

Abstract
Epithelial sheet spreading and fusion underlie important developmental processes. Well-characterized examples of such epithelial morphogenetic events have been provided by studies in Drosophila, and include embryonic dorsal closure, formation of the adult thorax and wound healing. All of these processes require the basic region-leucine zipper (bZIP) transcription factors Jun and Fos. Much less is known about morphogenesis of the fly abdomen, which involves replacement of larval epidermal cells (LECs) with adult histoblasts that divide, migrate and finally fuse to form the adult epidermis during metamorphosis. Here, we implicate Drosophila Activating transcription factor 3 (Atf3), the single ortholog of human ATF3 and JDP2 bZIP proteins, in abdominal morphogenesis. During the process of the epithelial cell replacement, transcription of the atf3 gene declines. When this downregulation is experimentally prevented, the affected LECs accumulate cell-adhesion proteins and their extrusion and replacement with histoblasts are blocked. The abnormally adhering LECs consequently obstruct the closure of the adult abdominal epithelium. This closure defect can be either mimicked and further enhanced by knockdown of the small GTPase Rho1 or, conversely, alleviated by stimulating ecdysone steroid hormone signaling. Both Rho and ecdysone pathways have been previously identified as effectors of the LEC replacement. To elicit the gain-of-function effect, Atf3 specifically requires its binding partner Jun. Our data thus identify Atf3 as a new functional partner of Drosophila Jun during development.
Keywords: Epithelial cell replacement, Cell adhesion, Epidermis, Metamorphosis, Abdomen, bZIP
INTRODUCTION
Metamorphosis of Drosophila larvae into pupae and adult flies provides remarkable examples of morphogenetic changes that involve replacement of entire cell populations. Epithelia that had served larval function undergo programmed cell death while imaginal cells proliferate and differentiate to take their position. The Drosophila abdomen is an attractive system for studying the developmental replacement of one epithelial cell population with another. Unlike the adult head and thorax with appendages, all forming from pre-patterned imaginal discs, the adult abdomen derives from histoblasts that reside in each abdominal segment (Madhavan and Madhavan, 1980; Madhavan and Schneiderman, 1977). Soon after the onset of metamorphosis, the diploid histoblasts undergo an initial phase of synchronized cell divisions; later the histoblasts expand while proliferating and replace the old polyploid larval epidermal cells (LECs) that cover the surface of the abdomen (Ninov et al., 2007; Bischoff and Cseresnyes, 2009). To free space for the histoblasts, LECs are extruded from the epithelial monolayer. In order to maintain integrity of the epithelia, changes in cell adhesion and cell migration must be precisely orchestrated during this tissue remodeling.
Ninov and colleagues have demonstrated that Rho kinase signaling, which stimulates constriction of the apical actomyosin cytoskeleton through myosin phosphorylation, is necessary for the extrusion and the ensuing apoptosis of LECs (Ninov et al., 2007). Perturbed myosin phosphorylation leaves the process of the epithelial exchange incomplete, with residual LECs obstructing closure of the adult abdominal epidermis at the dorsal midline. A similar defect results from compromised function of the ecdysone receptor (EcR), which is required for both the initial phase of histoblast proliferation (Ninov et al., 2009) and for the removal of LECs (Ninov et al., 2007). Other factors besides Rho signaling and EcR that regulate the epithelial cell replacement are unknown.
We have implicated Atf3 (A3-3 — FlyBase), the single Drosophila ortholog of the vertebrate Activating transcription factor 3 (ATF3) and Jun dimerization protein 2 (JDP2) in abdominal development. ATF3 and JDP2 belong among basic region-leucine zipper (bZIP) proteins, some of which play important roles in epithelial morphogenesis (Kockel et al., 2001; Xia and Karin, 2004). Particularly the functions of Jun (Jra — FlyBase) and Fos (Kayak — FlyBase) bZIP proteins in epithelial closure events during development are well understood owing to genetic studies in Drosophila (Hou et al., 1997; Kockel et al., 1997; Riesgo-Escovar and Hafen, 1997; Zeitlinger and Bohmann, 1999). By contrast, no morphogenetic function has yet been reported for Atf3 in Drosophila.
Mammalian ATF3 and JDP2 form homodimers but preferentially dimerize with members of the Jun subfamily (Aronheim et al., 1997; Hai et al., 1989; Hsu et al., 1991), functioning either as transcriptional activators (ATF3-Jun) or repressors (JDP2-Jun). Based mainly on cell-culture studies, multiple roles in cell proliferation, differentiation and apoptosis have been ascribed to ATF3 (Allan et al., 2001; Ishiguro and Nagawa, 2001; Iyengar et al., 2003; Kawauchi et al., 2002; Lu et al., 2006; Mashima et al., 2001; Nakagomi et al., 2003; Nobori et al., 2002; Tamura et al., 2005; Yin et al., 2008) and JDP2 (Aronheim et al., 1997; Heinrich et al., 2004; Jin et al., 2002; Ostrovsky et al., 2002; Piu et al., 2001). Atf3−/− mice are viable (Hartman et al., 2004) but suffer from altered glucose and immune homeostasis (Gilchrist et al., 2006; Qi et al., 2009; Rosenberger et al., 2008). Also Jdp2−/− mice survive but produce extra fat in their brown adipose tissue (Nakade et al., 2007). In vivo significance of the interaction between the ATF3 or JDP2 proteins and Jun remains unclear.
Here we show that Atf3 interacts biochemically and genetically with Jun in Drosophila. Temporal downregulation of atf3 transcription during metamorphosis is crucial, as sustained atf3 expression alters adhesive properties of LECs, thus preventing their extrusion and replacement by the adult epidermis. This effect of Atf3 requires the presence of Jun.
MATERIALS AND METHODS
Fly strains and preparation of atf3 mutants
Fly strains obtained from the Bloomington Drosophila Stock Center included w1118, arm-Gal4, Eip71CD-Gal4, Cg-Gal4, Lsp2-Gal4, T155-Gal4, P{EPgy2}A3-3EY02562, UAS-Rho1V14; UAS-p35, UAS-diap1, hsFlp; act5c-FRT-y+-FRT-Gal4, UAS-EGFP and UAS-myr mRFP. Additional lines were esg-Gal4 (National Institute of Genetics, Mishima, Japan), ppl-Gal4 (Colombani et al., 2003), sGMCA (Kiehart et al., 2000), C7-Gal4 (from B. Edgar, Seattle, USA) and GMR-Gal4 (from M. Mlodzik, New York, USA). UAS-α-Cat.T:GFP and ubi-p63E-shg::GFP (Oda and Tsukita, 2001) were from the Drosophila Genetic Resource Center (Kyoto, Japan), and UAS-Rho1RNAi (stock v12734) was from the Vienna Drosophila RNAi Center. The UAS-junRNAi and UAS-fosRNAi (Hyun et al., 2006) lines were a gift from C. Yanicostas (Paris, France). UAS-jun, UAS-fos, UAS-fosbZIP and UAS-fospanAla lines were described previously (Ciapponi et al., 2001; Eresh et al., 1997; Zeitlinger et al., 1997).
atf3 mutants were generated by imprecise excision of a P-element P{EPgy}A3-3EY02562 from the second intron of atf3, and deletions were detected by nested PCR with primers flanking the original insertion site of the P-element in crude genomic DNA extracts. Sequencing of genomic DNA amplified from atf3 mutants with primers 5′-GTGCGTGTAACCGTCAACGTCG-3′ and 5′-ATGCTGCTGGTCAATCACGTTG-3′ within the first and the third atf3 exons, respectively, determined the exact limits of the deletions.
atf3 misexpression
Transgenic flies were obtained by P-element mediated germline transformation (Genetic Services, Sudbury, USA). To express the entire Atf3 protein, the atf3 open reading frame (613 codons) was amplified with primers 5′-GTGGTACCGTCAACGTCGACC-3′ and 5′-GGTCTAGATCCCGCTCCCTGT-3′ from cDNA, and inserted into the pUAST vector. To express the Atf3 bZIP domain (amino acids 207-261), a cDNA fragment encoding residues 199-269 of Atf3 was subcloned using primers 5′-GTCACAGCATATGGGACTCACC-3′ and 5′-GGTCTAGATCCCGCTCCCTGT-3′. We used UAS-atf3 lines carrying single P-element insertions on the second chromosome. All crosses with Gal4 drivers were carried out at 25°C. To induce expression of atf3 in LECs, hsFlp; act-FRT-y+-FRT-Gal4, UAS-EGFP (or UAS-myr mRFP) homozygotes were crossed with UAS-atf3 flies and kept at 22°C. The progeny were exposed for 45 or 60 minutes to 37°C as mid-third instar larvae. Resulting atf3-expressing cells were marked with EGFP or myristoylated mRFP, respectively.
