

Abstract
Cyclooxygenase-2 (Cox-2) plays a critical role in TCDD-induced hydronephrosis in mouse neonates. In this study we found that induction of Cox-2 by TCDD in MMDD1, a mouse macula densa cell line, is accompanied with a rapid increase in the enzymatic activity of cytosolic phospholipase A2 (cPLA2) as well as activation of protein kinases. Calcium serves as a trigger for such an action of TCDD in this cell line. These observations indicate that the basic mode of action of TCDD to induce the rapid inflammatory response in MMDD1 is remarkably similar to those mediated by the nongenomic pathway of aryl hydrocarbon receptor (AhR) found in other types of cells. Such an action of TCDD to induce Cox-2 in MMDD1 was not affected by “DRE decoy oligonucleotides” treatment or by introduction of a mutation on the DRE site of Cox-2 promoter, suggesting that this route of action of TCDD is clearly different from that mediated by the classical genomic pathway. An in vivo study with Ahrnls mouse model has shown that TCDD-induces Cox-2 and renin expression in the kidneys of the Ahrnls mice as well as Ahr+/− mice, but not in the Ahr−/− mice, indicating that this initial action of TCDD in mouse kidney does not require the translocation of AhR into the nucleus, supporting our conclusion that induction of Cox-2 by TCDD in mouse kidney is largely mediated by the nongenomic pathway of TCDD-activated AhR.
Keywords: TCDD, cyclooxygenase-2, nongenomic, aryl hydrocarbon receptor
1. Introduction
The epidemiological evidence has been accumulating to indicate that there is an upward trend in the prevalence of chronic kidney diseases [1]. Inflammation is one of the major factors associate with those diseases [2]. It is known that TCDD, a potent environmental pollutant ubiquitously present in our environment, causes inflammation of the kidney in rats [3] and mice [4] at relatively low doses. One of the well-known toxic end-points of TCDD poisoning is “hydronephrosis” occurring in fetuses and neonates of rodents [5-7]. Recently it has been reported by our research team that hydronephrosis is induced in mouse pups by exposing them to TCDD through its ingestion through mothers’ milk [8]. We also found that the amount of cyclooxygenase-2 (Cox-2) mRNA and as well as that of prostaglandin E2 (PGE2) expressed were conspicuously up-regulated in an AhR-dependent manner in the TCDD-induced hydronephrotic kidney, which were accompanied with a subsequent down-regulation of the mRNA expressions of Na-K-2Cl co-transporter (NKCC2) and renal outer medullary potassium channel (ROMK). Daily administration of a Cox-2 selective inhibitor, indomethacin N-octylamide to newborns until PND 7 completely abrogated the TCDD-induced PGE2 synthesis and the expressions of inflammatory cytokines and electrolyte transporters, and thereby it prevented the onset of hydronephrosis [8]. These findings have illustrated the pivotal role of Cox-2 in mediating the TCDD action to induce hydronephrosis through impairment of those ion-transporting functions of kidney tubules.
This finding brought up the possibility of using this phenomenon for the purpose of “proving the concept” of the significance of the nongenomic action pathway of the ligand-activated aryl hydrocarbon receptor (AhR) in inducing inflammatory cell responses. The question whether the classical genomic pathway, based on the binding of ligand-activated AhR to the dioxin response element (DRE) sequence on the promoter region of the target genes with the help of ARNT, is robust enough to explain all of the toxic actions of TCDD, or not, is a hotly debated topic in toxicology today [9-12]. In this regard, this phenomenon of Cox-2 triggered hydronephrosis offers the potential to serve as a tool to elucidate the actual inflammatory pathway involved in toxic signaling of the TCDD-activated AhR. The reason for the need for proposing such a brand new pathway is that there has not been a clear-cut theoretical framework through which this type of inflammation-related toxic action of TCDD could be fully explained, despite the intense efforts in the past to understand the mechanistic bases of toxic actions of TCDD. This may be surprising to many scientists in view of the tremendous progress made in this field on the molecular basis of ligand-bound AhR activating its target genes through the formation of a dimer with ARNT. Yet, it has been very difficult to fully explain the phenomenon of TCDD induced inflammatory cell responses on the basis of this classical genomic action model alone [13].
Our previous studies on the phenomenon of early inflammatory responses of MCF10A, a line of mammary epithelial cells to the action of TCDD, have revealed that activation of its trigger, a rapid increase in the intracellular concentration of free calcium ions is quickly followed by up-regulation of the enzymatic activity of cytosolic phospholipase A2 (cPLA2), and then by activation of Src kinase and induction of Cox-2, all occurring within 15 to 30 min [10]. Subsequent studies aimed at clearly recognizing this route of inflammatory action of the ligand-activated AhR, apart from that of activation of detoxification enzymes (i.e. through the classical pathway), have shown that the former route is mostly mediated by protein kinases as shown by the effectiveness of a number of protein kinase inhibitors to antagonize the inflammatory actions of TCDD, but not through its action to induce CYP1A1. Furthermore, it was found that there are other distinct characteristics of this newly-found pathway that clearly delineate this route from the classical pathway, including the lack of the participation of its nuclear dimerization partner, aryl hydrocarbon receptor nuclear translocator (ARNT). Based on those observations, it has been proposed by us that this newly-defined route of action of TCDD is designated as an alternative, nongenomic inflammation pathway [11]. This proposal has raised a number of new questions, including its toxicological meaning (e.g. whether it can be related to manifestation some specific toxic end-point of affected animals), and how such initial nongenomic signaling could be made persistent enough to cause chronic toxicities of TCDD.
Thus, we have decided to extend our previous research endeavor on hydronephrosis [8] to address the molecular basis of TCDD-induced Cox-2 activation in the kidney tubular cells. For this purpose, our work has been aided by the availability of MMDD1 cell line. MMDD1 cells are a clonally derived a macula densa cell line from SV-40 transgenic mice [14]. MMDD1 cells express COX-2 and bNOS, as well as two transport proteins NKCC2 and ROMK, which are the specific markers of macula densa [15, 16]. As a result, MMDD1 cells have been recognized to closely mimic the known molecular expression pattern of native macula densa cells [14]. Additionally the in vivo models made available to us are the AhR knockout mouse (Ahr−/−) and AhR nuclear localization sequence mutant mouse (Ahrnls). In the Ahrnls mice, three amino acids (Arg37, His38, and Arg39) in the nuclear localization sequence of AhR were replaced by Ala, Gly, and Ser, respectively. As a result, the AhR protein in the Ahrnls mice lost its ability to translocate into the nucleus [17]. The original strain of the Ahrnls mouse was backcrossed to C57BL/6 mice (i.e. with the high affinity Ahrb) by Dr Bradfield’s group. However, the embryonic stem cells from which the Ahrnls mice have originally been derived carry the d allele of AhR, which encodes a receptor with a lower binding affinity for TCDD. As a result, the AhR of the Ahrnls mice is still less responsive to TCDD, but it can serve as a good in vivo model to study the nongenomic pathway of AhR.
