

Abstract
Mixed parasite infections are common in many parts of the world but little is known of the effects of concomitant parasite infections on the immune response or severity of clinical disease. We have used the non-lethal malaria infection model of Plasmodium chabaudi AS together with the gastro-intestinal nematode Heligmosomoides bakeri polygyrus to investigate the impact of nematode infections on malarial morbidity and anti-malarial immunity. The data demonstrate that wildtype (WT) C57BL/6 mice co-infected with both parasites simultaneously exhibit a striking increase in mortality, while mice deficient in interferon (IFN)-γ or IL-23 survive co-infection. The increase in mortality in WT mice was associated with severe liver pathology characterised by extensive coagulative necrosis and an increase in hepatic IFN-γ, IL-17 and IL-22 mRNA expression. This is the first demonstration of increased malaria-associated pathology associated with a switch towards a pro-inflammatory environment, involving not only IFN-γ but also the IL-17/IL-23 axis, as a result of co-infection with a gastro-intestinal helminth.
Keywords: parasitic-helminth, parasitic-protozoan, inflammation, cytokines
INTRODUCTION
Helminth infections are amongst the most common infections of man. Most helminth infections, such as gastro-intestinal nematode infections, are chronic infections, where the worms are long-lived and may survive within their host for many years. In order to survive such extended periods of time without provoking a strong inflammatory immune response, these organisms have developed sophisticated survival strategies, involving secretion of immune modulatory substances that induce anti-inflammatory and/or regulatory immune responses (1). This ability of helminth parasites to modulate immune responses and immune-responsiveness have recently generated a great deal of interest, and beneficial effects of helminth infections and/or products have been demonstrated using experimental models of conditions such as inflammatory bowel disease, diabetes and allergy (2-4). The majority of such studies have used the adult stages of helminth infections and little is known how the impact of infections with juvenile stages of helminths may differ from that of adult worms. Importantly, the immune response to larval stages of helminth infections can differ significantly from adult infections (5-9). This may be of importance in the human field situation during conditions such as migration where previously unexposed individuals may be exposed to primary infections with several pathogens simultaneously.
Experimental models of helminth-malaria co-infections have suggested a role for helminths in altering immune responsiveness and disease progression of malaria (10-12). In human helminth/malaria co-infections the picture is less clear (reviewed by (13, 14)). In order to investigate the effects of larval infections rather than established adult helminth infections, we have focused our studies on how a concurrent larval intestinal nematode infection may alter the course of a blood-stage malaria infection in vivo. We have used the non-lethal rodent malaria Plasmodium chabaudi chabaudi AS together with larval infection of the gastro-intestinal nematode Heligmosomoides bakeri polygyrus (=Nematospiroides dubius). The underlying immunological responses required to clear a blood-stage P. chabaudi AS infection have been well-described, and involve the sequential activation of NK cells and Th1 cells, followed by Th2 cells and antibody producing B cells (15-18). H. bakeri polygyrus is a well known murine model of gastrointestinal nematode infection. Established adult infections with this worm is characterised by production of Th2 cytokines, IL-10, TGF-β and expansion of T regulatory cells (1, 19-21), and this infection model has been commonly used to investigate the effects of chronic helminth infection on immune reactivity (12, 22-28). However, no previous studies have focused on the effects of early larval stage infections.
Our data show that when mice were co-infected with both pathogens at the same time a striking increase in mortality was detected. This was associated with severe liver pathology characterised by extensive necrosis with increased levels of pro-inflammatory cytokines and liver-associated transaminase in serum. Splenic cytokine responses were unaltered demonstrating that the dysregulated responses were organ-specific. Mice deficient in TNF-α or Lymphotoxin (LT)-α were as susceptible to co-infection as wildtype animals. However, IFN-γ, IL-12/23p40 and IL-23p19 deficient animals were resistant, demonstrating that the mortality was associated with organ-specific overproduction of Th1 and Th17 type cytokines. This is the first demonstration of increased malaria-associated pathology as a result from co-infection with larval stages of a gastro-intestinal helminth, and appears to be mediated by pro-inflammatory cytokine pathways involving IL-23/IL-17 and IFN-γ.
MATERIAL AND METHODS
Animals and infections
Six to eight week old female C57BL/6 mice were bred at the animal unit, LSHTM. Female mice deficient in IL-12/23p40 (29), TNF-α (30), LT-α (31) and IFN-γ (32) on a C57BL/6 background, were all originally obtained from the Jackson laboratory (Bar Harbour, Maine) and bred at the animal unit, LSHTM, under SPF conditions. IL-23p19 deficient (33) mice were originally obtained from Schering-Plough Biopharma (Palo Alto, CA), and kindly provided by Dr Brigitta Stockinger, NIMR London. All experiments were performed under the regulations of the Home Office Scientific Procedures Act (1986).
Experimental animals (8-20 per group) were infected with 106 asexual blood-stage P. chabaudi chabaudi AS malaria parasites i.p, and/or 200 H. polygyrus L3 larvae by oral gavage. Maintenance and infection of P. chabaudi AS were as described previously (10). Heligmosomoides bakeri polygyrus was originally obtained from Prof JM Behnke, Nottingham and maintained as stock infection in CD-1 mice. New stock was prepared every two months. Infective L3 larvae were cultured as described by (34). Control groups receiving single infections were infected in parallel for each experiment. Malaria parasitemia was determined on Giemsa stained thin blood smears and the levels of blood hemoglobin were analyzed using a Hemocue hemoglobin analyser (Hemocue, Dronfield, UK) according to the manufacturers instructions.
Cell culture
Mesenteric lymph node (MLN) and spleen cells were removed from uninfected and infected animals and resuspended in RPMI 1640 supplemented with 10% heat-inactivated FCS, 2mM L-glutamine, 0.05 mM β-mercaptoethanol (all from Invitrogen, Paisley, UK) and 25 μg/ml Gentamicin (Sigma, Gillingham, UK). Liver mononuclear cells were prepared by collagenase digestion and centrifugation over Ficoll. Cells were cultured at 37°C and 5% CO2 in flat-bottomed 96-well plates (Nunc, Roskilde, Denmark) at a final concentration of 5×106/ml in a final volume of 0.2 ml/well. Cells were stimulated with H. polygyrus crude worm antigen, P.chabaudi lysate (both at 25 μg/ml), or plate-bound anti-CD3 antibody (mAb145-2C11, 10 μg/ml, ATCC). Cell free supernatants were harvested after 48 hours and stored at −80°C.
Cytokine and LBP ELISA
Cytokine analyses were carried out using routine sandwich ELISAs for IL-4, IL-10 and IFN-γ (Mabtech AB, Nacka, Sweden). IL-13 and TNF-α was analysed using antibody pairs from R&D systems (Abingdon, UK). Lipopolysaccharide binding protein (LBP) in sera was analysed by a commercial ELISA kit (Hycult Biotechnology, Uden, Netherlands) according to the manufacturers instructions.
