PBN administered in the drinking water for 12 weeks while the rats were on the choline deficiency amino acid (CDAA) defined diet. At the end of this time period the rats were sacrificed and the livers examined to assess several parameters about preneoplastic nodule growth, size, etc. In this study we first noted that PBN in increasing amounts administered in the drinking water caused dramatic decreases in the size of preneoplastic nodules in the liver. In the same study we also noted a slight decrease, more so with increasing dosage, of the number of preneoplastic nodules in the liver. Additionally we also demonstrated that PBN at increased dosage, caused a decrease in the amount of oxidized DNA adducts as measured by 8-OHdG content in the livers [136]. The research results are summarized in Table 1.
Table 1.
PBN Effect on Choline Deficiency Induced Preneoplastic Lesions*
| GST-P-positive lesions | |||||
|---|---|---|---|---|---|
| Group | Treatment(s) | no./cm3 | Size(mm3) | 8-OHG (8-OHdG/106 dG) |
Av. PBN exposure (mg/kg body wt/day) |
| 1 | CDAA | 190 ± 10 | 1.92 ± 0.31 | 18.92 ± 1.96 | 0 |
| 2 | CDAA + PBN (low) | 170 ± 28 | 0.33 ± 0.20** | 13.10 ± 2.56** | 6.06 ± 1.07 |
| 3 | CDAA + PBN (middle) | 149 ± 15** | 0.17 ± 0.06** | 5.11 ± 1.24** | 32.86 ± 4.81 |
| 4 | CDAA + PBN (high) | 142 ± 10** | 0.10 ± 0.05** | 2.55 ± 0.83** | 61.76 ± 8.90 |
| 5 | CSAA | 0 | 1.00 ± 0.10 | 0 | |
| 6 | CSAA + PBN (high) | 0 | 1.00 ± 0.17 | 64.45 ± 9.53 | |
The dramatic decrease in the size of the preneoplastic nodules was evident even at very low levels (6 mg/kg-day) of PBN in the drinking water and this spurred us to explore the anti-cancer activity in more detail. Therefore we then followed up with a long term (70 week) study where rats were placed on a choline deficient amino acid defined diet (CDAA) or a control diet (choline sufficient amino acid defined diet (CSAA) and PBN was administered in the drinking water. The results of this study clearly demonstrated that PBN had anti-cancer activity. The results have been summarized in 3 publications [128–130] and also we were granted a patent on these observations [137]. These results clearly show that PBN when given in the drinking water for the first 26 weeks or for the last 44 weeks to the rats given the CDAA diet caused the complete prevention of hepatocellular carcinoma (HCC) formation assessed after 70 weeks. We also found that when PBN was given in the control diet (CSAA), no tumor formation was observed. Therefore PBN is not carcinogenic per se. This study with PBN was followed up by a new study testing the anti-cancer activity of 4-hydroxy-PBN i.e. 4-OHPBN, which is the natural p450 hydroxylated derivative of PBN. In this case 4-OHPBN was given in the diet as opposed to PBN which was given in the drinking water. A dietary administration route for 4-OHPBN was used simply because of convenience. The results of this 70 week study of 4-OHPBN demonstrated that 4-OHPBN had anti-cancer activity in this model also. Even though the activity of 4-OHPBN and PBN were not directly compared in this experiment they were judged to be approximately equal in efficacy.
The data presented in Table 1 suggested that PBN in some unknown manner caused decreased growth of the cells in the preneoplastic lesions. A clear demonstration that this was due to increased selective apoptosis of these cells was demonstrated in an experiment where PBN as well as some of its chemical derivatives (2-OHPBN, 3-OHPBN, 4-OHPBN and 2-Sulfate PBN) were tested in a short term study in the same model although in this case PBN and derivatives were administered in the CDAA diet [129]. Important data concerned with the effects of PBN and the two active derivatives on preneoplastic lesions are presented in Figure 2. This data is a summary of our published results [138]. Note that the highest levels of PBN as well as 4-OHPBN and 3-OHPBN caused significant decreases in the size of the preneoplastic lesions as well as the content of 8-OHdG in the DNA and the amount of lipid peroxidation as measured by thio barbituric acid reactive species (TBARS) in the tissue. As was noted in the earlier study [136] the numbers of preneoplastic lesions were decreased by the highest levels of PBN (as well as 4-OHPBN and 3-OHPBN in this study) although not as marked as that noted in the change in size of the preneoplastic lesions brought on by the nitrones. The data collected on apoptosis of cells within the lesions versus the surrounding “normal” tissue cells are presented in Table 2. These data show that treatment with the nitrones caused a large significant increase in the apoptosis of the cells within the preneoplastic lesions but had the opposite effect of the cells in the surrounding tissue. These results clearly show that the nitrones decreased the size of the preneoplastic lesions by enhancing the selective apoptosis of these cells. This is a remarkable finding and may be the key to understanding the anti-cancer activity of the nitrones in this model.
Figure 2.

The influence of PBN and its 3-hydroxy and 4-hydroxy derivatives on the parameters of preneoplastic lesions in the liver of rats administered these chemicals in a choline deficient and choline sufficient diet. The values were calculated from the data presented in Floyd et al [138].
Table 2.
Effect of PBN Derivations on Apoptotic Index of Perneoplastic Lesion Cells and Surrounding Tissue Cells*
| % of Cells in Apoptosis | ||
|---|---|---|
| Treatment | Preneoplastic Lesions |
Surrounding Cells |
| CDAA Diet Only | 3.24 | 6.48 |
| PBN Added | 12.65 | 2.40 |
| 4-OHPBN Added | 8.97 | 2.65 |
| 3-OHPBN Added | 10.90 | 2.40 |
| Control Diet Only | -** | 0.29 |
Experimental results obtained on liver tissue in the dietary choline deficiency rat model of hepatocellular carcinoma conducted by Floyd et al(133).
There were no preneoplastic lesions observed in the livers of rats fed the control (choline sufficient) diet.
Presently we do not understand the mechanisms involved as to why the active nitrones caused the enhanced selective apoptosis of the cells within the preneoplastic lesions. Many potential mechanisms can be envisioned. However a set of observations we made suggests that enhanced expression of iNOS and thus the enhanced level of NO produced could play an important role. Our observations and rationale implicating the role of iNOS in cancer development has been summarized previously [80,105] and therefore only the major points will be presented here. We observed that isolated hepatocytes from rats fed a choline deficiency diet had enhanced NO production [104] and that this was due to enhanced levels of iNOS expression and that PBN administration decreased NO production and iNOS expression by the isolated hepatocytes. These observations clearly implicate that iNOS and enhanced NO production is correlated with the growth of liver tumor in the choline deficiency model and that PBN administration caused the suppression of iNOS/NO production which may help to explain the anti-cancer activity of these compounds in this model. The scientific literature reinforces the notion that iNOS/NO is important in cancer development in several experimental models and in human studies as we summarized previously [105]. The mechanisms involved in the iNOS/NO mediated cancer development can be rationalized and are briefly summarized below.
Rigorous studies in the literature with several tumor cell lines [139–142], as well as isolated hepatocytes [143–149] demonstrated that NO prevents apoptosis by forming critical S-nitrosylation bonds with specific caspases, probably Caspase 8 [147] or Caspase 9 or Procaspase 9 [139] and thus preventing the initiation of the apoptosis processes. Recent studies have also demonstrated that NO mediates S-nitrosylation of Bcl-2 which prevents proteosomal degradation thereby enhancing the buildup of this protein and therefore preventing apoptosis of the cancer cells [150]. In addition to forming S-nitrosylation adducts of caspases and Bcl-2 thereby inactivating these enzymes and preventing apoptosis, NO also mediates the S-nitrosylation of OGG1 thus causing the inactivation of this DNA repair enzyme [151]. OGG1 inactivation is then expected to cause the buildup of 8-OHdG, as we observed [136]. The enhanced 8-OHdG is expected to then enhance cellular mutation events. Additionally NO mediates S-nitrosylation of PTEN inactivating this tumor suppressor protein [152] which then is expected to cause enhanced oncogenic cell cycle processes to proceed [153]. In effect then because NO is produced at sustained increased levels it causes the S-nitrosylation of OGG1, PTEN and specific caspases and Bcl-2 which then respectively causes the enhancement of cellular mutational rates and increased oncogenic cell growth as well as decreased apoptosis of preneoplastic and cancer cells. Therefore based on this rationale we postulate that PBN administration then shuts down NO production and this in effect causes cessation of these three very important carcinogenic processes by the mechanisms outlined above and therefore PBN slows down the development of cancer by enhancing the death of the preneoplastic cells and possible cancer cells. Although this rationale helps explain our results in this model, these notions have not been rigorously proven by direct experimental methods in this model. Additionally there may be other mechanisms involved in the anti-cancer action of the nitrones which supersede or encompass the iNOS/NO notions discussed above.
Studies on Gliomas
The efficacy of PBN as a potential anti-glioma therapeutic agent was assessed in a rat C6 intracerebral glioma model. Magnetic resonance (MR) imaging (MRI) and MR angiography (MRA) was used to follow tumor morphology, such as tumor heterogeneity, size and volume, and to determine alterations in tumor vascularity, such as associated with angiogenesis, respectively [154]. PBN was administered both as a prophylaxis and a post-tumor treatment agent. For prophylaxis, PBN treatment was administered in the drinking water (75 mg/kg rat/day or 0.065% w/w, for an estimated water uptake rate of 110–120 ml/kg rat/day) continuously starting 5 days before intracerebral implantation of C6 rat glioma cells (106 cells/ml in Dulbecco’s modified Eagle’s medium (DMEM)) [154]. For post-tumor treatment, PBN was administered in the drinking water (as above) starting 14 days after cell implantation, which corresponded to a tumor volume of ~50 mm3, as assessed by MRI. Prophylaxis PBN treatment was able to significantly decrease tumor growth (from 0.14±0.028 day−1 and a doubling time (Td) of 2.6 days to 0.05±0.007 day−1 and a Td of 7.4 days) and result in partial or even complete tumor regression over the course of 40 days [154]. Post-tumor PBN treatment was found to alter tumor growth and result in tumor regression in 40% of treated rats [154]. As shown in Fig. 3 treatment with PBN 5 days before implantation seems to clearly inhibit glioma formation, although post-tumor PBN treatment also seems to recede glioma formation in a significant proportion of the tumors. Histological examination of rat brain slices in animals with complete tumor regression indicated no evidence of tumor cells following PBN treatment.
Figure 3.
