

Abstract
Adaptor protein c-Abl SH3 domain-binding protein-2 (3BP2, also referred to SH3BP2) regulates immune receptor-mediated signal transduction. In this report we focused on the molecular mechanism of 3BP2 function in B cell receptor (BCR) signaling. Engagement of BCR induces tyrosine phosphorylation of 3BP2. Genetic analysis demonstrated that Syk is critical for BCR-mediated tyrosine phosphorylation of 3BP2. Mutational analysis of 3BP2 revealed that both Tyr183 and Src homology 2 (SH2) domain are necessary for 3BP2-mediated BCR-induced activation of nuclear factor of activated T cells (NFAT). Point mutation of Tyr183 or Arg486 in the SH2 domain of 3BP2 diminished BCR-mediated tyrosine phosphorylation of 3BP2. Endogenous 3BP2 forms a complex with tyrosine-phosphorylated cellular signaling molecules. Peptide binding experiments demonstrated that only phosphorylated Tyr183 in 3BP2 could form a complex with the SH2 domain(s) of phospholipase Cγ2 and Vav1 from B cell lysates. These interactions were represented by using bacterial glutathione S-transferase-phospholipase Cγ2 or -Vav1 SH2 domain. Furthermore, pulldown and Far Western experiments showed that the 3BP2-SH2 domain directly binds to B cell linker protein (BLNK) after BCR stimulation. These results demonstrated that 3BP2 induces the protein complex with cellular signaling molecules through phosphorylation of Tyr183 and SH2 domain leading to the activation of NFAT in B cells.
Introduction
Antigen-mediated cross-linking of BCR2 triggers a series of biochemical events in B cells by the activation of Src family protein-tyrosine kinase (PTK) Lyn to phosphorylate tyrosine residues on the immunoreceptor tyrosine-based activation motif within the receptor subunits Igα and Igβ. Phosphorylated immunoreceptor tyrosine-based activation motif provides docking sites to SH2 domain of Syk and then recruits and activates Syk in the plasma membrane to induce tyrosine phosphorylation of cellular signaling molecules including BLNK (also known as BASH or SLP-65), which forms a complex with Btk (1–3). Both Syk and Btk could phosphorylate and activate PLC-γ2, which catalyzes hydrolysis of phospholipids yielding inositol triphosphate and diacylglycerol (4). Inositol triphosphate mobilizes Ca2+ release from the intracellular store and leads to the Ca2+ influx from the extracellular milieu. Ca2+ mobilization activates the transcriptional activation of NFAT.
Syk was originally isolated from porcine spleen and is known to be activated through the immune receptor signaling in lymphocytes and myeloid lineage cells (5–7). Lack of Syk results in the perturbation of the development and maturation of B lymphocytes, dysfunction in the high affinity IgE receptor (FcϵRI)-mediated mast cell activation, and FcγR-mediated cellular events (8–14). Studies by using Syk-deficient DT40 B cells have demonstrated that Syk is essential for BCR-mediated tyrosine phosphorylation of PLC-γ2, production of inositol triphosphate, and Ca2+ mobilization (15, 16). Recent findings demonstrated that Syk has tumor suppressor function in breast cancer and melanoma cells and is presumably involved in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus (17–20).
Several adaptor proteins are the substrates of PTKs. These molecules lack catalytic activities but have tyrosine phosphorylation sites, multiple motifs, and domains that allow binding to the other signaling molecules to connect the receptor-activating PTKs to the downstream effectors and, therefore, act as positive or negative regulators controlling the immune receptor-mediated intracellular signaling (21, 22). 3BP2 was originally isolated as one of the c-Abl SH3 domain-binding proteins of unknown function (23). 3BP2 is composed of an N-terminal pleckstrin homology domain, central proline-rich regions, and C-terminal SH2 domain (24, 25). Point mutations in 3BP2 have been identified in human inherited disease cherubism, which is characterized by excessive bone resorption in the jaw bones (26, 27).