2. Material and Methods
2.1. Reagents and antibodies
TCDD (>99.99 % purity) was obtained from Dow Chemicals Co. (Midland, MI). 3′-methoxy-4′-nitroflavone (MNF) was a kind gift from Professor Josef Abel (University of Duesseldorf, Germany). 1-(p-chlorobenzoyl)-5-methoxy-2-methyl-1H-indole-3-acetic acid (indomethacin), 2′-Amino-3′-methoxyflavone (PD98059), 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), methylarachidonyl fluorophosphonate (MAFP), ethyleneglycol-bis(β-aminoethyl)-N,N,N’,N’-tetraacetoxymethyl ester (EGTA/AM) and Calcimycin (A23187), were purchased from Calbiochem (San Diego, CA). Rabbit polyclonal anti-Src, anti-ERK antibodies, and horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal anti-phospho-ERK1/2 (Thr 202/ Tyr 204) and anti-phospho-Src (Tyr 416) antibodies were purchased from Cell Signaling Technology (Danvers, MA).
2.2. Cell culture
MMDD1 cells were kindly provided by Dr. Jurgen Schnermann (National Institutes of Health, Bethesda, Maryland). They were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and antibiotics. Cells were routinely incubated at 37°C with 5% CO2 and medium was changed every two days.
2.3. Animal and treatment
Wild type C57BL/6J mice (Ahr+/+) and AhR knockout mice (Ahr−/−) were purchased from Jackson Laboratory (Bar Harbor, Maine). Heterozygous mice (Ahr+/−) were obtained by hybridization between Ahr+/+ and Ahr−/− mice. AhR nuclear localization sequence mutant mouse (Ahrnls) were kindly provided by Dr. Christopher Bradfield from the McArdle laboratory for Cancer Research at the University of Wisconsin (Madison, WI). Mice were housed (3 - 4 per cage) in a selective pathogen-free facility and humidity- and temperature-controlled room. The animals were maintained on a 12:12 h light/dark cycle and had free access to water and food according to the guidelines set by the University of California Davis. Mice were allowed to acclimate to the facility for at least one week. TCDD was administered via a single intraperitoneal injection of TCDD that was prepared from a stock solution and diluted in corn oil. Ahr+/− and Ahr−/− mice received a single dose of 15 μg/kg TCDD. Ahrnls mice carrying the lower affinity Ahrd allele received a single dose of 100 μg/kg TCDD. After 7 d, three to four animals from each group were scarified and the kidneys were excised and quickly frozen in liquid nitrogen and stored at −80°C for further analysis.
2.4. Quantitative reverse transcriptional PCR
Total RNA was extracted from cells using RNeasy Mini kit (Qiagen, Valencia, CA). Reverse transcription and qRT-PCR was carried out as described previously [18]. Briefly, 1 μg total RNA was mixed with and 40 pmol oligo-(dT)15 in a 10 μL total reaction volume. The mixture was run with an annealing program at 60°C for 5 min. After annealing, cDNA was synthesized using Omniscript Reverse Transcription kit (Qiagen) with an RT program (37°C for 60 min followed by 70°C for 10 min). To run PCR, 2 μL cDNA was mixed with 10 μL SYBRgreen (Qiagen, Valencia, CA) and 10 pmol of each primer in a 20 μL total reaction volume. PCR was then performed using a LightCycler (Roche Applied Science, Indianapolis, IN) with a PCR program (initial activation at 95°C for 15 min before first cycle, for each cycle, denaturation at 94°C for 15 sec, annealing at 59°C for 20 sec, and extension at 72°C for 20 sec). The data were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences for each gene are listed in Table 1.
Table 1.
primer sequences
| COX-2 | Forward | AGA AGG AAA TGG CTG CAG AA |
| Reverse | GCT CGG CTT CCA GTA TTG AG | |
| CYP1B1 | Forward | ATA GCC CTG GAT GAG GCT TT |
| Reverse | CCA AAT GAG GTT CGG CTT TA | |
| GAPDH | Forward | AAC TTT GGC ATT GTG GAA GG |
| Reverse | ACA CAT TGG GGG TAG GAA CA | |
| NKCC2 | Forward | CGT TTC CTT GCT CAG GTA GC |
| Reverse | GTG GTC TCC CAT GCA AAC TT | |
| ROMK | Forward | GGT AAG ACG GTG GAA GTG GA |
| Reverse | GCA GGA TGC TTC TGA ACA CA |
2.5. Prostaglandin E2 measurement
Cell culture medium was collected after treatment and centrifuged to remove floating cells. The Prostaglandin E2 (PGE2) level in cell culture medium was measured using a PGE2 EIA kit according to the manufacturer’s instructions (Cayman Chemical Co, Ann Arbor, MI).
2.6. DRE decoy oligonucleotides treatment
Cells were plated in 24-well culture plates. When the cells reached 60% confluence, they were transiently transfected with 0.25 μg DRE decoy oligonucleotides, containing the wild type DRE sequence (5′-GCCCCGGAGTTGCGTGAGAAGAGCCTGG-3′) or a mutant DRE sequence (5′-GCCCCGGAGTTGCGCGAGAAGAGCCTGG-3′) for 24 h using jetPEI (Qbiogene, Irvine, CA) according to the manufacturer’s instructions. After transfection, cells were incubated for another 2 h with 10 nM TCDD before RNA extraction and real-time RT-PCR analysis.
2.7. Plasmids and site-directed mutagenesis
The Cox-2 luciferase expression vector (pTIS10S) was kindly provided by Dr. Harvey Herschman (University of California, Los Angeles, CA). It contains the nucleotides −371 to +70 of the 5′-upstream regulatory sequence of the murine Cox-2 promoter and was cloned in a pXP2 plasmid (Promega, Madison, WI). The pTIS10S plasmid with the mutated C/EBP binding site 5′-CGGTTCTTGAGCACCTCACTGAA-3′ (A instead of C at −135 and C instead of A at −131) was kindly provided by Dr. Carol Pilbeam (University of Connecticut Health Center, Farmington, CT). A site-directed mutation in the DRE-binding site of the Cox-2 promoter was performed by a PCR-based technique using QuickChange Lightning site-directed mutagenesis kit (Stratagene, La Jolla, CA). Mutation of the core sequence to 5′-TGCGCG-3′ (C instead of T at −165) was confirmed by sequence analysis and shown to completely impair the AhR–ARNT binding.
2.8. Transient plasmid transfection
Cells were maintained in 24-well culture plates. After 16 h, standard medium was replaced with 1 mL medium without fetal calf serum. Cells were transiently transfected for 3 h using Transfectam (Promega, Madison, WI) with 1 μg of respective luciferase reporter constructs of the mouse Cox-2 promoter. After transfection, cells were incubated for another 6 h before treatment. Cells were washed twice with PBS and lysed with 250 μl lysis buffer (Promega, Madison, WI). Luciferase activities were measured with the Dual Luciferase Reporter Assay System (Promega, Madison, WI) using an automatic luminometer (Berthold Micro Lumat Plus). The luciferase activities were normalized to protein concentration.
2.9. Arachidonic acid release measurement
Arachidonic acid release was measured as described [19]. Cells were seeded in 12-well plates. When the cells reached 70% confluence, 0.2 μCi 3H-arachidonic acid (specific activity: 180 Ci/mmol) was added to each well and incubated overnight. The 3H-arachidonic-containing medium was discarded the next day and the cells were washed twice to remove residual 3H-arachidonic acid which was not incorporated into the cells. The cells were then treated with TCDD and/or additional chemical agents for specific time periods and thereafter the medium was collected and centrifuged to remove floating cells. The amount of arachidonic acid released from the cells into the medium was estimated by measuring the radioactivity of the medium using a liquid scintillation counter (Beckman Coulter Inc., Fullerton, CA).