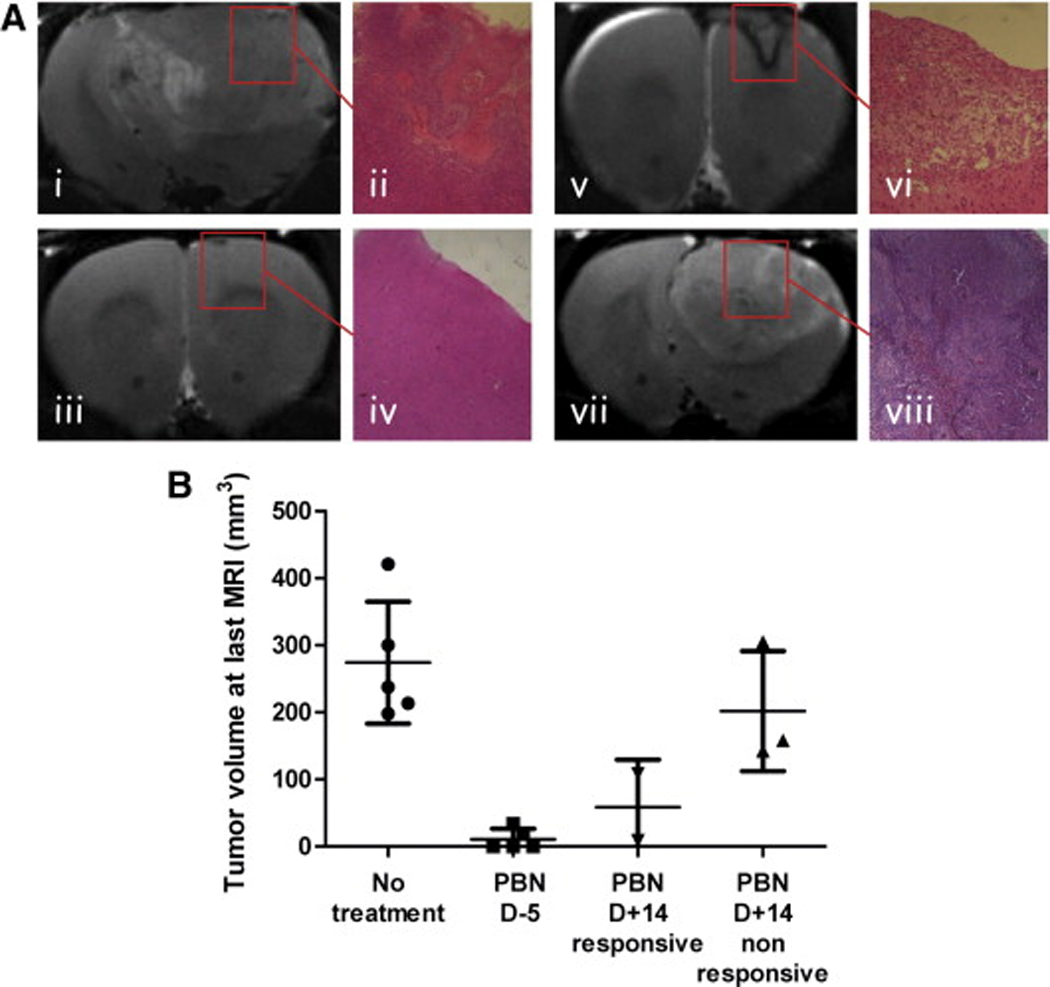
(A) Representative T2-weighted images and histology (H&E, x4) staining of C6 gliomas from the different treatment groups. (i–ii) Untreated C6 glioma at day 18 following cell implantation. (iii–iv) PBN pre-treated C6 glioma at day 27. (v–vi) Post-tumor PBN treated C6 glioma undergoing regression, shown at day 32. (vii–viii) Nonresponsive post-tumor PBN treated C6 glioma shown at day 16. (B) Tumor volumes (mm3) measured at the last MRI timepoint, represented as means ± standard deviation.
MRA analysis of rat brain vasculature, quantified using a Mathematica-based program developed in-house, indicated that PBN also decreased angiogenesis associated with the gliomas (see Figure 4) [154]. Non-treated C6 gliomas had a relative blood volume ratio (tumor blood volume at 23±3 days following cell implantation compared to the first day of assessment at 7 days following cell implantation when no tumor was visible by MRI) of 1.26±0.157 compared to 0.98±0.072 for rats implanted with non-neoplastic primary astrocytes [154]. Prophylaxis PBN treatment decreased the blood volume ratio to 0.79±0.144 (with an overall 50±8% decrease in blood vessel diameters), whereas post-tumor PBN treatment in responsive animals was found to decrease the blood volume ratio and vessel length by 20±7% [154]. Immunostaining for the proangiogenic factor VEGF (vascular endothelial growth factor) and a marker of endothelial cell proliferation, the von Willebrand factor (vWF), following PBN treatment indicated decreased expression of VEGF and vWF which was similar to normal brain tissue [154]. The neuroprotective [99], anti-inflammatory [100], anti-ischemic [155] and anti-carcinogenic [136] properties of PBN, coupled with its ability to freely cross the blood-brain-barrier, and also being both lipophilic and hydrophilic, makes PBN an interesting therapeutic candidate that may potentially be used for gliomas in humans, particularly following surgery and/or radiation to prevent tumor reoccurrence. PBN has been previously shown to decrease iNOS activity [102]. Increased iNOS levels have been found to occur in human brain tumors [156] and is strongly correlated with the degree of malignancy of a carcinoma [157]. Nitric oxide and VEGF tightly cooperate to promote angiogenesis. PBN has been found to down-regulate cytokines and NF-κB expression [45], which promotes iNOS expression [158], and therefore PBN may prevent or reduce angiogenesis in gliomas through NF-κB and/or suppression of iNOS, which would result in decreased NO production and subsequent reduced VEGF expression.
Figure 4.
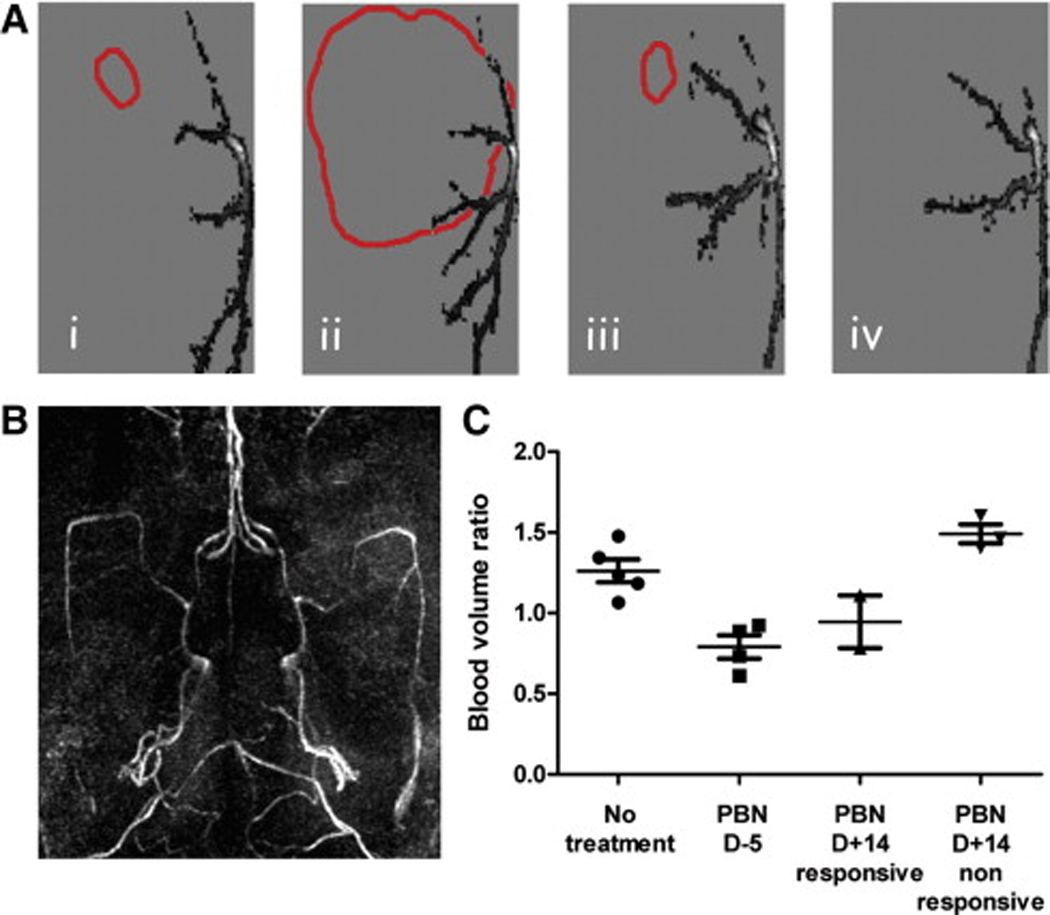
(A) 2D vasculature projections in the horizontal plane of a representative non-treated C6 glioma at day 8 (i) and 20 (ii) after cell implantation, and of a PBN pretreated C6 glioma at day 9 (iii) and 39 (iv). The tumor boundaries are also represented (red). (B) 3D maximum intensity projection (MIP) rendering of the brain vasculature. (C) Blood volume ratios for each treatment group at the last time point (days 17–21 for untreated rats and non-responsive rats (PBN D+14), and days 39–40 for treated rats (PBN D−5, PBN D+14 responsive), represented as means ± standard deviation.
Diffusion-weighted imaging (DWI) and diffusion tensor imaging (DTI) was also recently used to assess the white matter neuronal fiber tracts following glioma formation and treatment with PBN. In this study, we found that the displacement, loss of connectivity and disappearance of neuronal fiber tracts resulting from C6 glioma formation, was prevented if PBN treatment was implemented [159]. In some of these animals the C6 gliomas completely disappeared in 6 weeks after cell implantation, resulting in a total disappearance of the compressed neuronal fibers observed in non-treated rats [159]. In non-treated animals, the C6 glioma growth compressed surrounding tissues, inducing a hematogenous disorder such as ischemia, which led to destruction of the neuronal cells and fibers [159]. It is thought that perhaps PBN may be protecting the neuronal fibers from compression-induced ischemic injury [159]. PBN has been previously shown to increase cortical cerebral blood flow in rats by inhibiting the breakdown of NO [160].
Nitrones and Hearing Loss
Acute acoustic trauma (AAT) and oxidative stress
An abundance of scientific data is accumulating supporting the thesis that oxidative stress plays a substantial role in the induction of cochlear injury and permanent hearing loss after excessive acute noise exposure [161–167]. Acute acoustic trauma (AAT) has been associated with a variety of changes in mammalian cochleae, including excessive release of glutamate, ischemia-reperfusion, mitochondrial injury, glutathione depletion, and ionic fluxes leading to an increase in oxidative stress [165,167].
AAT induced by steady state noise and, presumably, a variety of other types of noises, is associated with the production of reactive oxygen species (ROS), reactive nitrogen species (RNS), and lipid peroxidation species both with the onset of the noise trauma as well as some days after cessation of noise exposure [168–172]. Additionally, recent evidence suggests that there is a significant upregulation of iNOS activity in the mammalian cochlea after intense steady state noise exposure. In one study mice were exposed to high level steady state noise and tissues of the lateral cochlear wall and stria vascularis were examined [173]. This region of the cochlea is highly vascularized and very metabolically active as it generates endocochlear potential responsible for sound transduction. The investigators found that in the noise exposure condition there was a greater expression of iNOS immunoreactivity as well as increased detection of NO production and ROS activity leading to apoptosis of cells in these tissues. These changes were attenuated with the administration of an iNOS inhibitor [173]. An additional study reported increased iNOS immunoreactivity in afferent nerve fibers, outer hair cells and a variety of supporting cells in the cochlear organ of Corti as well as cells of blood vessels and marginal cells of the stria vascularis in the lateral cochlear wall [174]. Importantly, one of the consequences of the excessive oxidative stress induced by excessive noise is the onset of programmed cell death associated with mitochondrial release of cytochrome c, activation of caspases and the c-jun NH2-terminal MAP kinase pathway [175,176].
