

Summary
Two genes are considered synthetic lethal if mutation of either alone allows cell viability, while simultaneous mutation leads to cell death. A synthetic lethal screen unveiled the dependency of Wnt/B-catenin-addicted colorectal cancer cells on vascular endothelial growth factor receptor-1 kinase activity and suggested a novel therapeutic approach for this malignancy.
Keywords: Synthetic lethality, Cell signaling, Receptor Tyrosine Kinase, Developmental Therapeutics, Gene Silencing
In this issue of Clinical Cancer Research, Naik and colleagues (1) provide the rationale for a synthetic lethal therapy theoretically based on targeted inhibition of VEGFR1-mediated kinase activity in Wnt/B-catenin-addicted human colorectal cancer (CRC) cells (Figure 1). Wnt is a cysteine rich secreted molecule and prototype of a 19-member family of proteins. It activates target genes primarily through binding and stabilization of the Frizzled/LRP6 membrane receptor complex. This complex sequestrates cytoplasmic Axin thus preventing it from mediating the ubiquitination and degradation of b-catenin. Stable b-catenin is phosphorylated and translocated to the nucleus, where it targets the TCF/LEF family of transcription factors and promotes expression of genes that regulate cell proliferation and cell polarity (2). A significant proportion of colorectal cancers have an activating mutation in the Wnt/b-catenin pathway resulting in the abnormal expression of proliferation mediators and growth factors (e.g. C-myc and VEGF) (3).
Figure 1.
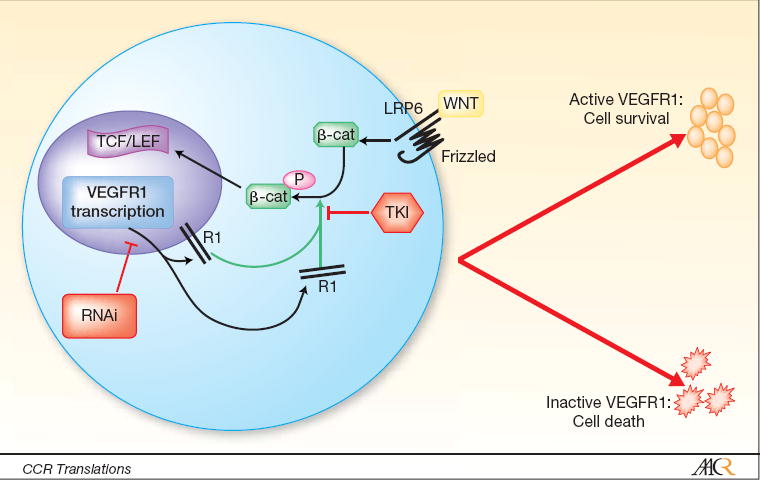
Wnt/b-catenin interaction with VEGFR1 (R1). Wnt binding to frizzled/LRP6 receptor complex induces accumulation of b-catenin, which is phosphorylated by VEGFR1. Phosphorylated b-catenin translocates to the nucleus and activates TCF/LEF transcription factor. Inhibition of VEGFR1 by RNA interference (RNAi) or tyrosine kinase inhibitors (TKI) in Wnt-addicted colon cancer cells results in cell death.
Naik and colleagues (1) expose a direct link between VEGFR1 function and the Wnt/b-catenin signaling pathway in CRC cells. To determine this linkage, they developed a Wnt/b-catenin screening protocol conceptually based on the phenomenon of synthetic lethality (4). The authors used a series of small inhibitory RNA (siRNA) and short hairpin RNA (shRNA) panels to interrogate a human embryonic kidney cell line (STF293) bearing a Wnt/b-catenin responsive reporter gene. RNA interference protocols have been introduced recently as synthetic lethal screens and have already begun to uncover other unexpected linkages between genes and survival pathways that show promise for therapeutic intervention (4, 5). Notably, this technique uncovers functional weaknesses of mutations specifically mediating survival of cancer cells thus allowing highly specific pharmacological targeting strategies.
The discoveries made by Naik and colleagues (1) shed new light on the complexities of VEGF receptor signaling and function. VEGF binds to both VEGFR1 and VEGFR2, the primary VEGF receptors involved with angiogenesis and vasculogenesis. VEGFR1 has weak tyrosine kinase activity compared to VEGFR2. Interestingly, VEGF binds to VEGFR2 with weaker affinity than to VEGFR1, but it results in far stronger VEGFR2 tyrosine kinase activity (6). The biologic relevance of the unique and reciprocal characteristics of these two receptors remains to be fully understood. We have recently reported that VEGF-dependent tumor angiogenesis involves the inverse and reciprocal regulation of VEGFR1 and VEGFR2 (7). VEGFR2 undergoes endocytosis, nuclear translocation, and downregulation via ubiquitination in response to VEGF induced signaling through the JNK/c-Jun pathway. In contrast, VEGF/VEGFR-1 signals through the Akt/ERK pathway to inhibit constitutive ubiquitination and induce rapid VEGFR1 protein accumulation in endothelial cells. Surprisingly, VEGFR1 is primarily localized intracellularly in endothelial cells (7, 8). VEGFR1 is strongly expressed in CRC cells where it also shows an intracellular localization (9) (LM Ellis, unpublished results). In light of the current observations by Naik and colleagues on the importance of VEGFR1 for the survival of CRC cells, these results bring to light the challenges in development of therapeutic agents targeting VEGFR1, particularly antibodies designed to inhibit VEGFR1 function.