2.10. Western blotting
Cells were rinsed twice with phosphate-buffered saline (PBS) and then lysed on ice with 200 μL cold RIPA buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM PMSF, 1 mM Na3VO4, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) for 30 min. The lysates were then centrifuged at 16,000 g at 4°C for 20 min and the supernatants were saved for electrophoresis. Proteins were then separated by 10% SDS-PAGE and transferred to PVDF membrane. The PVDF membrane was blocked with 5% non-fat milk in TBST (10 mM Tris-HCl, pH 8.0, 150 mM NaCl and 0.05% Tween-20) for 1 h at room temperature. The PVDF membrane was then incubated with primary antibody (1:500 dilution) in blocking buffer at 4°C for overnight. After incubation with horseradish peroxidase-conjugated secondary antibody (1:1000 dilution) in TBST for 3 h at room temperature, blots were developed using SuperSignal West Pico detection kit (Pierce, Rockford, IL). All western blottings were repeated at least three times for each experiment to confirm the reproducibility of the results.
2.11. Statistical Analysis
All data were obtained from at least three independent experiments and the results are given as the mean ± the standard error of the mean. To demonstrate statistical significance, the variables were examined for Student’s t test or ANOVA test. The level of significance was indicated by * (p<0.05) or ** (p<0.01).
3. Results
3.1. Comparison of the action of TCDD to induce Cox-2, CYP1B1 and IGFBP-1 expression in MMDD1 cells
To determine the sensitivity of this kidney tubular epithelial cell line to TCDD with respect to its response to activate Cox-2 mRNA expression, we first conducted a dose response study. The results showed that induction of Cox-2 by TCDD was dose-dependent and very sensitive (Fig. 1A). For instance, 1 nM TCDD could significantly induce Cox-2 expression after 24 h in this cell line. The induction of Cox-2 was higher when treated with 10 nM or 50 nM TCDD. A time course study has also been carried out to study the temporal change in the responsiveness of Cox-2 induction to TCDD in this cell line (Fig. 1B). It was found that induction of Cox-2 could be recognized as early as 1 h after 10 nM TCDD treatment. The expression of Cox-2 mRNA stayed elevated after 2 h, but started to decrease slightly after 6 h. However, its induction was still significant even 24 h after TCDD treatment. This pattern of induction of Cox-2 by TCDD was compared to two other genes, CYP1B1 and IGFBP-1. Although cytochrome P450s are well known to be induced by TCDD in a variety of cells and tissues, CYP1A1 was not detected in this cell line. Therefore CYP1B1 was chosen to compare with Cox-2. Additionally IGFBP-1 has been chosen to supplement CYP1B1 as the marker of genomic action of TCDD, since the former has been reported recently to be induced by TCDD through the classical action mechanism in mice [20]. Compared to Cox-2, induction of CYP1B1 by TCDD was slightly more sensitive, which was significantly induced by TCDD at the concentration of 0.1 nM (Fig. 1C). Otherwise, induction of IGFBP-1 showed a similar pattern of induction to TCDD comparable to Cox-2, both are significantly induced by TCDD at 1 nM (Fig. 1E). The temporal pattern of early induction of CYP1B1 and IGFBP-1 was also very similar to that of Cox-2 (Fig. 1D and 1F), starting 1 h after TCDD treatment, peaking at 2 h and started to decrease after 6 h. However, the temporal pattern of sustaining the state of elevated levels of IGFBP-1 expression at later time points was clearly different from other two in that the decrease after 6 h is much more pronounced in the case of IGFBP-1 induction.
Figure 1. Induction of Cox-2 after TCDD treatment and its effect on ROMK and NKCC2.

(A, C, E) MMDD1 cells were treated with TCDD of different concentrations as indicated for 24 h. (B, D, F) MMDD1 cells were treated with 10 nM TCDD for different time periods as indicated. The relative mRNA expression levels for Cox-2 (A, B), CYP1B1 (C, D) and IGFBP-1 (E, F) after TCDD treatment were shown and the statistically significant differences between control and TCDD treatment are indicated by * (p<0.05) or ** (p<0.01).
3.2. Effects of a Cox-2 inhibitor on the action of TCDD in MMDD1 cells
The importance of Cox-2 in TCDD-induced hydronephrosis has already been implicated in vivo [8, 21]. One proposed mechanism of pathogenesis of hydronephrosis is that the activated Cox-2 in the kidney tubular cells leads to down-regulation of the renal outer medullary potassium channel (ROMK) and the Na-K-2Cl co-transporter (NKCC2) expressions, since inhibition of Cox-2 did block their actions in vivo [8]. In this regard, the responsiveness of MMDD1 to sodium chloride has been considered to serve as a positive control in vitro for the purpose of illustrating the causal relationship between elevated Cox-2 and suppression of ROMK and NKCC2. Indeed, the pattern of NaCl-induced up-regulation of Cox-2 and down-regulation of ROMK and NKCC2 (Fig. 2A) appears to be qualitatively similar to the results we have seen in kidneys of TCDD-exposed neonates in vivo [8]. Interestingly, the expression of ROMK and NKCC2 in MMDD1 cells could also be down-regulated by TCDD (Fig. 2B-C), which supports the idea that MMDD1 cells can be served as a good in vitro model to study the hydronephrosis induced by TCDD. More importantly, the ability of TCDD to down-regulate the expression of ROMK and NKCC2 in this cell line disappeared when MMDD1 cells were pre-incubated with indomethacin, a compound known to inhibit Cox-2 activity (Fig. 2B-C). Although indomethacin is known to also inhibit Cox-1, it has been reported that Cox-2 is responsible for over 90% of prostaglandins release in MMDD1 cells [14]. Therefore, a more direct piece of evidence was procured to show that indomethacin did indeed block the 24 h action of 10 nM TCDD to increase the release prostaglandin E2 (PGE2) release from MMDD1 cells (Fig. 2D).
Figure 2. Effects of indomethacin on the action of TCDD on ROMK, NKCC2 and PGE2 release.

(A) MMDD1 cells were treated with 150 mM NaCl for 24 h and the relative mRNA expression levels were shown. (B, C) MMDD1 cells were pre-incubated with 50 μM indomethacin for 30 min followed by 10 nM TCDD for 24 h and the relative mRNA expression levels were shown. (D) MMDD1 cells were pre-incubated with 50 μM indomethacin for 1 h followed by 10 nM TCDD for 16 h and the amount of PGE2 released into culture medium was measured. The statistically significant differences between control and treatment (NaCl or TCDD) are indicated by * (p<0.05) or ** (p<0.01).