20E feeding
20-hydroxyecdysone (20E) extracted from plant material in the laboratory of Dr P. Simek (Ceske Budejovice, Czech Republic) and purified to 99% 20E was dissolved in 10% ethanol and added to the diet of mid-third instar larvae at a final concentration of 1 mg/ml (Gaziova et al., 2004).
mRNA expression analysis
Northern blots with 5 μg of total RNA per lane were hybridized with an atf3 antisense cRNA digoxigenin-labeled probe. For RT-PCR, cDNA was prepared with the Superscript II system (Invitrogen, Carlsbad, USA) from 2 μg of total DNase-treated RNA. cDNA samples were subjected to standard PCR reactions. Quantitative RT-PCR was performed with the iQ SYBR Green kit (BioRad, Hercules, USA) using the RotorGene RG 3000 cycler (Corbett Research, Sydney, Australia). In situ hybridization of atf3 mRNA was performed using a digoxigenin-labeled atf3 antisense probe as described (Tautz and Pfeifle, 1989).
Immunostaining and confocal microscopy
Dorsal pupal epidermis attached to the cuticle was fixed for 30 minutes with 4% formaldehyde in PBS, blocked in 0.1% BSA, 0.3% Triton X-100 in PBS for 1 hour, and incubated overnight at 4°C with primary antibodies against Drosophila Dlg (4F3; 1:1000), Armadillo (N27A1; 1:100) or Rho1 (1D9; 1:100) from the Developmental Studies Hybridoma Bank (Iowa, USA). The tissue was washed with PBS, 0.3% Triton X-100, incubated for 2 hours with Cy3- or Alexa fluor 488-conjugated antibodies, counterstained with 4 μg/ml DAPI, and analyzed under the Olympus FV1000 confocal microscope. For imaging of live pupae, animals were removed from the puparia, placed into a drop of halocarbon oil and analyzed using a Leica SP2 confocal microscope. The images were processed with IMARIS (Bitplane) and ImageJ (NIH) software.
Electrophoresis mobility shift assay
The cDNA sequence encoding residues 199-269 encompassing the bZIP domain of Atf3 was subcloned into the pET-28a vector (Novagen) using NdeI and BamHI restriction sites. Using an N-terminal histidine tag, the bZIP domains of Atf3 and of the Jun and Fos proteins were purified from Escherichia coli and used in DNA-binding assays as described (Jindra et al., 2004). Double-stranded oligonucleotides comprising the ATF/CRE element (underlined) (Fawcett et al., 1999; Montminy et al., 1986), TGAGCCGCAAGTGACGTCACGCGGGGCGTGTGCAGG, or the AP-1 consensus site (Eresh et al., 1997; Perkins et al., 1990) were used as probes.
Immunoprecipitation
The full-length atf3 open reading frame was cloned into the pMT/V5-His B vector (Invitrogen) using XhoI and ApaI restriction sites. pMT-dATF3-His-V5 (5 μg) or the empty plasmid was co-transfected with a vector expressing a C-terminally histidine-tagged Fos protein into Drosophila S2 cells. Cells were harvested 30 hours after induction with 0.5 mM CuSO4 and processed as described (Jindra et al., 2004). The lysates (100 μl per assay) were incubated for 2 hours at 4°C with 20 μl of Protein G-conjugated Dynabeads (Invitrogen), combined with 1 μg of the mouse anti-V5 antibody (Invitrogen). Bound proteins were analyzed on western blots with anti-Jun (1:1500) (Bohmann et al., 1994) and anti-Fos (1:1000) (Zeitlinger et al., 1997) rabbit antibodies.
RESULTS
The Drosophila ortholog of ATF3/JDP2 interacts with Jun
Among Drosophila bZIP proteins, the predicted product of the CG11405 gene (also referred to as a3-3), located on the X chromosome, shows the closest similarity to the mammalian ATF3 and JDP2 proteins. The DNA-binding/dimerization bZIP domains of the human ATF3 and Drosophila Atf3 proteins are identical in 60% of their amino acids (Fig. 1A); there is 58% identity between Atf3 and JDP2 in this region.
Fig. 1.

The Drosophila Atf3 protein interacts with Jun. (A) Alignment between bZIP domains of human ATF3 and Drosophila Atf3 shows identical (black shading) and similar (gray) amino acids. (B) Atf3 immunoprecipitated with Jun but not with Fos. S2 cells were transfected with Fos-His alone or together with V5-tagged Atf3; Jun was endogenous. Proteins bound to the anti-V5 antibody were analyzed by western blot (WB) with anti-V5, anti-Jun and anti-Fos antibodies. Cell lysates are shown as input. (C) Electrophoresis mobility shift assay shows binding of isolated bZIP domains of the Atf3 (A), Jun (J) and Fos (F) proteins, single or in combination, to the AP-1 and ATF/CRE consensus elements.
Dimerization between Atf3 and Jun in Drosophila has been theoretically predicted (Fassler et al., 2002) and confirmed by results of a yeast two-hybrid screen (Giot et al., 2003). To demonstrate direct binding, we conducted co-immunoprecipitation experiments. The endogenous Jun protein from Drosophila S2 cells co-precipitated with a transiently expressed Atf3 whereas Fos did not (Fig. 1B). Next, we performed a DNA mobility-shift assay with recombinant bZIP domains of Atf3, Jun and Fos to test for their DNA-binding properties. We found that Atf3 specifically bound an ATF/CRE consensus element but not the AP-1 site, which was recognized by the Jun-Fos (AP-1) complex (Fig. 1C). Although Atf3 bound DNA by itself, presumably as a homodimer, the binding was enhanced in the presence of Jun (compare lanes 7 and 8 in Fig. 1C). Fos did not synergize with Atf3 in DNA binding (lane 9). Excess unlabeled DNA bearing the ATF/CRE binding site competed for the Atf3 bandshift activity whereas the AP-1 binding element did not (data not shown). These results have shown that, like ATF3 or JDP2 in mammals, Atf3 in Drosophila selectively dimerizes with Jun, with which it cooperatively and specifically binds the ATF/CRE DNA element.
Atf3 cooperates with Jun in vivo
To test whether Atf3 and Jun interact in vivo, we conducted experiments in the Drosophila compound eye, the precise structure of which sensitively reflects genetic interactions. Overexpression of atf3 under the GMR-Gal4 driver disrupted the ommatidial arrangement, resulting in smaller eyes with a glossy appearance (Fig. 2). This atf3 misexpression phenotype could be completely suppressed by simultaneous RNAi-mediated knockdown of jun (Fig. 2) but not of fos (data not shown). Conversely, the phenotype was exacerbated when jun was overexpressed in the eye together with atf3 (Fig. 2), suggesting that it is Atf3 in a complex with Jun that derails the normal eye development. Neither RNAi nor overexpression of jun alone had any effect on eye morphology. Interestingly, like depletion of Jun, co-expression of fos under the GMR-Gal4 driver completely averted the atf3 misexpression phenotype, restoring the normal appearance of the eye (Fig. 2). Expression of fos (data not shown) or its mutant versions (see Fig. S1 in the supplementary material) alone had no effect. These data can be explained by the ability of the surplus Fos to bind Jun and thus reduce its availability for interaction with the Atf3 protein. This interpretation is further supported by experiments showing that expression of the truncated bZIP domain of Fos was sufficient to suppress the Atf3 gain-of-function phenotype, whereas its transcription activation domain or phosphorylation sites were dispensable (see Fig. S1 in the supplementary material).