PBN and toxin-potentiated, noise-induced hearing loss (NIHL)
Further evidence for the importance of oxidative stress in producing the cochlear injury associated with AAT includes the extensive experimental therapeutic benefit demonstrated with compounds having antioxidant properties. For example, free radical scavengers, such as mannitol, salicylate and ebselen, have been reported to decrease permanent hearing loss due to acoustic overexposure [167]. Also, approaches to enhance glutathione replenishment have been successful strategies to reduce NIHL in animal models [171,172,177–179]. Glutathione replenishment with NAC (N-acetylcysteine) has been reported to be effective in the prevention and treatment of NIHL in several animal models from several different independent laboratories and has been studied in an initial pilot study in humans [178].
A variety of early experiments were conducted with PBN. Fechter and colleagues reported that several compounds, such as carbon monoxide (CO), hydrogen cyanide (HCN) and acrylonitrile (ACN), can potentiate the hearing loss associated with acoustic overexposure [63,180–183]. Fechter and colleagues hypothesized that the mechanism of the potentiation of NIHL by these toxins involved increasing cochlear oxidative stress. They noted that PBN given systemically before and after high level steady state noise was able to decrease the toxin-induced potentiation of the noise-induced threshold shifts [181,182]. However, PBN was unable to reduce the auditory threshold shifts induced by noise alone [181,182].
4-OHPBN and AAT
Surprisingly, a compound closely related to PBN, 4-hydroxy phenyl-n-tert-butyl-nitrone (4-OHPBN), has recently been reported to decrease permanent hearing loss induced by AAT in a chinchilla model [184]. The compound 4-OHPBN is a major metabolite of PBN. This was surprising based on the previously noted work by Fechter and colleagues that PBN was ineffective in decreasing noise-induced auditory threshold shifts. In the study reported by Choi et al., chinchilla were exposed to high level steady state noise for 6 hours and treated with intraperitoneal injections of 4-OHPBN alone or in combination with NAC or NAC plus acetyl-L-carnitine (ALCAR) four hours after cessation of noise exposure and continuing twice daily for another 48 hours. Treatment with 4-OHPBN alone reduced permanent hearing threshold shifts and outer hair cell loss in a dose-dependent manner. A summary of these recently published results is presented in Figure 5. The results show that when 4-OHPBN is combined with either NAC or NAC plus ALCAR permanent hearing loss was reduced from 35 dB to 5 dB or less, and outer hair cell loss was reduced from 60% to 10% or less. Current studies suggest that substantial reduction in permanent hearing loss can still be obtained using the combination of 4-OHPBN plus NAC and ALCAR when given even 24 hours after the noise exposure (Choi et al, unpublished results). Utilizing these compounds in combination with 4-OHPBN allowed for a substantial reduction in doses of compounds required while still affecting a maximal reduction in hearing and hair cell loss.
Figure 5.

Figure 5A. Auditory brainstem response (ABR) threshold shifts averaged at frequencies of 2–8 kHz for control group, different 4-OHPBN dosage group, and different drug combination group. The threshold shifts of control group were reduced with increases of 4-OHPBN dosage and the number of drug combination. Asterisks of * and *** represent statistically significant differences in ABR threshold shifts between control group and each experimental group at p<0.05 and p<0.001, respectively. Asterisks of ***10, **20, ***20, **50, and ***50 indicate statistically significant differences in ABR threshold shifts between each experimental group (75 mg/kg, two, and three) and each different dosage of 4-OHPBN (10, 20, and 50 mg/kg) at p<0.001, p<0.01, p<0.001, p<0.01, and p<0.001, respectively. This figure has been published previously by Choi et al [184] and is reproduced with the permission of the publisher.
Figure 5B. Percentage of missing OHC averaged at cochlear frequency regions corresponding to 2–8 kHz for four 4-OHPBN dosage groups, different drug combination groups, and control group. Outer hair cell (OHC) losses of control group were reduced with increases of 4-OHPBN dosage and the number of drug combination. Asterisks of ** and *** represent statistically significant differences in OHC loss between each experimental group and control group at p<0.01, and p<0.001, respectively. Asterisks of *10–20 indicate statistically significant differences in OHC loss between each experimental group and 4-OHPBN of 10–20 mg/kg at p<0.05. This figure has been published previously by Choi et al [184] and is reproduced with the permission of the publisher.
Figure 5C. Auditory brainstem response (ABR) threshold shifts averaged at frequencies of 2–8 kHz for different antioxidant drugs and their combinations. Asterisks of ** and *** indicate statistically significant differences between control group and each experimental group at p<0.01 and p<0.001, respectively. The symbol # shown in the two-drug combination group indicates significant differences in averaged ABR threshold shifts between the two-drug combination and other experimental groups [4-OHPBN(20), NAC(325), ALCAR(100)] at p<0.001, respectively while the symbol # shown in the three-drug combination groups shows significant differences between the three-drug combination and other experimental groups [4-OHPBN(20), NAC(325), ALCAR(100)] at p<0.001 and the three-drug combination and an experimental group [4-OHPBN(50)] at p<0.05, respectively. Data for NAC (325 mg/kg) and ALCAR (100mg/kg) were excerpted from Coleman et al [189]. This figure has been published previously by Choi [184] and is reproduced with the permission of the publisher.
The exact mechanisms by which 4-OHPBN reduces cochlear injury associated with AAT are still unknown. Possibilities include free radical scavenging, inhibition of iNOS activation, suppression of ROS and RNS formation, decreased mitochondrial ROS production and reduced neuroinflammation and activation of MAP kinase cascades [185]. Preclinical testing to further define toxicity, mechanism of action, pharmacokinetics, and oral dosing parameters are now underway.
Conclusions and Perspective on Antioxidant Therapeutics
What is the Candidate Drug-Therapeutic Target?
Our experience with the PBN-related nitrones has taught us many lessons in terms of the development of therapeutics that are generally considered antioxidants. We have focused on nitrones simply because the PBN-related nitrones have been shown to have potent biologic activity in preclinical models of several pathologic conditions and in age-related diseases where exacerbated oxidative stress is generally considered an etiological factor. Even though PBN and related compounds are generally considered antioxidants, the truth is they are poor antioxidants in several in vitro systems but in fact generally they do act in biological systems to decrease oxidative stress at least in those systems where the condition has been evaluated. Nitrones became widely known because of their ability to spin-trap free radicals yet the biologic action of PBN, especially, is most likely not due to its ability to spin-trap radicals in biological systems. It should be noted that in the case of NXY-059 it might act as a spin-trap simply because its level in blood is so high (260 µm) under treatment conditions that it could act as a spin-trap. But there certainly was no agreement as to its mechanism of action in preclinical models [39,106]. Therefore there is considerable uncertainty and confusion about the mechanism of action of the nitrones.
Our research has shown that the nitrones have potent anti-inflammatory properties and this is probably important. Therefore, at first glance, in the case of the nitrones there is considerable confusion regarding the drug-therapeutic target when compared to other well known drugs such as the statin’s for instance. In truth, however, many well known drugs such as aspirin, the statins and the non steroidal anti-inflammation drugs have action on many other processes and biological parameters other than those that they are widely considered to act upon. Conversely some well known drugs are also very potent antioxidants. This is the case for the well known β blocker D-propranolol which has very potent antioxidant activity and this property is considered to be an important reason for its therapeutic mechanism of action [186–188].
Confusion over Antioxidants and Their Action
There is another widely unrecognized problem with defining the mechanism of action or drug-therapeutic target for antioxidants. Simply stated, oxidative stress as well as the activity of antioxidants in quelling oxidative stress is an area of active research and as such, a well defined drug target is difficult to present at the present time when the therapeutic under question is categorized as an antioxidant. It is therefore clear from the foregoing discussion that much development research effort is necessary to understand and then clearly define the candidate drug-therapeutic target for a specific “antioxidant” therapeutic. The case of alpha-tocopherol (vitamin E) is instructive. Vitamin E was considered an essential vitamin since the 1920’s and only later found to possess antioxidant properties. But its true mechanism of action, especially how its position in the membrane and inferfaces with ascorbate in the cytosolic phase to quell oxidative stress and oxidative damage was only elucidated in the last ten years. It is probable that in the case of many so-called antioxidants that the therapeutic target may be very different than what is generally considered an antioxidant property. As noted above this task is probably harder for “antioxidant drugs” than many other drugs. Therefore in many cases the best that can be done is to precisely define which biological processes are of vital importance in the development of the pathologic indication where the therapeutic agent acts and then use this as a first line screening tool. In most cases the basic science underling the processes involved in causing the pathologic condition may be a major limitation as for instance in the case of stroke. Also as in the case of stroke, several different processes combine to play a synergistic role leading to the neuronal damage that occurs. Blocking only one of these processes is probably not enough. In this case there is strong rationale for using a combination of compounds as was pointed out as a suggestion for stroke [127]. Our observations on the synergistic effectiveness of NAC combined with 4-OHPBN is a good illustration of this point in the case of acute acoustical trauma-induced hearing loss [184]. It is possible that the active plant constituents could be useful in this regard.
Acknowledgements
Funding for the early observations in the anti-cancer activity of the nitrones was in part supported by NIH RO1 CA-82506 and OARS AR05-041. Funding for the observations related to the anti-glioma activity of nitrones was in part by NIH P20RR016478 and OCAST OARS AR071-063. We are very grateful to many colleagues who helped generate the data for the original publications and to Donna Howell who helped with the manuscript preparation and graphics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures
RAF has been active in the commercial development of NXY-059 in stroke. He’s a cofounder of Centaur Pharmaceuticals, Inc. and then a consultant of Renovis, Inc. He and RAT are cofounders of Onconos, Inc. a start up company involved in cancer therapeutics.