We and others have demonstrated the biological relevance of 3BP2 in immune system (27, 28). In mast cells 3BP2 is rapidly tyrosine-phosphorylated by the aggregation of FcϵRI, and the overexpression of the SH2 domain of 3BP2 results in the dramatic suppression of IgE-mediated tyrosine phosphorylation of PLC-γ and Ca2+ mobilization and degranulation (29). FcϵRI-activating PTKs such as Lyn, Syk, and Btk could phosphorylate 3BP2 on Tyr174, Tyr183, and Tyr446 (30). Phosphorylation of Tyr446 induces the binding to the SH2 domain of Lyn and enhances its kinase activity (30). In B lymphocytes, engagement of BCR triggers tyrosine phosphorylation of 3BP2 (25). Analysis of the tissue distribution of 3BP2 reveals the predominant expression of 3BP2 mRNA in B cells (24). Suppression of the 3BP2 expression by small interfering RNA results in the inhibition of BCR and T cell receptor (TCR)-mediated activation of NFAT (25, 31). Analysis of 3BP2-deficient mice demonstrated that 3BP2 is required for B cell proliferation and cell cycle progression (32, 33). Lack of 3BP2 results in the abnormal phenotype in splenic marginal-zone B cells and peritoneal B1 B cells and diminished thymus-independent type 2 antigen response (33). 3BP2 binds to CD19 through its SH2 domain in a tyrosine-phosphorylated-dependent manner (33). This suggests that 3BP2 forms a co-stimulatory complex for the optimum BCR signaling and plays an important role in innate immunity.
The present study has demonstrated that tyrosine phosphorylation of 3BP2 regulates BCR-mediated activation of NFAT. Syk is responsible for tyrosine phosphorylation of 3BP2. Both phosphorylation of Tyr183 and SH2 domain are required for BCR-mediated tyrosine phosphorylation of 3BP2 and activation of NFAT by associating specific signaling molecules. Phosphorylated Tyr183 interacts with the SH2 domains of PLC-γ2 and Vav1, whereas the C-terminal SH2 domain binds to BLNK. These 3BP2 signaling complexes may contribute to BCR-mediated activation of NFAT.
EXPERIMENTAL PROCEDURES
Antibodies and cDNAs
Anti-phosphotyrosine (Tyr(P)) (4G10) and anti-glyceraldehyde-3-phosphate dehydrogenase mAbs were purchased from Millipore (Bedford, MA). Anti-hemagglutinin epitope (HA) mAb was obtained from Covance (Princeton, NJ). Anti-PLC-γ2, anti-CD19, anti-Vav1, and anti-BLNK antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal anti-3BP2 antibody raised against an amino acid sequence from Leu359 to Ser462 of 3BP2, which is capable for the immunoprecipitation of 3BP2, was a gift from Dr. Raif S. Geha (Harvard Medical School) (32). Anti-glutathione S-transferase (GST) mAb was from Nacalai (Kyoto, Japan). Anti-phospho-PLC-γ2 antibody was from Cell Signaling Technology (Danvers, MA). Anti-chicken IgM mAb M4 was kindly provided by Dr. Tomohiro Kurosaki (RIKEN Research Center for Allergy and Immunology). Anti-human IgM mAb was from Zymed Laboratories Inc. (San Francisco, CA).
The HA-tagged expression constructs pMT3-HA-3BP2 cDNA were kindly provided by Dr. Amnon Altman (La Jolla Institute, La Jolla, CA) (24). All the other mutant forms of pMT3-HA-3BP2 cDNAs were described previously (30). For the immunoprecipitation study, a Myc tag inserted form of pMT3 vector was generated and confirmed by the DNA sequencing. Reporter plasmid pNFAT-luc was a gift from Dr. Gerald R. Crabtree (Stanford University). Control plasmid phRL-TK (Renilla-luc) was from Toyobo (Osaka, Japan).
Cell Culture and Transfection
DT40, Ramos B, and Daudi cells were maintained in RPMI 1640 medium (Sigma), 100 units/ml penicillin, and 10% heat-inactivated fetal calf serum. Lyn-deficient (Lyn−) and Syk-deficient (Syk−) DT40 cells were from Dr. Tomohiro Kurosaki (15). Ramos B cells expressing simian virus 40 T antigen (Ramos T cells) were provided by Dr. Hamid Band (Harvard Medical School, Boston, MA) and maintained in the same manner as the parental Ramos B cells (34, 35).
For the transient transfection of DT40 cells, 10 μg of pMT3-HA-3BP2 was transiently transfected into 5 × 106 of wild type, Lyn−, or Syk− DT40 cells by electroporation using Nucleofector device (Amaxa GmbH, Cologne, Germany). For the transient transfection of Ramos-T cells, 10 μg of pMT3-HA-3BP2 and its mutant forms of cDNAs were transfected into 107 cells by electroporation (310 V, 950 microfarads) (35). After 24 h cells were utilized for the experiments.