Serum aspartate aminotransferase assay
To measure the liver-associated enzyme aspartate aminotransferase (AST) in serum a modified protocol of the standard colorimetric end-point method was used. Briefly, 20 ul of serum was added to 100ul of 0.2M DL-aspartate and 1.8M α-ketoglutaric acid in PBS, mixed and incubated (37C, 1h), followed by addition of 100 ul 2,4-dinitrophenylhydrazine. The mixture was incubated for a further 20 minutes at room temperature. The reaction was stopped by addition of 1 ml 0.4M NaOH and absorbance was read at 490 nm after 5 minutes.
Histopathological analyses
Livers were fixed in neutral buffered formalin, histologically processed using standard methods, and sections were stained with hematoxylin-eosin.
Real -time PCR
Tissues were harvested and stored in RNAlater (Qiagen, Crawley, UK) at −80C until processing. RNA was purified using an RNeasy mini kit from Qiagen according to the manufacturers instructions using the additional DNAse treatment step (Qiagen). Reverse transcription was performed using the Omniscript RT kit (Qiagen). Real time PCR was performed in an ABI 7000 sequence detection system (Applied Biosystem, Warrington, UK) using the SYBR Green PCR Master Mix (Qiagen). Results were normalized to the housekeeping gene HPRT and expressed as fold increase compared to tissue from naïve, uninfected controls (given an arbitrary value of 1).
Statistical analyses
Significant differences (P<0.05) between experimental groups were determined using the Students t-test.
RESULTS
Early stage H. polygyrus infection increases the virulence of non-lethal P. chabaudi malaria
To investigate if an intestinal nematode infection can alter the course of a malaria infection in vivo we infected female C57BL/6 mice (10-20 per group) with 200 H. polygyrus L3 larvae by oral gavage and/or 106 asexual blood-stage P. chabaudi chabaudi AS malaria parasites i.p. Control groups receiving single infections were infected in parallel for each experiment. Malaria parasitemia was determined on Giemsa stained thin blood smears and blood hemoglobin levels were analyzed using a Hemocue® hemoglobin analyser. The results in Figure 1 show that simultaneous co-infection of H polygyrus and P. chabaudi malaria dramatically increased malaria mortality. P. chabaudi AS is normally a non-lethal malaria infection in most strains of mice, including the C57BL/6 strain, which was used in these experiments. Results in Figure 1D, show that the survival rate of mice infected with either H. polygyrus or P. chabaudi alone was 100%. However, mice co-infected with H. polygyrus and P. chabaudi developed severe symptoms of disease and by day 7 post infection (p.i) 12 out of 20 animals (60%) succumbed to the infection. By day 9 p.i the mortality rate had risen to 70%. The experiment was repeated five times with similar outcome (data not shown). When the experiment was performed in BALB/c mice the mortality reached 100% in the co-infected mice by day 9 p.i (Figure 1E).
FIGURE 1. Malaria and helminth co-infection results in high mortality without increasing malaria parasitemia.

Malaria parasitemia (A), hemoglobin (B), body weight (C), and survival (D) during P. chabaudi/H. polygyrus co-infection in C57BL/6 mice. Mice were infected orally with 200 H. polygyrus L3 larvae (black triangles), 106 asexual blood-stage P. chabaudi AS parasites i.p (black squares), or both (white squares), at day 0. Mean of 10-20 mice per group and SEM are shown. Asterisks indicate statistically significant difference between groups of mice with P. chabaudi or P. chabaudi plus H. polygyrus infection (P<0.05). Data from one representative experiment out of five is shown. Mortality (E) during H. polygyrus/P. chabaudi co-infection in BALB/c mice infected with P. chabaudi AS (squares), or H. polygyrus plus P. chabaudi (triangles), at day 0 (8 mice per group). Data from one representative experiment out of two is shown. Mortality (F) during H. polygyrus/P. chabaudi co-infection in C57BL/6 mice infected with P. chabaudi AS only at day 0 (squares), or P. chabaudi at day 0 plus H. polygyrus at the indicated timepoints (day 0, or 7 or 14 days before P. chabaudi) (5-7 mice per group). Data from one representative experiment out of three is shown.
The development of malaria parasitemia was similar in the single and co-infected groups before (day 3-5 p.i) and at the peak of parasitemia (day 6-7 p.i) (Figure 1A). However, the malaria parasitemia was higher in the small proportion of co-infected mice that survived infection, after the peak of parasitemia (at days 9 and 10 p.i) than in mice infected with malaria alone (Figure 1A).
As a measure of the severity of disease, we also measured the levels of hemoglobin in blood and changes in bodyweight during the course of infection. The co-infected group developed anemia at the same rate as the control group but the small group of surviving co-infected animals had significantly lower hemoglobin levels than the singly-infected animals at day 9 and 12 p.i (Fig 1B), correlating with the higher parasitemia detected in this group at these time points (Fig 1A). Weight loss was similar in single infected and surviving co-infected animals and was maximal at times of peak parasitemia (Fig 1C). Surviving co-infection animals regained weight at a similar rate to the malaria alone-infected group (Fig 1C). As expected, mice infected with H. polygyrus alone developed neither anemia nor weight loss and survived indefinitely.
We further investigated the importance of the timing of H. polygyrus infection. When mice were inoculated with H. polygyrus 7 or 14 days before P. chabaudi infection there was only a minor, non-significant, effect on P. chabaudi infection (Figure 1F) demonstrating that it was the larval stages rather than adult stages of the helminth infection that was responsible for the increased malaria mortality.
Co-infection does not alter cytokine responses in the spleen
In order to investigate if the increased mortality was due to an imbalance in cytokine production in the spleen, the main organ for anti-malarial immune responses, we analysed cytokine mRNA expression by quantitative real time PCR. The data presented in Figure 2 show that P. chabaudi infection alone strongly increased the expression of IFN-γ by day 4 post infection, and to a lesser degree IL-12p40, IL-12p35, TNF-α, IL-10, IL-13 and IL-4. The expression of IL-10 continued to increase by day 7 post infection and a concomitant decrease in the mRNA expression of all other cytokines at this timepoint was observed in animals infected with malaria only, in agreement with other studies showing the switch away from a Th1 response around the time of peak parasitaemia (15-18). P. chabaudi infection alone did not increase expression of IL-23p19 at any timepoint. Interestingly, H. polygyrus infection alone strongly increased IFN-γ and IL-23p19 already at day 4 post infection, and this was followed by an increased expression of IL-4 and IL-13 by day 7 post infection. H. polygyrus alone did not increase expression of IL-12p35 or TNF-α and only induced a very moderate increase in IL-10. Spleens from co-infected animals showed a similar cytokine profile to P. chabaudi infected animals demonstrating that the concurrent helminth infection did not inhibit or enhance the cytokine response to the malaria infection. We also measured protein levels of IL-4, IFN-γ and IL-10 from in vitro restimulated spleen cell cultures by ELISA, and found no differences in the malaria-specific response between malaria only infected and helminth co-infected animals (Figure 3). The ELISA data also confirm that the malaria-specific response had little effect on worm-specific responses on day 4, but by day 7 the malaria-specific responses had come to dominate, resulting in downregulation of worm-specific IFN-γ and IL-4 responses (but not worm-specific IL-10) in the spleens of co-infected animals (Figure 3).