Fig. 2.

Genetic interaction among Drosophila (d) atf3, jun and fos during compound eye development. The GMR-Gal4 driver was used to express the indicated transgenes in the eye-imaginal disc. jun RNAi did not affect eye morphology but was able to suppress the effect of overexpressed Atf3. Overexpression of Fos also precluded the gain-of-function effect of Atf3. Conversely, co-expression of Jun with Atf3 aggravated the phenotype.
Taken together, our results show that Atf3 cooperates with Jun, as Jun is specifically required for an effect caused by overexpression of Atf3 in the developing eye. Given the capacity of both Atf3 and Fos to bind Jun, and based on the ability of Jun to enhance and of Fos to suppress the Atf3 gain-of-function phenotype, we suggest that Atf3 and Fos compete for their common partner Jun in vivo.
atf3 is an essential gene with transcription dynamically regulated during metamorphosis
To find out whether Atf3 is required for Drosophila development and whether its absence might resemble a phenotype caused by loss of its partner Jun, we generated atf3 mutant flies. The longest deletion (line atf376) obtained by imprecise excision of a P element (see Fig. S2A in the supplementary material), removed the entire bZIP domain of Atf3, and atf376 hemizygous (male) larvae lacked detectable atf3 mRNA (see Fig. S2B in the supplementary material). Thus, atf376 probably represents a null allele. Most atf376 larvae died soon after hatching and during all three larval stages. Only a few (approximately 2%) reached the third instar but died before metamorphosis as defective pseudopuparia (see Fig. S2C in the supplementary material). Expression of atf3 cDNA under the ubiquitous armadillo (arm-Gal4) driver rescued some atf376 hemizygotes to adults, confirming that loss of atf3 was the cause of the lethal phenotype. Interestingly, the moribund atf376 larvae abnormally enlarged lipid droplets in their fat body (see Fig. S2D in the supplementary material), thus displaying a phenotype reminiscent of that in mice lacking one of the Atf3 orthologs, JDP2 (Nakade et al., 2007). However, in contrast to viable Jdp2 or Atf3 (Hartman et al., 2004) knockout mice, atf3 is an essential gene in Drosophila.
Fly embryos lacking the function of Jun or Fos die because of the failed dorsal closure (Hou et al., 1997; Kockel et al., 1997; Riesgo-Escovar and Hafen, 1997). However, atf376 embryos develop normally, without the dorsal open defect, even when derived from atf3-deficient germline clones induced in atf376/ovoD1 mothers (data not shown). Thus, unlike its partner Jun, Atf3 is not required for dorsal closure, suggesting that dorsal closure is regulated by Jun-Fos dimers and that the Atf3-Jun complex has another function later in development.
Consistent with the vital requirement for Atf3 during larval stages, atf3 mRNA was expressed in embryos and larvae (Fig. 3A). Expression then sharply declined by the late-third larval instar, and no atf3 mRNA was detected by northern blot hybridization in wandering larvae and during metamorphosis from the time of puparium formation until the second day of pupal development. Detailed RT-PCR analysis showed that atf3 downregulation coincided with the cessation of feeding and the onset of metamorphosis [0 hours after puparium formation (APF)] (Fig. 3B). A pulse of expression occurred at 6 hours APF. RT-PCR from isolated fat body and abdominal integuments, together with in situ hybridization performed on puparia at this stage, showed that atf3 mRNA was primarily present in the larval epidermis (LECs) during the expression peak at 6 hours APF (Fig. 3C,D). From the time of head eversion (12 hours APF) the mRNA level remained low until the second day of pupal development, and then it grew steadily during morphogenesis of the adult (Fig. 3B). Quantitative RT-PCR revealed a 4.3-fold difference in atf3 mRNA abundance between 0 and 72 hours APF (see Fig. S3A in the supplementary material). In contrast to the tight regulation of atf3, the mRNAs of fos and jun fluctuated little during the examined period (Fig. 3B). Therefore, unlike Jun or Fos, Atf3 was dynamically regulated during metamorphosis at the level of transcription.
Fig. 3.
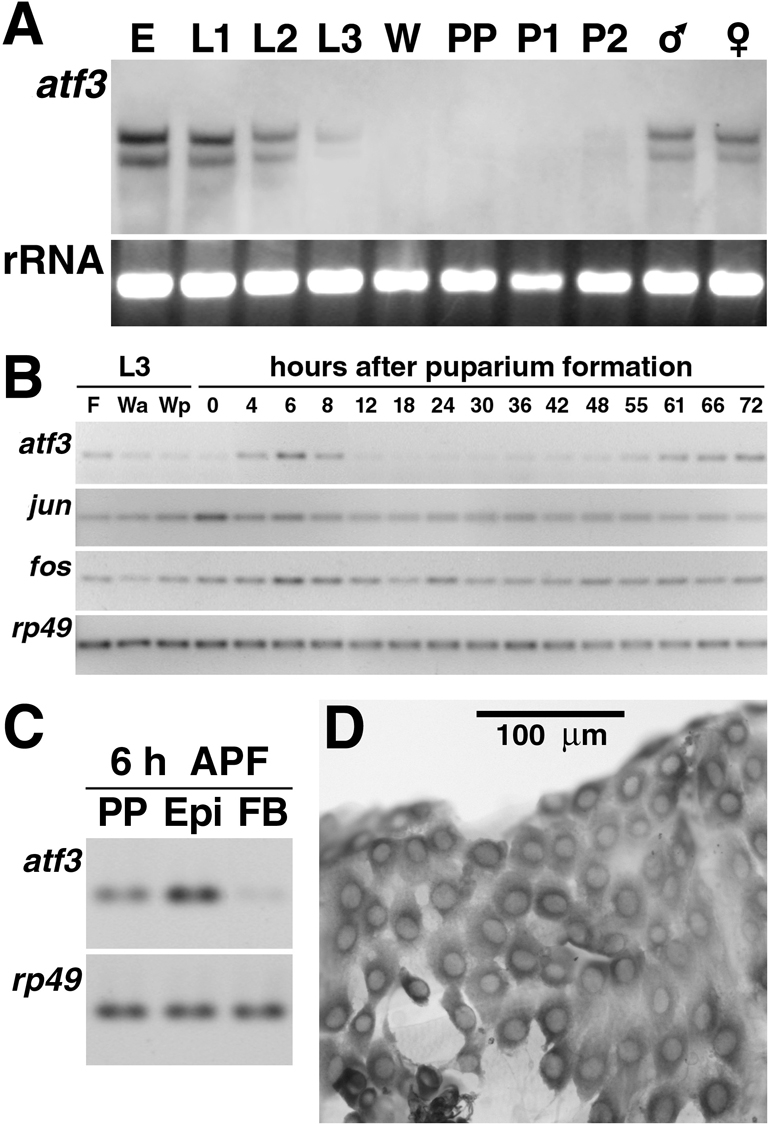
Expression of atf3 mRNA is developmentally regulated. (A) Northern blot of total RNA from mixed-stage embryos (E), the three larval instars (L1-L3), wandering larvae (W), white puparia (prepupae; PP), pupae 24 hours (P1) and 48 hours (P2) after puparium formation, and male and female adults. Ribosomal RNA serves as loading control. (B) Temporal profiles of atf3, jun and fos mRNAs during metamorphosis as assessed with RT-PCR (26 cycles) in total RNA from whole animals (for quantitative results see Fig. S3 in the supplementary material). The atf3 profile was verified three times with two different sets of primers. F, feeding late-third instar larvae; Wa, wandering larvae with anterior gut clear; Wp, wandering larvae with posterior gut clear. Expression of rp49 (23 cycles) serves for control. (C,D) At the peak of expression at 6 hours APF (see panel B), atf3 mRNA occurs in the larval epidermis as shown by RT-PCR (C) performed on isolated integuments (Epi), fat bodies (FB) and whole prepupae (PP). In situ hybridization (D) shows atf3 mRNA in LECs. Scale bar: 100 μm.