Reference List
- 1.Iwamura M, Inamoto N. Novel formation of nitroxide radicals by radical addition to nitrones. Bull. Chem. Soc. Jpn. 1967;40:703. [Google Scholar]
- 2.Janzen EG, Blackburn BJ. Detection and identification of short-lived free radicals by electron spin resonance trapping techniques (spin trapping). Photolysis of organolead, -tin, and -mercury compounds. J. Am. Chem. Soc. 1969;91:4481–4490. [Google Scholar]
- 3.Janzen EG. Spin trapping. Acc. Chem. Res. 1971;4:31–40. [Google Scholar]
- 4.Harbour JR, Bolton JR. Superoxide formation in spinach chloroplasts: Electron spin resonance detection by spin trapping. Biochem. Biophys. Res. Commun. 1975;64:803–807. doi: 10.1016/0006-291x(75)90118-7. [DOI] [PubMed] [Google Scholar]
- 5.McCay PB, Poyer JL, Floyd RA, Fong K-L, Lai EK. Spin-trapping of radicals produced during enzymic NADPH-dependent and CCl4-dependent microsomal lipid peroxidation. Fed. Proc. 1976;35:421. [Google Scholar]
- 6.Barr DP, Gunther MR, Deterding LJ, Tomer KB, Mason RP. ESR spin-trapping of a protein-derived tyrosyl radical from the reaction of cytochrome c with hydrogen peroxide. J. Biol. Chem. 1996;271:15498–15503. doi: 10.1074/jbc.271.26.15498. [DOI] [PubMed] [Google Scholar]
- 7.Chamulitrat W, Mason RP. Alkyl free radicals from the beta scission of fatty acid alkoxyl radicals as detected by spin trapping in a lipoxygenase system. Arch. Biochem. Biophys. 1990;282:65–69. doi: 10.1016/0003-9861(90)90087-f. [DOI] [PubMed] [Google Scholar]
- 8.Fischer V, Harman LS, West PR, Mason RP. Direct electron spin resonance detection of free radical intermediates during the peroxidase catalyzed oxidation of phenacetin metabolites. Chem. Biol. Interact. 1986;60:115–127. doi: 10.1016/0009-2797(86)90021-9. [DOI] [PubMed] [Google Scholar]
- 9.Kalyanaraman B, Mottley C, Mason RP. A direct electron spin resonance and spin-trapping investigation of peroxyl free radical formation by hematin/hydroperoxide systems. J. Biol. Chem. 1983;258:3855–3858. [PubMed] [Google Scholar]
- 10.Kalyanaraman B, Perez-Reyes E, Mason RP. Spin-trapping and direct electron spin resonance investigations of the redox metabolism of quinone anticancer drugs. Biochim. Biophys. Acta. 1980;630:119–130. doi: 10.1016/0304-4165(80)90142-7. [DOI] [PubMed] [Google Scholar]
- 11.Saprin AN, Piette LH. Spin trapping and its application in the study of lipid peroxidation and free radical production with liver microsomes. Arch. Biochem. Biophys. 1977;180:480–492. doi: 10.1016/0003-9861(77)90063-7. [DOI] [PubMed] [Google Scholar]
- 12.Yamazaki I, Piette LH. EPR spin-trapping study on the oxidizing species formed in the reaction of the ferrous ion with hydrogen peroxide. J. Am. Chem. Soc. 1991;113:7588–7593. [Google Scholar]
- 13.Yamazaki I, Piette LH. ESR spin trapping studies on the reaction of Fe 2+ ions with H2O2 reactive species in oxygen toxicity in biology. J. Biol. Chem. 1990;265:13589–13594. [PubMed] [Google Scholar]
- 14.Yamazaki I, Piette LH, Grover TA. Kinetic studies on spin trapping of superoxide and hydroxyl radicals generated in NADPH-cytochrome P-450 reductase-paraquat systems. Effect of iron chelates. J. Biol. Chem. 1990;265:652–659. [PubMed] [Google Scholar]
- 15.Kalyanaraman B, Felix CC, Sealy RC. Photoionization of Melanin precursors: an electron spin resonance investigation using the spin trap 5,5-dimethyl-1-pyrroline-1-oxide (DMPO) Photochem. Photobiol. 1982;36:5–12. [Google Scholar]
- 16.Kalyanaraman B, Karoui H, Singh RJ, Tordo P, Hogg N. Spin trapping of superoxide and carbon-centered radicals formed during autoxidation of tetrahydrobiopterin (abstract) Ann. Mtg. Oxygen Soc. 1995:32. [Google Scholar]
- 17.Kalyanaraman B, Joseph J, Parthasarathy S. The Spin Trap, α-phenyl N-tert-butylnitrone, inhibits the Oxidative Modification of Low Density Lipoprotein. FEBS Lett. 1991;280:17–20. doi: 10.1016/0014-5793(91)80194-8. [DOI] [PubMed] [Google Scholar]
- 18.Sheng P-G, Feix J, Kalyanaraman B. Charaterization of radical adducts formed during photochemical spin trapping in liposomes. Photochem. Photobiol. 1990;52:323–331. doi: 10.1111/j.1751-1097.1990.tb04188.x. [DOI] [PubMed] [Google Scholar]
- 19.Floyd RA, Wiseman BB. Spin-trapping free radicals in the autooxidation of 6-hydroxydopamine. Biochim. Biophys. Acta. 1979;586:196–207. [Google Scholar]
- 20.Floyd RA, Soong LM. Spin trapping in biological systems. Oxidation of the spin trap 5,5-dimethyl-1-pyrroline-1-oxide by a hydroperoxide-hematin system. Biochem. Biophys. Res. Commun. 1977;74:79–84. doi: 10.1016/0006-291x(77)91377-8. [DOI] [PubMed] [Google Scholar]
- 21.Floyd RA, Soong LM, Stuart MA, Reigh DL. Spin trapping of free radicals produced from nitrosoamine carcinogens. Photochem. Photobiol. 1978;28:857–862. doi: 10.1111/j.1751-1097.1978.tb07032.x. [DOI] [PubMed] [Google Scholar]
- 22.Poyer JL, Floyd RA, McCay PB, Janzen EG, Davis ER. Spin trapping of the trichloromethyl radical produced during enzymic NADPH oxidation in the presence of carbon tetrachloride or carbon bromotrichloromethane. Biochim. Biophys. Acta. 1978;539:402–409. doi: 10.1016/0304-4165(78)90044-2. [DOI] [PubMed] [Google Scholar]
- 23.Wong PK, Poyer JL, DuBose CM, Floyd RA. Hydralazine-dependent carbon dioxide free radical formation by metabolizing mitochondria. J. Biol. Chem. 1988;263:11296–11301. [PubMed] [Google Scholar]
- 24.Zs.-Nagy I, Floyd RA. Hydroxyl free radical reactions with amino acids and proteins studies by electron spin resonance spectroscopy and spin trapping. Biochim. Biophys. Acta. 1984;790:238–250. doi: 10.1016/0167-4838(84)90028-1. [DOI] [PubMed] [Google Scholar]
- 25.Lai EK, McCay PB, Noguchi T, Fong K-L. In vivo spin-trapping of trichloromethyl radicals formed from CCl4. Biochem. Pharmacol. 1979;28:2231–2235. doi: 10.1016/0006-2952(79)90212-0. [DOI] [PubMed] [Google Scholar]
- 26.Lee PY, McCay PB, Hornbrook KR. Evidence for CCl4-induced lipid peroxidation in mouse liver. Biochem. Pharmacol. 1982;31:405–409. doi: 10.1016/0006-2952(82)90189-7. [DOI] [PubMed] [Google Scholar]
- 27.McCay PB, Reinke LA. Detection of reactive free radicals in livers of ethanolfed rats: Potentiating effect of high fat diets. Drugs Affecting Lipid Metab. 1987:181–192. [Google Scholar]
- 28.Lai EK, Crossley C, Sridhar R, Misra HP, Janzen EG, McCay PB. In vivo spin trapping of free radicals generated in brain, spleen, and liver during gamma radiation of mice. Arch. Biochem. Biophys. 1986;244(1):156–160. doi: 10.1016/0003-9861(86)90104-9. [DOI] [PubMed] [Google Scholar]
- 29.Bolli R, Patel BS, Jeroudi MO, Lai EK, McCay PB. Demonstration of free radical generation on "stunned" myocardium of intact dogs with the use of the spin trap α-phenyl N-tert-butyl nitrone. J. Clin. Invest. 1988;82:476–485. doi: 10.1172/JCI113621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R, McCay PB. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc. Natl. Acad. Sci. USA. 1989;86:4695–4699. doi: 10.1073/pnas.86.12.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolli R, Patel BS, Jeroudi MO, Li X-Y, Triana JF, Lai EK, McCay PB. Iron-mediated radical reactions upon reperfusion contribute to myocardial "stunning". Am. J. Physiol. 1990:H1901–H1911. doi: 10.1152/ajpheart.1990.259.6.H1901. [DOI] [PubMed] [Google Scholar]
- 32.Bolli R, Jeroudi MO, Patel BS, Aruoma OI, Halliwell B, Lai EK, McCay PB. Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Circ. Res. 1989;65:607–622. doi: 10.1161/01.res.65.3.607. [DOI] [PubMed] [Google Scholar]
- 33.Bolli R, McCay PB. Use of spin traps in intact animals undergoing myocardial ischemia/reperfusion: A new approach to assessing the role of oxygen radicals in myocardial "stunning". Free Radic. Biol. Med. 1990;9:3–6. doi: 10.3109/10715769009145674. [DOI] [PubMed] [Google Scholar]
- 34.Chen G, Bray TM, Janzen EG, McCay PB. Excretion, metabolism and tissue distribution of a spin trapping agent, α-phenyl-N-tert-butyl-nitrone (PBN) in rats. Free Radic. Res. Commun. 1990;9:317–323. doi: 10.3109/10715769009145690. [DOI] [PubMed] [Google Scholar]
- 35.Chen G, Janzen EG, Bray TM, McCay PB. PBN and its applications in biology. In: Davies KJA, Ursini F, editors. The Oxygen Paradox. Padova, Italy: Cleup University Press; 1995. pp. 790–800. [Google Scholar]
- 36.Floyd RA. Protective action of nitrone-based free radical traps against oxidative damage to the central nervous system. Adv. Pharmacol. 1997;38:361–378. doi: 10.1016/s1054-3589(08)60991-6. [DOI] [PubMed] [Google Scholar]
- 37.Kotake Y. Pharmacologic properties of phenyl N-tert-butylnitrone. Antioxidants & Redox Signaling. 1999;1:481–499. doi: 10.1089/ars.1999.1.4-481. [DOI] [PubMed] [Google Scholar]
- 38.Floyd RA, Hensley K, Forster MJ, Kelleher-Anderson JA, Wood PL. Nitrones as neuroprotectants and antiaging drugs. Ann. N. Y. Acad. Sci. 2002;959:321–329. doi: 10.1111/j.1749-6632.2002.tb02103.x. [DOI] [PubMed] [Google Scholar]
- 39.Maples KR, Green AR, Floyd RA. Nitrone-related therapeutics: potential of NXY-059 for the treatment of acute ischaemic stroke. CNS. Drugs. 2004;18:1071–1084. doi: 10.2165/00023210-200418150-00003. [DOI] [PubMed] [Google Scholar]
- 40.Novelli G, Angiolini P, Cansales G, Lippi R, Tani R. Anti-shock action of phenyl-t-butyl-nitrone, a spin trapper. In: Novelli GP, Ursini F, editors. Oxygen Free Radicals in Shock. Florence: Karger, Basel; 1986. pp. 119–124. [Google Scholar]