FIGURE 2. Normal splenic cytokine mRNA expression during P. chabaudi/H. polygyrus co-infection.

C57BL/6 mice were infected orally with 200 H. polygyrus L3 larvae (black triangles), or 106 asexual blood-stage P. chabaudi AS parasites i.p (black squares), or both (white squares), at day 0. RNA was prepared from freshly isolated spleen tissue at various time points post infection, reverse transcribed and cytokine mRNA expression was analysed by real-time PCR. Mean of 4-6 mice per group and SEM are shown. Data from one representative experiment out of three is shown.
FIGURE 3.

Antigen-specific splenic cytokine secretion during P. chabaudi/H. polygyrus co-infection. C57BL/6 mice were infected orally with 200 H. polygyrus L3 larvae (black triangles), or 106 asexual blood-stage P. chabaudi AS parasites i.p (black squares), or both (white squares), at day 0. Spleen cell suspensions were prepared at the indicated time-points and re-stimulated with H. polygyrus antigen (H.poly Ag) or lysate of P. chabaudi infected red blood cells (pRBC) for 48 hours. IFN-γ, IL-4 and IL-10 levels in supernatants were analysed by sandwich ELISA. Mean of 4-6 mice per group and SEM are shown. Data from one representative experiment out of three is shown.
Nematode-malaria co-infection increases serum cytokine, liver enzyme and LBP levels
We next investigated whether the appearance of inflammatory mediators in serum, which normally accompanies acute P. chabaudi infection, was altered by the presence of larval nematode infection. Animals infected with P. chabaudi showed a moderate, but significant, increase in serum IFN-γ (day 4 p.i) and TNF-α (day 4 and 7 p.i) compared to sera from uninfected animals (Figure 4A and B). No such increase could be detected in animals with only H. polygyrus infection. Co-infected animals, however, had significantly higher levels of serum IFN-γ (day 4 and 7 p.i) and TNF-α (day 7 p.i) (Fig 4A and B) than animals infected with malaria alone. Mice with only P. chabaudi infection had significantly elevated levels of IL-10 in the circulation already from day 2 post infection. The levels of IL-10 then continued to increase as the infection progressed (Figure 4C). The co-infected group, however, had significantly lower levels of circulating IL-10 throughout the infection (Fig 4C), which were similar to the modest upregulation of IL-10 seen in animals infected with H. polygyrus alone. When expressing the data as the ratio of IFN-γ versus IL-10 in serum it is clear that the co-infected mice had a very strong bias towards circulating IFN-γ, both at day 4 and day 7, compared to the groups with single malaria or nematode infections (Fig 4D).
FIGURE 4. Increased levels of serum cytokines, LBP and liver enzyme during malaria/helminth co-infection.

C57Bl/6 mice were infected orally as above. Serum levels of IFN-γ (A), TNF-α (B), IL-10 (C) and LBP (F) were measured by sandwich ELISA and AST (E) by colorimetric assay. Mean of 4-8 mice per group and SEM are shown. Data from one representative experiment out of three is shown. Asterisks indicate statistically significant difference between groups of mice with P. chabaudi or P. chabaudi plus H. polygyrus infection (P<0.05).
The liver is well known as the site of pre-erythrocytic development of Plasmodium parasites. However, the liver also participates in the killing and removal of infected red blood cells from the circulation (35, 36) and signs of T cell mediated liver injury are present during blood-stage infection (35, 37-39). We therefore measured serum levels of the liver-associated enzyme aspartate aminotransferase (AST) in mice with single and co-infection (Figure 4E). AST is normally contained within hepatocytes, but when liver damage occurs, AST is released, resulting in elevated levels of the enzyme in the circulation. The data in Figure 4E show that co-infected mice displayed greatly increased levels of AST in serum already at day 2 post malaria infection. The levels increased further on day 4 and day 7 p.i (Figure 4E), indicating severe liver damage during H. polygyrus/P. chabaudi co-infection but not during single infection with either pathogen.
Lipopolysaccharide binding protein (LBP) act to amplify the cellular response to LPS and can be used as a marker for acute phase responses and sepsis-like syndromes (40). We analysed the levels of LBP in serum and found that the co-infected mice had greatly enhanced LBP levels at day 7 p.i compared to the singly infected mice, again suggesting a severe, acute endotoxin-like response during the malaria/nematode infection (Figure 4F).
Severe liver pathology in nematode-malaria co-infection
Since high levels of AST in serum are indicative of liver injury we performed histological analysis on the livers from the different groups of mice. As can be seen in Figure 5, severe liver damage characterised by centrilobular necrosis, inflammatory infiltration and hemorrhaging was evident in the co-infected animals at day 7 p.i (Figure 5D and F). H. polygyrus infection alone did not alter liver histology and there was no evidence of inflammation or eosinophil infiltration (Figure 5B). P. chabaudi infection alone resulted in inflammatory cell infiltration but no overt hepatic damage or necrosis was observed (Figure 5C and E). These results clearly demonstrate that malaria-H. polygyrus co-infection results in severe hepatic liver injury with increased levels of serum AST, hepatic inflammatory infiltrates and centrilobular necrosis.
FIGURE 5. Severe liver damage during malaria/helminth co-infection.

Liver histology at day 7 post infection. C57BL/6 mice were infected orally as above. Livers were fixed in neutralbuffered formalin, processed and stained with hematoxylin/eosin using standard methodology. Liver from uninfected control (A). H. polygyrus alone day 7 p.i (B), P. chabaudi alone day 7 p.i (C and E), H. polygyrus plus P. chabaudi day 7 p.i (D and F) A-D, x100 magnification. E-F x200 magnification. Data from one representative experiment out of three is shown.
Severely altered cytokine responses in the liver during malaria-nematode co-infection
Since co-infected animals displayed inflammatory infiltrates and severe pathology of the liver we analysed hepatic cytokine mRNA expression by quantitative real-time PCR. The data presented in Figure 6 show that liver cytokine expression during P. chabaudi infection alone closely mirrored that of the mRNA expression in the spleen. There was a strong increase in the expression of IFN-γ and TNF-α already at day 4 post-infection. However, by day 7 both IFN-γ and TNF-α expression were downregulated. This downregulation coincided with a strong upregulation of IL-10. P. chabaudi infection alone did not increase liver expression of IL-23p19, IL-12p35, IL-4, IL-13 or the Th17 cytokines IL-17 and IL-22. H. polygyrus infection alone increased IL-4 and IL-13 expression, and to a lesser degree IL-12p40, IL-23p19 and IFN-γ. H. polygyrus infection alone did not increase expression of IL-10, IFN-γ, TNF-α, IL-12p35, IL-17 or IL-22. However, livers from co-infected animals expressed high levels of IFN-γ, TNF-α, IL-12/23p40, IL-23p19, IL-17, IL-22 and IL-4 mRNA with little or no increase in IL-12p35, IL-13 and IL-10 expression. Thus, P. chabaudi infection alone results in strong inflammatory cytokine responses in the liver, similar to the cytokine profile observed in the spleen, and this pro-inflammatory cytokine response appears to be controlled by IL-10. In contrast, the low levels of IL-10 and high levels of inflammatory cytokines in the livers of H. polygyrus/P. chabaudi co-infected animals suggest that co-infection may result in an uncontrolled pro-inflammatory response leading to enhanced liver pathology. The lack of IL-12p35 expression during both P. chabaudi infection alone and during co-infection, in conjunction with increased IL-12p40 and IL-23p19 mRNA expression, strongly suggests that the pro-inflammatory response is driven by IL-23 rather than IL-12. This is further supported by the increase in IL-17 and IL-22, cytokines produced by IL-23 driven Th17 cells (41).