3.3. Effects of DRE decoy oligonucleotides treatment on the action of TCDD to induce Cox-2, CYP1B1 and IGFBP-1 expression in MMDD1 cells
To differentiate the routes of induction of Cox-2 from IGFBP-1 and/or CYP1B1 we have carried out a “DRE (dioxin response element) decoy oligonucleotides” treatment approach that has been developed in our laboratory [22]. Briefly, this approach aims at blocking only the classical genomic route of action of the ligand-activated AhR, by introducing excess amounts of oligonucleotides containing the DRE sequence, “GCGTG”, but leaving the alternative nongenomic pathway of action intact. As shown in Figure 3, pretreatment of MMDD1 with a large quantity of double-stranded oligonucleotides, containing the consensus DRE sequence did not eliminate the statistically significant action of TCDD to up-regulate Cox-2 (Fig. 3A) or CYP1B1 (Fig. 3B) mRNA expression. However, this DRE decoy oligonucleotides treatment clearly suppressed the action of TCDD to induce IGFBP-1 mRNA expression to the extent that in its presence the inducing effect of TCDD on this marker was no longer significant (Fig. 3C). In contrast, a mutated and therefore inactive DRE oligonucleotides (mDRE, GCGCG instead of GCGTG) treatment did not affect the action of TCDD to induce Cox-2 (Fig. 3A) or CYP1B1 (Fig. 3B) mRNA expression. To be sure, the mRNA expression of IGFBP-1 after TCDD treatment was reduced by 47% in the case of mDRE decoy treatment compared to no decoy treatment (Fig. 3C). However, the important point is that the induction of IGFBP-1 after TCDD treatment was still significant is the case of mDRE decoy treatment, unlike the case of DRE decoy treatment (Fig. 3C).
Figure 3. Effects of DRE decoy oligonucleotides treatment on the on the induction of Cox-2, CYP1B1 and IGFBP-1 by TCDD.

Cells were transfected with 0.25 μg DRE decoy oligonucleotides or mutant DRE decoy oligonucleotides for 24 h followed by 10 nM TCDD for 2 h. The gene expression of Cox-2 (A), CYP1B1 (B) and IGFBP-1 (C) were measured. The results are expressed as relative mRNA expression levels and the statistically significant differences between control and TCDD treatment are indicated by * (p<0.05) or ** (p<0.01).
3.4. Mutation studies on the mouse Cox-2 promoter in MMDD1 cells
The effectiveness of DRE decoy oligonucleotides to inhibit induction of IGFBP-1 by TCDD, but not induction of Cox-2, suggests that in this in vitro model the induction of Cox-2 is clearly different from that of IGFBP-1, which is known to be mostly dependent on the DRE binding site, i.e. the classic genomic pathway, as shown by other research groups [20, 23]. Since the promoter region of the mouse Cox-2 gene is known to contain a few responsive elements including a putative DRE site [24], we conducted another experiment to further study the induction of Cox-2 by TCDD in this cell line. The result of the luciferase reporter assay based on Cox-2 promoter analysis showed that introduction of a mutation to the DRE site of the mouse Cox-2 promoter did not affect the induction of Cox-2 by TCDD in this cell line (Fig. 4). In contrast, a mutation on the C/EBP site clearly inhibited the induction of Cox-2 mRNA. This result shows that binding of C/EBP proteins to the C/EBP site is a more important factor controlling the induction of Cox-2 by TCDD than AhR/ARNT dimer binding to DRE in this cell line.
Figure 4. Mutation studies on the mouse Cox-2 promoter.

MMDD1 cells were transiently transfected with wild type of the pTIS10S reporter plasmid containing the murine Cox-2 promoter sequences, as well as the DRE mutant and C/EBP mutant. Cells were treated with 10 nM TCDD or with dioxane vehicle (control) for 24 h. Results are expressed as luciferase activity normalized to protein concentration and the statistically significant differences between control and TCDD treatment are indicated by * (p<0.05) or ** (p<0.01).
3.5. Effects of selected inhibitory agents on the action of TCDD to induce of Cox-2, CYP1B1 and IGFBP-1 expression in MMDD1 cells
Next, we examined possible differences among the induction of Cox-2, CYP1B1 and IGFBP-1 using selected inhibitory agents. The action of MNF, an AhR antagonist, was unequivocal in all cases as expected (Fig. 5A-C), indicating the essential role of AhR in mediating these actions of TCDD. However, the effects of PD98059 (an MEK inhibitor), PP2 (a Src kinase inhibitor) and MAFP (a cPLA2 inhibitor) on the TCDD-induced mRNA expression of those three genes were clearly different. All three inhibitors successfully abolished the action of TCDD to up-regulate Cox-2 significantly (Fig. 5A). In contrast, MAFP did not affect the induction of CYP1B1 at all (Fig. 5B). In addition it was found that, although both PD98059 and PP2 pretreatment reduced the CYP1B1 expression in control and TCDD-treated samples, , neither PD98059 nor PP2 could totally abolish the ability of TCDD to induce CYP1B1 (Fig. 5B), unlike the case of Cox-2. Moreover, in the case of IGFBP-1, none of the three inhibitors abrogated the action of TCDD to induce this gene although PD98059 pretreatment evenly reduced the IGFBP-1 expression with or without TCDD treatment by 60% (Fig. 5C). The above results again illustrate the difference between the route of action of the ligand-activated AhR in terms of inducing the mRNA expression of Cox-2 mRNA and that of CYP1B1 or IGFBP-1.
Figure 5. Effects of selected inhibitory agents on the induction of Cox-2, CYP1B1 and IGFBP-1 by TCDD.

MMDD1 cells were pre-incubated with 10 μM MNF, 2 μM PD98059, 1 μM PP2 or 20 μM MAFP for 30 min followed by 10 nM TCDD for 2 h. The gene expression of Cox-2 (A), CYP1B1 (B) and IGFBP-1 (C) were measured. The results are expressed as relative mRNA expression levels and the statistically significant differences between control and TCDD treatment are indicated by * (p<0.05) or ** (p<0.01).
3.6. TCDD-induced rapid activation of cPLA2 in MMDD1 cells as measured by the release of arachidonic acid release in MMDD1 cells
The effectiveness of MAFP (Fig. 5) in antagonizing the early action of TCDD to induce Cox-2 mRNA expression implies the involvement of cytosolic phospholipase A2 (cPLA2) in the action of TCDD in this cell line. The activation of cPLA2 after TCDD treatment was directly measured by the release of free arachidonic acid. The results showed that TCDD did activate the enzymatic activity of cPLA2 in MMDD1 cells (Fig. 6). The activation of cPLA2, as assessed by this approach, took place as early as 15 min after TCDD treatment. MNF treatment abolished such an action of TCDD to activate cPLA2 (Fig. 6), which suggests that this TCDD-induced release of arachidonic acid requires AhR as expected.
Figure 6. Increased arachidonic acid release after TCDD treatment.
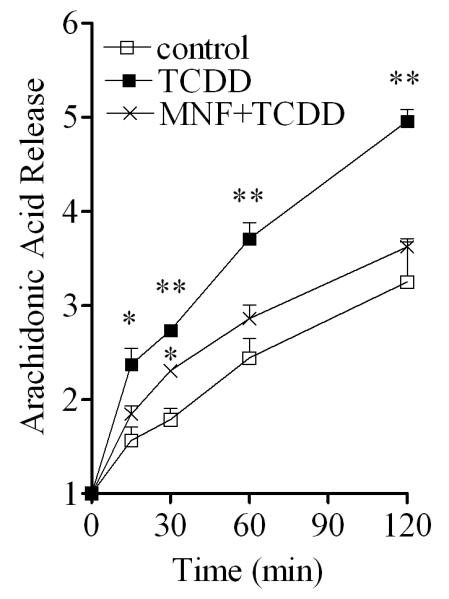
MMDD1 cells were pre-incubated with or without 10 μM MNF for 30 min followed by 10 nM TCDD for 2 h. Control was treated with vehicle solvent (p-dioxane) only. The amount of 3H-arachidonic acid release was measured at different time points after treatment. The results are expressed as relative values to the initial (0 min) background radioactivity for each treatment group. The statistically significant differences between the control and treatment (TCDD or MNF+TCDD) at the same time point are indicated by * (p<0.05) or ** (p<0.01).