Sustained expression of atf3 in LECs disrupts abdominal development
The precise temporal control of atf3 expression (Fig. 3) suggested that the rise and subsequent fall of Atf3 during metamorphosis might be critical for the complex morphogenesis occurring in fly pupae. We tested this possibility by means of sustained expression of the full-length Atf3 protein using the UAS-Gal4 system with various drivers. A striking, fully penetrant metamorphic defect was observed with the pumpless (ppl) Gal4 driver. Although ppl>atf3 animals developed normally until the pupal stage, they failed to complete fusion of the adult abdominal epidermis (Fig. 4A). A dorsal cleft in the abdomen remained that could not be covered with the adult cuticle, and consequently 86% of the flies (n=913) died inside the puparium. All of the ppl>atf3 adults that did eclose showed abdominal lesions filled with the old pupal cuticle lacking adult pigmentation and bristles, often with a clot covering a bleeding wound (Fig. 4B). Adults with the same abdominal cleft (but otherwise normal) also emerged when atf3 was moderately and ubiquitously misexpressed under the arm-Gal4 driver (Fig. 4C), suggesting that abdominal morphogenesis was the process most sensitive to ectopic Atf3.
Fig. 4.

Misexpression of atf3 causes incomplete fusion of abdominal epidermis. (A) ppl>atf3 pharate adults usually die inside puparia with a dorsally open abdominal cleft in the adult cuticle. The scar is outlined with a brown line of necrotic tissue, and it is filled with the old pupal cuticle. Other body parts metamorphose normally and the unaffected regions of the abdomen produce adult cuticle with sensory bristles. (B) Few ppl>atf3 flies eclose, invariantly bearing the scar, often with signs of bleeding. (C) Effect of atf3 expressed under the ubiquitous (arm) driver. (D) Expression of UAS-α-Catenin::GFP in posterior abdominal segments of a normally developing pupa at 27 hours APF shows that the ppl-Gal4 driver is active in LECs but not in the surrounding histoblasts that occupy the areas around and between LECs at this time. The punctate GFP signals probably come from LECs that had already been extruded and phagocytosed. (E) The Eip71CD driver is also expressed in LECs (large GFP-positive cells) but not in histoblasts. DAPI staining (magenta) shows cell nuclei. (F) Abdominal lesion in a rarely eclosing Eip71CD>atf3 adult. (G) Flp-out induction of atf3 in the polyploid larval cells reproduces the abdominal closure phenotype (for more severe defects see Fig. S4 in the supplementary material). The LECs that express atf3 and GFP persist in the adult abdomen and mainly localize to the dorsal cleft. Scale bars: 100 μm in D; 50 μm in E.
The adult fly abdomen derives from histoblasts that proliferate, replace LECs and finally differentiate, giving rise to the adult cuticle. Therefore, the observed abdominal defect suggested a compromised function of the epidermis, either LECs, histoblasts or both cell types. To distinguish between these possibilities, we first examined expression of the ppl-Gal4 driver in the epidermis. We found that ppl-Gal4 was active in LECs but not in histoblasts (Fig. 4D). Second, we used another driver, Eip71CD-Gal4, which was inactive in histoblasts but strongly expressed in LECs (Fig. 4E). Eip71CD-Gal4-driven misexpression of atf3 mostly produced lethal pupae lacking adult cuticle (data not shown), but it occasionally yielded adults with a dorsal abdominal cleft (Fig. 4F). In addition to being active in LECs, both ppl-Gal4 (Colombani et al., 2003) (see Fig. S3B in the supplementary material) and Eip71CD-Gal4 (data not shown) were also expressed in the fat body. However, no abdominal defects occurred when atf3 was misexpressed under either of three fat-body-specific Gal4 drivers, Lsp2, Cg or C7 (data not shown). Third, to rule out the possibility that ectopic Atf3 affected the imaginal epidermis, we directed its expression to histoblasts by using the escargot (esg) and T155 Gal4 drivers (Harrison et al., 1995; Hayashi et al., 1993); in neither case the fusion of the adult abdominal epidermis was affected (data not shown).
To finally confirm that abdominal morphogenesis was disrupted by sustained atf3 activity in LECs, we induced atf3 by using the flp-out technique. Owing to the timing of heat-shock induction to the mid-third instar, this method triggers expression in the polyploid larval cells but not in the diploid histoblasts (Ninov et al., 2007) (see Fig. S6 in the supplementary material). Misexpression of atf3 under the actin promoter following the flp-out event invariantly led to an abdominal cleft (Fig. 4G). The lesions were often more severe than those observed in ppl>atf3 animals, affecting also lateral and ventral parts of the abdomen (see Fig. S4 in the supplementary material). Together, the above data demonstrate that the sustained expression of atf3 prevents fusion of the adult abdominal epidermis by acting upon LECs, suggesting that the replacement of these obsolete larval cells by adult histoblasts requires the developmental downregulation of atf3 expression.
Deregulated Atf3 prevents extrusion of LECs
To understand the cellular events underlying the incomplete epithelial closure in ppl>atf3 animals, we visualized cell membranes by antibody staining of the septate junction component, Discs large 1 (Dlg1), or used a transgenic DE-cadherin::GFP fusion protein (shg::gfp). In wild-type animals 24 hours APF, LECs covering the surface of the abdomen gave way to the rapidly expanding nests of histoblasts (Fig. 5A) that began to fuse laterally and ventrally (Bischoff and Cseresnyes, 2009; Ninov et al., 2007). In ppl>atf3 pupae the histoblast nests also spread (Fig. 5D), and at least at 16 hours APF, before their fusion, they comprised normal numbers of histoblasts (see Fig. S5 in the supplementary material). By 48 hours APF a control abdomen was fully covered with adult epidermis consisting exclusively of histoblasts, now forming sensory bristles (Fig. 5B). Histoblasts in ppl>atf3 abdomens also differentiated the adult cuticle with sensory bristles, although polarity of the bristles near the dorsal cleft was altered (Fig. 4A and Fig. 5H; see Fig. S4C in the supplementary material). However, in contrast to the control, a large population of LECs remained in the dorsal abdomen of ppl>atf3 animals at 48 hours APF (Fig. 5E,G). The membranes of the persisting LECs accumulated the Dlg protein (Fig. 5F), and although these cells became severely deformed they survived throughout metamorphosis (Fig. 5E-I) to the adult stage (Fig. 4G). When visualized in live ppl>atf3 pupae, the apical junctions of the remaining LECs displayed interdigitation and accumulation of DE-cadherin::GFP (Fig. 5G,H). Another adherens junction component, the Drosophila β-catenin Armadillo, was also enriched in atf3-expressing LECs (see Fig. S6 in the supplementary material).
Fig. 5.

