

Abstract
The majority of known human tumor-associated antigens derive from non-mutated self proteins. T cell tolerance, essential to prevent autoimmunity, must therefore be cautiously circumvented to generate cytotoxic T cell responses against these targets. Our strategy uses DNA fusion vaccines to activate high levels of peptide-specific CTL. Key foreign sequences from tetanus toxin activate tolerance-breaking CD4+ T cell help. Candidate MHC class I binding tumor peptide sequences are fused to the C terminus for optimal processing and presentation. To model performance against a leukemia-associated antigen in a tolerized setting, we constructed a fusion vaccine encoding an immunodominant CTL epitope derived from Friend murine leukemia virus gag protein (FMuLVgag) and vaccinated tolerant FMuLVgag-transgenic (gag-Tg) mice. Vaccination with the construct induced epitope-specific IFN-γ-producing CD8+ T cells in normal and gag-Tg mice. The frequency and avidity of activated cells were reduced in gag-Tg mice, and no autoimmune injury resulted. However, these CD8+ T cells did exhibit gag-specific cytotoxicity in vitro and in vivo. Also, epitope-specific CTL killed FBL-3 leukemia cells expressing endogenous FMuLVgag antigen and protected against leukemia challenge in vivo. These results demonstrate a simple strategy to engage anti-microbial T cell help to activate epitope-specific polyclonal CD8+ T cell responses from a residual tolerized repertoire.
Keywords: T cells, Tolerance, Transgenic mice models, Tumor immunology, Vaccination
Introduction
There is clear clinical evidence from recipients of allogeneic hematopoietic transplants that CD8+ T cells reactive with leukemic cells can be curative [1]. To build on this success, passive transfer of autologous, allogeneic or engineered CD8+ T cells against a range of target antigens is now being tested [2]. Such studies should provide proof of principle and validation of selected targets, but the technical demands of this approach are high, and questions still remain regarding the nature of the cell to be transferred and the ability of cells extensively expanded in vitro to survive, divide, and migrate in vivo [2]. An alternative approach is to actively induce an immune response in the tumor-bearing host, but the ability to achieve this has been questioned [3]. A major obstacle derives from the limited capacity of patients to respond if the available repertoire has been weakened by the processes of tolerance. However, the fact that cytotoxic CD8+ T cells (CTL) can be induced in vitro against a range of normally expressed peptides, and that these can kill tumor cells, suggests that deletional tolerance is incomplete in humans [4]. In addition, there is clinical evidence that tumor-specific T cells can be induced in vivo against self proteins, although whether the magnitude and avidity of the responses will be sufficient to control tumor growth is less clear [5].
For candidate tumor antigens that are self proteins, it remains likely that the highest-avidity cells have been deleted and the remaining potentially reactive repertoire is subject to peripheral tolerance mechanisms [6-8]. Therefore, the challenge is to design vaccines that can overcome the tolerogenic pressure on the residual repertoire. Once rescued from a tolerant state, it is essential that these T cells, likely of intermediate or low avidity, can recognize and kill tumor cells expressing the targeted antigen. Numerous vaccination strategies are being actively pursued [9]. We have focused on genebased vaccines since they provide a flexible platform for expressing antigens together with molecules able to direct and amplify immunity [10]. Clinical use of optimized DNA vaccines could follow from murine studies but translation to human subjects has had to address two problems. The first was a concern that stimulation of innate immunity via backbone CpG sequences might fail due to differences in expression of TLR9 between mice and humans [11]. However, DNA vaccines retain immunogenicity in TLR9-knockout mice [12], pointing to TLR9-independent pathways of immune activation. The second obstacle reflected the dependence of transfection level on the volume injected, which posed problems for scaling up from mice to human subjects [13-15]. Adequate antigen levels are required especially at boosting, and difficulty in achieving high transfection efficiencies could fail to boost responses in larger hosts [15]. To overcome these problems, and to add an inflammatory stimulus, viral or bacterial vectors are being examined [16]. New physical methods (e.g. electroporation) also appear to be effective and have the advantage, compared to pathogen-based vectors, of not inducing blocking immunity against the vector [13, 14, 17]. Electroporation is highly effective in large animals [18] and we are currently testing it in the clinic, with no ill effects noted. Importantly, DNA vaccines appear safe, and, based on their potential and safety profile, are now in pilot clinical trials [19, 20]. We have developed DNA fusion vaccines which encode two functional elements: one to stimulate CD4+ T cell help using a microbial sequence, in our case a domain (DOM) of fragment C (FrC) from tetanus toxin (TT), and the other consisting of a candidate tumor-specific MHC class I-binding peptide, fused to the C terminus (p.DOM-peptide) [21]. The microbial component (DOM) was minimized to reduce the potential for inclusion of immunodominant CTL epitopes that could compete with the tumor-specific peptide [22]. Crucially, this design engages CD4+ T cells from the non-tolerized anti-microbial repertoire. Induced responses against the fused MHC class I-binding epitope were shown to depend on T cell help [23], a key requirement for the induction and maintenance of effective CD8+ T cell immunity [24-26]. The ability of the cellular machinery to dissect out candidate tumor epitopes from the backbone sequence of the encoded antigen presents a limiting factor for presentation and immune induction [27]. We have overcome this by cutting out the tumor-derived CTL epitope, placing it downstream of the microbial DOM sequence to ensure optimal processing and presentation [27]. We have demonstrated that this strategy activates high levels of epitope-specific CD8+ T cells and establishes a durable memory response, leading to protective immunity in pre-clinical tumor models [22, 27-29]. These studies also revealed that it was possible to mobilize CD8+ T cells reactive with a male (H-Y)- specific epitope from a profoundly tolerized repertoire in male mice [28]. However, the induced CD8+ T cells were unable to kill male splenocytes [28].
To model the performance of this strategy against a leukemia-associated antigen which is also a self protein, we have focused on the immunodominant CTL epitope (gag85–93) derived from the Friend murine leukemia virus (FMuLV) gag protein (FMuLVgag) expressed by FBL-3, an FMuLV-transformed mouse leukemia [30, 31]. Previously we generated a transgenic mouse model in which FMuLVgag is expressed in the liver as a normal self protein (gag-Tg mice). Tolerance to gag was evident from the fact that, in contrast to normal C57BL/6 (B6) mice, “conventional” vaccination with FBL-3 tumor cells failed to generate a CD8+ T cell response to FMuLVgag or to induce resistance to tumor [32]. In the current study, we used the tolerant transgenic mice to assess the ability of a novel DNA fusion gene vaccine to induce effective immunity against a tumor antigen that is also expressed by normal cells. For disseminated leukemia, the goal can be limited to induction of CTL, since, once induced, CD8+ T cells should be able to find and destroy accessible targets. Here we show that a DNA fusion vaccine containing an epitope from the targeted tumor antigen and heterologous helper epitopes can break tolerance and activate low-avidity gag85–93-specific CTL in mice expressing gag protein as a self antigen, with no evidence of autoimmune injury. The induced CTL acquired cytotoxic function in vivo and provided some protection against leukemia in vaccinated transgenic mice. This model of continuous expression of a leukemia-associated antigen in normal tissues has relevance for the clinic and reveals that DNA vaccination can activate surviving epitope-specific CTL for focused immune attack against leukemic cells.
Results
Expression of gag RNA in liver and thymus of gag-Tg mice and leukemic cells
In the gag-Tg model the antigen is expressed not only in the liver but also in the thymus, leading to tolerance in the CD8+ T cell repertoire [32, 33]. However, the level of gag antigen expression in these transgenic tissues compared to FBL-3 tumor cells is unknown. As a measure of expression, we used quantitative RT-PCR (qRT-PCR). Surprisingly, both thymus and liver biopsies from gag-Tg mice expressed higher levels of gag RNA compared to FBL-3 tumor cells (about twofold and threefold, respectively) (Fig. 1). Although this is at the RNA level only, it is indicative of higher levels of antigen expression. No amplification was detected in control reactions lacking cDNA (data not shown). No gag transcript amplification was detected using cDNA from EL4 cells or liver/thymus biopsies from WT mice (data not shown).