3.7. Activation of ERK and Src kinase by TCDD or arachidonic acid inMMDD1 cells
The effectiveness of PD98059 and PP2 in attenuating the effect of TCDD to induce Cox-2 mRNA expression (Fig. 5A) implies the likely involvement of ERK and Src kinase in the action of TCDD. To confirm this interpretation at the protein level, Antibodies against the phosphorylated and therefore the activated forms of ERK and Src proteins were used. The effect of TCDD on ERK has been recognized by the increase of the intensity of the phosphorylated upper band (p44 or ERK 1) between 15 and 30 min, which was followed by the increase of the intensity of the phosphorylated lower band (p42 or ERK2) between 30 min and 60 min (Fig. 7A). In a similar manner TCDD was found to up-regulate the intensity of the active tyrosine phosphorylated (Try 416) form of Src kinase, which can be detected in 30 min and is sustained during this 120 min period of action of TCDD (Fig. 7A). More specifically, the TCDD-induced activation of ERK could be inhibited by MNF or PD98059, but not by PP2. In contrast, the action of TCDD to activate Src kinase was antagonized by all these three inhibitors (Fig. 7B). Interestingly, compared to TCDD, exogenously added arachidonic acid has a very similar effect to activate both ERK and Src kinase (Fig. 7C). These results, together with the previous one that TCDD activated cPLA2 shortly after TCDD treatment (Fig. 6), support our interpretation that TCDD-induced functional activation of both ERK and Src kinase in MMDD1 cells is mediated by arachidonic acid that is released by TCDD-activated cPLA2, and that subsequent activation of Cox-2 is mediated by these two protein kinases, which explains why the effect of TCDD to induce mRNA expression of Cox-2 is antagonized by both PD98059 and PP2.
Figure 7. Activation of ERK and Src kinase by TCDD.
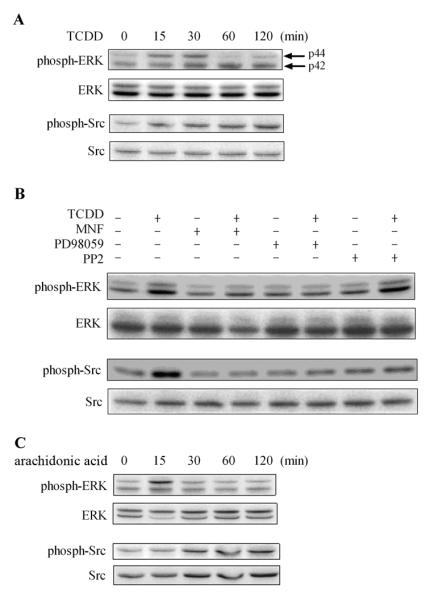
(A) MMDD1 cells were treated with 10 nM TCDD for different time periods as indicated. (B) MMDD1 cells were pre-incubated with 10 μM MNF, 2 μM PD98059 or 1 μM PP2 for 30 min followed by 10 nM TCDD for 30 min. (C) MMDD1 cells were treated with 10 μg/mL arachidonic acid for different time periods as indicated. The titer of phosphorylated form of the protein kinases in comparison to those of total protein kinases was measured by western blotting.
3.8. Sensitivity of the action of TCDD EGTA/AM in MMDD1 cells
Calcium has been shown to be involved in the action of TCDD through the nongenomic pathway in other types of cells [10, 12, 22]. Here, the involvement of calcium in the early nongenomic action of TCDD in this cell line was further tested using EGTA/AM, a permeable calcium chelator that is able to penetrate into the cell and binding to free calcium. The result showed that blocking calcium signaling by EGTA/AM totally abolished the action of TCDD to induce Cox-2 (Fig. 8A), as well as CYP1B1 (Fig. 8B). In the case of IGFBP-1, EGTA/AM pretreatment reduced the IGFBP-1 expression with or without TCDD. However, the induction of IGFBP-1 was still significant after TCDD even with EGTA/AM pretreatment (Fig. 8C). In fact, the induction ratios of IGFBP-1 by TCDD with or without EGTA/AM pretreatment were comparable (3.6 folds vs. 3.8 folds). Moreover, the activation of ERK and Src kinase induced by TCDD was also inhibited by EGTA/AM as expected, (Fig. 8D), indicating that the increase in the intracellular calcium concentration is likely the triggering event for activation of the entire nongenomic pathway as in other types of cells [25].
Figure 8. Effect of EGTA/AM on the action of TCDD.

MMDD1 cells were pre-incubated with 5 μM EGTA/AM for 30 min. (A-C) For mRNA measurement, cells were then treated with 10 nM TCDD for 2 h. The results are expressed as relative mRNA expression levels and the statistically significant differences between control and TCDD treatment are indicated by * (p<0.05) or ** (p<0.01). (D) For western blotting, cells were then treated with 10 nM TCDD for 30 min. The titer of phosphorylated form of the protein kinases in comparison to those of total protein kinases was measured by western blotting.
3.9. Induction of Cox-2 and activation of protein kinases by calcium ionophore in MMDD1 cells
In accordance with the above interpretation of the initial triggering role of calcium in the nongenomic action of TCDD in MMDD1 cells, the effect of artificial increase in intracellular calcium by treating the cells with A23187 (a calcium ionophore) was examined. This treatment produced similar effects to those induced by TCDD as expected (Fig. 9A). The temporal pattern of this induction was also very similar to that induced by TCDD except that the action of A23187 was stronger as compared to that of TCDD. Nevertheless A23187 was also able to activate ERK and Src kinase (Fig. 9B). The activation of ERK could be observed as early as 15 min after treatment and disappeared by 2 h after treatment. The activation of Src kinase lasted for a longer time span and the increase of the band intensity was still recognized 2 h after treatment.
Figure 9. Induction of Cox-2 and activation of ERK and Src kinase by A23187.

MMDD1 cells were treated with 1 μM A23187 for different time periods as indicated. (A) For mRNA measurement, the results are expressed as relative mRNA expression levels and the statistically significant differences between control and A23187 treatment are indicated by *(p<0.05) or ** (p<0.01). (B) For western blotting, the titer of phosphorylated form of the protein kinases in comparison to those of total protein kinases was measured by western blotting.