Ectopic Atf3 prevents removal and death of LECs. (A-C) In control pupae, LECs that still reside in the abdomen at 24 hours APF (A) become replaced by histoblasts. By 48 hours APF (B), histoblasts cover the entire abdomen and differentiate sensory bristles (arrowhead). The bristles have been formed and adult cuticle deposited at 72 hours APF (C). (D-I) Abdominal epidermis in ppl>atf3 pupae. Although histoblasts spread and differentiate (bristle formation indicated with arrowheads), many LECs persist, posing a barrier to histoblasts progressing from the lateral sides. The affected LECs display warping of membranes (G, arrow) and accumulate the cell junction components Dlg and DE-cadherin (arrows in F and H, respectively). (I) A large lesion affecting most of the dorsolateral abdomen of a pharate adult fly (96 hours APF) is populated by persisting LECs, the membranes of which are extremely deformed. The arrow points to a thick wall separating the scar from the adult cuticle on the left. DAPI (magenta) is used for staining nuclei; cell membranes are visualized either with anti-Dlg antibody staining (green) or by ubiquitous expression of DE-cadherin::GFP fusion protein in live pupae (G-I). Anterior is to the top in all panels. Images are z-stacks of confocal slices. Scale bars: 20 μm in A-C,F; 50 μm in D,E,G,H; 100 μm in I.
Cooperation between adherens junctions and the apical ring of actomyosin cytoskeleton is required for basal extrusion of LECs (Ninov et al., 2007). The altered pattern of DE-cadherin and β-catenin therefore suggests that excessive Atf3 might prevent LEC extrusion through stabilization of the cell-cell adhesion complex. To examine the effect of Atf3 on LECs in further detail, we employed the flp-out technique, which allows comparisons of atf3-misexpressing and control LECs within one tissue. Fig. 6 shows that membrane interdigitation occurred between atf3-positive LECs already at 18 and 24 hours APF, even in areas where the LECs had no contact with histoblasts. At 48 hours APF only LECs expressing atf3 persisted, apparently being squeezed by the expanding histoblasts. The membrane-associated DE-cadherin::GFP signal was stronger in adjacent atf3-positive LECs compared with non-induced LECs (Fig. 6), and quantitative analysis of confocal images acquired at 18 hours APF (see Fig. S7 in the supplementary material) and at 24 hours APF (data not shown) both revealed a statistically significant 1.4-fold increase of the DE-cadherin::GFP signal intensity upon atf3 induction. Enrichment of DE-cadherin on apical membranes of atf3-expressing LECs was further confirmed on confocal cross sections (Fig. 7).
Fig. 6.

Behavior of LECs upon atf3 misexpression. Images of LECs with membranes marked by DE-cadherin::GFP (ubi-shg::gfp) were captured in live pupae at indicated times. Cells expressing mRFP (magenta, bottom row) were induced to express atf3 with the flp-out system. Compared with their uninduced neighbors, which have smooth membranes (blue arrowheads), the atf3-positive LECs show membrane interdigitation (yellow arrowheads) and enrichment of DE-cadherin (arrows) on apical junctions. By 48 hours APF only Atf3-positive LECs survive, being completely surrounded and apparently pressured by histoblasts. Note that junctions between atf3-expressing neighbors are thicker compared with their boundaries with uninduced LECs or with histoblasts. Images are z-stacks of confocal slices. Scale bar: 20 μm.
Fig. 7.

LECs expressing atf3 are unable to complete extrusion. Shown are images from live pupae with cell membranes marked with DE-cadherin::GFP at 24 hours APF. The flp-out system was employed to induce expression of mRFP alone (control) or together with atf3. Lines indicate three confocal cross sections, depicted below each panel. Arrowheads in control (left) point to extruding LECs that have apically constricted and detached from the epidermal layer. By contrast, although LECs expressing atf3 can apically constrict, they continue to adhere to the epithelium (arrowheads on the right) even when completely surrounded by histoblasts. Note that DE-cadherin colocalizes with the mRFP signal that marks control LECs, whereas it remains associated with the apical surface of atf3-expressing LECs that attempt to delaminate. Arrows show atf3-expressing LECs with membranes that interdigitate and contain more DE-cadherin compared with their uninduced neighbors. Scale bar: 20 μm.
Although some atf3-positive LECs began the extrusion process, they could not detach from the apical surface even when entirely surrounded by histoblasts, possibly being tethered to it by the excessive adhesion protein (Fig. 7). By contrast, control LECs did completely separate from the epithelium (Fig. 7). In addition, LECs overexpressing atf3 displayed apical enrichment of moesin, an actin-binding protein of the ERM (ezrin, radixin, moesin) family, which links transmembrane proteins to cortical actin filaments (see Fig. S8 in the supplementary material). Interestingly, prominent accumulation of DE-cadherin was also observed in atf3-expressing clones of epithelial cells within the hinge region of wing discs that form the adult thorax (see Fig. S9 in the supplementary material), indicating that the effect of Atf3 on cell adhesion components may not be limited to larval epithelia.
In summary, our results show that deregulation of atf3 expression causes marked changes of cell membranes, including interdigitation and accumulation of cell adhesion molecules, suggesting that LEC adhesiveness might be increased. Although some of the affected LECs initiate extrusion, this process stays incomplete. Consequently, the adhering LECs present a physical barrier for the migrating histoblasts.
Effects of Rho1 signaling and 20-hydroxyecdysone
Rho kinase (Rok)-dependent phosphorylation of myosin regulatory light chain was shown to be required for LEC extrusion (Ninov et al., 2007). To examine a possible relationship between the Rok-dependent cytoskeletal regulation and Atf3, we interfered with the function of the GTPase Rho1 (also called RhoA), which acts immediately upstream of Rok. RNAi silencing of Rho1 using the ppl-Gal4 driver produced a phenocopy of atf3 misexpression, causing a dorsal abdominal cleft in 100% of ppl>Rho1(RNAi) adults (n=172), of which most died in the puparium and about 12% eclosed (Fig. 8A,B), similar to ppl>atf3 animals (Fig. 4A,B). However, when Rho1 RNAi and misexpression of atf3 in LECs were combined, the abdominal defect became more severe (Fig. 8C), not allowing any pharate adults to eclose (n=77). Conversely, co-expression of a dominantly active Rho1V14 protein suppressed the otherwise fully penetrant abdominal defect in some ppl-atf3 flies (Fig. 8D). Surprisingly, we found that the endogenous Rho1 protein was mislocalized in atf3-misexpressing LECs, showing a diffuse cytoplasmic signal, compared with membrane localization in control LECs (Fig. 8E). These results suggest a genetic interaction between Rho signaling and atf3, and support the idea that excess Atf3 prevents extrusion of LECs by altering their cell adhesion properties.
Fig. 8.

Misexpression of atf3 enhances the effect of Rho1 silencing and leads to mislocalization of the Rho1 protein. (A-C) Rho1 RNAi alone (A,B) caused abdominal defects similar in extent to those in ppl>atf3 animals (see Fig. 4A,B), whereas combined with misexpression of atf3 (C) it led to broader, lethal scars (compare arrows). (D) The effect of ectopic atf3 was averted by co-expression of active Rho1V14. All transgenes were induced under the ppl-Gal4 driver. (E,E′) Rho1 is redistributed in GFP-marked LECs (28 hours APF) that express atf3 upon flp-out induction. DAPI (magenta) is used for staining nuclei. Scale bar: 50 μm.
Disturbed function of the ecdysone receptor (EcR) has been shown to prevent extrusion of LECs, causing a dorsal abdominal cleft that closely resembles the Atf3 gain-of-function phenotype (Ninov et al., 2007). We therefore tested whether stimulating EcR-dependent signaling by addition of the natural agonist 20E might overcome the defect caused by sustained atf3 expression. Indeed, supplying third-instar ppl>atf3 larvae with dietary 20E increased the number of eclosing adults (from 14 to 62%, n=760), the abdominal scars of which were in 22% of the cases partially or completely sealed with normal adult cuticle (Fig. 9).
Fig. 9.