- 41.Novelli GP, Angiolini P, Tani R, Consales G, Bordi L. Phenyl-T-butyl-nitrone is active against traumatic shock in rats. Free Radic. Res. Commun. 1985;1:321–327. doi: 10.3109/10715768609080971. [DOI] [PubMed] [Google Scholar]
- 42.Novelli GP, Anglolini P, Tani R. The Spin Trap Phenyl Butyl Nitrone Prevents Lethal Shock in the Rat. In: Poll G, Cheeseman KH, Dianzani MU, Slater TF, editors. Free Radicals in Liver Injury. Oxford: IRL Press Limited; 1986. pp. 225–228. [Google Scholar]
- 43.McKechnie K, Furman BL, Parratt JR. Modification by oxygen free radical scavengers of the metabolic and cardiovascular effects of endotoxin infusion in conscious rats. Circ. Shock. 1986;19:429–439. [PubMed] [Google Scholar]
- 44.Hamburger SA, McCay PB. Endotoxin-induced mortality in rats is reduced by nitrones. Circ. Shock. 1989;29:329–334. [PubMed] [Google Scholar]
- 45.Pogrebniak HW, Merino MJ, Hahn SM, Mitchell JB, Pass HI. Spin trap salvage from endotoxemia: The role of cytokine down-regulation. Surgery. 1992;112:130–139. [PubMed] [Google Scholar]
- 46.Floyd RA. Role of oxygen free radicals in carcinogenesis and brain ischemia. FASEB J. 1990;4:2587–2597. [PubMed] [Google Scholar]
- 47.Carney JM, Starke-Reed PE, Oliver CN, Landrum RW, Chen MS, Wu JF, Floyd RA. Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spacial memory by chronic administration of the spin-trapping compound N-tert-butyl-α-phenylnitrone. Proc. Natl. Acad. Sci. USA. 1991;88:3633–3636. doi: 10.1073/pnas.88.9.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carney JM, Floyd RA. Phenyl butyl nitrone compositions and methods for treatment of oxidative tissue damage. # 5,025,032 Filed: 10-07-89. [U.S. Patent] 1991 Jan 18;
- 49.Cao W, Carney JM, Duchon A, Floyd RA, Chevion M. Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci. Lett. 1988;88:233–238. doi: 10.1016/0304-3940(88)90132-2. [DOI] [PubMed] [Google Scholar]
- 50.Phillis JW, Clough-Helfman C. Protection from cerebral ischemic injury in gerbils with the spin trap agent N-tert-butyl-α-phenylnitrone (PBN) Neurosci. Lett. 1990;116:315–319. doi: 10.1016/0304-3940(90)90093-o. [DOI] [PubMed] [Google Scholar]
- 51.Cao X, Phillis JW. α-Phenyl-tert-butyl-nitrone reduces cortical infarct and edema in rats subjected to focal ischemia. Brain Res. 1994;644:267–272. doi: 10.1016/0006-8993(94)91689-6. [DOI] [PubMed] [Google Scholar]
- 52.Mori H, Arai T, Ishii H, Adachi T, Endo N, Makino K, Mori K. Neuroprotective effects of pterin-6-aldehyde in gerbil global brain ischemia: comparison with those of alpha-phenyl-N-tert-butyl nitrone. Neurosci. Lett. 1998;241:99–102. doi: 10.1016/s0304-3940(98)00010-x. [DOI] [PubMed] [Google Scholar]
- 53.Schulz JB, Henshaw DR, Siwek D, Jenkins BG, Ferrante RJ, Cipolloni PB, Kowall NW, Rosen BR, Beal MF. Involvement of free radicals in excitotoxicity in vivo. J. Neurochem. 1995;64:2239–2247. doi: 10.1046/j.1471-4159.1995.64052239.x. [DOI] [PubMed] [Google Scholar]
- 54.He Q-P, Smith M-L, Li P-A, Siesjo BK. Necrosis of the substantia nigra, pars reticulate in flurothys-induced status epilepticus is ameliorated by the spin trap a phenyl-N-butyl nitrone. Free Radical Biology & Medicine. 1997;22:917–922. doi: 10.1016/s0891-5849(96)00478-9. [DOI] [PubMed] [Google Scholar]
- 55.Floyd RA, Hensley K, Bing G. Evidence for enhanced neuro-inflammatory processes in neurodegenerative diseases and the action of nitrones as potential therapeutics. J. Neural. Trans. 2000;60:337–364. doi: 10.1007/978-3-7091-6301-6_28. [DOI] [PubMed] [Google Scholar]
- 56.Leib SL, Kim YS, Chow LL, Sheldon RA, Tauber MG. Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J. Clin. Invest. 1996;98:2632–2639. doi: 10.1172/JCI119084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marklund N, Hillered L. PhD Thesis Disertation of UPPSALA University. 2001. The role of reactive oxygen species in traumatic brain injury. Experimental studies in the rat. [Google Scholar]
- 58.Pitari G, Zingman L, Hodgson D, Alekseev A, Kazerounian S, Bienengraeber M, Hajniczky G, Terzic A, Waldman S. Bacterial enterotoxins are associated with resistance to colon cancer. Proc. Natl. Acad. Sci. USA. 2003;100:2695–2699. doi: 10.1073/pnas.0434905100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marklund N, Lewander T, Clausen F, Hillered L. Effects of the nitrone radical scavengers PBN and S-PBN on in vivo trapping of reactive oxygen species after traumatic brain injury in rats. J Cereb. Blood Flow Metab. 2001;21:1259–1267. doi: 10.1097/00004647-200111000-00002. [DOI] [PubMed] [Google Scholar]
- 60.Marklund N, Sihver S, Langstrom B, Bergstrom M, Hillered L. Effect of traumatic brain injury and nitrone radical scavengers on relative changes in regional cerebral blood flow and glucose uptake in rats. J Neurotrauma. 2002;19:1139–1153. doi: 10.1089/08977150260337958. [DOI] [PubMed] [Google Scholar]
- 61.Marklund N, Clausen F, Lewen A, Hovda DA, Olsson Y, Hillered L. alpha-Phenyl-tert-N-butyl nitrone (PBN) improves functional and morphological outcome after cortical contusion injury in the rat. Acta Neurochir. (Wien. ) 2001;143:73–81. doi: 10.1007/s007010170141. [DOI] [PubMed] [Google Scholar]
- 62.Lewen A, Skoglosa Y, Clausen F, Marklund N, Chan PH, Lindholm D, Hillered L. Paradoxical increase in neuronal DNA fragmentation after neuroprotective free radical scavenger treatment in experimental traumatic brain injury. J Cereb. Blood Flow Metab. 2001;21:344–350. doi: 10.1097/00004647-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 63.Fechter LD, Liu Y, Pearce TA. Cochlear protection from carbon monoxide exposure by free radical blockers in the guinea pig. Toxicol. Appl. Pharmacol. 1997;142:47–55. doi: 10.1006/taap.1996.8027. [DOI] [PubMed] [Google Scholar]
- 64.Tabatabaie T, Kotake Y, Wallis G, Jacob JM, Floyd RA. Spin trapping agent phenyl N-tert-butylnitrone protects against the onset of drug-induced insulin-dependent diabetes mellitus. FEBS Lett. 1997;407:148–152. doi: 10.1016/s0014-5793(97)00327-x. [DOI] [PubMed] [Google Scholar]
- 65.Parman T, Wiley MJ, Wells PG. Free radical-mediated oxidative DNA damage in the mechanism of thalidomide teratogenicity. Nature Med. 1999;5:582–585. doi: 10.1038/8466. [DOI] [PubMed] [Google Scholar]
- 66.Pedraza-Chaverri J, Tapia E, Bobadilla N. Ischemia-reperfusion induced acute renal failure in the rat is ameliorated by the spin-trapping agent α-phenyl-N-tert-butyl nitrone (PBN) Renal Failure. 1992;14:467–471. doi: 10.3109/08860229209047654. [DOI] [PubMed] [Google Scholar]
- 67.Ranchon I, LaVail MM, Kotake Y, Anderson RE. Free radical trap phenyl-N-tert-butylnitrone protects against light damage but does not rescue P23H and S334ter rhodopsin transgenic rats from inherited retinal degeneration. J. Neurosci. 2003;23:6050–6057. doi: 10.1523/JNEUROSCI.23-14-06050.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ranchon I, Chen S, Alvarez K, Anderson RE. Systemic administration of phenyl-n-tert-butylnitrone protects the retina from light damage. Invest. Ophthalmol. Vis. Sci. 2001;42:1375–1379. [PubMed] [Google Scholar]
- 69.Tomita H, Kotake Y, Anderson RE. Mechanism of protection from light-induced retinal degeneration by the synthetic antioxidant phenyl-N-tert-butylnitrone. Invest Ophthalmol. Vis. Sci. 2005;46:427–434. doi: 10.1167/iovs.04-0946. [DOI] [PubMed] [Google Scholar]
- 70.Saito K, Yoshioka H, Cutler RG. A spin trap, N-tert-butyl-α-phenylnitrone extends the life span of mice. BIOSCI. BIOTECHNOL. BIOCHEM. 1998;62:792–794. doi: 10.1271/bbb.62.792. [DOI] [PubMed] [Google Scholar]
- 71.Edamatsu R, Mori A, Packer L. The spin-trap N-tert-α-phenyl-butylnitrone prolongs the life span of the senescence accelerated mouse. Biochem. Biophys. Res. Commun. 1995;211:847–849. doi: 10.1006/bbrc.1995.1889. [DOI] [PubMed] [Google Scholar]
- 72.Sack CA, Socci DJ, Crandall BM, Arendash GW. Antioxidant treatment with phenyl-α-tert-butyl nitrone (PBN) improves the cognitive performance and survival of aging rats. Neurosci. Lett. 1996;205:181–184. doi: 10.1016/0304-3940(96)12417-4. [DOI] [PubMed] [Google Scholar]
- 73.Floyd RA. Oxidative damage to behavior during aging. Science. 1991;254:1597. doi: 10.1126/science.1684251. [DOI] [PubMed] [Google Scholar]
- 74.Miller RA, Harrison DE, Astle CM, Floyd RA, Flurkey K, Hensley KL, Javors MA, Leeuwenburgh C, Nelson JF, Ongini E, Nadon NL, Warner HR, Strong R. An aging Interventions Testing Program: study design and interim report. Aging Cell. 2007:565–575. doi: 10.1111/j.1474-9726.2007.00311.x. [DOI] [PubMed] [Google Scholar]
- 75.Strong R, Miller RA, Astle CM, Floyd RA, Flurkey K, Hensley KL, Javors MA, Leeuwenburgh C, Nelson JF, Ongini E, Nadon NL, Warner HR, Harrison DE. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell. 2008 doi: 10.1111/j.1474-9726.2008.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fan LW, Tien LT, Mitchell HJ, Rhodes PG, Cai Z. Alpha-phenyl-n-tert-butyl-nitrone ameliorates hippocampal injury and improves learning and memory in juvenile rats following neonatal exposure to lipopolysaccharide. Eur J Neurosci. 2008;27:1475–1484. doi: 10.1111/j.1460-9568.2008.06121.x. [DOI] [PubMed] [Google Scholar]
- 77.Zhao L, Chen YH, Wang H, Ji YL, Ning H, Wang SF, Zhang C, Lu JW, Duan ZH, Xu DX. Reactive oxygen species contribute to lipopolysaccharide-induced teratogenesis in mice. Toxicol Sci. 2008;103:149–157. doi: 10.1093/toxsci/kfn027. [DOI] [PubMed] [Google Scholar]