FIGURE 6. Altered liver cytokine mRNA expression during P. chabaudi/H. polygyrus co-infection.

C57BL/6 mice were infected orally as above. RNA was prepared from freshly isolated liver tissue, reverse transcribed and cytokine mRNA expression was analysed by real-time PCR. Mean of 4-6 mice per group and SEM are shown. Data from one representative experiment out of three is shown.
Cytokine secretion from purified liver mononuclear cells analysed by ELISA confirmed the high IFN-γ and low IL-10 response in livers from co-infected animals (IFN-γ: P.c infected 3.42 +/− 0.78 ng/ml vs. P.c+H.p infected 6.46 +/− 0.46 ng/ml, IL-10: P.c infected 2.15 +/− 0.09 ng/ml vs. P.c+H.p infected 0.61 +/− 0.10 ng/ml).
Thus, our data suggest that a larval intestinal nematode infection can disrupt the delicate and critical balance of pro- and anti-inflammatory responses needed to prevent excessive liver pathology during acute blood-stage malaria infection.
Increased mortality and liver damage during H. polygyrus/P. chabaudi co-infection is dependent on the IL-23 and IFN-γ pathways but not on TNF-α or LT̃α
In view of the fact that the co-infected mice expressed high levels of IFN-γ, TNF-α, IL-12p40 and IL-23p19 in the liver, we investigated the importance of these cytokines in the increased mortality seen during co-infection. Groups of IFN-γ, TNF-α, LT-α, IL-12p40, IL-23p19 knockout and C57BL/6 wildtype mice were infected with P. chabaudi alone, H. polygyrus alone or were co-infected. The data in Figure 7A show that the C57BL/6 WT mice developed severe pathology and over 80% of the animals had died by day 9 post infection. Surprisingly, the TNF-α and LT-α KO mice succumbed to the co-infection at a similar rate as the WT mice demonstrating that the mortality associated with co-infection is not mediated by TNF-α or LT-α (Figure 7A). Importantly, the majority of the IL-12p40, IL-23p19 and IFN-γ KO mice survived co-infection demonstrating that the mortality was dependent on IL-12/23 and IFN-γ (Figure 7A). None of the mice died of malaria infection or H. polygyrus infection alone during the time course studied (21 days) (data not shown). There were no major differences in malaria parasitemia between malaria only and co-infected animals for each strain of knockout mice used (Figure 7B). IFN-γ and IL-12p40 KO mice had increased peak parasitemias and delayed clearance from bloodstream as compared to WT mice, in agreement with other studies using female mice (42, 43), and co-infection did not alter this (Figure 7B). TNF-α and LT-α knockout mice did not exhibit altered parasitemias compared to WT mice, again in agreement with previous studies (44).
FIGURE 7. Increased mortality during H. polygyrus/P. chabaudi co-infection is dependent on IL-12p40, IFN-γ and IL-23p19, but not on TNF-α or LT-α.

Survival rates (A) and malaria parasitemias (B) in various KO strains co-infected with P. chabaudi and H. polygyrus. C57BL/6 WT mice, IFN-γ, IL-12p40, TNF-α, LT-α and IL-23p19KO mice were infected as above (5-8 mice per group). Data from one representative experiment out of three (two for p19KO experiment) is shown.
Serum cytokine analysis at day 7 post infection demonstrated that the susceptible WT and TNF-α KO mice had high ratios of IFN-γ versus IL-10 in serum, while the resistant IL-12p40 KO mice managed to retain balanced levels of these two cytokines (Figure 8A). Measurement of AST levels in serum confirmed that the liver damage was dependent on IL-12p40, IL-23p19 and IFN-γ but not on TNF-α (Figure 8B). Furthermore, liver histology at day 7 post infection again confirmed that WT and TNF-α KO mice developed extensive centrilobular necrosis and inflammatory infiltrates while no such histopathology could be detected in IFN-γ and IL-12p40 KO mice (Figure 9).
FIGURE 8. Liver damage during H. polygyrus/P. chabaudi co-infection is dependent on IL-12p40, IFN-γ and IL-23p19, but not on TNF-α or LT-α.

Serum cytokines (A) and AST levels in serum (B) in various KO strains co-infected with P. chabaudi and H. polygyrus. C57BL/6 WT mice, IFN-γ, IL-12p40, TNF-α, LT-α and IL-23p19KO mice were infected as above (5-8 mice per group). Data from one representative experiment out of three (two for p19KO experiment) is shown. ND: not detectable. Asterisks indicate statistically significant difference between groups of mice with P. chabaudi or P. chabaudi plus H. polygyrus infection (P<0.05).
FIGURE 9. Liver histology at day 7 post infection.
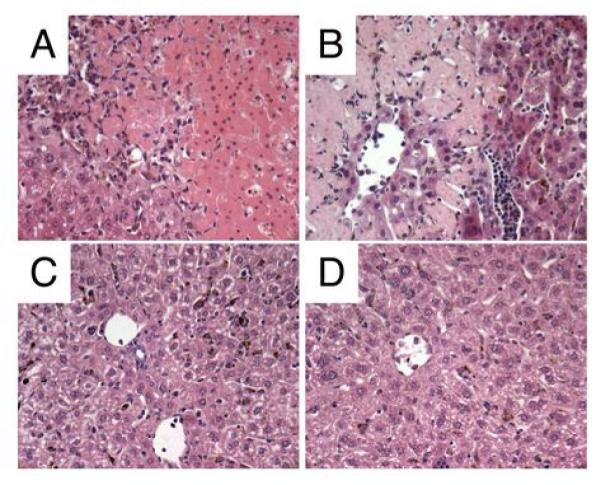
C57BL/6 WT mice, IFN-γ, IL-12p40 and TNF-α KO mice were co-infected as above. Livers were fixed in neutralbuffered formalin, processed and stained with hematoxylin/eosin using standard methodology. C57BL/6 WT (A), TNF-α KO (B), IFN-γ KO (C), IL-12p40 KO (D). A-D, x200 magnification. Data from one representative experiment out of three is shown.