Figure 1.
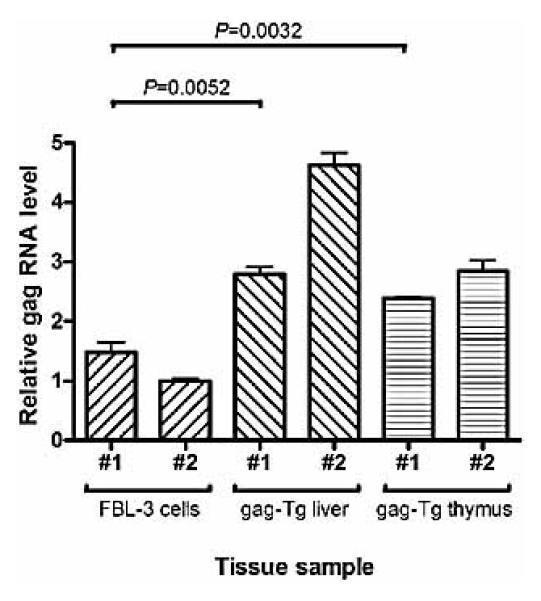
Relative levels of gag RNA expression in FBL-3 tumor cells compared to liver and thymus cells from gag-Tg mice. Total RNA was extracted from FBL-3 cells, thymus or liver biopsies from gag-Tg mice; samples were isolated from either two independent FBL-3 cell cultures (#1, #2) or two individual mice (#1, #2). Following cDNA synthesis, qRT-PCR was used to compare the relative levels of gag gene expression between samples; qRT-PCR reactions were run in triplicate for each sample and combined; error bars: SEM. -actin transcript amplification was detected in all cDNA samples and was used to normalize gene expression levels.
DNA vaccination induces gag85–93-specific CD8+ T cells in WT and gag-Tg mice
In order to investigate whether our vaccination strategy could overcome immune tolerance in this model, we constructed a DNA fusion vaccine encoding the first domain (DOM) of FrC from TT linked to the immunodominant H-2Db-restricted gag85–93 peptide (p.DOM-gag). DNA vaccine design is indicated in Fig. 2. The ability of the p.DOM-gag DNA vaccine to induce CD8+ T cell responses to gag85–93 was assessed by vaccinating WT and gag-Tg mice. For comparison, control groups were immunized with irradiated FBL-3 cells, known to induce a readily detectable protective CD8+ T cell response specific for the gag85–93 epitope in WT mice [30, 31]. T cell responses in the spleen were measured immediately ex vivo by ELISPOT assay on day 36. Vaccination of B6 mice with p.DOM-gag induced gag85–93- specific T cell responses as measured by ELISPOT assay for the production of IFN-γ (Fig. 3A), with frequencies similar to those observed in FBL-3-vaccinated mice. The control DNA vaccine (p.DOM) generated no gag85–93-specific responses (Fig. 3A). CD4+ Th cell responses against the ‘promiscuous’ MHC class II binding peptide p30, embedded in the FrC domain (DOM), were also detected in mice vaccinated with either DNAvaccine (p.DOMgag or p.DOM), but not in mice immunized with irradiated FBL-3 cells, in which the low frequency of IFN-γ-producing cells detected was equivalent to background levels without peptide stimulation (Fig. 3A).
Figure 2.
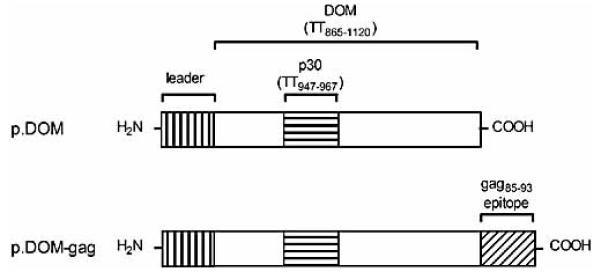
Schematic diagram indicating DNA fusion vaccine design. The control vaccine p.DOM contains the sequence encoding the N-terminal domain (DOM) of FrC from TT, including the p30 CD4+ Th epitope. Sequence coordinates relative to the original TT sequence are indicated. p.DOM-gag contains DNA sequence encoding the immunodominant H-2Db-restricted gag85-93 CTL epitope derived from FMuLVgag ligated in-frame to the C terminus of DOM. Each vaccine includes a leader sequence at the N terminus. Vaccine sequences were assembled and inserted into the commercial vector pcDNA3 using Hind III and Not I restriction sites.
Figure 3.

Ex vivo detection of gag-specific T cells in WT and gag-Tg mice following DNA vaccination. WT and gag-Tg mice were immunized with p.DOM-gag or p.DOM DNA vaccines, or irradiated FBL-3 cells, as indicated, on days 0 and 28; DNA booster injections at day 28 were administered, together with electroporation. Splenocytes were harvested from individual mice at day 36. (A, B) The numbers of SFC secreting IFN- were assessed ex vivo by ELISPOT assay. Splenocytes were incubated with gag85-93 peptide to assess the epitope-specific CD8+ T cell response, and with the p30 peptide to assess CD4+ T cell responses to the DOM component of the vaccine. Test samples were set up in triplicate; baseline responses without peptides are indicated. The data indicate mean SFC/million splenocytes and the SEM (error bars). Representative data from one of two identical experiments are shown. (C) The mean number of gag85-93 epitope-specific SFC was compared between WT and gag-Tg mice. Responses to 1 M gag85-93 peptide were pooled from individual mice; data are expressed as mean SFC/million splenocytes, together with the SEM. The number (n) of animals pooled per group is indicated. (D) The frequency of gag85-93-specific CD8+ T cells producing IFN- was assessed ex vivo by intracellular cytokine staining. Representative flow cytometric plots from individual mice are shown (upper panel). Data were pooled from individual mice (lower panel) to indicate the mean percentage of CD8+ T cells producing IFN- in response to gag85-93 peptide; error bars: SEM.
Gag-Tg mice were also tested for the ability to respond to the p.DOM-gag DNA vaccine. This vaccine induced robust gag85–93- specific T cell responses, as monitored by the ex vivo IFN-γ ELISPOT assay (Fig. 3B), although the frequency of responding cells was ~2.5-fold lower than in WT mice (Fig. 3C). By contrast, immunization with irradiated FBL-3 tumor cells failed to induce detectable gag85–93-specific T cell responses in gag-Tg mice (Fig. 3B), as previously reported [32]. A non-specific background response of cells producing IFN-γ was observed in mice after immunization with FBL-3, but this response was detected independent of stimulation with any peptide. It may be the consequence of an inflammatory response resulting from the injection of 1×107 irradiated tumor cells 8 days prior to obtaining the spleen, as a non-specific response of similar strength was also observed in WT mice immunized with the cellular vaccine (Fig. 3A).
The control p.DOM vaccine again induced no gag85–93-specific responses, although CD4+ Th cell responses to the p30 peptide were detected in these animals, as well as those immunized with p.DOM-gag (Fig. 3B). Since the CD4+ T cell response to p30 is derived from the unmanipulated mouse repertoire, this was used as an overall indicator of the DNA vaccine performance, which appeared effective in both WT and gag-Tg mice (Table 1). A survey of larger numbers of individual mice was then carried out, which demonstrated that although p.DOM-gag activated a lower frequency of gag85–93-specific T cells in gag-Tg mice compared to WT mice (Fig. 3C), the number of animals responding was comparable between strains: 92% (12 of 13) of gag-Tg mice and 100% (11 of 11) of WT mice (Table 1). The efficacy of the vaccine was further assessed by flow cytometric analysis to directly quantify the number of gag85–93-specific CD8+ T cells present in splenic lymphocytes, using anti-IFN-γ(intracellular) and anti-CD8 (surface) antibodies (Fig. 3D). Responses were again clearly evident in WT and gag-Tg mice, and confirmed the lower frequency of responding cells in the latter (Fig. 3D).
Table I.