20E suppresses the effect of ectopic Atf3. (A-C) Dorsal abdominal cleft in ppl>atf3 adults (A) was partially (B, arrow points to a residual narrow scar), or completely (C) repaired by supplementing mid-third instar larvae with dietary 20E; control larvae received solvent (ethanol) only (A).
Jun is required for Atf3 effect on abdominal morphogenesis
Atf3 interacts with Jun to form a DNA-binding complex (Fig. 1B,C) and genetically when overexpressed in the developing compound eye (Fig. 2). To see if this interaction is biologically relevant during abdominal morphogenesis, we tested whether Atf3 relies on the presence of Jun to cause the dorsal cleft phenotype. First, we confirmed that Jun is indeed expressed in LECs during metamorphosis (see Fig. S10 in the supplementary material). RNAi-mediated depletion of Jun in animals that misexpressed atf3 under the ppl-Gal4 driver restored viability of adults from 14% (atf3 alone) to 100% (n=565). Strikingly, 87% of the ppl>atf3, jun(RNAi) adults eclosed with a completely normal abdomen (Fig. 10A,B). By contrast, RNAi knockdown of Fos in ppl>atf3 background did not improve the abdominal defect (Fig. 10C). RNAi silencing of either jun or fos alone under the ppl-Gal4 driver had no effect on the abdomen (data not shown). These results demonstrate that Atf3 requires its partner Jun but not Fos to disrupt abdominal morphogenesis. Similar to the situation in the compound eye (Fig. 2 and Fig. S1 in the supplementary material), the effect of misexpressed atf3 can be neutralized by simultaneously expressing Fos or its truncated bZIP domain under the ppl-Gal4 driver (data not shown). Therefore, the model in which Atf3 and Fos compete for their common partner Jun may be extended to the developing abdomen.
Fig. 10.

Jun is specifically required for Atf3 to disrupt abdominal development. (A-C) The severe abdominal cleft caused by misexpression of atf3 (A) was completely suppressed by Jun RNAi knockdown (B), whereas depletion of Fos had no such effect (C). Males on the left, females on the right.
DISCUSSION
Functional interaction between Atf3 and the AP-1 proteins
We identified Atf3 as a new partner of Jun in Drosophila. Previously, Jun has only been known to dimerize with itself and with the Drosophila homolog of Fos (Perkins et al., 1988; Perkins et al., 1990). Functional analysis of Atf3 has not yet been reported. Our biochemical data show that, similar to mammalian ATF3 and JDP2 (Aronheim et al., 1997; Chu et al., 1994; Hai and Curran, 1991; Hsu et al., 1991), the Atf3 protein selectively binds Jun but not Fos. Also consistent with the properties of ATF3 and JDP2 is the ability of Atf3 to bind the ATF/CRE response element alone or synergistically with Jun (Aronheim et al., 1997; Hsu et al., 1991). In contrast to its mammalian counterparts, however, neither Atf3 alone nor in complex with Jun bound to the AP-1 element under the same conditions. The selective interactions of Atf3 point to distinct biological roles for the Atf3-Jun and the Fos-Jun dimers, respectively.
Using the Drosophila model, we show a genetic interaction between Atf3 and Jun. The evidence is based on the ability of ectopic Atf3 to disturb morphogenesis of the adult abdomen and the compound eye, which strictly depends on the availability of Jun. Importantly, none of the Atf3 gain-of-function phenotypes could be induced by misexpression of the truncated bZIP domain of Atf3 (data not shown), suggesting that the functional Atf3 protein in complex with Jun is required. Based on the selectivity of Atf3 in our DNA-binding assay, we predict that the Atf3-Jun complex regulates specific target genes distinct from those targeted by Fos-Jun dimers.
Our data also reflect a relationship between the AP-1 and Atf3-Jun complexes. Although Fos does not dimerize or bind DNA with Atf3, its ability to suppress the Atf3 misexpression phenotype in the eye suggests that Fos and Atf3 compete in vivo for their common partner Jun. The fact that the same suppression can be achieved by overexpressing either the truncated Fos bZIP domain or Fos lacking phosphorylation sites indicates that the suppression does not rely on a transcriptional function of Fos but probably occurs through sequestering of Jun, even by a transcriptionally inactive Fos protein. Early in vitro studies have proposed a competition model for the AP-1 and Atf3 proteins to explain a temporal regulation of gene expression in the regenerating liver (Hsu et al., 1992). However, to date such a relationship among Fos, Jun and Atf3 has not been supported with direct genetic evidence.
Deregulated Atf3 increases adhesiveness of LECs
Removal of LECs is normally complete by 36 hours APF, at which time the sheets of histoblasts reach the dorsal midline (Bischoff and Cseresnyes, 2009; Ninov et al., 2007). Our data strongly support the argument that the temporal downregulation of atf3 expression during abdominal morphogenesis is necessary for LECs to be replaced by the adult epidermis. When experimentally sustained, atf3 activity in LECs interfered with this exchange by blocking extrusion and death of the LECs. This was evident as the atf3-expressing LECs survived within the epithelial layer for days after their scheduled destruction.
Interdigitation of cell membranes and accumulation of adherens junction proteins in LECs suggested that ectopic Atf3 caused adjacent LECs to reinforce their mutual contacts. This probably resulted from altered distribution of the proteins, as levels of the shg (DE-cadherin) mRNA remained unchanged in LECs of ppl>atf3 animals (data not shown). By contrast, junctions between atf3-expressing LECs and their normal neighbors or histoblasts were smooth and presumably less rigid. DE-cadherin was similarly enriched in clones of imaginal disc cells (see Fig. S9 in the supplementary material). These observations suggested that differential adhesion of atf3-expressing cells might have led to their sorting out from the surrounding epithelium. Even modest differences in cadherin levels have been shown to cause segregation of cells within a population by altering their adhesiveness (Duguay et al., 2003).
Recent live imaging data (Bischoff and Cseresnyes, 2009) have revealed that migrating histoblasts push the LECs ahead of themselves towards the dorsal midline, where histoblasts fuse last. The atf3-expressing LECs that adhered to each other were probably moved and pressed by the expanding histoblasts to the dorsal side, whereas non-induced LECs were eliminated. This explains why the abdominal lesions primarily occurred at the dorsal midline, although our flp-out experiments showed that atf3 misexpression could affect LECs in other areas as well (see Fig. S4 in the supplementary material). Strengthened contacts among persisting LECs probably blocked invasion of histoblasts in between them and inhibited LEC extrusion, eventually causing gaps in the adult epidermis.
In accord with the notion that extrusion from the epithelium is a prerequisite for LECs to undergo apoptosis (Ninov et al., 2007), we assume that sustained presence of Atf3 primarily enhanced adhesiveness of LECs, which only consequently prevented their death. This view is supported by the observation that membranes of atf3-expressing LECs interdigitated and accumulated DE-cadherin as early as 18-24 hours APF, even in areas of the larval epidermis that were far from histoblasts and where control LECs did not yet extrude (Fig. 6 and Fig. S7 in the supplementary material). In addition, the Atf3 gain-of-function phenotype was stronger than abdominal closure defects caused by caspase mutation or inhibition (Muro et al., 2006; Ninov et al., 2007). When we misexpressed the anti-apoptotic proteins p35 or DIAP1 (Thread — FlyBase) under the ppl-Gal4 driver, the resulting dorsal lesions were not lethal and were clearly milder than the broad, mostly fatal scars in ppl>atf3 animals (see Fig. S11A in the supplementary material). Compared with the large contiguous populations of persisting LECs in ppl>atf3 pupae (Fig. 5), inhibiting apoptosis with p35 only allowed small islands of LECs to survive (see Fig. S11B in the supplementary material).