- 78.Hobbs CE, Oorschot DE. Neonatal Rat Hypoxia-Ischemia: Long-Term Rescue of Striatal Neurons and Motor Skills by Combined Antioxidant-Hypothermia Treatment. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Floyd RA, Hensley K, Forster MJ, Kelleher-Anderson JA, Wood PL. Nitrones, their value as therapeutics and probes to understand aging. Mech. of Ageing and Development. 2007;123:1021–1031. doi: 10.1016/s0047-6374(01)00385-2. [DOI] [PubMed] [Google Scholar]
- 80.Floyd RA. Nitrones as therapeutics in age-related diseases. Aging Cell. 2006;5:51–57. doi: 10.1111/j.1474-9726.2006.00189.x. [DOI] [PubMed] [Google Scholar]
- 81.Maples KR, Jordan SJ, Mason RP. In vivo rat hemoglobin thiyl free radical formation following administration of phenylhydrazine and hydrazine-based drugs. Drug Metab. Dispos. 1988;16:799–803. [PubMed] [Google Scholar]
- 82.Detweiler GD, Deterding LJ, Tomer KB, Chignell CF, Germolec D, Mason RP. Immunological identification of the heart myoglobin radical formed by hydrogen peroxide. Free Radic. Biol. Med. 2002;33:364–369. doi: 10.1016/s0891-5849(02)00895-x. [DOI] [PubMed] [Google Scholar]
- 83.Deterding LJ, Ramirez DC, Dubin JR, Mason RP, Tomer KB. Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. J. Biol Chem. 2004;279:11600–11607. doi: 10.1074/jbc.M310704200. [DOI] [PubMed] [Google Scholar]
- 84.Detweiler CD, Lardinois OM, Deterding LJ, de Montellano PR, Tomer KB, Mason RP. Identification of the myoglobin tyrosyl radical by immuno-spin trapping and its dimerization. Free Radic. Biol. Med. 2005;38:969–976. doi: 10.1016/j.freeradbiomed.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 85.Bhattacharjee S, Deterding LJ, Jiang J, Bonini MG, Tomer KB, Ramirez DC, Mason RP. Electron transfer between a tyrosyl radical and a cysteine residue in hemoproteins: spin trapping analysis. J Am. Chem Soc. 2007;129:13493–13501. doi: 10.1021/ja073349w. [DOI] [PubMed] [Google Scholar]
- 86.Lardinois OM, Detweiler CD, Tomer KB, Mason RP, Deterding LJ. Identifying the site of spin trapping in proteins by a combination of liquid chromatography, ELISA, and off-line tandem mass spectrometry. Free Radic. Biol. Med. 2008;44:893–906. doi: 10.1016/j.freeradbiomed.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramirez DC, Mejiba SE, Mason RP. Immuno-spin trapping of DNA radicals. Nat. Methods. 2006;3:123–127. doi: 10.1038/nmeth852. [DOI] [PubMed] [Google Scholar]
- 88.Bonini MG, Siraki AG, Atanassov BS, Mason RP. Immunolocalization of hypochlorite-induced, catalase-bound free radical formation in mouse hepatocytes. Free Radic. Biol. Med. 2007;42:530–540. doi: 10.1016/j.freeradbiomed.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ehrenshaft M, Mason RP. Protein radical formation on thyroid peroxidase during turnover as detected by immuno-spin trapping. Free Radic. Biol. Med. 2006;41:422–430. doi: 10.1016/j.freeradbiomed.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 90.Janzen EG, West MS, Poyer JL. Comparison of antioxidant activity of PBN with hindered phenols in initiated rat liver microsomal lipid peroxidation. In: Asada K, Toshikawa T, editors. Frontiers of Reactive Oxygen Species in Biology and Medicine. Elsevier Science; 1994. pp. 431–446. [Google Scholar]
- 91.Barclay LRC, Vinqvist MR. Do spin traps also act as classical chain-breaking antioxidants? A quantitative kinetic study of phenyl-tert-butylnitrone (PBN) in solution and in liposomes. Free Radic. Biol. Med. 2000;28:1079–1090. doi: 10.1016/s0891-5849(00)00197-0. [DOI] [PubMed] [Google Scholar]
- 92.Anderson DE, Yuan X-J, Tseng C-M, Rubin LJ, Rosen GM, Tod ML. Nitrone spin-traps block calcium channels and induce pulmonary artery relaxation independent of free radicals. Biochem. Biophys. Res. Commun. 1993;193:878–885. doi: 10.1006/bbrc.1993.1707. [DOI] [PubMed] [Google Scholar]
- 93.Milatovic D, Radic Z, Zivin M, Dettbarn W-D. Atypical effect of some spin trapping agents: reversible inhibition of acetylcholinesterase. Free Radic. Biol. Med. 2000;28:597–603. doi: 10.1016/s0891-5849(99)00270-1. [DOI] [PubMed] [Google Scholar]
- 94.Zivin M, Milatovic D, Dettbarn W-D. Nitrone spin trapping compound N-tert-butyl-α-phenylnitrone prevents seizures induced by anticholinesterases. Brain Res. 1999;850:63–72. doi: 10.1016/s0006-8993(99)02101-0. [DOI] [PubMed] [Google Scholar]
- 95.Milatovic D, Zivin M, Hustedt E, Dettbarn W-D. Spin trapping agent phenyl-N-tert-butylnitrone prevents diisopropylphosphorofluoridate-induced excitotoxicity in skeletal muscle of the rat. Neurosci. Lett. 2000;278:25–28. doi: 10.1016/s0304-3940(99)00904-0. [DOI] [PubMed] [Google Scholar]
- 96.Biegon A, Alvarado M, Budinger TF, Grossman R, Hensley K, West MS, Kotake Y, Ono M, Floyd RA. Region-selective effects of neuroinflammation and antioxidant treatment on peripheral benzodiazepine receptors and NMDA receptors in the rat brain. J. Neurochem. 2002;82:924–934. doi: 10.1046/j.1471-4159.2002.01050.x. [DOI] [PubMed] [Google Scholar]
- 97.Kotake Y, Sang H, Miyajima T, Wallis GL. Inhibition of NF-κB, iNOS mRNA, COX2 mRNA, and COX catalytic activity by phenyl-N-tert-butylnitrone (PBN) Biochim. Biophys. Acta. 1998;1448:77–84. doi: 10.1016/s0167-4889(98)00126-8. [DOI] [PubMed] [Google Scholar]
- 98.Miyajima T, Kotake Y. Spin trapping agent, phenyl N-tert-butyl nitrone, inhibits induction of nitric oxide synthase in endotoxin-induced shock in mice. Biochem. Biophys. Res. Commun. 1995;215:114–121. doi: 10.1006/bbrc.1995.2440. [DOI] [PubMed] [Google Scholar]
- 99.Endoh H, Kato N, Fujii S, Suzuki Y, Sato S, Kayama T, Kotake Y, Yoshimura T. Spin trapping agent, phenyl N-tert-butylnitrone, reduces nitric oxide production in the rat brain during experimental meningitis. Free Radic. Res. 2001;35:583–591. doi: 10.1080/10715760100301591. [DOI] [PubMed] [Google Scholar]
- 100.Sang H, Wallis GL, Stewart CA, Kotake Y. Expression of cytokines and activation of transcription factors in lipopolysaccharide-administered rats and their inhibition by phenyl N-tert-butylnitrone (PBN) Arch. Biochem. Biophys. 1999;363:341–348. doi: 10.1006/abbi.1998.1086. [DOI] [PubMed] [Google Scholar]
- 101.Cuevas P, Garcia-Calvo M, Carceller F, Reimers D, Zazo M, Cuevas B, Munoz-Willery I, Martinez-Coso V, Lamas S, Gimenez-Gallego G. Correction of hypertension by normalization of endothelial levels of fibroblast growth factor and nitric oxide synthase in spontaneously hypertensive rats. Proc. Natl. Acad. Sci. USA. 1996;93:11996–12001. doi: 10.1073/pnas.93.21.11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tabatabaie T, Graham KL, Vasquez AM, Floyd RA, Kotake Y. Inhibition of the cytokine-mediated inducible nitric oxide synthase expression in rat insulinoma cells by phenyl N-tert-butylnitrone. Nitric Oxide: Biology & Chemistry. 2000;4:157–167. doi: 10.1006/niox.2000.0281. [DOI] [PubMed] [Google Scholar]
- 103.Stewart CA, Hyam K, Wallis G, Sang H, Robinson KA, Floyd RA, Kotake Y, Hensley K. Phenyl-N-tert-butylnitrone demonstrates broad-spectrum inhibition of apoptosis-associated gene expression in endotoxin-treated rats. Arch. Biochem. Biophys. 1999;365:71–74. doi: 10.1006/abbi.1999.1159. [DOI] [PubMed] [Google Scholar]
- 104.Kotake Y, Kishida H, Nakae D, Floyd RA. Nitric Oxide Production by Primary Liver Cells Isolated from Amino Acid Diet Fed Rats. Methods Enzymol. 2005 doi: 10.1016/S0076-6879(05)96045-X. In Press. [DOI] [PubMed] [Google Scholar]
- 105.Floyd RA, Kotake Y, Towner RA, Guo W-X, Nakae D, Konishi Y. Nitric Oxide and Cancer Development. J. Toxicol Pathol. 2007;20:77–92. [Google Scholar]
- 106.Green AR, Ashwood T, Odergren T, Jackson DM. Nitrones as neuroprotective agents in cerebral ischemia, with particular reference to NXY-059. Pharmacology & Therapeutics. 2003;100:195–214. doi: 10.1016/j.pharmthera.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 107.Maples KR, Ma F, Zhang Y-K. Comparison of the radical trapping ability of PBN, S-PBN and NXY-059. Free Radic. Biol. Med. 2001;34:417–426. doi: 10.1080/10715760100300351. [DOI] [PubMed] [Google Scholar]
- 108.Marshall JW, Green AR, Ridley RM. Comparison of the neuroprotective effect of clomethiazole, AR-R15896AR and NXY-059 in a primate model of stroke using histological and behavioural measures. Brain Res. 2003;972:119–126. doi: 10.1016/s0006-8993(03)02511-3. [DOI] [PubMed] [Google Scholar]
- 109.Marshall JW, Cummings RM, Bowes LJ, Ridley RM, Green AR. Functional and histological evidence for the protective effect of NXY-059 in a primate model of stroke when given 4 hours after occlusion. Stroke. 2003;34:2228–2233. doi: 10.1161/01.STR.0000087790.79851.A8. [DOI] [PubMed] [Google Scholar]
- 110.Dehouck M-P, Cecchelli R, Green AR, Renftel M, Lundquist S. In vitro blood-brain barrier permeability and cerebral endothelial cell uptake of the neuroprotective nitrone compound NXY-059 in normoxic, hypoxic and ischemic conditions. Brain Res. 2002;995:229–235. doi: 10.1016/s0006-8993(02)03469-8. [DOI] [PubMed] [Google Scholar]
- 111.Edenius C, Strid S, Borga O, Breitholtz-Emanuelsson A, Vallen KL, Fransson B. Pharmacokinetics of NXY-059, a nitrone-based free radical trapping agent, in healthy young and elderly subjects. J Stroke Cerebrovasc. Dis. 2002;11:34–42. doi: 10.1053/jscd.2002.123973. [DOI] [PubMed] [Google Scholar]
- 112.Nilsson D, Wemer J, Cheng YF, Reinholdsson I, Englund G, Egberg N, Schulman S. NXY-059 does not affect bleeding time in healthy volunteers: a randomized, double-blind, placebo-controlled, 3-period crossover phase I study. J Clin. Pharmacol. 2007;47:264–272. doi: 10.1177/0091270006293752. [DOI] [PubMed] [Google Scholar]