Ex vivo detection of vaccine-specific T lymphocytes in wt and gag-Tg mice by IFNγ ELISpot assay.
| Vaccine | Peptide* | Vaccine-specific responses: † | |
|---|---|---|---|
| wt mice | gag-Tg mice | ||
| p.DOM-gag | gag85-93 | 11/11 (100%) | 12/13 (92%) |
| p30 | 10/11 (91%) | 13/13 (100%) | |
| p.DOM | gag85-93 | 0/7 (0%) | 0/8 (0%) |
| p30 | 7/7 (100%) | 6/8 (75%) | |
| Irradiated FBL-3 cells |
gag85-93 | 10/10 (100%) | 0/8 (0%) |
| p30 | 0/10 (0%) | 0/8 (0%) | |
| None | gag85-93 | 0/2 (0%) | 0/3 (0%) |
| p30 | 0/2 (0%) | 0/3 (0%) | |
Splenocytes incubated with 0.1μM gag85-93 peptide or 1μM p30 peptide.
Data presented as: number of positive responders/total mice tested (% responding).
Avidity of induced gag85–93-specific CD8+ T cells in WT and gag-Tg mice
The frequency of gag85–93-specific cells elicited in gag-Tg mice is low compared to WT mice, probably due to central and peripheral tolerance mechanisms which could potentially have deleted a fraction of the repertoire, particularly the high-avidity gag85–93- specific CD8+ T cells. To address the question of avidity, we compared the frequency of CD8+ T cells from WT and gag-Tg mice that responded to varying concentrations of gag85–93 peptide in an IFN-γ ELISPOT assay. WT and gag-Tg mice were vaccinated with either p.DOM-gag or the control DNA vaccine and ELISPOT responses were measured at day 36, as described above. Gag85–93-specific CD8+ T cells elicited in the gag-Tg mice had about tenfold lower avidity compared to those from WT mice when tested against a range of gag85–93 peptide concentrations ex vivo, with responses first detectable at 1 log higher peptide concentration (0.01 lM vs. 0.001 lM) and peak responses attained at 1 log higher peptide concentration (1 lM vs. 0.1 lM) (Fig. 4). This difference, observed in two independent experiments comparing 12 gag-Tg and 11 WT mice, was reproducible and highly statistically significant (p≤0.001).
Figure 4.
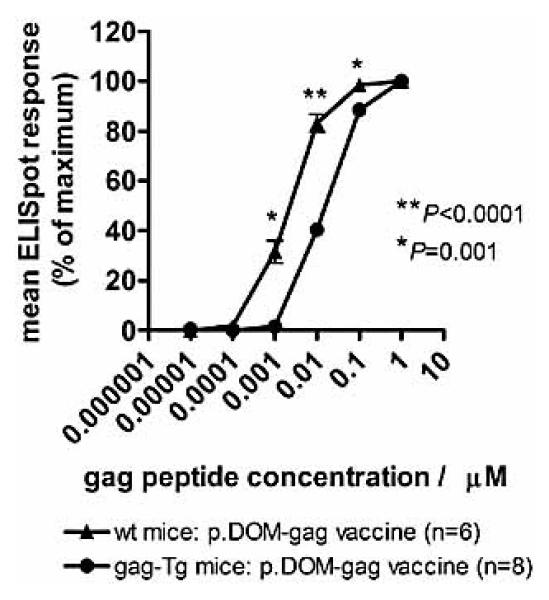
DNA vaccination induces lower-avidity gag85-93-specific CTL in gag-Tg mice compared to WT mice. WT and gag-Tg mice were immunized with p.DOM-gag on days 0 and 28; booster injections at day 28 were administered, together with electroporation. Splenocytes were harvested from individual mice at day 36 and the numbers of SFC secreting IFN- were assessed ex vivo by ELISPOT assay. Splenocytes were incubated with varying concentrations of gag85-93 peptide to assess the epitope-specific CD8+ T cell response; test samples were set up in triplicate for each peptide concentration. Baseline responses without peptide were subtracted and the number of SFC/million splenocytes was calculated as a percentage of the maximum number of SFC/million splenocytes responding per mouse; data were then pooled for each experimental group to calculate the mean ELISPOT response, together with the SEM (error bars). Representative data from one of two identical experiments are shown.
Cytotoxic activity in vitro of gag85–93-specific CD8+ T cells
The ability of the lower-avidity CD8+ T cell response induced by the p.DOM-gag DNA vaccine in gag-Tg mice to exhibit lytic activity against targets expressing the gag85–93 epitope was tested. At day 36 after vaccination, splenocytes from WT or gag-Tg mice were stimulated in vitro with peptide for 6 days and lytic activity assessed in a 51Cr-release assay. CTL from either WT or gag-Tg mice, primed with p.DOM-gag, lysed EL4 target cells pulsed with gag85–93 peptide but not a control peptide (Fig. 5A). CTL from either host also lysed FBL-3 leukemia cells expressing endogenous FMuLVgag antigen (Fig. 5A). No specific CTL activity was generated by culture of splenocytes in vitro with gag85–93 peptide following vaccination with the control vaccine, p.DOM, and no lytic activity was observed against the NK-susceptible cell line YAC-1 (Fig. 5A).
Figure 5.
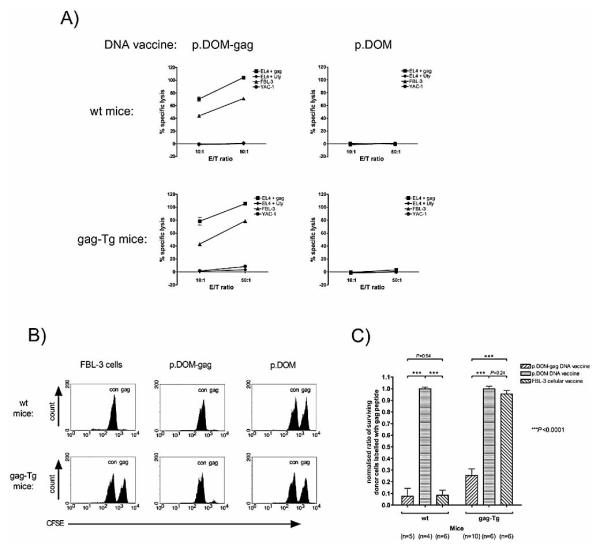
DNA vaccination of gag-Tg mice induces gag85-93-specific effector CTL capable of in vitro and in vivo cytolytic activity. WT and gag-Tg mice were immunized with p.DOM-gag or p.DOM DNA vaccines, or irradiated FBL-3 cells, as indicated, on days 0 and 28. (A) At day 36, splenocytes were cultured with 0.01 M gag85-93 peptide for 6 days in vitro, prior to measuring CTL activity by standard 51Cr-release assay. Targets were EL4 cells pulsed with gag85-93 peptide or control peptide (Uty), FBL-3 leukemia cells expressing endogenous FMuLVgag antigen, or the control cell line YAC-1. Representative data from individual mice are shown; data from one of two identical experiments are shown; error bars: SEM. (B, C) At day 36 following vaccination, mice were infused, by intravenous injection, with a 1:1 mixture of target splenocytes: one population of cells had been pulsed with gag85-93 (gag) peptide while the other had been pulsed with control (con) peptide (SNWYFNHL); each population was labeled with a different concentration of CFSE (gag85-93 peptide: CFSEhi; control peptide: CFSElow). Splenocytes were harvested 20 h later and the frequency of each CFSE-labeled target cell population determined by flow cytometric analysis. (B) Representative flow cytometric profiles from individual mice. (C) The ratio of gag85-93- to control peptide-pulsed cells surviving in each recipient was calculated. The ratios were normalized, by assuming that the mean ratio was 1:1 in mice given the control vaccine (p.DOM), and pooled to calculate the mean proportion of surviving donor cells pulsed with the gag85-93 peptide, together with the SEM (error bars). Data are pooled from two of two identical experiments, each giving similar results; the number (n) of animals pooled per group is indicated.