DISCUSSION
Its is well established that the control and clearance of a non-lethal blood-stage P. chabaudi AS infection is dependent on a strong pro-inflammatory response involving NK cells and Th1 cells (15-18) and that this pro-inflammatory response is under the critical control of the anti-inflammatory cytokines IL-10 and TGF-β (45-47). While the spleen is of central importance in the immune response to blood-stage malaria, the liver is well known as the site of pre-erythrocytic development of Plasmodium parasites. However, the liver is also an important organ during blood-stage infection and participates in the killing and removal of infected red blood cells from the circulation (35, 36). Adoptive transfer experiments have shown that lymphomyeloid cells isolated from infected livers can confer protection to naïve recipients (35), and there is an increase in the number of active Kupffer cells (48) during blood-stage infection. Furthermore, elevated levels of IFN–γ, IL-12p40, IL-18 and IL-10 in the liver have been demonstrated during blood-stage malaria (37, 49). As with the need for a critically balanced pro- and anti-inflammatory response in the spleen, the response in the liver needs to be similarly carefully balanced. Lethal strains of murine malaria (e.g. P. berghei NK65) induce signs of severe hepatotoxicity, mediated by cytotoxic T cells and NK cells (50), while non-lethal strains (e.g P. chabaudi AS) induce mild but clear signs of T cell mediated liver injury (35, 37-39). Importantly, in vivo blockade of cytotoxic T-lymphocyte-associated protein (CTLA)-4 results in a more severe liver injury and increased mortality, suggesting that regulatory activity is crucial in controlling hepatotoxicity during blood-stage infection (38). This is in agreement with our data showing that at the point of peak parasitemia in the bloodstream, the pro-inflammatory response in both liver and spleen is downregulated and a concomitant increase in IL-10 is detected, representing an attempt to balance the pro-inflammatory response and prevent immune-mediated pathology. However, despite the fact that H. polygyrus infection induced IL-10 production in the spleen in both singly and co-infected mice, the regulatory IL-10 response was absent from the liver in co-infected animals and a strong increase in pro-inflammatory cytokines was detected instead. This was in contrast to the cytokine profile in the spleen, where the pro- and anti-inflammatory cytokine response remained balanced during co-infection. Using gene-deficient mice we have demonstrated that liver injury and mortality in this model is dependent on IL-12p40, IL-23p19 and IFN-γ but not on TNF-α or LT-α. The IL-12p40 chain, together with IL-12p35, forms bioactive IL-12, and together with IL-23p19 forms IL-23 (41). The observed upregulation of IL-17 and IL-22 mRNA in the livers of co-infected mice further support a role for IL-23 in this system. IL-17 and IL-22 are both secreted by IL-23-driven Th17 cells and participate in inflammatory reactions (51, 52). Interestingly, a recent report show that IL-22 plays a protective role in preventing hepatic tissue injury (53), and it is possible that the hepatic upregulation of IL-22 detected in our model represent a host-protective response. This is to our knowledge the first demonstration of the involvement of IL-23-driven responses in the pathogenesis of malaria infection. The inherent difficulties with co-infection models makes it complicated to pinpoint the exact functional mechanisms behind these observations, as experiments aimed at functional intervention often results in alterations of the course of the single infections, making data interpretation difficult. Be that as it may, future work will shed more light on the relative contribution of these particular cytokine responses in the development of immunity and immunopathology associated with protozoan and/or helminth infections.
Most studies aimed at investigating the immunomodulatory effects of concurrent H. polygyrus infection have focused on the adult phase of infection, i.e. > 2 weeks post infection. In most cases the worm infection has been initiated several weeks, or even months, before co-infection or co-administration of antigen, thus allowing for the development of the strong Th2/regulatory response that is the hallmark of chronic H. polygyrus infection. By using this approach, studies have demonstrated the immunomodulatory effects of H. polygyrus on colitis, Citrobacter, Helicobacter and malaria infection, airway hyperreactivity and diabetes (12, 22-26). As such, there is no doubt that an adult H. polygyrus infection is strongly immunomodulatory towards the Th2/regulatory spectrum of response. Previous experimental helminth/malaria co-infection studies have all focussed on the possible counter-regulatory anti-parasite cytokine responses, where the Th1-dependent control of malaria parasitemia might be impaired by the Th2-dominated environment of an animal carrying a pre-existing helminth infection. Indeed, several studies using different types of helminth infections have shown that an established dominant Th2 response can alter the disease progression of a murine malaria infection by diminishing the Th1 response (10-12, 54-58). However, the consequences of the early larval stages of helminth infections have received little attention. Our data show that a larval H. polygyrus infection on its own is sufficient to selectively induce the up-regulation of certain cytokines in the liver and spleen, including pro-inflammatory cytokines. Previous studies have reported Th1 responses in the intestine, mesenteric lymph nodes and/or spleen during larval infections with several intestinal nematodes (5-9). Thus, our data are in agreement with previous studies showing that different life stages of helminth parasites may affect immune responsiveness differently depending on timing and anatomical site, a fact that is often overlooked in the literature.
The life cycle of H. polygyrus, a trichostrongylid nematode commonly occurring in wild small rodents, is direct and involves both free-living and parasitic stages. The host is infected by the ingestion of infective third stage larvae, which penetrate the anterior half of the small intestinal mucosa within 24 hours of infection. The developing larvae forms cysts in the intestinal wall, surrounded by mild inflammatory infiltrates (histotrophic phase) (59). Around 7-8 days post infection the larvae finally molt to adult stages and move out to the gut lumen and egg production can be detected in faeces around 10 days post infection (34). After the adult worms have left the cysts and migrated to the lumen, an inflammatory response involving neutrophils and macrophages develop around the empty cysts (59, 60). Since the early stages of H. polygyrus larval infection are invasive and induce localized inflammation it is reasonable to believe that any secreted antigens will be transported directly from the intestine to the liver via the portal system, and once in the liver may affect T cell responses directly, or indirectly by affecting antigen presentation. Studies have shown that during the development from L3 larvae to adult worms the parasite express a number of stage specific antigens (61, 62), including several secreted larval stage antigens (63). The identity and functions of these antigens are largely unknown but several have been suggested to act as immunosuppressors, facilitating the establishment and survival of the worm (64, 65). Considering that larval stages of nematodes are susceptible to Th2-induced immune-mediated resistance mechanisms (66), it would be beneficial for the larvae to interfere with the development of such immune responses at the earliest possible stage of infection, and this may result in the promotion of pro-inflammatory responses. Further detailed investigations into the identities and characteristics of stage specific antigens and their effects on the immune system are needed in order to better understand how helminths can manipulate the immune system.
Another potential stimulus for the liver damage observed in the co-infected animals, may be the leakage of bacteria or bacterial endotoxin (LPS) from the injured intestine to the portal system. The timing of mortality in the co-infected mice coincides with the emergence of adult worms from the intestinal cysts to the gut lumen. The inflammatory response that develops around the empty larval cysts in the intestinal wall (59, 60), suggest that bacteria may be involved, and it is possible that the bacteria itself, or LPS and/or LPS-like products, may reach the liver and cause some degree of inflammation. During H. polygyrus infection alone this may not be sufficient to induce liver injury, but in combination with another liver-associated pathogen it may result in enhanced organ-specific pathology. Previous studies have shown that as a murine malaria infection progress the mice become more resistant to systemic endotoxin challenge, probably due to the continuous stimulation by hemozoin through Toll-like receptor (TLR) 9 (67, 68). Nevertheless, it is possible that LPS leakage from the intestine, coinciding with an early inflammatory malaria response in the liver, could act synergistically to promote liver injury. Moreover, liver injury during blood-stage malaria infection is dependent on the MyD88 adapter molecule (69), a molecule intrinsic to the signalling pathway of TLRs, suggesting that increased stimulation through TLRs may trigger inflammation. Increased levels of pathology caused by intestinal “leakage” and reduced epithelial barrier function have previously been implicated in other models of helminth infection (70, 71).