Cell Activation, Immunoprecipitation, and Immunoblotting
Cultured B cells were washed twice with serum-free medium and activated by 4 μg/ml anti-chicken IgM mAb (M4) (DT40 cells) or 10 μg/ml anti-human IgM mAb (Ramos-T and Ramos B cells) in the same medium. For immunoprecipitation studies, cells were washed twice with ice-cold phosphate-buffered saline and then solubilized in 1% Triton lysis buffer (1% Triton X-100, 50 mm Tris, pH 7.4, 150 mm NaCl, 10 mm EDTA, 100 mm NaF, 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, and 2 μg/ml aprotinin) on ice. For the immunoprecipitation of HA-3BP2 from Ramos-T cells, cells were solubilized in the denature buffer (1% Triton lysis buffer containing 0.1% SDS and 0.5% deoxycholic acid). For the immunoprecipitation of endogenous 3BP2, 1% digitonin (Wako, Osaka, Japan) was used instead of Triton in 1% Triton lysis buffer. Precleared cell lysates were incubated with the indicated antibodies prebound to protein A-agarose beads (Sigma) or anti-HA mAb prebound to the beads (Anti-HA Affinity Matrix, Roche Applied Science). After rotation for 90 min at 4 °C, the beads were washed 4 times with the lysis buffer, and the immunoprecipitated proteins were eluted by the heat treatment for 5 min at 100 °C with 2× sampling buffer. Immunoprecipitated proteins and cell lysates were separated by SDS-PAGE and electronically transferred to polyvinylidene difluoride transfer membrane (Millipore). After blocking in 5% milk in TBST (25 mm Tris, pH 8.0, 150 mm NaCl, and 0.1% Tween 20), the blots were reacted with the indicated primary antibodies and then horseradish peroxidase-conjugated second antibodies in TBST. Proteins were visualized by the enhanced chemiluminescence reagent (Western Lightning, PerkinElmer Life Sciences) (29, 36).
Reporter Gene Assay
The NFAT-luc reporter (15 μg), Renilla-luc reporter (1 μg), and pMT3-HA-3BP2 cDNAs (15 μg) were transiently cotransfected into DT40 cells (107) by electroporation (300 V, 960 microfarads). 48 h after the transfection, cells were stimulated without or with 4 μg/ml M4 for 6 h at 37 °C. For the control experiments, cells were stimulated with the mixture of 50 ng/ml phorbol 12-myristate 13-acetate and 1 μm of ionomycin for 6 h at 37 °C (37). The luciferase activities were determined by the luciferase assay system (Promega, Madison, WI) using the universal plate reader (Packard Fusion, PerkinElmer Life Sciences). The activities of NFAT reporter gene were normalized by those of control Renilla-luc and expressed as -fold increase compared to the activity in unstimulated control cells. Total cell lysates were generated by the direct addition of 2× sampling buffer, separated by SDS-PAGE, and analyzed by the immunoblotting with anti-HA and anti-glyceraldehyde-3-phosphate dehydrogenase mAbs.
Affinity Purification by Peptide Binding Experiments
For affinity purification of proteins specifically bound to phosphorylated Tyr174, Tyr183, or Tyr446 of 3BP2, phosphorylated and non-phosphorylated 3BP2 peptides 166–182 (NH2-biotin-SYPMDNEDpYEHEDEDDS-COOH), 177–193 (NH2-biotin-EDEDDSpYLEPDSPGPMK-COOH), and 438–453 (NH2-biotin-GEEDSDEDpYEKVPLPNS-COOH) (p indicated phosphorylated) were synthesized, respectively. All these peptides were biotinylated. 1 nmol of each peptide was bound to streptavidin beads (Pierce, Thermo Fisher Scientific, Rockford, IL) and incubated with Daudi cell lysate (equivalent to 5 × 106 cells, solubilized in 1% Triton lysis buffer). The beads were washed 4 times with the same buffer, and proteins interacting with synthesized peptides were eluted by heat treatment for 5 min at 100 °C with 2× sampling buffer, separated by SDS-PAGE, and analyzed by the immunoblotting.