Cytotoxic activity in vivo of gag85–93-specific CD8+ T cells
Clearly, gag85–93-specific lytic activity could be elicited following in vitro stimulation of CD8+ T cells that had been induced in gag-Tg mice. However, it was unknown whether the cells were capable of expressing such lytic activity within the in vivo tolerizing environment. To address this, WT and gag-Tg mice were vaccinated with either p.DOM-gag, the control DNA vaccine (p.DOM), or irradiated FBL-3 cells. At day 36, mice were injected intravenously with sex-matched, syngeneic splenocyte targets pulsed with either gag85–93 peptide or control peptide that had been differentially labeled with CFSE to permit distinction between the two targets by flow cytometry. Twenty hours later, mice were sacrificed and the spleens analyzed for persistence of CFSE-labeled targets. WT mice that had previously been vaccinated with either p.DOM-gag or irradiated FBL-3 cells rapidly and efficiently lysed target splenocytes pulsed with the gag85–93 peptide, as reflected by the clearance of >90% of the CFSEhi targets by 20 h, but not those from the co-transferred population pulsed with the control peptide (CFSElow) (Fig. 5B). Both populations of CFSE-labeled target cells survived in WT mice given the control DNA vaccine, p.DOM (Fig. 5B).
Gag-Tg mice vaccinated with p.DOM-gag also specifically eliminated the gag85–93 peptide-pulsed target cells (Fig. 5B). The specific lytic activity in gag-Tg mice immunized with this DNA vaccine was slightly less than that observed in WT mice, but the difference did not reach statistical significance (p=0.073; Fig. 5C). Thus, for target cells pulsed with high concentrations of the peptide epitope, the reduced avidity of the CTL elicited in the transgenic mice did not markedly affect their performance. In contrast to WT mice, gag-Tg mice vaccinated with irradiated FBL-3 cells failed to lyse target cells labeled with the gag85–93 peptide (Fig. 5B and C), confirming the inability of this approach to induce gag85–93-specific CTL in these animals. Both populations of CFSE labeled target cells survived in gag-Tg mice that had received the control DNA vaccine (Fig. 5B and C).
Induction of gag85–93-specific CTL by p.DOM-gag protects against FBL-3 leukemia growth in vivo
Although the lower-avidity gag85–93-specific CTL induced in gag-Tg mice demonstrated the ability to lyse peptide-pulsed targets in vivo, their capacity to recognize and protect mice from leukemia in vivo was unknown. To address this, WT or gag-Tg mice were vaccinated with p.DOM-gag or the control vaccine (p.DOM) and then challenged at day 36 by intraperitoneal injection of 5×104 FBL-3 leukemia cells. Immunization with p.DOM-gag afforded significant protection from leukemia in WT mice, with ~95% surviving, compared to naive animals or those given the control vaccine (Fig. 6A). A low level of resistance (≤25%) was occasionally observed in control WT groups, suggesting that injection of this dose of FBL-3 leukemia cells alone can lead to the spontaneous induction of natural protective immunity in WT mice (Fig. 6A).
Figure 6.
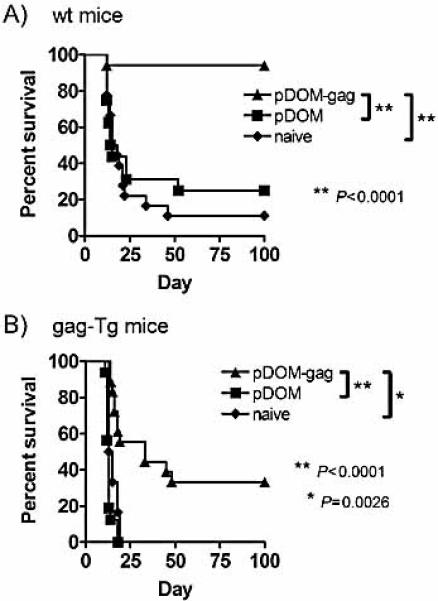
DNA vaccination of gag-Tg mice induces gag85-93-specific effector CTL able to protect mice from FBL-3 leukemia. (A) WT and (B) gag-Tg mice were immunized with p.DOM-gag or p.DOM DNA vaccines as indicated, on days 0 and 28; DNA booster injections at day 28 were administered, together with electroporation. Naive (unimmunized) mice served as controls. At day 36 mice were challenged by intraperitoneal injection of FBL-3 leukemia cells, and tumor development monitored. Survival data in each panel are pooled from two of two identical experiments, each giving similar results.
Vaccination with p.DOM-gag also led to significant protection in gag-Tg mice (Fig. 6B). In these mice, there was no evidence for spontaneous induction of immunity following injection of FBL-3 cells, since all naive or control-vaccinated mice succumbed by day 19 (Fig. 6B). Thus, the lower-avidity gag85–93-specific CTL response induced in gag-Tg mice by the p.DOM-gag vaccine is capable of mediating demonstrable protection from challenge with live FBL-3 leukemia cells in vivo. However, this response appears suboptimal compared to the responses elicited in WT mice, with only about one third of the gag-Tg mice vaccinated with p.DOM-gag exhibiting prolongation of survival (Fig. 6B). It is unclear whether this detectable but reduced protection reflects primarily a qualitative or quantitative defect. One possibility is that the response might be further amplified by adding novel adjuvants.
Assessment of autoimmunity in vaccinated gag-Tg mice
Despite the significant degree of protection against leukemia afforded by vaccination of gag-Tg mice with p.DOM-gag, the presence of a population of activated gag85–93-specific CD8+ T cells following DNA immunization could potentially result in autoimmune injury to hepatocytes expressing the FMuLVgag protein. To assess this, blood was drawn from gag-Tg mice at day 36 following vaccination, and serum levels of the liver enzymes aspartate aminotransferase and alanine aminotransferase measured as indicators of liver injury. Mice were also sacrificed at this time point for blinded histological analysis of liver tissue. All animals appeared healthy with no evidence of increased levels of aspartate aminotransferase or alanine aminotransferase in the serum (data not shown). Histologic analysis of liver sections in vaccinated gag-Tg animals indicated that all portal tracts were normal, with normal biliary structures (Fig. 7). There was no inflammation in portal tracts or parenchyma, no hepatocyte degeneration or loss, and the normal sinusoidal architecture was present (Fig. 7). No autoimmune hepatocyte injury was evident, despite the relatively high levels of gag transcript expression observed in gag-Tg liver compared to FBL-3 tumor cells (Fig. 1).
Figure 7.
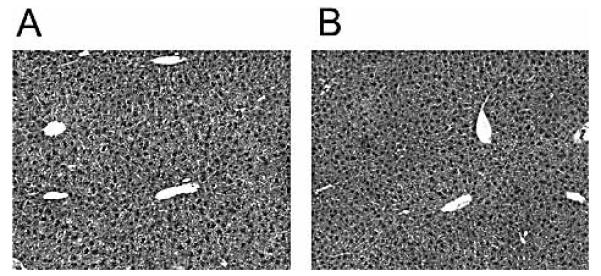
Absence of autoimmune liver injury following DNA vaccination of gag-Tg mice. Gag-Tg mice were immunized with p.DOM-gag on days 0 and 28; DNA booster injections at day 28 were administered, together with electroporation. Unimmunized mice served as naive controls. Mice were euthanized at day 36 and liver samples were fixed in formaldehyde, paraffin-embedded, sectioned and stained with hematoxylin/eosin. Representative sections from gag-Tg mice immunized with p.DOM-gag (A) or naive gag-Tg controls (B) are shown; magnification, ×10.
Discussion
Effective vaccination of patients with cancer using tumor-associated antigens has several major problems to overcome. These include restrictions placed on the immune repertoire by expression of the tumor antigen on normal tissues and the obstacles to activating the residual repertoire to attack cancer cells and avoid autoimmunity.
The issue of tolerogenic pressure acting on the T cell repertoire has been widely investigated in mouse models. The most common strategy has been to combine two transgenic strains expressing either a targeted antigen or a T cell receptor (TCR) specific for an MHC class I-associated peptide derived from the antigen. Although these studies are confined to assessing the effects of antigen on a selected single-clonal T cell population, useful insights regarding the obstacles to inducing T cell responses have been derived. Many models involve T cells with high-affinity TCR, for which deletion is the usual outcome [34]. However, it is becoming clear that low-avidity T cells can survive both central and peripheral tolerance mechanisms provided that antigen expression is modest [7]. Development of autoimmune attack on tissues expressing the target antigen appears to follow the same rules of dependence on level of expression and T cell avidity [7]. Ideally, as is evident from clinical transplantation, the focus should be on antigens that are highly expressed by tumor cells but weakly expressed by normal cells.