3.10. Studies on the increase of selected mRNA expression after TCDD treatment in mouse kidney samples from Ahrnls mice in comparison to that from Ahr+/− and Ahr−/− mice
To assess the significance of this newly-found alternative nongenomic pathway of the action of TCDD in kidney in vivo, we examined the induction of mRNA expression of several genes including CYP1A1, CYP1B1, COX-2 and renin in kidney samples from AhR nuclear localization sequence mutant mice (Ahrnls) in comparison to the heterozygous (Ahr+/−) as well as AhR knockout C57BL/6J mice (Ahr−/−). The TCDD-induced up-regulation of CYP1A1 mRNA expression in the kidney samples from Ahr+/− mice was very significant (more than 100-fold induction) 7 d after TCDD treatment. In contrast, kidney samples from Ahr−/− mice did not show any CYP1A1 induction and there was only a 2-fold induction of CYP1A1 in the kidney samples from Ahrnls mice (Fig. 10A). This set of results is not surprising, because the classical genomic pathway of ligand-activated AhR responsible for the induction of CYP1A1 is blocked in both AhR−/− and Ahrnls mice. However, the pattern is different in the case of CYP1B1 (Fig. 10B), Cox-2 (Fig. 10C) and renin (Fig. 10D). As expected, expressions of those mRNAs were significantly increased in the kidney samples from AhR+/− mice 7 d after TCDD, while no increase was seen in the kidney samples from AhR−/− mice. Interestingly, unlike the case of induction of CYP1A1, the levels of induction of CYP1B1, COX-2 and renin in the kidney samples from Ahrnls mice were qualitatively comparable to that from the Ahr+/− mice, although the Ahrnls mice received 6.67-fold higher dose (i.e. 100 μg/kg) than Ahr+/− mice did. The reason why the former received a higher dose of TCDD was because the Ahrnls mice carry the lower affinity Ahrd allele as compared to the high affinity Ahrb allele operating in the Ahr+/− strain.
Figure 10. Induction of selected genes in mouse kidney samples after 7-d TCDD treatment.

Heterozygous C57BL/6J mice (Ahr+/−), AhR knockout mice (Ahr−/−) and AhR nuclear localization sequence mutant mice (Ahrnls) were treated with TCDD for 7 d as described in material and methods. The genes expression levels of CYP1A1 (A), CYP1B1 (B), Cox-2 (C) and renin (D) in the kidney samples were tested. The results are expressed as relative mRNA expression levels and the statistically significant differences between control and TCDD treatment are indicated by * (p<0.05) or ** (p<0.01).
4. Discussion
The main reason why we have selected this study topic, to critically examine the molecular mechanism through which TCDD causes inflammation of kidney, has been that this C57BL/6 mouse model was successful in demonstrating, at least in the case of their neonates, Cox-2 is involved in the pathogenesis of TCDD-induced hydronephrosis. It is very important to re-emphasize here that the critical role of Cox-2 has already been demonstrated in the above in vivo model by the effectiveness of a Cox-2 specific inhibitor indomethacin N-octylamide to completely abrogate this action of TCDD to induce hydronephrosis. Furthermore, the critical role of AhR in this regard has been also shown by the lack of action of TCDD to cause hydronephrosis as well as inducing Cox-2 activation in neonates of AhR-null mice [8]. Therefore, in studying this current in vitro model, we have taken the strategy of maximally utilizing our previous discovery on TCDD-induced hydronephrosis being mediated by Cox-2 activation [8]. As for the likely mechanism through which Cox-2 induces hydronephrosis our main hypothesis has been that the down-regulation of ROMK and NKCC2 induced by the increased Cox-2 activity is likely to be the cause. In support of this hypothesis, previously it has been shown by other scientists that ROMK-deficient (i.e. knockout) mouse neonates develop the syndrome of hydronephrosis (i.e. Bartter’s syndrome) [26, 27]. Moreover, it was also shown that NKCC2-deficiency also serves as the cause for the etiology of hydronephrosis in mouse neonates [28]. Taken together, these findings clearly provide the basic information needed for this current study of ours to address the molecular mechanisms of this particular action of TCDD in that the impairment induced by TCDD through the activation of Cox-2 could indeed serve as the main cause for the etiology of hydronephrosis. In this regard, it is noteworthy that in the current study we could demonstrate that TCDD indeed causes significant down-regulation of the mRNA expression of both ROMK and NKCC2, and that indomethacin clearly antagonizes this action of TCDD (Fig. 2). It must be added that, although we used indomethacin, a non-selective inhibitor of Cox-2 and Cox-1, the dominance of the former in the production and release of prostaglandins in MMDD1 over the latter has been well documented [14]. Thus, together with the information on the similar action of NaCl to that of TCDD on ROMK and NKCC2, it is safe to conclude that Cox-2 is the likely the major target of TCDD in this cell model.
It is also important to point out that the previous in vivo study has not provided the sufficient information on the molecular mechanism how Cox-2 itself is induced by TCDD. This is the very reason why we have undertaken this study in vitro in an effort to gain the information on the action mechanism of TCDD directly in its proposed target cells in vitro, particularly from a specific viewpoint of recognizing the contribution of the alternative nongenomic pathway of action of TCDD-activated AhR as compared to that of its classical genomic actions [11, 25]. One of the advantages of having the appropriate in vitro model is that one can study the intricate process of cellular responses to the given toxicant directly in its target cells without the involvement of other types of cells such as macrophages as well as the effects of other factors such as endocrine hormones as well as cytokines and peptides that are known to influence the cellular state of inflammation. As for the appropriateness of macula densa cells as the possible target of action of TCDD, we have shown in our previous publication that TCDD causes an increase of prostaglandin E2, which can be produced by Cox-2 enzyme in the kidney tubular cells including macula densa cells in TCDD-treated neonates [8]. Indeed, in this in vitro model, we have found that TCDD, as well as high concentration of sodium chloride, clearly induces Cox-2 mRNA expression in this cell line (Fig. 1 and 2). More importantly, we could demonstrate that TCDD causes significant down-regulation of the expressions of ROMK as well as NKCC2, and that such an action of TCDD on these two markers could be antagonized by a Cox-2 inhibitor. Such an observation, reproducing the in vivo end-points of the action of TCDD in vitro, at least qualitatively, assured of the appropriateness of the MMDD1 as an in vitro model in studying the molecular mechanisms of the action of TCDD to induce inflammation through activation of Cox-2. According to Bell et al. [29], macula densa cells are renal sensor elements detecting changes in distal tubular fluid composition and transmitting signals to the glomerular vascular elements. To do so macula densa cells detect changes in luminal sodium chloride concentration through a complex series of ion transport-related intracellular events. Transport of sodium and chlorine ions via NKCC2 co-transporter leads to cell depolarization and increase in cytosolic calcium. Thus the action of TCDD to rapidly increase the intracellular concentration of free calcium as well as activation of Cox-2 in MMDD1 cells, as shown in other types of cells [25], is logically related to the suppression of ROMK and NKCC2.
Having ascertained the overall adequacy of the use of MMDD1 in our current investigation, we must discuss the meaning of the results of our current studies from the specific viewpoint of delineating the action of TCDD to activate Cox-2 through the alternative nongenomic pathway from that mediated by its classical action pathway (i.e. the DRE-based genomic action). This is not a trivial question for toxicologists considering the currently held view that most of actions of TCDD, if not all, are mediated by the classical pathway as attested to by the widely used toxic equivalence (TEQ) approach in assessing toxic actions of dioxin-like chemicals comprising many congeners, which are presumably act in an AhR-dependent manner.