Ecdysone signaling promotes replacement of the abdominal epithelia by stimulating both the early histoblast proliferation and the extrusion of LECs (Ninov et al., 2007; Ninov et al., 2009). As atf3 misexpression affected LECs but did not impair early histoblast proliferation (see Fig. S5 in the supplementary material), we are left with the latter possibility that added 20E counteracted the effect of ectopic Atf3 by facilitating the extrusion process. As we detected normal 20E titers in ppl>atf3 larvae or prepupae (data not shown), the failure of LEC extrusion was not a result of steroid deficiency. Also, 20E had no effect on atf3 mRNA levels, at least in Drosophila S2 cells or third-instar larvae (data not shown). Atf3 and ecdysone signaling therefore probably influence LEC extrusion by acting independently.
Although the mechanism through which ecdysone contributes to LEC removal is unknown, one attractive possibility is that it might cooperate with Rho signaling, which is required for LEC extrusion as well (Ninov et al., 2007). It has been demonstrated that genetic interaction between the 20E-response gene broad and components of the Rho pathway including RhoGEF2, Rho1 and myosin II is important for ecdysone-dependent epithelial cell elongation during Drosophila leg morphogenesis (Bayer et al., 2003; Ward et al., 2003). Our data show that Rho1 becomes mislocalized in LECs upon atf3 misexpression and that Rho1 silencing enhances the abdominal gain-of-function phenotype of atf3. The exact relationship between Atf3, Rho1 and ecdysone remains to be determined. However, Atf3 clearly represents a new intrinsic regulator of epithelial cell replacement during Drosophila metamorphosis.
Supplementary Material
Acknowledgements
We appreciate receiving fly stocks from C. Yanicostas, M. Mlodzik, B. Edgar, C. Mirth and from the Bloomington (USA), the NIG (Mishima, Japan), the DGRC (Kyoto, Japan) and the VDRC (Vienna, Austria) Drosophila stock centers and antibodies from the DSHB (Iowa, USA). We thank C. Sommers for technical assistance, A. Trojanova for fly maintenance, A. Pospech for photographing pupae, P. Simek for 20E and A. Schauss for advice on quantitative image analysis. This work was supported by the Sofja Kovalevskaja Award and CRC 832 to M.U., projects 2B06129 and 600766580 from the Czech Ministry of Education to M.J., and by NIH grant R03-CA123591 to D.B. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.037861/-/DC1
References
- Allan A. L., Albanese C., Pestell R. G., LaMarre J. (2001). Activating transcription factor 3 induces DNA synthesis and expression of cyclin D1 in hepatocytes. J. Biol. Chem. 276, 27272-27280 [DOI] [PubMed] [Google Scholar]
- Aronheim A., Zandi E., Hennemann H., Elledge S. J., Karin M. (1997). Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol. Cell. Biol. 17, 3094-3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer C. A., Halsell S. R., Fristrom J. W., Kiehart D. P., von Kalm L. (2003). Genetic interactions between the RhoA and Stubble-stubbloid loci suggest a role for a type II transmembrane serine protease in intracellular signaling during Drosophila imaginal disc morphogenesis. Genetics 165, 1417-1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M., Cseresnyes Z. (2009). Cell rearrangements, cell divisions and cell death in a migrating epithelial sheet in the abdomen of Drosophila. Development 136, 2403-2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmann D., Ellis M. C., Staszewski L. M., Mlodzik M. (1994).. Drosophila Jun mediates Ras-dependent photoreceptor determination. Cell 78, 973-986 [DOI] [PubMed] [Google Scholar]
- Chu H. M., Tan Y., Kobierski L. A., Balsam L. B., Comb M. J. (1994). Activating transcription factor-3 stimulates 3′,5′-cyclic adenosine monophosphate-dependent gene expression. Mol. Endocrinol. 8, 59-68 [DOI] [PubMed] [Google Scholar]
- Ciapponi L., Jackson D. B., Mlodzik M., Bohmann D. (2001). Drosophila Fos mediates ERK and JNK signals via distinct phosphorylation sites. Genes Dev. 15, 1540-1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J., Raisin S., Pantalacci S., Radimerski T., Montagne J., Leopold P. (2003). A nutrient sensor mechanism controls Drosophila growth. Cell 114, 739-749 [DOI] [PubMed] [Google Scholar]
- Duguay D., Foty R. A., Steinberg M. S. (2003). Cadherin-mediated cell adhesion and tissue segregation: qualitative and quantitative determinants. Dev. Biol. 253, 309-323 [DOI] [PubMed] [Google Scholar]
- Eresh S., Riese J., Jackson D. B., Bohmann D., Bienz M. (1997). A CREB-binding site as a target for decapentaplegic signalling during Drosophila endoderm induction. EMBO J. 16, 2014-2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler J., Landsman D., Acharya A., Moll J. R., Bonovich M., Vinson C. (2002). B-ZIP proteins encoded by the Drosophila genome: evaluation of potential dimerization partners. Genome Res. 12, 1190-1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. W., Martindale J. L., Guyton K. Z., Hai T., Holbrook N. J. (1999). Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem. J. 339, 135-141 [PMC free article] [PubMed] [Google Scholar]
- Gaziova I., Bonnette P. C., Henrich V. C., Jindra M. (2004). Cell-autonomous roles of the ecdysoneless gene in Drosophila development and oogenesis. Development 131, 2715-2725 [DOI] [PubMed] [Google Scholar]
- Gilchrist M., Thorsson V., Li B., Rust A. G., Korb M., Roach J. C., Kennedy K., Hai T., Bolouri H., Aderem A. (2006). Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173-178 [DOI] [PubMed] [Google Scholar]
- Giot L., Bader J. S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y. L., Ooi C. E., Godwin B., Vitols E., et al. (2003). A protein interaction map of Drosophila melanogaster. Science 302, 1727-1736 [DOI] [PubMed] [Google Scholar]
- Hai T., Curran T. (1991). Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. USA 88, 3720-3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T. W., Liu F., Coukos W. J., Green M. R. (1989). Transcription factor ATF cDNA clones: an extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 3, 2083-2090 [DOI] [PubMed] [Google Scholar]
- Harrison D. A., Binari R., Nahreini T. S., Gilman M., Perrimon N. (1995). Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 14, 2857-2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman M. G., Lu D., Kim M. L., Kociba G. J., Shukri T., Buteau J., Wang X., Frankel W. L., Guttridge D., Prentki M., et al. (2004). Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell. Biol. 24, 5721-5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S., Hirose S., Metcalfe T., Shirras A. D. (1993). Control of imaginal cell development by the escargot gene of Drosophila. Development 118, 105-115 [DOI] [PubMed] [Google Scholar]
- Heinrich R., Livne E., Ben-Izhak O., Aronheim A. (2004). The c-Jun dimerization protein 2 inhibits cell transformation and acts as a tumor suppressor gene. J. Biol. Chem. 279, 5708-5715 [DOI] [PubMed] [Google Scholar]
- Hou X. S., Goldstein E. S., Perrimon N. (1997). Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes Dev. 11, 1728-1737 [DOI] [PubMed] [Google Scholar]
- Hsu J. C., Laz T., Mohn K. L., Taub R. (1991). Identification of LRF-1, a leucine-zipper protein that is rapidly and highly induced in regenerating liver. Proc. Natl. Acad. Sci. USA 88, 3511-3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J. C., Bravo R., Taub R. (1992). Interactions among LRF-1, JunB, c-Jun, and c-Fos define a regulatory program in the G1 phase of liver regeneration. Mol. Cell. Biol. 12, 4654-4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun J., Becam I., Yanicostas C., Bohmann D. (2006). Control of G2/M transition by Drosophila Fos. Mol. Cell. Biol. 26, 8293-8302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro T., Nagawa H. (2001). ATF3 gene regulates cell form and migration potential of HT29 colon cancer cells. Oncol. Res. 12, 343-346 [DOI] [PubMed] [Google Scholar]