A key challenge for the clinic is how best to activate the surviving lower-avidity T cells. In a transgenic model in which influenza hemagglutinin (HA) is expressed in pancreatic islet β-cells, low-avidity HA-specific CTL could be activated by immunization/infection with HA-expressing virus [35]. Such CTL were capable of recognizing a tumor expressing high levels of the HA antigen without injuring the pancreas. Further study of the fate of low-avidity transgenic CD8+ T cells in this and a related model has revealed that the level of antigen is a critical determinant, and that passively transferred CD8+ T cells ignored low levels of antigen in normal pancreatic tissue, but could recognize higher levels of antigen expressed in malignant pancreatic islet β-cells. However, these T cells exhibited limited anti-tumor activity unless antigen-specific CD4+ Th cells were provided and both T cell subsets were activated together by vaccination/infection with influenza virus [36].
Unfortunately, the repeated use of viral vectors is not possible due to induction of blocking immunity against viral proteins [37], a key disadvantage given that repeated vaccination is likely to be required for cancer patients. Even if blocking antibody can be avoided, viral vectors can lead to domination of the CTL hierarchy by virally derived epitope-specific responses, to the detriment of immune induction against lower-affinity tumor-derived epitopes encoded within the vaccine [38, 39]. Considering these requirements, we have explored a DNA fusion vaccine format that affords concurrent heterologous T cell help for effective CTL induction in vivo, avoiding the use of viral vectors or adoptive transfer. The design focuses immunity onto the encoded epitope and, when combined with electroporation at boosting, can achieve high levels of CTL [14].
Using this strategy, our aim was to induce CTL against a model tumor antigen that was also a self protein continually expressed in a large peripheral organ, but in which the T cell repertoire was unmanipulated and therefore polyclonal, as this represents a typical setting for many human cancers. We chose the gag-Tg mice since the antigen is mainly expressed in the liver, but lower levels are detectable in the thymus, and there is clear evidence for tolerance [32, 33]. Furthermore, in a double-transgenic mouse expressing both gag in the liver and a TCR in CD8+ T cells specific for the gag85–93 epitope [33], T cell numbers were reduced and those remaining failed to proliferate or produce IL-2 in response to antigen [40]. Although our DNA vaccine was operating within a polyclonal T cell repertoire, it is likely that it had to contend with a similar combination of deletional and regulatory influences in the gag-Tg mice, and the challenge was to activate the residual repertoire.
The DNA vaccine was able to mobilize an epitope-specific CD8+ T cell response capable of proliferation, cytokine production, and killing of target FBL-3 leukemia cells in vitro and in vivo. The provision of heterologous T cell help within the vaccine was aimed to circumvent tolerance to tumor antigens, which may be more profound in the CD4+ T cell arm. Interestingly the cellular vaccine failed to induce a detectable CD8+ T cell response against this dominant epitope in the transgenic mice. Since the potential response is clear, the weakening of the cellular vaccine is likely due to an inability to generate a tumor-derived CD4+ T cell response, possibly due to the activity of Treg. It is known that effective CD4+ T cell help is critical for inducing and maintaining an effective CTL response [24-26]. One question is whether true tumor-specific T cell help might be required for long-term maintenance of CD8+ T cells [41]. In fact, our experience has been that challenge with tumor cells can expand tumor-specific CD8+ T cells that have previously been primed by DNA fusion gene vaccination [27]. Nonetheless, we are investigating the effect on CD8+ T cell priming of also encoding tumor-derived CD4+ T cell epitopes within our DNA vaccine and exploring the role of Treg. However, the main point of our current study is that, in contrast to the cellular vaccine, the DNA fusion vaccine is able to activate the residual CD8+ T cell repertoire and that these T cells can carry out their effector functions in vivo.
Epitope-specific CTL appear able to kill target tumor cells effectively, although targeting multiple epitopes might be even more potent [42], and avoid escape via epitope loss [43]. Vaccines encoding whole antigen cDNA appear to offer this potential, with the added advantage of being suitable for all patients regardless of MHC class I haplotype. However, placing an antigenic epitope at the C terminus of the FrC domain (DOM) promotes more efficient induction of CD8+ T cells compared with using native cDNA [27], and immunodominance argues against delivering multiple epitopes within the same vaccine [44-46]. Instead, multiple vaccines could be used, each targeting a distinct tumor epitope, with delivery of the second vaccine into a separate site to avoid competition [45, 46]. This integrated approach could compensate for the loss of T cell repertoire against individual epitopes.
Following vaccination, the residual gag-specific CD8+ T cell repertoire surviving in the tolerized mice clearly retain the capacity to kill tumor cells in vivo. Unfortunately, the highly aggressive nature of the FBL-3 tumor makes it unsuitable to model therapeutic efficacy since even a dose of 100 FBL-3 tumor cells per gag-Tg animal leads to death at day 12–14 (data not shown). However, we have established previously that the vaccine design can be used therapeutically, using a less aggressive tumor model that allows the immune system time to respond to vaccination [14].
Once effector CTL are induced, the balance between tumor destruction and autoimmunity depends at least in part on the relative levels of peptide expressed [7], and on the accessibility of each target cell population to effector CTL infiltration [47]. Leukemic cells tend to be in blood or lymphoid tissues and access to effector CTL seems assured. Our data indicate that in this tolerance model, where expression of antigen in the thymus appears to cause deletion of the repertoire [32, 33], our DNA vaccine can activate residual gag-specific CTL able to lyse leukemia cells exhibiting a relatively low level of gag transcript expression. The vaccine design elicits no tumor-specific CD4+ T cells, thereby potentially protecting antigen-expressing organs from entry of CTL, which is likely to depend on cytokine-producing CD4+ T cells at the site [48]. Also, since these CD4+ T cells are targets for regulation, the vaccine may circumvent suppression first by utilizing the microbial repertoire and then by evading tumor-specific regulatory mechanisms. It is possible that the cellular vaccine containing irradiated tumor cells could elicit CD4+ T cell responses, but this was not investigated due to the lack of well-characterized gag-derived Th cell epitopes. For antigens expressed in the liver, there may also be other factors operating, such as an intrinsic resistance of the liver to autoimmune damage [32, 47, 49]. Previous attempts to promote autoreactivity in this model by combining viral infection of the liver with transfer of gag-specific CTL were unsuccessful.
Control of tumor may require continuing surveillance by anti-tumor CTL. Once induced, their ability to survive will be influenced by the level, mode and persistence of expression of tumor antigen, together with the presence or absence of inflammatory mediators. Each model can provide only a partial view, and the best test of performance of the DNA fusion vaccines will therefore be in patients with cancer, with clinical trials now underway [21].
Materials and Methods
Cells
FBL-3 is a Friend virus-induced erythroleukemia of B6 origin that expresses FMuLV gag- and env-encoded products and MHC class I molecules [50]. EL4 is a T cell lymphoma derived from C57BL/6N mice, and YAC-1 is an NK-susceptible T cell lymphoma originating from the A/Sn strain. All cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS (Life Technologies, Paisley, UK), 1 mM sodium pyruvate, 2 mM L-glutamine, non-essential amino acids (1% of 100x stock), 25 mM HEPES buffer and 50 μM 2-mercaptoethanol (complete medium).
Peptides
The H-2Db-restricted gag peptide gag85–93 derived from FMuLVgag (CCLCLTVFL) and the FrC-derived Th peptide p30 (TT947–967; FNNFTVSFWLRVPKVSASHLE) have been described previously [31, 51]. Peptide controls included the H-2Db-restricted HY peptide (WMHHNMDLI) derived from the Uty gene [52] and a TT-derived H-2Kb-restricted peptide (SNWYFNHL) which is not encoded within these DNA vaccines [22]. Peptides were synthesized commercially and supplied at >95% purity (PPRL, Southampton, UK). Gag85–93 peptide stocks (5 mM) were dissolved in DMSO, and all other peptide stocks (1 mM) were dissolved in water. Stocks were stored at −20°C.