- 113.Mutch NJ, Moore NR, Mattsson C, Jonasson H, Green AR, Booth NA. The use of the Chandler loop to examine the interaction potential of NXY-059 on the thrombolytic properties of rtPA on human thrombi in vitro. Br. J Pharmacol. 2008;153:124–131. doi: 10.1038/sj.bjp.0707543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cheng YF, Strid S, Borga O, Nilsson D, Wemer J. Active renal secretion of NXY-059, a novel neuroprotectant, is mediated via an organic acid transporter. J Clin. Pharmacol. 2007;47:909–914. doi: 10.1177/0091270007301803. [DOI] [PubMed] [Google Scholar]
- 115.Lees KR, Zivin JA, Ashwood T, Davalos A, Davis SM, Diener HC, Grotta J, Lyden P, Shuaib A, Hardemark HG, Wasiewski WW. NXY-059 for acute ischemic stroke. N. Engl. J. Med. 2006;354:588–600. doi: 10.1056/NEJMoa052980. [DOI] [PubMed] [Google Scholar]
- 116.Saver JL. Clinical impact of NXY-059 demonstrated in the SAINT I trial: derivation of number needed to treat for benefit over entire range of functional disability. Stroke. 2007;38:1515–1518. doi: 10.1161/01.STR.0000263135.47779.9e. [DOI] [PubMed] [Google Scholar]
- 117.Koziol JA, Feng AC. On the analysis and interpretation of outcome measures in stroke clinical trials: lessons from the SAINT I study of NXY-059 for acute ischemic stroke. Stroke. 2006;37:2644–2647. doi: 10.1161/01.STR.0000241106.81293.2b. [DOI] [PubMed] [Google Scholar]
- 118.Lees KR, Davalos A, Davis SM, Diener HC, Grotta J, Lyden P, Shuaib A, Ashwood T, Hardemark HG, Wasiewski W, Emeribe U, Zivin JA. Additional outcomes and subgroup analyses of NXY-059 for acute ischemic stroke in the SAINT I trial. Stroke. 2006;37:2970–2978. doi: 10.1161/01.STR.0000249410.91473.44. [DOI] [PubMed] [Google Scholar]
- 119.Shuaib A, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Diener HC, Ashwood T, Wasiewski WW, Emeribe U. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J Med. 2007;357:562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- 120.Diener HC, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Shuaib A, Ashwood T, Wasiewski W, Alderfer V, Hardemark HG, Rodichok L. NXY-059 for the treatment of acute stroke: pooled analysis of the SAINT I and II Trials. Stroke. 2008;39:1751–1758. doi: 10.1161/STROKEAHA.107.503334. [DOI] [PubMed] [Google Scholar]
- 121.Green AR. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br. J Pharmacol. 2008;153 Suppl 1:S325–S338. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ford GA. Clinical pharmacological issues in the development of acute stroke therapies. Br. J Pharmacol. 2008;153 Suppl 1:S112–S119. doi: 10.1038/sj.bjp.0707654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Feuerstein GZ, Zaleska MM, Krams M, Wang X, Day M, Rutkowski JL, Finklestein SP, Pangalos MN, Poole M, Stiles GL, Ruffolo RR, Walsh FL. Missing steps in the STAIR case: a Translational Medicine perspective on the development of NXY-059 for treatment of acute ischemic stroke. J Cereb. Blood Flow Metab. 2008;28:217–219. doi: 10.1038/sj.jcbfm.9600516. [DOI] [PubMed] [Google Scholar]
- 124.Savitz SI, Fisher M. Future of neuroprotection for acute stroke: in the aftermath of the SAINT trials. Ann. Neurol. 2007;61:396–402. doi: 10.1002/ana.21127. [DOI] [PubMed] [Google Scholar]
- 125.Savitz SI. A critical appraisal of the NXY-059 neuroprotection studies for acute stroke: a need for more rigorous testing of neuroprotective agents in animal models of stroke. Exp. Neurol. 2007;205:20–25. doi: 10.1016/j.expneurol.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 126.Savitz SI, Schabitz WR. A Critique of SAINT II: wishful thinking, dashed hopes, and the future of neuroprotection for acute stroke. Stroke. 2008;39:1389–1391. doi: 10.1161/STROKEAHA.107.504415. [DOI] [PubMed] [Google Scholar]
- 127.Wang CX, Shuaib A. Neuroprotective effects of free radical scavengers in stroke. Drugs Aging. 2007;24:537–546. doi: 10.2165/00002512-200724070-00002. [DOI] [PubMed] [Google Scholar]
- 128.Hsiao K, Chapman P, Nilsen S, Eckman C, Haigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 129.Nakae D, Kishida H, Enami T, Konishi Y, Hensley KL, Floyd RA, Kotake Y. Effects of phenyl N-tert-butyl nitrone and its derivatives on the early phase of hepatocarcinogenesis in rats fed a choline-deficient, l-amino acid-defined diet. Cancer Sci. 2003;94:26–31. doi: 10.1111/j.1349-7006.2003.tb01347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rattazzi M, Puato M, Faggin E, Bertipaglia B, Zambon A, Pauletto P. Creactive protein and interleukin-6 in vascular disease: culprits or passive bystanders? J Hypertens. 2003;21:1787–1803. doi: 10.1097/00004872-200310000-00002. [DOI] [PubMed] [Google Scholar]
- 131.Ghoshal AK, Farber E. Short communication: The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis. 1984;5:1367–1370. doi: 10.1093/carcin/5.10.1367. [DOI] [PubMed] [Google Scholar]
- 132.Rushmore TH, Ghazarian DM, Subrahmanyan V, Farber E, Grotta J. Probable free radical effects on rat liver nuclei during early hepatocarcinogenesis with a choline-devoid low methionine diet. Cancer Res. 1987;47:6731–6740. [PubMed] [Google Scholar]
- 133.Ghoshal AK, Rushmore TH, Lim YP, Farber E. Early detection of lipid peroxidation in the hepatic nuclei of rats fed a diet deficient in choline and methionine (CMD) Proc. Am. Assoc. Cancer Res. 1984;25:94. [Google Scholar]
- 134.Ghoshal AK, Laconi E, Willemsen F, Ghoshal A, Rushmore TH, Farber E. Modulation by Calcium of the Carcinogenic Process in the Liver Induced by a Choline-Deficient Diet. Can. J. Physiol. Pharmacol. 1987;65:478–482. doi: 10.1139/y87-082. [DOI] [PubMed] [Google Scholar]
- 135.Ghoshal AK, Rushmore TH, Buc-Calderon P, Roberfroid M, Farber E. Prevention by Free Radical Scavenger AD5 of Prooxidant Effects of Choline Deficiency. Free Radic. Biol. Med. 1990;8:3–7. doi: 10.1016/0891-5849(90)90137-8. [DOI] [PubMed] [Google Scholar]
- 136.Nakae D, Kotake Y, Kishida H, Hensley KL, Denda A, Kobayashi Y, Kitayama W, Tsujiuchi T, Sang H, Stewart CA, Tabatabaie T, Floyd RA, Konishi Y. Inhibition by phenyl N-tert-butyl nitrone on early phase carcinogenesis in the livers of rats fed a choline-deficient, L-amino acid-defined diet. Cancer Res. 1998;58:4548–4551. [PubMed] [Google Scholar]
- 137.Floyd RA, Kotake Y, Hensley K, Nakae D. Nitrone inhibition of cancer development. Oklahoma Medical Research Foundation. 819570[6,569,902] Oklahoma. 2003 Mar 28;
- 138.Floyd RA, Kotake Y, Hensley K, Nakae D, Konishi Y. Reactive oxygen pecies in choline deficiency induced carcinogenesis and nitrone inhibition. Mol. Cell. Biochem. 2002;234/235:195–203. [PubMed] [Google Scholar]
- 139.Rincavage HL, McDonnell DP, Kuhn CM. Expression of functional estrogen receptor β in locus coeruleus-derived cath.a cells. Endocrinology. 2003;144:2829–2835. doi: 10.1210/en.2002-221120. [DOI] [PubMed] [Google Scholar]
- 140.Torok NJ, Higuchi H, Bronk S, Gores GJ. Nitric oxide inhibits apoptosis downstream of cytochrome c release by nitrosylating caspase 9. Cancer Res. 2002;62:1648–1653. [PubMed] [Google Scholar]
- 141.Radisavljevic Z. Nitric oxide suppression triggers apoptosis through the FKHRL1 (FOXO3A)/ROCK kinase pathway in human breast carcinoma cells. Cancer. 2003;97:1358–1363. doi: 10.1002/cncr.10081. [DOI] [PubMed] [Google Scholar]
- 142.Salvucci O, Carsana M, Bersani I, Tragni G, Anichini A. Antiapoptotic role of endogenous nitric oxide in human melanoma cells. Cancer Res. 2001;61:318–326. [PubMed] [Google Scholar]
- 143.Kim Y-M, deVera ME, Watkins SC, Billiar TR. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-α-induced apoptosis by inducing heat shock protein 70 expression. J. Biol. Chem. 1997;272:1402–1411. doi: 10.1074/jbc.272.2.1402. [DOI] [PubMed] [Google Scholar]
- 144.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 145.Tzeng E, Billiar TR, Williams DL, Li J, Lizonova A, Kovesdi I, Kim YM. Adenovirus-mediated inducible nitric oxide synthase gene transfer inhibits hepatocyte apoptosis. Surgery. 1998;124:278–283. [PubMed] [Google Scholar]
- 146.Li J, Bombeck CA, Yang S, Kim Y-M, Billiar TR. Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J. Biol. Chem. 1999;274:17325–17333. doi: 10.1074/jbc.274.24.17325. [DOI] [PubMed] [Google Scholar]
- 147.Kim YM, Kim TH, Chung HT, Talanian RV, Yin XM, Billiar TR. Nitric oxide prevents tumor necrosis factor alpha-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. 2000;32:770–778. doi: 10.1053/jhep.2000.18291. [DOI] [PubMed] [Google Scholar]
- 148.Kim YM, Chung HT, Simmons RL, Billiar TR. Cellular non-heme iron content is a determinant of nitric oxide-mediated apoptosis, necrosis, and caspase inhibition. J. Biol Chem. 2000;275:10954–10961. doi: 10.1074/jbc.275.15.10954. [DOI] [PubMed] [Google Scholar]
- 149.Stolz DB, Zamora R, Vodovotz Y, Loughran PA, Billiar TR, Kim YM, Simmons RL, Watkins SC. Peroxisomal localization of inducible nitric oxide synthase in hepatocytes. Hepatology. 2002;36:81–93. doi: 10.1053/jhep.2002.33716. [DOI] [PubMed] [Google Scholar]
- 150.Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006;281:34124–34134. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- 151.Jaiswal M, LaRusso NF, Nishioka N, Nakabeppu Y, Gores GJ. Human Ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001;61:6388–6393. [PubMed] [Google Scholar]