- Iyengar P., Combs T. P., Shah S. J., Gouon-Evans V., Pollard J. W., Albanese C., Flanagan L., Tenniswood M. P., Guha C., Lisanti M. P., et al. (2003). Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 22, 6408-6423 [DOI] [PubMed] [Google Scholar]
- Jin C., Li H., Murata T., Sun K., Horikoshi M., Chiu R., Yokoyama K. K. (2002). JDP2, a repressor of AP-1, recruits a histone deacetylase 3 complex to inhibit the retinoic acid-induced differentiation of F9 cells. Mol. Cell. Biol. 22, 4815-4826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindra M., Gaziova I., Uhlirova M., Okabe M., Hiromi Y., Hirose S. (2004). Coactivator MBF1 preserves the redox-dependent AP-1 activity during oxidative stress in Drosophila. EMBO J. 23, 3538-3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi J., Zhang C., Nobori K., Hashimoto Y., Adachi M. T., Noda A., Sunamori M., Kitajima S. (2002). Transcriptional repressor activating transcription factor 3 protects human umbilical vein endothelial cells from tumor necrosis factor-alpha-induced apoptosis through down-regulation of p53 transcription. J. Biol. Chem. 277, 39025-39034 [DOI] [PubMed] [Google Scholar]
- Kiehart D. P., Galbraith C. G., Edwards K. A., Rickoll W. L., Montague R. A. (2000). Multiple forces contribute to cell sheet morphogenesis for dorsal closure in Drosophila. J. Cell Biol. 149, 471-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockel L., Zeitlinger J., Staszewski L. M., Mlodzik M., Bohmann D. (1997). Jun in Drosophila development: redundant and non-redundant functions, and regulation by two MAPK signal transduction pathways. Genes Dev. 11, 1748-1758 [DOI] [PubMed] [Google Scholar]
- Kockel L., Homsy J. G., Bohmann D. (2001). Drosophila AP-1: lessons from an invertebrate. Oncogene 20, 2347-2364 [DOI] [PubMed] [Google Scholar]
- Lu D., Wolfgang C. D., Hai T. (2006). Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J. Biol. Chem. 281, 10473-10481 [DOI] [PubMed] [Google Scholar]
- Madhavan M. M., Madhavan K. (1980). Morphogenesis of the epidermis of adult abdomen of Drosophila. J. Embryol. Exp. Morphol. 60, 1-31 [PubMed] [Google Scholar]
- Madhavan M. M., Schneiderman H. A. (1977). Histological analysis of the dynamics of growth of imaginal discs and histoblast nests during the larval development of Drosophila melanogaster. Wilhem Roux' Arch. Dev. Biol. 183, 269-305 [DOI] [PubMed] [Google Scholar]
- Mashima T., Udagawa S., Tsuruo T. (2001). Involvement of transcriptional repressor ATF3 in acceleration of caspase protease activation during DNA damaging agent-induced apoptosis. J. Cell. Physiol. 188, 352-358 [DOI] [PubMed] [Google Scholar]
- Montminy M. R., Sevarino K. A., Wagner J. A., Mandel G., Goodman R. H. (1986). Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc. Natl. Acad. Sci. USA 83, 6682-6686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muro I., Berry D. L., Huh J. R., Chen C. H., Huang H., Yoo S. J., Guo M., Baehrecke E. H., Hay B. A. (2006). The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development 133, 3305-3315 [DOI] [PubMed] [Google Scholar]
- Nakade K., Pan J., Yoshiki A., Ugai H., Kimura M., Liu B., Li H., Obata Y., Iwama M., Itohara S., et al. (2007). JDP2 suppresses adipocyte differentiation by regulating histone acetylation. Cell Death Differ. 14, 1398-1405 [DOI] [PubMed] [Google Scholar]
- Nakagomi S., Suzuki Y., Namikawa K., Kiryu-Seo S., Kiyama H. (2003). Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J. Neurosci. 23, 5187-5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninov N., Chiarelli D. A., Martin-Blanco E. (2007). Extrinsic and intrinsic mechanisms directing epithelial cell sheet replacement during Drosophila metamorphosis. Development 134, 367-379 [DOI] [PubMed] [Google Scholar]
- Ninov N., Manjon C., Martin-Blanco E. (2009). Dynamic control of cell cycle and growth coupling by ecdysone, EGFR, and PI3K signaling in Drosophila histoblasts. PLoS Biol. 7, E1000079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobori K., Ito H., Tamamori-Adachi M., Adachi S., Ono Y., Kawauchi J., Kitajima S., Marumo F., Isobe M. (2002). ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: a novel cardioprotective role of ATF3. J. Mol. Cell. Cardiol. 34, 1387-1397 [DOI] [PubMed] [Google Scholar]
- Oda H., Tsukita S. (2001). Real-time imaging of cell-cell adherens junctions reveals that Drosophila mesoderm invagination begins with two phases of apical constriction of cells. J. Cell Sci. 114, 493-501 [DOI] [PubMed] [Google Scholar]
- Ostrovsky O., Bengal E., Aronheim A. (2002). Induction of terminal differentiation by the c-Jun dimerization protein JDP2 in C2 myoblasts and rhabdomyosarcoma cells. J. Biol. Chem. 277, 40043-40054 [DOI] [PubMed] [Google Scholar]
- Perkins K. K., Admon A., Patel N., Tjian R. (1990). The Drosophila Fos-related AP-1 protein is a developmentally regulated transcription factor. Genes Dev. 4, 822-834 [DOI] [PubMed] [Google Scholar]
- Perkins K. K., Dailey G. M., Tjian R. (1988). Novel Jun- and Fos-related proteins in Drosophila are functionally homologous to enhancer factor AP-1. EMBO J. 7, 4265-4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piu F., Aronheim A., Katz S., Karin M. (2001). AP-1 repressor protein JDP-2: inhibition of UV-mediated apoptosis through p53 down-regulation. Mol. Cell. Biol. 21, 3012-3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L., Saberi M., Zmuda E., Wang Y., Altarejos J., Zhang X., Dentin R., Hedrick S., Bandyopadhyay G., Hai T., et al. (2009). Adipocyte CREB promotes insulin resistance in obesity. Cell Metab. 9, 277-286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesgo-Escovar J. R., Hafen E. (1997). Common and distinct roles of DFos and DJun during Drosophila development. Science 278, 669-672 [DOI] [PubMed] [Google Scholar]
- Rosenberger C. M., Clark A. E., Treuting P. M., Johnson C. D., Aderem A. (2008). ATF3 regulates MCMV infection in mice by modulating IFN-gamma expression in natural killer cells. Proc. Natl. Acad. Sci. USA 105, 2544-2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Hua B., Adachi S., Guney I., Kawauchi J., Morioka M., Tamamori-Adachi M., Tanaka Y., Nakabeppu Y., Sunamori M., et al. (2005). Stress response gene ATF3 is a target of c-myc in serum-induced cell proliferation. EMBO J. 24, 2590-2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D., Pfeifle C. (1989). A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 98, 81-85 [DOI] [PubMed] [Google Scholar]
- Ward R. E., Evans J., Thummel C. S. (2003). Genetic modifier screens in Drosophila demonstrate a role for Rho1 signaling in ecdysone-triggered imaginal disc morphogenesis. Genetics 165, 1397-1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Karin M. (2004). The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol. 14, 94-101 [DOI] [PubMed] [Google Scholar]
- Yin X., Dewille J. W., Hai T. (2008). A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 27, 2118-2127 [DOI] [PubMed] [Google Scholar]
- Zeitlinger J., Bohmann D. (1999). Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 126, 3947-3956 [DOI] [PubMed] [Google Scholar]
- Zeitlinger J., Kockel L., Peverali F. A., Jackson D. B., Mlodzik M., Bohmann D. (1997). Defective dorsal closure and loss of epidermal decapentaplegic expression in Drosophila fos mutants. EMBO J. 16, 7393-7401 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.