DNA vaccines
DNA vaccine design is indicated in Fig. 2. Construction of a DNA vaccine (p.DOM) containing the gene encoding the first domain (DOM; TT865–1120, 256 amino acids plus leader sequence of 19 amino acids) of FrC (TT865–1316) from TT has been described [22]. The p.DOM vaccine was used as a template to construct p.DOM-gag, which encodes the immunodominant H-2Db-restricted FMuLVgag CTL motif (gag85–93) fused to the C terminus of DOM. This was achieved by PCR amplification of DOM using the forward primer 50-TTTTAAGCTTGCCGCCACCATGGGTTGGAGC-3′ and the reverse primer 5′-TTTTGCGGCCGCTTACAGAAAAACAGTCAAACAGAGACAACAGTTACCCCAGAAGTCACGCAGGAA-3′, which fuses the gag85–93-encoding sequence to the 3′ terminus of DOM. The resulting PCR fragment was cloned into the expression vector pcDNA3 (Invitrogen, San Diego, CA). Both DNA constructs encode a leader sequence derived from the VH of the IgM idiotype of the BCL1 tumor. DNA vaccine stocks were prepared and verified as previously described [29].
Mice and vaccination protocol
The B6 gag-Tg model, in which the gag protein from FMuLV is expressed under the control of the mouse albumin promoter in the liver, has been described previously [32, 33, 40]. For DNA immunization, WT B6 or gag-Tg mice, bred in house, were vaccinated at 6–12 wk of age with a total of 50 μg DNA in saline injected into two sites in the quadriceps muscles on day 0; mice were anesthetized and administered booster injections together with electroporation at day 28, as described previously [14]. For cellular immunization, mice were injected with 1×107 irradiated (10 000 rad) FBL-3 leukemia cells intraperitoneally on days 0 and 28. Animal experimentation was conducted within UK Coordinating Committee for Cancer Research (London, UK) guidelines, under Governmental license.
ELISPOT assay
Following priming (day 0) and booster injections (day 28), splenocytes were harvested on day 36 and vaccine-specific responses were assessed ex vivo by ELISPOT assay for IFN-γ secretion (BD ELISPOT Set; BD PharMingen, San Diego, CA), as described previously [28]. Splenocytes were incubated with either the H-2Db-restricted gag85–93 peptide for 24 h to assess CD8+ Tcell responses or the p30 peptide (derived from the FrC fusion domain, DOM) to assess vaccine-induced CD4+ T cell responses. Triplicate wells were tested with a range of gag85–93 peptide concentrations; control samples were incubated without peptide. The reducing agent tris(2-carboxyethyl) phosphine hydrochloride (TCEP; Pierce Biotechnology, Rockford, IL), which has been shown to enhance the antigenicity of cysteine-containing synthetic peptides [53], was included in each microtiter well (200 μM) during the incubation stage, as the gag85–93 epitope with three cysteines is otherwise very insoluble in vitro.
To compare the frequency of T cells responding to different concentrations of the gag85–93 peptide, as a measure of T cell avidity, baseline ELISPOT responses without peptide were subtracted and the number of spot-forming cells (SFC)/million splenocytes calculated as a percentage of the maximum SFC/million splenocytes for each individual mouse. The data were pooled within each experimental group to calculate the mean ELISPOT response as a percentage of the maximum response observed for each peptide concentration.
Generation and assay of gag85–93-specific CTL
To assess gag85–93-specific CTL responses, vaccinated mice were sacrificed at day 36 and single-cell suspensions made from individual spleens in complete medium. Splenocytes were washed and resuspended in 15 mL complete medium at 3×106 cells/mL, together with recombinant human IL-2 (20 IU/mL; Perkin-Elmer, Foster City, CA), gag85–93 peptide (0.01 μM) and 200 μM TCEP [53]. Following 6 days stimulation in vitro (37°C, 5% CO2), cytolytic activity of the T cell cultures was assessed by standard 5-h 51Cr-release assay as previously described [22], with target cells that were labeled with peptide/51Cr in the presence of 200 μM TCEP for 1 h at 37°C. Targets included EL4 cells pulsed with gag85–93 peptide or control peptide (Uty), FBL-3 leukemia cells or NK-sensitive YAC-1 cells.
In vivo cytotoxicity assay
Splenocytes were harvested from WT and gag-Tg mice (2×107/mL in PBS) and cells from each strain were pulsed with 5 μM gag85–93 peptide or control peptide (SNWYFNHL) for 30 min at 37°C in the presence of 200 μM TCEP and washed in PBS. The gag- and control peptide-pulsed cells were then incubated with 5 μM or 0.5 μM CFSE (Molecular Probes, Invitrogen), respectively, at room temperature for 8 min in the dark, and FBS (final concentration 20%) was added to quench the labeling reaction. After washing, syngeneic cells were mixed together, resuspended in PBS, and 2×107 cells in 0.1 mL injected intravenously to each sex-matched syngeneic recipient. Splenocytes were harvested from individual recipients after 20 h and, following lysis of RBC, cells expressing CFSE analyzed by FACSCalibur (BD Biosciences, San Diego, CA).
Tumor challenge
Mice were challenged 8 days after the booster immunization (day 36 following priming) by intraperitoneal injection of 5×104 FBL-3 leukemia cells in PBS. Mice were monitored daily and euthanized on detection of tumor development, in accordance with UK Coordinating Committee for Cancer Research guidelines.
Ex vivo intracellular IFN-γ assay
To assess priming of gag85–93-specific CD8+ T cells, mice were culled at day 36 following immunization, and splenocytes processed for detection of intracellular IFN-γ. Viable splenocytes were selected by density centrifugation and B cells removed using mouse pan B DynabeadsJ (Invitrogen). Cells were incubated for 4 h at 37°C in 96-well plates (1×106 cells/well) in complete medium together with 200 μM TCEP, 10 IU/well human recombinant IL-2, 1 μM gag85–93 peptide or control peptide (SNWYFNHL) and 1 μL/well Golgi Plug (BD Biosciences). Following incubation, samples were processed to label surface CD8 and intracellular IFN-γ, as previously described [27], and analyzed by FACSCalibur.
Analysis of autoimmune injury
Following priming (day 0) and booster vaccinations (day 28), groups of B6 and gag-Tg mice were euthanized on day 36 to assess autoimmune injury. Control, naive groups received no vaccinations. Liver samples were fixed in formaldehyde, paraffinembedded, sectioned and stained with hematoxylin/eosin. Coded specimens were analyzed by a liver pathologist in a blinded manner for inflammation and lymphocyte infiltration using a Zeiss Axioskop 2 MOT microscope (Carl Zeiss, Oberkochen, Germany) and Zeiss Plan-NEOFLUAR 10x/0.30 objective lens. Images were recorded using a Zeiss AxioCam camera and Zeiss Axiovision 4 software with white balance correction provided by GNU Image Manipulation Program (http://www.gimp.org) and processed with CorelDraw Graphics Suite 12 (Corel, Ottawa, Canada).
Quantitative analysis of gag RNA expression
The FMuLVgag open reading frame encodes two alternative translational products: Pr65gag and gPr75gag. The higher-molecular- weight protein gPr75gag has an additional 98-amino-acid N-terminal signal sequence [54, 55] within which the target immunodominant gag85–93 epitope is located [31]. The major protein species generated in FBL-3 tumor cells is Pr65gag [32, 54]. The gag transgene present in gag-Tg mice was designed to optimize expression of only gPr75gag [32, 54].