In the current study our initial test results produced in vitro showed that the time course of TCDD-induced up-regulation of Cox-2 mRNA expression are remarkably similar to those of CYP1B1 and IGFBP-1 (Fig. 1). The message here is that the sensitivity of the nongenomic response to TCDD, at least in this cell material, is not less than that of classical, genomic response, clearly refuting a generally held view that the former may require higher doses of TCDD than the latter. It must be added that the latter two markers have been selected as the markers representing the end-result of the classical, genomic action of TCDD in view of the absence of the expression of CYP1A1 in MMDD1 cells. To this end, the differential inhibitory actions of DRE decoy oligonucleotides as well as chemical inhibitors including EGTA/AM, MAFP, PD98059 and PP2 on the action of TCDD to induce IGFBP-1 versus Cox-2 expression suggest the similarity of the temporal pattern is only superficial. While all those inhibitors totally abolished the effect of TCDD to cause up-regulation of the Cox-2 mRNA expression, none of those inhibitors could totally abolish the action of TCDD to induce CYP1B1 or IGFBP-1 (Fig. 5 and 8). Knowing that the induction of IGFBP-1 is largely dependent on the DRE site, i.e. the classical, genomic pathway [20, 23], it is understandable why its induction was not affected by those protein kinase inhibitors. Although it is known that induction of CYP1B1 also heavily depends on the DRE site, its induction is also known to be influenced by PKA [30]. That may be the reason why the induction of CYP1B1 is partially suppressed by some of kinase inhibitors (Fig. 5B) as well as being affected by EGTA-AM (Fig. 8B) unlike the case of IGFBP-1 expression (Fig. 5C and 8C). In contrast, the effectiveness of all the kinase inhibitors as well as EGTA-AM to block the induction of Cox-2 (Fig. 5A and 8A) suggests that the action of TCDD to induce Cox-2 is mediated by the nongenomic pathway in contrast to the case of IGFBP-1, of which induction is clearly mediated by the classical, genomic action. This conclusion is also supported by the observation that the mutation on the C/EBP site, not the DRE site of the promoter region, could abolish the action of TCDD to induce Cox-2 in MMDD1 cells (Fig. 4). Also, the rapid activation of cPLA2 as well as protein kinases including ERK and Src kinase by TCDD also supports the nongenomic theory because it is not possible for the genomic action to activate these kinases within 15 min.
Another common feature found throughout our studies on the characterization of this nongenomic pathway is the non-involvement of ARNT in this nongenomic pathway of ligand-activated AhR [25]. This lack of the involvement of ARNT has been confirmed in vivo using the Ahrnls mouse model. Compared to the Ahr+/− mice, which shows significant Cox-2 and CYP1A1 induction in the kidney after 7 d TCDD treatment, the induction of Cox-2 in the Ahrnls mice still exists after TCDD treatment while the induction of CYP1A1 disappears. This Ahrnls mice lack the capability to translocate AhR into the nucleus due to the mutation in the nuclear translocation sequence, and therefore it is not possible that the induction of Cox-2 is through the classical genomic pathway. Although there is a report showing that the Ahrnls mice are resistant to the toxic effects of TCDD and hence the nuclear translocation is needed in mediating the toxic effects of TCDD [17], our current results suggest that at least the initial phase of TCDD-induced activation of the alternative nongenomic pathway takes place without the translocation of the ligand-activated AhR into the nucleus and binding to ARNT.
The most important point of these in vitro findings is that they clearly support our main conclusion: i.e. qualitatively these data obtained from TCDD-treated MMDD1 cells are essentially identical to what found in MCF10A cells at least in terms of the overall patterns of the activation sequence of several key components [10], especially the activation of protein kinases and the involvement of calcium and cPLA2 in the early time. This overall similarity in the pattern of activation of the nongenomic, inflammatory pathway among several cell types [24] clearly supports our conclusion that the nongenomic pathway is the main route of action of TCDD to induce Cox-2 in kidney tubular cells. Our in vitro and in vivo study approaches in the current investigation have clearly helped us to establish that Cox-2 activation in the tubular cells of macula densa is due directly to the action of TCDD to activate this nongenomic pathway.
In conclusion, the current study results clearly provide the necessary pieces of evidence supporting the view that the nongenomic pathway of the TCDD-activated AhR plays an indispensable role in inducing inflammatory responses in kidney tubular cells through the induction of Cox-2 and renin and subsequent suppression of ROMK and NKCC2 in kidney, leading the final expression of the pathological development of hydronephrosis in affected mouse neonates. In view of the long held view that the concept of the classical action pathway is sufficient to explain most of toxic actions of TCDD, the evidence provided by the current study on the significance of the nongenomic pathway in inflammatory response of mouse kidney tubular cells merits the attention of toxicologists, who are interested in the whole spectrum of toxic actions of this class of enigmatic environmental pollutants.
Acknowledgements
This work was supported by research grants R01-ES05233 from the National Institute of Environmental Health Sciences, and in part by research grant, FAS0703859, helping our studies on MCF breast cancer cells, which served as the valuable model for this study.
We would like to thank Dr. Jurgen Schnermann (National Institutes of Health, Bethesda, MD) for providing the MMDD1 cells and Dr. Christopher Bradfield (University of Wisconsin, Madison, WI) for providing the Ahr−/− and Ahrnls mice. We also thank Dr. Harvey Herschman (University of California, Los Angeles, CA) for providing the Cox-2 luciferase expression vector (pTIS10S), and Dr. Carol Pilbeam (University of Connecticut Health Center, Farmington, CT) for providing the pTIS10S plasmid with the mutated C/EBP binding site.
Abbreviations
- AhR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor nuclear translocator
- Cox-2
cyclooxygenase-2
- cPLA2
cytosolic phospholipase A2
- CYP1A1
cytochrome P450 1A1
- CYP1B1
cytochrome P450 1B1
- DRE
dioxin responsive element
- ERK
extracellular signal-regulated kinase
- IGFBP-1
insulin-like growth factor binding protein-1
- NKCC2
Na-K-2Cl co-transporter
- ROMK
renal outer medullary potassium channel
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Chalmers L, Kaskel FJ, Bamgbola O. The role of obesity and its bioclinical correlates in the progression of chronic kidney disease. Adv Chronic Kidney Dis. 2006;13:352–64. doi: 10.1053/j.ackd.2006.07.010. [DOI] [PubMed] [Google Scholar]