Naik and colleagues (1) identify VEGFR1, amongst others, as a synthetic lethal partner to the Wnt signaling pathway. In confirmation of the screen data, VEGFR tyrosine kinase inhibitors were effective at inhibiting growth of Wnt/b-catenin addicted CRC cells but not control cells bearing normal Wnt function. Does this new observation fit in with any currently recognized VEGF receptor related mechanism? VEGFR2 is primarily expressed on endothelial cells whereas VEGFR1 has been detected on a variety of normal cell types but also on tumor cells including, notably, CRC cells (9). In endothelial cells, the pro-survival effect of VEGF depends on VEGFR1-mediated upregulation of Bcl-2 and inhibition of caspase-3 activity (7). Until Naik and colleagues demonstrated that inhibition of VEGFR1 leads to cell death via direct disruption of the Wnt/b-catenin survival pathway, the Wnt/bcatenin pathway was not known to be directly related to signaling by any VEGF receptor. However, Wnt has been shown to upregulate VEGF secretion in CRC cells in vitro (10). We perhaps begin to glimpse a possible autocrine survival strategy involving elements of a network traditionally thought to be primarily related to angiogenesis but exploited here by Wnt/b-catenin addicted CRC cells.
Current clinical studies of angiogenesis inhibitors, particularly VEGF targeted agents, have lead to variable results. Two classes of therapeutic approach warrant discussion: antibody-based drugs and non-peptidic small molecule inhibitors (11). The main benefit of antibody-based therapies is clear, in that they are highly specific and therefore, as a class, may have fewer off-target effects or clinical side effects than other drug types. Bevacizumab, a humanized monoclonal anti-VEGF antibody has been approved by the FDA after showing efficacy in combination with traditional chemotherapeutics (11) as well as when studied as a single agent in select cancers. However, for the inhibition of an intracellular target, antibodies suffer from physical chemistry, being too large to enter cells. If the very recent observations of VEGFR1 intracellular localization in endothelial cells and CRC cells are confirmed in vivo, anti-VEGFR1 antibodies will likely not be effective in CRC, unless they act through other cellular components of the tumor microenvironment (e.g. cancer associated fibroblasts or immune cells). In contrast, small molecule tyrosine kinase inhibitors may achieve direct inhibition of intracellular kinase activity, as demonstrated by Naik and colleagues. These low molecular weight drugs are readily modifiable to increase bioavailability, specificity and efficacy. However, off-target effects have been frequently observed with tyrosine kinase inhibitors, which hinders one’s ability to clearly understand their exact mechanisms of action.
For high specificity, we may ultimately seek gene silencing methodologies for effective and precise inhibition of VEGFR-1 pathway function. Some questions do exist regarding off-target effects of RNA based gene silencing methodologies although providentially these effects may be anti-angiogenic in their own right (12). The stringency of nucleotide complementarity is high, however tissue specific delivery is inherently absent in these molecules. Fortunately, RNAi is compatible with a number of modifiable clinically relevant and specific delivery systems, in particular viral packaging and liposome encapsulation. Notably, it is conceptually attractive to be able to identify intimately reliant tumor cell survival pathways using the same technology and mechanism, in this instance RNAi, that can be directly translated in vivo and potentially into clinical settings.
Everything being equal, inhibition of VEGF pathways is expected to attenuate angiogenesis and impair tumor growth. However, clinical experience is showing that this is frequently not the case. In light of the data described by Naik and colleagues (1) rational treatment with anti-VEGFR-1 antibodies may require the detailed study of cellular localization of the target receptors, as several receptors that are targets for therapy have been found to have intracellular functions independent of their kinase activities (13). Naik and colleagues (1) perhaps provide one additional explanation for the observed variable effects of anti-VEGF therapy, by linking the effect of VEGFR1 inhibition to the presence/absence of aberrant Wnt/B-catenin signaling. These exciting new observations reveal a new facet for selectivity of anti-tumor agents masquerading as anti-angiogenic agents, and underline the urgent need for deeper understanding of the biology of VEGF receptor signaling.
References
- 1.Naik S, Dothager RS, Marasa J, Lewis CL, Piwnica-Worms D. RNA Interference Screen Identifies VEGFR1 as Potentially Synthetic Lethal to Aberrant Wnt/β-catenin Activation in Colon Cancer. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-09-0336. in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang D, Du X. Crosstalk between tumor cells and microenvironment via wnt pathway in colorectal cancer dissemination. World J Gastroenterol. 2008;14:1823–7. doi: 10.3748/wjg.14.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scholl C, Frohling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 6.Roskoski R., Jr Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007;62:179–213. doi: 10.1016/j.critrevonc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z, Neiva K, Lingen MW, Ellis LM, Nör JE. VEGF-dependent tumor angiogenesis requires the inverse and reciprocal regulation of VEGFR1 and VEGFR2. Cell Death Diff. doi: 10.1038/cdd.2009.152. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mittar S, Ulyatt C, Howell GJ, et al. VEGFR1 receptor tyrosine kinase localization to the golgi apparatus is calcium-dependent. Exp Cell Res. 2009;315:877–89. doi: 10.1016/j.yexcr.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 9.Fan F, Wey JS, McCarty MF, et al. Expression and function of vascular endothelial growth factor receptor-1 on human colorectal cancer cells. Oncogene. 2005;24:2647–53. doi: 10.1038/sj.onc.1208246. [DOI] [PubMed] [Google Scholar]
- 10.Zhang X, Gaspard JP, Chung DC. Regulation of vascular endothelial growth factor by the wnt and K-ras pathways in colonic neoplasia. Cancer Res. 2001;61:6050–4. [PubMed] [Google Scholar]
- 11.Ellis LM, Hicklin DJ. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–91. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 12.Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008;452:591–7. doi: 10.1038/nature06765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weihua Z, Tsan R, Huang WC, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–93. doi: 10.1016/j.ccr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]