To assess gPr75gag RNA expression, total RNA was extracted from FBL-3 cells, EL4 cells, liver or thymus biopsies from gag-Tg and WT mice using RNeasy Plus mini kit (QIAGEN, Valencia, CA). Total RNAwas reverse-transcribed using an oligo(dT) primer and a First-Strand cDNA Synthesis kit (Amersham Pharmacia Biotech). The cDNA was used for all subsequent qRT-PCR assays, using Taqman Universal PCR master mix (Applied Biosystems, Foster City, CA) together with a primer/probe combination to detect either gPr75gag gene expression (forward primer: 5′- GGCCGACTAGCTCTGTACCT- 3′; reverse primer: 50-GGTGCACCAAAGAGTCCAA- 3′; probe 5′-FAM-CGGGTGTCTCGAACCCGTCA-TAMRA-3′) or endogenous mouse β-actin gene expression (Applied Biosystems). The gag primers were designed to only amplify product from gPr75gag, containing the gag85–93 epitope. All reactions were performed in triplicate using the ABI PRISM 7500 Sequence detection System (Applied Biosystems) according to the following thermal cycle protocol: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 90 s. The β-actin transcript level was used to normalize gene expression levels.
Statistical analysis
Experimental groups were compared using an unpaired, two-tailed t-test. Survival curves were compared using the χ2 log-rank test. Experimental groups were considered significantly different from control groups if p<0.05.
Acknowledgements
We thank Dr. Iacob Mathiesen for supplying the electroporation equipment, and Dr. Abigail Lapham and Dr. Melanie Howell for technical assistance with qRT-PCR. This study was supported by Leukaemia Research Fund (LRF) grant 0306 (J.R., F.K.S. and S.L.B.), Tenovus grant 43333 and Cancer Research UK project grant 7576 (S.N.D.). M.L.D. was supported by a Poncin Scholarship.
Abbreviations
- B6
C57BL/6
- FMuLV
Friend murine leukemia virus
- FMuLVgag
Friend murine leukemia virus gag protein
- FrC
fragment C
- gag-Tg
FMuLVgag-transgenic
- qRT-PCR
quantitative RT-PCR
- SFC
spot-forming cells
- TCEP
tris(2-carboxyethyl) phosphine hydrochloride
- TT
tetanus toxin
Footnotes
Conflict of interest: F.K.S. and J.R. hold a patent relating to the DNA vaccine design described in the article and have declared a financial interest in a company which holds an exclusive license relating to this design.
References
- 1.Goldman JM, Gale RP, Horowitz MM, Biggs JC, Champlin RE, Gluckman E, Hoffmann RG, et al. Bone marrow transplantation for chronic myelogenous leukemia in chronic phase. Increased risk for relapse associated with T-cell depletion. Ann. Intern. Med. 1988;108:806–814. doi: 10.7326/0003-4819-108-6-806. [DOI] [PubMed] [Google Scholar]
- 2.Gattinoni L, Powell DJ, Jr., Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: Building on success. Nat. Rev. Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Degiovanni G, Hainaut P, Lahaye T, Weynants P, Boon T. Antigens recognized on a melanoma cell line by autologous cytolytic T lymphocytes are also expressed on freshly collected tumor cells. Eur. J. Immunol. 1990;20:1865–1868. doi: 10.1002/eji.1830200835. [DOI] [PubMed] [Google Scholar]
- 5.Coulie PG, Karanikas V, Colau D, Lurquin C, Landry C, Marchand M, Dorval T, et al. A monoclonal cytolytic T-lymphocyte response observed in a melanoma patient vaccinated with a tumor-specific antigenic peptide encoded by gene MAGE-3. Proc. Natl. Acad. Sci. USA. 2001;98:10290–10295. doi: 10.1073/pnas.161260098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity. 2005;22:275–284. doi: 10.1016/j.immuni.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Zehn D, Bevan MJ. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity. 2006;25:261–270. doi: 10.1016/j.immuni.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyewski B, Klein L. A central role for central tolerance. Annu. Rev. Immunol. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 9.Kast WM, Levitsky H, Marincola FM. Synopsis of the 6thWalker’s Cay Colloquium on Cancer Vaccines and Immunotherapy. J. Transl. Med. 2004;2:20. doi: 10.1186/1479-5876-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevenson FK, Ottensmeier CH, Johnson P, Zhu D, Buchan SL, McCann KJ, Roddick JS, et al. DNA vaccines to attack cancer. Proc. Natl. Acad. Sci. USA. 2004;101(Suppl 2):14646–14652. doi: 10.1073/pnas.0404896101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klinman DM, Currie D, Gursel I, Verthelyi D. Use of CpG oligodeoxynucleotides as immune adjuvants. Immunol. Rev. 2004;199:201–216. doi: 10.1111/j.0105-2896.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- 12.Spies B, Hochrein H, Vabulas M, Huster K, Busch DH, Schmitz F, Heit A, Wagner H. Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J. Immunol. 2003;171:5908–5912. doi: 10.4049/jimmunol.171.11.5908. [DOI] [PubMed] [Google Scholar]
- 13.Dupuis M, Denis-Mize K, Woo C, Goldbeck C, Selby MJ, Chen M, Otten GR, et al. Distribution of DNA vaccines determines their immunogenicity after intramuscular injection in mice. J. Immunol. 2000;165:2850–2858. doi: 10.4049/jimmunol.165.5.2850. [DOI] [PubMed] [Google Scholar]
- 14.Buchan S, Gronevik E, Mathiesen I, King CA, Stevenson FK, Rice J. Electroporation as a “prime/boost” strategy for naked DNA vaccination against a tumor antigen. J. Immunol. 2005;174:6292–6298. doi: 10.4049/jimmunol.174.10.6292. [DOI] [PubMed] [Google Scholar]
- 15.Gurunathan S, Klinman DM, Seder RA. DNA vaccines: Immunology, application, and optimization. Annu. Rev. Immunol. 2000;18:927–974. doi: 10.1146/annurev.immunol.18.1.927. [DOI] [PubMed] [Google Scholar]
- 16.McConkey SJ, Reece WH, Moorthy VS, Webster D, Dunachie S, Butcher G, Vuola JM, et al. Enhanced T-cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nat. Med. 2003;9:729–735. doi: 10.1038/nm881. [DOI] [PubMed] [Google Scholar]
- 17.Widera G, Austin M, Rabussay D, Goldbeck C, Barnett SW, Chen M, Leung L, et al. Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J. Immunol. 2000;164:4635–4640. doi: 10.4049/jimmunol.164.9.4635. [DOI] [PubMed] [Google Scholar]
- 18.Babiuk S, Baca-Estrada ME, Foldvari M, Storms M, Rabussay D, Widera G, Babiuk LA. Electroporation improves the efficacy of DNA vaccines in large animals. Vaccine. 2002;20:3399–3408. doi: 10.1016/s0264-410x(02)00269-4. [DOI] [PubMed] [Google Scholar]
- 19.Tacket CO, Roy MJ, Widera G, Swain WF, Broome S, Edelman R. Phase 1 safety and immune response studies of a DNA vaccine encoding hepatitis B surface antigen delivered by a gene delivery device. Vaccine. 1999;17:2826–2829. doi: 10.1016/s0264-410x(99)00094-8. [DOI] [PubMed] [Google Scholar]
- 20.Stevenson FK, Rice J, Ottensmeier CH, Thirdborough SM, Zhu D. DNA fusion gene vaccines against cancer: From the laboratory to the clinic. Immunol. Rev. 2004;199:156–180. doi: 10.1111/j.0105-2896.2004.00145.x. [DOI] [PubMed] [Google Scholar]
- 21.Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: Precision tools for activating effective immunity against cancer. Nat. Rev. Cancer. 2008;8:108–120. doi: 10.1038/nrc2326. [DOI] [PubMed] [Google Scholar]
- 22.Rice J, Elliott T, Buchan S, Stevenson FK. DNA fusion vaccine designed to induce cytotoxic Tcell responses against defined peptide motifs: Implications for cancer vaccines. J. Immunol. 2001;167:1558–1565. doi: 10.4049/jimmunol.167.3.1558. [DOI] [PubMed] [Google Scholar]
- 23.Radcliffe JN, Roddick JS, Friedmann PS, Stevenson FK, Thirdborough SM. Prime-boost with alternating DNA vaccines designed to engage different antigen presentation pathways generates high frequencies of peptide-specific CD8+ T cells. J. Immunol. 2006;177:6626–6633. doi: 10.4049/jimmunol.177.10.6626. [DOI] [PubMed] [Google Scholar]