- [2].Keller C, Katz R, Cushman M, Fried LF, Shlipak M. Association of kidney function with inflammatory and procoagulant markers in a diverse cohort: a cross-sectional analysis from the Multi-Ethnic Study of Atherosclerosis (MESA) BMC Nephrol. 2008;9:9. doi: 10.1186/1471-2369-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].National Toxicology Program (U.S.), United States. Dept. of Health and Human Services., United States. Public Health Service., National Institutes of Health (U.S.) NTP technical report on the toxicology and carcinogenesis studies of a mixture of 2,3,7,8-tetrachlorodibenzo- rho -dioxin (TCDD) (CAS no. 1746-01-6), 2,3,4,7,8-pentachlorodibenzofuran (PeCDF) (CAS no. 57117-31-4), and 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) (CAS no. 57465-28-8) in female Harlan Sprague-Dawley rats (gavage studies) National Toxicology Program National Institutes of Health Public Health Service U.S. Dept. of Health and Human Services; Research Triangle Park, N.C.: 2006. [Google Scholar]
- [4].Vogel CF, Nishimura N, Sciullo E, Wong P, Li W, Matsumura F. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch Biochem Biophys. 2007;461:169–75. doi: 10.1016/j.abb.2007.01.015. [DOI] [PubMed] [Google Scholar]
- [5].Abbott BD. Review of the interaction between TCDD and glucocorticoids in embryonic palate. Toxicology. 1995;105:365–73. doi: 10.1016/0300-483x(95)03234-7. [DOI] [PubMed] [Google Scholar]
- [6].Couture LA, Abbott BD, Birnbaum LS. A critical review of the developmental toxicity and teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin: recent advances toward understanding the mechanism. Teratology. 1990;42:619–27. doi: 10.1002/tera.1420420606. [DOI] [PubMed] [Google Scholar]
- [7].Gibson JE. Perinatal nephropathies. Environ Health Perspect. 1976;15:121–30. doi: 10.1289/ehp.7615121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nishimura N, Matsumura F, Vogel CF, Nishimura H, Yonemoto J, Yoshioka W, et al. Critical role of cyclooxygenase-2 activation in pathogenesis of hydronephrosis caused by lactational exposure of mice to dioxin. Toxicol Appl Pharmacol. 2008;231:374–83. doi: 10.1016/j.taap.2008.05.012. [DOI] [PubMed] [Google Scholar]
- [9].Dong B, Matsumura F. The conversion of rapid TCCD nongenomic signals to persistent inflammatory effects via select protein kinases in MCF10A cells. Mol Endocrinol. 2009;23:549–58. doi: 10.1210/me.2008-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dong B, Matsumura F. Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-tetrachlorodibenzo-p-dioxin through a nongenomic pathway in MCF10A cells. Molecular pharmacology. 2008;74:255–63. doi: 10.1124/mol.107.044669. [DOI] [PubMed] [Google Scholar]
- [11].Matsumura F, Puga A, Tohyama C. Biological functions of the arylhydrocarbon receptor: beyond induction of cytochrome P450s. Introduction to this special issue. Biochem Pharmacol. 2009;77:473. doi: 10.1016/j.bcp.2008.10.037. [DOI] [PubMed] [Google Scholar]
- [12].Sciullo EM, Vogel CF, Li W, Matsumura F. Initial and extended inflammatory messages of the nongenomic signaling pathway of the TCDD-activated Ah receptor in U937 macrophages. Arch Biochem Biophys. 2008;480:143–55. doi: 10.1016/j.abb.2008.09.017. [DOI] [PubMed] [Google Scholar]
- [13].Matsumura F. On the significance of the role of cellular stress response reactions in the toxic actions of dioxin. Biochem Pharmacol. 2003;66:527–40. doi: 10.1016/s0006-2952(03)00157-6. [DOI] [PubMed] [Google Scholar]
- [14].Yang T, Park JM, Arend L, Huang Y, Topaloglu R, Pasumarthy A, et al. Low chloride stimulation of prostaglandin E2 release and cyclooxygenase-2 expression in a mouse macula densa cell line. The Journal of biological chemistry. 2000;275:37922–9. doi: 10.1074/jbc.M006218200. [DOI] [PubMed] [Google Scholar]
- [15].Nielsen S, Maunsbach AB, Ecelbarger CA, Knepper MA. Ultrastructural localization of Na-K-2Cl cotransporter in thick ascending limb and macula densa of rat kidney. Am J Physiol. 1998;275:F885–93. doi: 10.1152/ajprenal.1998.275.6.F885. [DOI] [PubMed] [Google Scholar]
- [16].Xu JZ, Hall AE, Peterson LN, Bienkowski MJ, Eessalu TE, Hebert SC. Localization of the ROMK protein on apical membranes of rat kidney nephron segments. Am J Physiol. 1997;273:F739–48. doi: 10.1152/ajprenal.1997.273.5.F739. [DOI] [PubMed] [Google Scholar]
- [17].Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, et al. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J Biol Chem. 2003;278:17767–74. doi: 10.1074/jbc.M209594200. [DOI] [PubMed] [Google Scholar]
- [18].Park S, Dong B, Matsumura F. Rapid activation of c-Src kinase by dioxin is mediated by the Cdc37-HSP90 complex as part of Ah receptor signaling in MCF10A cells. Biochemistry. 2007;46:899–908. doi: 10.1021/bi061925f. [DOI] [PubMed] [Google Scholar]
- [19].Viu E, Zapata A, Capdevila JL, Fossom LH, Skolnick P, Trullas R. Glycine site antagonists and partial agonists inhibit N-methyl-D-aspartate receptor-mediated [3H]arachidonic acid release in cerebellar granule cells. The Journal of pharmacology and experimental therapeutics. 1998;285:527–32. [PubMed] [Google Scholar]
- [20].Murray IA, Perdew GH. Omeprazole stimulates the induction of human insulin-like growth factor binding protein-1 through aryl hydrocarbon receptor activation. J Pharmacol Exp Ther. 2008;324:1102–10. doi: 10.1124/jpet.107.132241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Seibert K, Masferrer JL, Needleman P, Salvemini D. Pharmacological manipulation of cyclo-oxygenase-2 in the inflamed hydronephrotic kidney. Br J Pharmacol. 1996;117:1016–20. doi: 10.1111/j.1476-5381.1996.tb16691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li W, Matsumura F. Significance of the nongenomic, inflammatory pathway in mediating the toxic action of TCDD to induce rapid and long-term cellular responses in 3T3-L1 adipocytes. Biochemistry. 2008;47:13997–4008. doi: 10.1021/bi801913w. [DOI] [PubMed] [Google Scholar]
- [23].Marchand A, Tomkiewicz C, Marchandeau JP, Boitier E, Barouki R, Garlatti M. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces insulin-like growth factor binding protein-1 gene expression and counteracts the negative effect of insulin. Mol Pharmacol. 2005;67:444–52. doi: 10.1124/mol.104.004010. [DOI] [PubMed] [Google Scholar]
- [24].Vogel C, Boerboom AM, Baechle C, El-Bahay C, Kahl R, Degen GH, et al. Regulation of prostaglandin endoperoxide H synthase-2 induction by dioxin in rat hepatocytes: possible c-Src-mediated pathway. Carcinogenesis. 2000;21:2267–74. doi: 10.1093/carcin/21.12.2267. [DOI] [PubMed] [Google Scholar]
- [25].Matsumura F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem Pharmacol. 2009;77:608–26. doi: 10.1016/j.bcp.2008.10.013. [DOI] [PubMed] [Google Scholar]
- [26].Lorenz JN, Baird NR, Judd LM, Noonan WT, Andringa A, Doetschman T, et al. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome. J Biol Chem. 2002;277:37871–80. doi: 10.1074/jbc.M205627200. [DOI] [PubMed] [Google Scholar]
- [27].Lu M, Wang T, Yan Q, Yang X, Dong K, Knepper MA, et al. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. J Biol Chem. 2002;277:37881–7. doi: 10.1074/jbc.M206644200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Takahashi N, Chernavvsky DR, Gomez RA, Igarashi P, Gitelman HJ, Smithies O. Uncompensated polyuria in a mouse model of Bartter’s syndrome. Proc Natl Acad Sci U S A. 2000;97:5434–9. doi: 10.1073/pnas.090091297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bell PD, Lapointe JY, Peti-Peterdi J. Macula densa cell signaling. Annu Rev Physiol. 2003;65:481–500. doi: 10.1146/annurev.physiol.65.050102.085730. [DOI] [PubMed] [Google Scholar]
- [30].Zheng W, Jefcoate CR. Steroidogenic factor-1 interacts with cAMP response element-binding protein to mediate cAMP stimulation of CYP1B1 via a far upstream enhancer. Mol Pharmacol. 2005;67:499–512. doi: 10.1124/mol.104.005504. [DOI] [PubMed] [Google Scholar]