- 152.gado-Esteban M, Martin-Zanca D, ndres-Martin L, Almeida A, Bolanos JP. Inhibition of PTEN by peroxynitrite activates the phosphoinositide-3-kinase/Akt neuroprotective signaling pathway. J Neurochem. 2007;102:194–205. doi: 10.1111/j.1471-4159.2007.04450.x. [DOI] [PubMed] [Google Scholar]
- 153.Yu CX, Li S, Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol. 2005;68:847–854. doi: 10.1124/mol.104.010504. [DOI] [PubMed] [Google Scholar]
- 154.Doblas S, Saunders D, Tesiram Y, Kshirsager P, Pye Q, Oblander J, Gordon B, Kosanke S, Floyd RA, Towner RA. Phenyl-tert-butyl-nitrone induces tumor regression and decreases angiogenesis in a C6 rat glioma model. Free Radical Biology & Medicine. 2007 doi: 10.1016/j.freeradbiomed.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 155.Vergely C, Renard C, Moreau D, Sarrado C, Roubaud V, Tuccio B, Rochette L. Effect of two new PBN-derived phosphorylated nitrones against postischaemic ventricular dysrhythmias. Fundam. Clin. Pharmacol. 2003;17:433–442. doi: 10.1046/j.1472-8206.2003.00158.x. [DOI] [PubMed] [Google Scholar]
- 156.Cobbs CS, Brenman JE, Aldape KD, Bredt DS, Israel MA. Expression of nitric oxide synthase in human central nervous system tumors. Cancer Res. 1995;55:727–730. [PubMed] [Google Scholar]
- 157.Lala PK, Chakraborty C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001;2:149–156. doi: 10.1016/S1470-2045(00)00256-4. [DOI] [PubMed] [Google Scholar]
- 158.Xie Q, Whisnant R, Nathan C. Promoter of the mouse gene encoding calciumin-dependent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med. 1993;177:1779–1784. doi: 10.1084/jem.177.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Asanuma T, Tesiram Y, Doblas S, Saunders D, Cranford R, Pearson J, Oblander J, Smith N, Kotake Y, Floyd RA, Towner RA. Assessment of the therapeutic effect of a free-radical trapping compound against C6 and F98 gliomas in rats with diffusion-tensor neuronal fiber tractography. J. Magn. Reson. 2008 doi: 10.1002/jmri.21474. In Press. [DOI] [PubMed] [Google Scholar]
- 160.Inanami O, Kuwabara M. alpha-Phenyl N-tert-butyl nitrone (PBN) increases the cortical cerebral blood flow by inhibiting the breakdown of nitric oxide in anesthetized rats. Free Radic. Res. 1995;23:33–39. doi: 10.3109/10715769509064017. [DOI] [PubMed] [Google Scholar]
- 161.Yamane H, Nakai Y, Takayama M, Iguchi H, Nakagawa T, Kojima A. Appearance of free radicals in the guinea pig inner ear after noise-induced acoustic trauma. Eur Arch. Otorhinolaryngol. 1995;252:504–508. doi: 10.1007/BF02114761. [DOI] [PubMed] [Google Scholar]
- 162.Seidman MD, Shivapuja BG, Quirk WS. The protective effects of allopurinol and superoxide dismutase on noise-induced cochlear damage. Otolaryngol. Head Neck Surg. 1993;109:1052–1056. doi: 10.1177/019459989310900613. [DOI] [PubMed] [Google Scholar]
- 163.Hu BH, Zheng XY, McFadden SL, Kopke RD, Henderson D. R-phenylisopropyladenosine attenuates noise-induced hearing loss in the chinchilla. Hear. Res. 1997;113:198–206. doi: 10.1016/s0378-5955(97)00143-3. [DOI] [PubMed] [Google Scholar]
- 164.Jacono AA, Hu B, Kopke RD, Henderson D, Van De Water TR, Steinman HM. Changes in cochlear antioxidant enzyme activity after sound conditioning and noise exposure in the chinchilla. Hear. Res. 1998;117:31–38. doi: 10.1016/s0378-5955(97)00214-1. [DOI] [PubMed] [Google Scholar]
- 165.Kopke R, Allen KA, Henderson D, Hoffer M, Frenz D, Van de WT. A radical demise. Toxins and trauma share common pathways in hair cell death. Ann. N. Y. Acad. Sci. 1999;884:171–191. doi: 10.1111/j.1749-6632.1999.tb08641.x. [DOI] [PubMed] [Google Scholar]
- 166.Evans P, Halliwell B. Free radicals and hearing. Cause, consequence, and criteria. Ann. N. Y. Acad. Sci. 1999;884:19–40. doi: 10.1111/j.1749-6632.1999.tb08633.x. [DOI] [PubMed] [Google Scholar]
- 167.Henderson D, Bielefeld EC, Harris KC, Hu BH. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006;27:1–19. doi: 10.1097/01.aud.0000191942.36672.f3. [DOI] [PubMed] [Google Scholar]
- 168.Ohlemiller KK, Wright JS, Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol. Neurootol. 1999;4:229–236. doi: 10.1159/000013846. [DOI] [PubMed] [Google Scholar]
- 169.Ohinata Y, Miller JM, Altschuler RA, Schacht J. Intense noise induces formation of vasoactive lipid peroxidation products in the cochlea. Brain Res. 2000;878:163–173. doi: 10.1016/s0006-8993(00)02733-5. [DOI] [PubMed] [Google Scholar]
- 170.Yamashita D, Jiang HY, Schacht J, Miller JM. Delayed production of free radicals following noise exposure. Brain Res. 2004;1019:201–209. doi: 10.1016/j.brainres.2004.05.104. [DOI] [PubMed] [Google Scholar]
- 171.Kopke RD, Coleman JK, Liu J, Campbell KC, Riffenburgh RH. Candidate's thesis: enhancing intrinsic cochlear stress defenses to reduce noise-induced hearing loss. Laryngoscope. 2002;112:1515–1532. doi: 10.1097/00005537-200209000-00001. [DOI] [PubMed] [Google Scholar]
- 172.Kopke R, Bielefeld E, Liu J, Zheng J, Jackson R, Henderson D, Coleman JK. Prevention of impulse noise-induced hearing loss with antioxidants. Acta Otolaryngol. 2005;125:235–243. doi: 10.1080/00016480410023038. [DOI] [PubMed] [Google Scholar]
- 173.Shi X, Nuttall AL. Upregulated iNOS and oxidative damage to the cochlear stria vascularis due to noise stress. Brain Res. 2003;967:1–10. doi: 10.1016/s0006-8993(02)04090-8. [DOI] [PubMed] [Google Scholar]
- 174.Shi X, Dai C, Nuttall AL. Altered expression of inducible nitric oxide synthase (iNOS) in the cochlea. Hear. Res. 2003;177:43–52. doi: 10.1016/s0378-5955(02)00796-7. [DOI] [PubMed] [Google Scholar]
- 175.Wang J, Pignol B, Chabrier PE, Saido T, Lloyd R, Tang Y, Lenoir M, Puel JL. A novel dual inhibitor of calpains and lipid peroxidation (BN82270) rescues the cochlea from sound trauma. Neuropharmacology. 2007;52:1426–1437. doi: 10.1016/j.neuropharm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 176.Cheng AG, Cunningham LL, Rubel EW. Mechanisms of hair cell death and protection. Curr. Opin. Otolaryngol. Head Neck Surg. 2005;13:343–348. doi: 10.1097/01.moo.0000186799.45377.63. [DOI] [PubMed] [Google Scholar]
- 177.Kopke RD, Weisskopf PA, Boone JL, Jackson RL, Wester DC, Hoffer ME, Lambert DC, Charon CC, Ding DL, McBride D. Reduction of noise-induced hearing loss using L-NAC and salicylate in the chinchilla. Hear. Res. 2000;149:138–146. doi: 10.1016/s0378-5955(00)00176-3. [DOI] [PubMed] [Google Scholar]
- 178.Kopke RD, Jackson RL, Coleman JK, Liu J, Bielefeld EC, Balough BJ. NAC for noise: from the bench top to the clinic. Hear. Res. 2007;226:114–125. doi: 10.1016/j.heares.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 179.Hight NG, McFadden SL, Henderson D, Burkard RF, Nicotera T. Noise-induced hearing loss in chinchillas pre-treated with glutathione monoethylester and R-PIA. Hear. Res. 2003;179:21–32. doi: 10.1016/s0378-5955(03)00067-4. [DOI] [PubMed] [Google Scholar]
- 180.Fechter LD, Gearhart C, Shirwany NA. Acrylonitrile potentiates noise-induced hearing loss in rat. JARO. 2004;5:90–98. doi: 10.1007/s10162-003-4028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fechter LD, Klis SF, Shirwany NA, Moore TG, Rao DB. Acrylonitrile produces transient cochlear function loss and potentiates permanent noise-induced hearing loss. Toxicol. Sci. 2003;75:117–123. doi: 10.1093/toxsci/kfg169. [DOI] [PubMed] [Google Scholar]
- 182.Rao D, Fechter LD. Protective effects of phenyl-N-tert-butylnitrone on the potentiation of noice-induced hearing loss by carbon monoxide. Toxicol. Appl. Pharmacol. 2000;167:125–131. doi: 10.1006/taap.2000.8995. [DOI] [PubMed] [Google Scholar]
- 183.Pouyatos B, Gearhart CA, Fechter LD. Acrylonitrile potentiates hearing loss and cochlear damage induced by moderate noise exposure in rats. Toxicol Appl. Pharmacol. 2005;204:46–56. doi: 10.1016/j.taap.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 184.Choi CH, Chen K, Vasquez-Weldon A, Jackson RL, Floyd RA, Kopke RD. Effectiveness of 4-hydroxy phenyl N-tert-butylnitrone (4-OHPBN) alone and in combination with other antioxidant drugs in the treatment of acute acoustic trauma in chinchilla. Free Radic. Biol. Med. 2008 doi: 10.1016/j.freeradbiomed.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 185.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc. Soc. Exp. Biol. Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 186.Mak IT, Weglicki WB. Potent antioxidant properties of 4-hydroxylpropranolol. J Pharmacol. Exp. Ther. 2004;308:85–90. doi: 10.1124/jpet.103.058032. [DOI] [PubMed] [Google Scholar]
- 187.Mak IT, Chmielinska JJ, Nedelec L, Torres A, Weglicki WB. D-propranolol attenuates lysosomal iron accumulation and oxidative injury in endothelial cells. J Pharmacol. Exp. Ther. 2006;317:522–528. doi: 10.1124/jpet.105.097709. [DOI] [PubMed] [Google Scholar]
- 188.Kramer JH, Murthi SB, Wise RM, Mak IT, Weglicki WB. Antioxidant and lysosomotropic properties of acute D-propranolol underlies its cardioprotection of postischemic hearts from moderate iron-overloaded rats. Exp. Biol. Med. (Maywood. ) 2006;231:473–484. doi: 10.1177/153537020623100413. [DOI] [PubMed] [Google Scholar]
- 189.Coleman JK, Kopke RD, Liu J, Ge X, Harper EA, Jones GE, Cater TL, Jackson RL. Pharmacological rescue of noise induced hearing loss using N-acetylcysteine and acetyl-L-carnitine. Hear. Res. 2007;226:104–113. doi: 10.1016/j.heares.2006.08.008. [DOI] [PubMed] [Google Scholar]