The data from the current study, demonstrate the striking and unanticipated results that concurrent exposure to the larval stages of an intestinal helminth and malaria infection may result in increased severity of malaria in a mouse model. Certainly, in the human field situation individuals often carry chronic helminth infections from an early age and the likelihood of being exposed to concurrent primary larval stage helminth infections and malaria may not be very high. There are situations however, when this may occur, such as during transmigration from non-endemic to endemic areas, or from urban to rural areas. The high incidence of severe malaria reported in non-immune adults during the first years of transmigration from Java to Irian Jaya (72) is particularly striking in this aspect. It is certainly possible that these adults were not only exposed to malaria for the first time but also to other parasitic infections, including intestinal helminths. Furthermore, with the current emphasis of anti-helminthic mass treatment in sub-Saharan Africa (73), re-infection situations where the host is exposed to repeated larval infections, may coincide with malarial infection. As such, it will be of interest to investigate how drug-abbreviated infections, as well as trickle infections, function in this aspect of co-infection in the murine model.
Taken together, the data presented here demonstrate that an early stage intestinal nematode infection can enhance malaria-associated pathology without necessarily altering immune reactivity in the spleen. Larval infections may also alter disease progression in different ways to established adult worm infections. It is therefore of importance to consider a number of aspects of co-infections such as the stage of the infection, the dose and the species involved, when interpreting data obtained from animal and human studies. Moreover, it is clear that further work is needed to clarify the exact role of cytokines – especially the IL-23/Th17 axis - and other mediators that play a role in the pathogenesis of nematode/malaria co-infections.
Acknowledgments
I would like to thank Dr. Eleanor Riley and Dr. Quentin Bickle for data discussions and critical reading of the manuscript, Ms. Jenna Murdoch for technical support, Dr Daniel Cua for the p19KO mice and to Dr Brigitta Stockinger for helpful discussions, critical evaluation of the manuscript and provision of mice.
3Abbreviations
- WT
wildtype
- p.i.
post infection
- MLN
mesenteric lymph node
- LT
Lymphotoxin
- AST
aspartate aminotransferase
- LBP
lipopolysaccharide binding protein
Footnotes
This study was funded by the Wellcome Trust (GR067320).
The author has no conflicting financial interests.
REFERENCES
- 1.Maizels RM, Balic A, Gomez-Escobar N, Nair M, Taylor MD, Allen JE. Helminth parasites--masters of regulation. Immunol. Rev. 2004;201:89–116. doi: 10.1111/j.0105-2896.2004.00191.x. [DOI] [PubMed] [Google Scholar]
- 2.Elliott DE, Summers RW, Weinstock JV. Helminths and the modulation of mucosal inflammation. Curr. Opin. Gastroenterol. 2005;21:51–58. [PubMed] [Google Scholar]
- 3.Yazdanbakhsh M, Wahyuni S. The role of helminth infections in protection from atopic disorders. Curr Opin Allergy Clin Immunol. 2005;5:386–391. doi: 10.1097/01.all.0000182541.52971.eb. [DOI] [PubMed] [Google Scholar]
- 4.Zaccone P, Fehervari Z, Phillips JM, Dunne DW, Cooke A. Parasitic worms and inflammatory diseases. Parasite Immunol. 2006;28:515–523. doi: 10.1111/j.1365-3024.2006.00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly EA, Cruz ES, Hauda KM, Wassom DL. IFN-γ- and IL-5-producing cells compartmentalize to different lymphoid organs in Trichinella spiralis-infected mice. J.Immunol. 1991;147:306–311. [PubMed] [Google Scholar]
- 6.Svetic A, Madden KB, Zhou XD, Lu P, Katona IM, Finkelman FD, Urban JF, Gause WC. A primary intestinal helminthic infection rapidly induces a gut-associated elevation of Th2-associated cytokines and IL-3. J.Immunol. 1993;150:3434–3441. [PubMed] [Google Scholar]
- 7.Ishikawa N, Goyal PK, Mahida YR, Li K-F, Wakelin D. Early cytokine responses during intestinal parasitic infections. Immunology. 1998;93:257–263. doi: 10.1046/j.1365-2567.1998.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart GL, Na H, Smart L, Seelig LLJ. The temporal relationship among anti-parasite immune elements expressed during the early phase of infection of the rat with Trichinella spiralis. Parasitol. Res. 1999;85:672–684. doi: 10.1007/s004360050613. [DOI] [PubMed] [Google Scholar]
- 9.Helmby H, Grencis RK. Interleukin-18 regulates intestinal mastocytosis and Th2 cytokine production independently of IFN-γ during Trichinella spiralis infection. J.Immunol. 2002;169:2553–2560. doi: 10.4049/jimmunol.169.5.2553. [DOI] [PubMed] [Google Scholar]
- 10.Helmby H, Kullberg M, Troye-Blomberg M. Altered immune responses in mice with concomitant Schistosoma mansoni and Plasmodium chabaudi infection. Infect.Immun. 1998;66:5167–5174. doi: 10.1128/iai.66.11.5167-5174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham AL, Lamb TJ, Read AF, Allen JE. Malaria-filaria coinfection in mice makes malarial disease more severe unless filarial infection achieves patency. J.Infect.Dis. 2005;191:410–421. doi: 10.1086/426871. [DOI] [PubMed] [Google Scholar]
- 12.Su Z, Segura M, Morgan K, Loredo-Osti JC, Stevenson MM. Impairment of protective immunity to blood-stage malaria by concurrent nematode infection. Infect. Immun. 2005;73:3531–3539. doi: 10.1128/IAI.73.6.3531-3539.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Druilhe P, Tall A, Sokhna C. Worms can worsen malaria: towards a new means to roll back malaria? Trends Parasitol. 2005;21:359–362. doi: 10.1016/j.pt.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Hartgers FC, Yazdanbakhsh M. Co-infection of helminths and malaria: modulation of the immune responses to malaria. Parasite Immunol. 2006;28:497–506. doi: 10.1111/j.1365-3024.2006.00901.x. [DOI] [PubMed] [Google Scholar]
- 15.Langhorne J, Meding SJ, Eichmann K, Gillard SS. The response of CD4+ T cells to Plasmodium chabaudi chabaudi. Immunol.Rev. 1989;112:71–94. doi: 10.1111/j.1600-065x.1989.tb00553.x. [DOI] [PubMed] [Google Scholar]