- 24.Shedlock DJ, Shen H. Requirement for CD4 Tcell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 25.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 27.Rice J, Buchan S, Stevenson FK. Critical components of a DNA fusion vaccine able to induce protective cytotoxic T cells against a single epitope of a tumor antigen. J. Immunol. 2002;169:3908–3913. doi: 10.4049/jimmunol.169.7.3908. [DOI] [PubMed] [Google Scholar]
- 28.Rice J, Buchan S, Dewchand H, Simpson E, Stevenson FK. DNA fusion vaccines induce targeted epitope-specific CTLs against minor histocompatibility antigens from a normal or tolerized repertoire. J. Immunol. 2004;173:4492–4499. doi: 10.4049/jimmunol.173.7.4492. [DOI] [PubMed] [Google Scholar]
- 29.Rice J, Dunn S, Piper K, Buchan SL, Moss PA, Stevenson FK. DNA fusion vaccines induce epitope-specific cytotoxic CD8(+) T cells against human leukemia-associated minor histocompatibility antigens. Cancer Res. 2006;66:5436–5442. doi: 10.1158/0008-5472.CAN-05-3130. [DOI] [PubMed] [Google Scholar]
- 30.Klarnet JP, Kern DE, Okuno K, Holt C, Lilly F, Greenberg PD. FBL-reactive CD8+ cytotoxic and CD4+ helper T lymphocytes recognize distinct Friend murine leukemia virus-encoded antigens. J. Exp. Med. 1989;169:457–467. doi: 10.1084/jem.169.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen W, Qin H, Chesebro B, Cheever MA. Identification of a gagencoded cytotoxic T-lymphocyte epitope from FBL-3 leukemia shared by Friend, Moloney, and Rauscher murine leukemia virus-induced tumors. J. Virol. 1996;70:7773–7782. doi: 10.1128/jvi.70.11.7773-7782.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohlen C, Kalos M, Hong DJ, Shur AC, Greenberg PD. Expression of a tolerizing tumor antigen in peripheral tissue does not preclude recovery of high-affinity CD8+ T cells or CTL immunotherapy of tumors expressing the antigen. J. Immunol. 2001;166:2863–2870. doi: 10.4049/jimmunol.166.4.2863. [DOI] [PubMed] [Google Scholar]
- 33.Ohlen C, Kalos M, Cheng LE, Shur AC, Hong DJ, Carson BD, Kokot NC, et al. CD8(+) T cell tolerance to a tumor-associated antigen is maintained at the level of expansion rather than effector function. J. Exp. Med. 2002;195:1407–1418. doi: 10.1084/jem.20011063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu. Rev. Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 35.Morgan DJ, Kreuwel HT, Fleck S, Levitsky HI, Pardoll DM, Sherman LA. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J. Immunol. 1998;160:643–651. [PubMed] [Google Scholar]
- 36.Lyman MA, Nugent CT, Marquardt KL, Biggs JA, Pamer EG, Sherman LA. The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J. Immunol. 2005;174:2563–2572. doi: 10.4049/jimmunol.174.5.2563. [DOI] [PubMed] [Google Scholar]
- 37.Kundig TM, Kalberer CP, Hengartner H, Zinkernagel RM. Vaccination with two different vaccinia recombinant viruses: Long-term inhibition of secondary vaccination. Vaccine. 1993;11:1154–1158. doi: 10.1016/0264-410x(93)90079-d. [DOI] [PubMed] [Google Scholar]
- 38.Bos R, van Duikeren S, van Hall T, Lauwen MM, Parrington M, Berinstein NL, McNeil B, et al. Characterization of antigen-specific immune responses induced by canarypox virus vaccines. J. Immunol. 2007;179:6115–6122. doi: 10.4049/jimmunol.179.9.6115. [DOI] [PubMed] [Google Scholar]
- 39.Smith CL, Mirza F, Pasquetto V, Tscharke DC, Palmowski MJ, Dunbar PR, Sette A, et al. Immunodominance of poxviral-specific CTL in a human trial of recombinant-modified vaccinia Ankara. J. Immunol. 2005;175:8431–8437. doi: 10.4049/jimmunol.175.12.8431. [DOI] [PubMed] [Google Scholar]
- 40.Teague RM, Sather BD, Sacks JA, Huang MZ, Dossett ML, Morimoto J, Tan X, et al. Interleukin-15 rescues tolerant CD8+ Tcells for use in adoptive immunotherapy of established tumors. Nat. Med. 2006;12:335–341. doi: 10.1038/nm1359. [DOI] [PubMed] [Google Scholar]
- 41.Ossendorp F, Mengede E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J. Exp. Med. 1998;187:693–702. doi: 10.1084/jem.187.5.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heiser A, Maurice MA, Yancey DR, Wu NZ, Dahm P, Pruitt SK, Boczkowski D, et al. Induction of polyclonal prostate cancer-specific CTL using dendritic cells transfected with amplified tumor RNA. J. Immunol. 2001;166:2953–2960. doi: 10.4049/jimmunol.166.5.2953. [DOI] [PubMed] [Google Scholar]
- 43.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive Tcell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred Tcells. Proc. Natl. Acad. Sci. USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmowski MJ, Choi EM, Hermans IF, Gilbert SC, Chen JL, Gileadi U, Salio M, et al. Competition between CTL narrows the immune response induced by prime-boost vaccination protocols. J. Immunol. 2002;168:4391–4398. doi: 10.4049/jimmunol.168.9.4391. [DOI] [PubMed] [Google Scholar]
- 45.Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- 46.Chen W, McCluskey J. Immunodominance and immunodomination: Critical factors in developing effective CD8+ T-cell-based cancer vaccines. Adv. Cancer Res. 2006;95:203–247. doi: 10.1016/S0065-230X(06)95006-4. [DOI] [PubMed] [Google Scholar]
- 47.Limmer A, Sacher T, Alferink J, Kretschmar M, Schonrich G, Nichterlein T, Arnold B, Hammerling GJ. Failure to induce organspecific autoimmunity by breaking of tolerance: Importance of the microenvironment. Eur. J. Immunol. 1998;28:2395–2406. doi: 10.1002/(SICI)1521-4141(199808)28:08<2395::AID-IMMU2395>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 48.Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, Scott B. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J. Immunol. 2000;165:6047–6055. doi: 10.4049/jimmunol.165.11.6047. [DOI] [PubMed] [Google Scholar]
- 49.Lang KS, Georgiev P, Recher M, Navarini AA, Bergthaler A, Heikenwalder M, Harris NL, et al. Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J. Clin. Invest. 2006;116:2456–2463. doi: 10.1172/JCI28349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenberg PD, Kern DE, Cheever MA. Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+,2− T cells. Tumor eradication does not require participation of cytotoxic T cells. J. Exp. Med. 1985;161:1122–1134. doi: 10.1084/jem.161.5.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Panina-Bordignon P, Tan A, Termijtelen A, Demotz S, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes: Promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur. J. Immunol. 1989;19:2237–2242. doi: 10.1002/eji.1830191209. [DOI] [PubMed] [Google Scholar]
- 52.Greenfield A, Scott D, Pennisi D, Ehrmann I, Ellis P, Cooper L, Simpson E, Koopman P. An H-YDb epitope is encoded by a novel mouse Y chromosome gene. Nat. Genet. 1996;14:474–478. doi: 10.1038/ng1296-474. [DOI] [PubMed] [Google Scholar]
- 53.Chen W, Yewdell JW, Levine RL, Bennink JR. Modification of cysteine residues in vitro and in vivo affects the immunogenicity and antigenicity of major histocompatibility complex class I-restricted viral determinants. J. Exp. Med. 1999;189:1757–1764. doi: 10.1084/jem.189.11.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berlioz C, Darlix JL. An internal ribosomal entry mechanism promotes translation of murine leukemia virus gag polyprotein precursors. J. Virol. 1995;69:2214–2222. doi: 10.1128/jvi.69.4.2214-2222.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Prats AC, De Billy G, Wang P, Darlix JL. CUG initiation codon used for the synthesis of a cell surface antigen coded by the murine leukemia virus. J. Mol. Biol. 1989;205:363–372. doi: 10.1016/0022-2836(89)90347-1. [DOI] [PubMed] [Google Scholar]