- 16.Stevenson MM, Tam M-F. Differential induction of helper T cell subsets during blood-stage Plasmodium chabaudi AS infection in resistant and susceptible mice. Clin.Exp.Immunol. 1993;92:77–83. doi: 10.1111/j.1365-2249.1993.tb05951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langhorne J, Cross C, Seixas E, Li C, Von Der Weid T. A role for B cells in the development of T cell helper function in a malaria infection in mice. Proc.Natl.Acad.Sci.USA. 1998;95:1730–1734. doi: 10.1073/pnas.95.4.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helmby H, Kullberg M, Troye-Blomberg M. Expansion of IL-3-responsive IL-4-producing non-B non-T cells correlates with anemia and IL-3 production in mice infected with blood-stage Plasmodium chabaudi malaria. Eur.J.Immunol. 1998;28:2559–2570. doi: 10.1002/(SICI)1521-4141(199808)28:08<2559::AID-IMMU2559>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 19.Finney CA, Taylor MD, Wilson MS, Maizels RM. Expansion and activation of CD4(+)CD25(+) regulatory T cells in Heligmosomoides polygyrus infection. Eur. J. Immunol. 2007;37:1874–1886. doi: 10.1002/eji.200636751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Setiawan T, Metwali A, Blum AM, Ince MN, Urban JFJ, Elliott DE, Weinstock JV. Heligmosomoides polygyrus promotes regulatory T-cell cytokine production in the murine normal distal intestine. Infect. Immun. 2007;75:4655–4663. doi: 10.1128/IAI.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ince MN, Elliott DE, Setiawan T, Blum A, Metwali A, Wang Y, Urban JFJ, Weinstock JV. Heligmosomoides polygyrus induces TLR4 on murine mucosal T cells that produce TGFbeta after lipopolysaccharide stimulation. J.Immunol. 2006;176:726–729. doi: 10.4049/jimmunol.176.2.726. [DOI] [PubMed] [Google Scholar]
- 22.Fox JG, Beck P, Dangler CA, Whary MT, Wang TC, Shi HN, Nagler-Anderson C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat. Med. 2000;6:536–542. doi: 10.1038/75015. [DOI] [PubMed] [Google Scholar]
- 23.Elliott DE, Setiawan T, Metwali A, Blum A, Urban JFJ, Weinstock JV. Heligmosomoides polygyrus inhibits established colitis in IL-10-deficient mice. Eur. J. Immunol. 2004;34:2690–2698. doi: 10.1002/eji.200324833. [DOI] [PubMed] [Google Scholar]
- 24.Chen CC, Louie S, McCormick B, Walker WA, Shi HN. Concurrent infection with an intestinal helminth parasite impairs host resistance to enteric Citrobacter rodentium and enhances Citrobacter-induced colitis in mice. Infect. Immun. 2005;73:5468–5481. doi: 10.1128/IAI.73.9.5468-5481.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson MS, Taylor MD, Balic A, Finney CA, Lamb JR, Maizels RM. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J.Exp.Med. 2005;202:1199–1212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saunders KA, Raine T, Cooke A, Lawrence CE. Inhibition of autoimmune type 1 diabetes by gastrointestinal helminth infection. Infect. Immun. 2007;75:397–407. doi: 10.1128/IAI.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitagaki K, Businga TR, Racila D, Elliott DE, Weinstock JV, Kline JN. Intestinal helminths protect in a murine model of asthma. J.Immunol. 2006;177:1628–1635. doi: 10.4049/jimmunol.177.3.1628. [DOI] [PubMed] [Google Scholar]
- 28.Khan IA, Hakak R, Eberle K, Sayles P, Weiss LM, Urban JFJ. Coinfection with Heligmosomoides polygyrus fails to establish CD8+ T-cell immunity against Toxoplasma gondii. Infect. Immun. 2008;76:1305–1313. doi: 10.1128/IAI.01236-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu C.-y., Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN-γ production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 30.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J.Exp.Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, Smith SC, Carlson R, Shornick LP, Strauss-Schoenberger J, Russell JH, Karr R, Chaplin DD. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 32.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1745. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 33.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 34.Bryant V. The life cycle of Nematospiroides dubius, Baylis, 1926 (Nematoda: Heligmosomidae) J.Helminthol. 1973;47:263–268. doi: 10.1017/s0022149x00026535. [DOI] [PubMed] [Google Scholar]
- 35.Balmer P, Alexander J, Phillips RS. Protective immunity to erythrocytic Plasmodium chabaudi AS infection involves IFNgamma-mediated responses and a cellular infiltrate to the liver. Parasitology. 2000;121:473–482. doi: 10.1017/s0031182099006757. [DOI] [PubMed] [Google Scholar]
- 36.Mannoor MK, Halder RC, Morshed SR, Ariyasinghe A, Bakir HY, Kawamura H, Watanabe H, Sekikawa H, Abo T. Essential role of extrathymic T cells in protection against malaria. J.Immunol. 2002;169:301–306. doi: 10.4049/jimmunol.169.1.301. [DOI] [PubMed] [Google Scholar]
- 37.Singh RP, Kashiwamura S, Rao P, Okamura H, Mukherjee A, Chauhan VS. The role of IL-18 in blood-stage immunity against murine malaria Plasmodium yoelii 265 and Plasmodium berghei ANKA. J.Immunol. 2002;168:4674–4681. doi: 10.4049/jimmunol.168.9.4674. [DOI] [PubMed] [Google Scholar]
- 38.Jacobs T, Plate T, Gaworski I, Fleischer B. CTLA-4-dependent mechanisms prevent T cell induced-liver pathology during the erythrocyte stage of Plasmodium berghei malaria. Eur. J. Immunol. 2004;34:972–980. doi: 10.1002/eji.200324477. [DOI] [PubMed] [Google Scholar]
- 39.Krucken J, Dkhil MA, Braun JV, Schroetel RM, El-Khadragy M, Carmeliet P, Mossmann H, Wunderlich F. Testosterone suppresses protective responses of the liver to blood-stage malaria. Infect. Immun. 2005;73:436–443. doi: 10.1128/IAI.73.1.436-443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zweigner J, Schumann RR, Weber JR. The role of lipopolysaccharide-binding protein in modulating the innate immune response. Microbes Infect. 2006;8:946–952. doi: 10.1016/j.micinf.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 41.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 42.Su Z, Stevenson MM. Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect. Immun. 2000;68:4399–4406. doi: 10.1128/iai.68.8.4399-4406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su Z, Stevenson MM. IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J.Immunol. 2002;168:1348–1355. doi: 10.4049/jimmunol.168.3.1348. [DOI] [PubMed] [Google Scholar]
- 44.Clark K, Kulk N, Amante F, Haque A, Engwerda C. Lymphotoxin alpha and tumour necrosis factor are not required for control of parasite growth, but differentially regulate cytokine production during Plasmodium chabaudi chabaudi AS infection. Parasite Immunol. 2007;29:153–158. doi: 10.1111/j.1365-3024.2006.00930.x. [DOI] [PubMed] [Google Scholar]
- 45.Omer FM, Riley EM. Transforming growth factor beta production is inversely correlated with severity of murine malaria infection. J.Exp.Med. 1998;188:39–48. doi: 10.1084/jem.188.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C, Corraliza I, Langhorne J. A defect in interleukin-10 leads to enhanced malarial disease in Plasmodium chabaudi chabaudi infection in mice. Infect. Immun. 1999;67:4435–4442. doi: 10.1128/iai.67.9.4435-4442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li C, Sanni LA, Omer F, Riley EM, Langhorne J. Pathology of Plasmodium chabaudi chabaudi infection and mortality in interleukin-10-deficient mice are ameliorated by anti-tumor necrosis factor alpha and exacerbated by anti-transforming growth factor beta antibodies. Infect. Immun. 2003;71:4850–4856. doi: 10.1128/IAI.71.9.4850-4856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dockrell HM, de Souza JB, Playfair JH. The role of the liver in immunity to blood-stage murine malaria. Immunology. 1980;41:421–430. [PMC free article] [PubMed] [Google Scholar]
- 49.Tan RS, Kara AU, Feng C, Asano Y, Sinniah R. Differential interleukin-10 expression in interferon regulatory factor-1 deficient mice during Plasmodium berghei blood-stage infection. Parasite Immunol. 2000;22:425–435. doi: 10.1046/j.1365-3024.2000.00312.x. [DOI] [PubMed] [Google Scholar]
- 50.Adachi K, Tsutsui H, Seki E, Nakano H, Takeda K, Okumura K, Van Kaer L, Nakanishi K. Contribution of CD1d-unrestricted hepatic DX5+ NKT cells to liver injury in Plasmodium berghei-parasitized erythrocyte-injected mice. Int.Immunol. 2004;16:787–798. doi: 10.1093/intimm/dxh080. [DOI] [PubMed] [Google Scholar]
- 51.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J.Exp.Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 53.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but Not Interleukin-17 Provides Protection to Hepatocytes during Acute Liver Inflammation. Immunity. 2007;27:647–659. doi: 10.1016/j.immuni.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan Y, Inuo G, Akao N, Tsukidate S, Fujita K. Down-regulation of murine susceptibility to cerebral malaria by inoculation with third-stage larvae of the filarial nematode Brugia pahangi. Parasitology. 1997;114:333–338. doi: 10.1017/s0031182096008566. [DOI] [PubMed] [Google Scholar]
- 55.Yoshida A, Maruyama H, Kumagai T, Amano T, Kobayashi F, Zhang M, Himeno K, Ohta N. Schistosoma mansoni infection cancels the susceptibility to Plasmodium chabaudi through induction of type 1 immune responses in A/J mice. Int. Immunol. 2000;12:1117–1125. doi: 10.1093/intimm/12.8.1117. [DOI] [PubMed] [Google Scholar]
- 56.Legesse M, Erko B, Balcha F. Increased parasitaemia and delayed parasite clearance in Schistosoma mansoni and Plasmodium berghei co-infected mice. Acta Trop. 2004;91:161–166. doi: 10.1016/j.actatropica.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 57.de Souza B, Helmby H. Concurrent gastro-intestinal nematode infection does not alter the development of experimental cerebral malaria. Microbes Infect. 2008;10:916–921. doi: 10.1016/j.micinf.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Noland GS, Urban JFJ, Fried B, Kumar N. Counter-regulatory anti-parasite cytokine responses during concurrent Plasmodium yoelii and intestinal helminth infections in mice. Exp. Parasitol. 2008;119:272–278. doi: 10.1016/j.exppara.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cypess RH, Lucia HL, Dunsford HA, Enriquez FJ. The tissue reactions of mice to infection with Heligmosmoides polygyrus. J.Helminthol. 1988;62:69–76. doi: 10.1017/s0022149x0001124x. [DOI] [PubMed] [Google Scholar]
- 60.Morimoto M, Morimoto M, Whitmire J, Xiao S, Anthony RM, Mirakami H, Star RA, Urban JFJ, Gause WC. Peripheral CD4 T cells rapidly accumulate at the host: parasite interface during an inflammatory Th2 memory response. J.Immunol. 2004;172:2424–2430. doi: 10.4049/jimmunol.172.4.2424. [DOI] [PubMed] [Google Scholar]
- 61.Pritchard DI, Maizels RM, Behnke JM, Appleby P. Stage-specific antigens of Nematospiroides dubius. Immunology. 1984;53:325–335. [PMC free article] [PubMed] [Google Scholar]
- 62.Adams JH, East IJ, Monroy F, Washington EA, Dobson C. Stage-specific antigens of Nematospiroides dubius Baylis, 1926 (Nematoda: Heligmosomides) J.Parasitol. 1987;73:1164–1168. [PubMed] [Google Scholar]
- 63.Ey PL. Heligmosomoides polygyrus: retarded development and stunting of larvae by antibodies specific for excretory/secretory antigens. Exp.Parasitol. 1988;65:232–243. doi: 10.1016/0014-4894(88)90127-0. [DOI] [PubMed] [Google Scholar]
- 64.Pritchard DI, Lawrence CE, Appleby P, Gibb IA, Glover K. Immunosuppressive proteins secreted by the gastrointestinal nematode parasite Heligmosomoides polygyrus. Int.J.Parasitol. 1994;24:495–500. doi: 10.1016/0020-7519(94)90140-6. [DOI] [PubMed] [Google Scholar]
- 65.Telford G, Wheeler DJ, Appleby P, Bowen JG, Pritchard DI. Heligmosomoides polygyrus immunomodulatory factor (IMF), targets T-lymphocytes. Parasite Immunol. 1998;20:601–611. doi: 10.1046/j.1365-3024.1998.00190.x. [DOI] [PubMed] [Google Scholar]
- 66.Anthony RM, Urban JFJ, Alem F, Hamed HA, Rozo CT, Boucher JL, Van Rooijen N, Gause WC. Memory T(H)2 cells induce alternatively activated macrophages to mediate protection against nematode parasites. Nat. Med. 2006;12:955–960. doi: 10.1038/nm1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perry JA, Olver CS, Burnett RC, Avery AC. Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J.Immunol. 2005;174:5921–5925. doi: 10.4049/jimmunol.174.10.5921. [DOI] [PubMed] [Google Scholar]
- 68.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J.Exp.Med. 2005;201:19–25. doi: 10.1084/jem.20041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adachi K, Tsutsui H, Kashiwamura S, Seki E, Nakano H, Takeuchi O, Takeda K, Okumura K, Van Kaer L, Okamura H, Akira S, Nakanishi K. Plasmodium berghei infection in mice induces liver injury by an IL-12- and toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J.Immunol. 2001;167:5928–5934. doi: 10.4049/jimmunol.167.10.5928. [DOI] [PubMed] [Google Scholar]
- 70.Schopf LR, Hoffmann KF, Cheever AW, Urban JF, Wynn TA. IL-10 is critical for host resistance and survival during gastrointestinal helminth infection. J.Immunol. 2002;168:2383–2392. doi: 10.4049/jimmunol.168.5.2383. [DOI] [PubMed] [Google Scholar]
- 71.Herbert DR, Hölscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, Kirsch R, Hall P, Mossmann H, Claussen B, Förster I, Brombacher F. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 72.Baird JK. Age-dependent characteristics of protection v. susceptibility to Plasmodium falciparum. Ann Trop Med Parasitol. 1998;92:367–390. doi: 10.1080/00034989859366. [DOI] [PubMed] [Google Scholar]
- 73.WHO . Deworming for health and development. Report of the third global meeting of the partners for parasite control. World Health Organization; Geneva: 2005. p. 51. Geneva. [Google Scholar]