For further analysis, 1 nmol of phosphorylated and non-phosphorylated 3BP2 peptides were bound to streptavidin beads and incubated with each 2.5 μg of GST, GST-PLC-γ2-SH2 domains (N-terminal and C-terminal SH2 domains: N+C) or GST-Vav1-SH2 domain solubilized in 1% Triton lysis buffer. The cDNA for PLC-γ2-SH2 (N+C) (Gly528 to Leu739) were isolated by reverse transcription-PCR from Ramos B cells using gene-specific primer (GSP1) (5′-TAGAGGGAGTTTATATCTCTTTCC-3′), GSP2, (5′-CCGGAATTCCGGGGAGAAATGGTTCCACAAG-3′), and GSP3 (5′-GGCCGCTCGAGCTCGGGGGTCACGGGGTA-3′). GSP1 was used as a primer for reverse transcription, and nested GSP2 and GSP3 were used as primers for PCR. The resulting PCR fragment was subcloned into pGEX4T.3 (GE Healthcare). The pGEX4T.3-Vav1-SH2 was described previously (38). GST fusion proteins were expressed in bacteria and purified by glutathione-Sepharose 4B beads (GE Healthcare). Preparation of GST fusion proteins was confirmed by the SDS-PAGE and Coomassie Brilliant Blue staining. After the incubation, the beads were washed 4 times with the same buffer, and co-precipitated proteins were separated by SDS-PAGE and analyzed by the immunoblotting.
Pulldown Assay
The cDNA for mouse 3BP2-SH2 (Ser454-Arg559) was amplified by PCR using two primers, 5′-CGGGATCCTCGGTGTTTGTCAACACGACA-3′ and 5′-CGGAATTCTCACCTGGGCCCAGCGTAGCC-3′, from template pMT3-HA 3BP2 wild type and subcloned into the pGEX-4T.3 (GE Healthcare). The point mutation of Arg486 of pGEX-4T.3–3BP2-SH2 to Lys (R486K) was prepared by the site-directed mutagenesis kit from Stratagene (La Jolla, CA) using two primers, 5′-TGGGCTGTATTGCATTAAGAACTCCTCTACCAAG-3′ and 5′-CTTGGTAGAGGAGTTCTTAATGCAATACAGCCCA-3′. Preparation of GST fusion proteins from bacteria was confirmed by the SDS-PAGE and Coomassie Brilliant Blue staining. Ramos B cells (108) were either unstimulated or stimulated with 10 μg/ml anti-IgM for 3 min at 37 °C and then solubilized in the binding buffer (1% Nonidet P-40, 25 mm Hepes, pH 7.5, 150 mm NaCl, 10 mm MgCl2, 1 mm EDTA, 2% glycerol, 1 mm phenylmethylsulfonyl fluoride, and 2 μg/ml aprotinin). After the centrifugation the resulted supernatants were reacted with 20 μg of GST fusion proteins (GST, GST-3BP2-SH2, or GST-3BP2-SH2 R486K) prebound to glutathione-Sepharose 4B beads for 90 min at 4 °C. Τhe beads were washed 4 times with the binding buffer. Proteins interacting with GST fusion proteins were separated by SDS-PAGE and analyzed by the immunoblotting.
Far Western Experiments
Anti-BLNK immunoprecipitates from unstimulated or anti-IgM-stimulated Ramos B cells (3 × 107) were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. After blocking, the membranes were incubated with 2.5 μg/ml GST, GST-3BP2-SH2, or GST-3BP2-SH2 R486K for 60 min at 4 °C. After extensive washing, membranes were reacted with anti-GST mAb and then horseradish peroxidase-conjugated mAb and subjected to ECL detection (39).
RESULTS
Syk Phosphorylates 3BP2 after BCR Stimulation
Recently, Foucault et al. (25) demonstrated that stimulation of BCR causes rapid tyrosine phosphorylation of 3BP2. We previously demonstrated that adaptor protein 3BP2 is tyrosine-phosphorylated by a non-receptor type of PTKs, Syk, Lyn, or Btk, which are known to be activated after BCR stimulation (30). Among those PTKs, Syk predominantly phosphorylates 3BP2 in COS cells (30). Moreover, Syk phosphorylates 3BP2 on Tyr174, Tyr183, and Tyr446, and those residues are located within the sequence preferred to be phosphorylated by Syk (30). Point mutation of Tyr183 or Tyr446 resulted in the suppression of TCR-induced 3BP2-mediated activation of NFAT in T cells (31). These findings suggested that tyrosine phosphorylation of 3BP2 may occur in the early events in BCR signaling pathway. Thus, first we examined the essential PTK required for BCR-mediated tyrosine phosphorylation of 3BP2 using chicken DT40 B cells lacking Lyn or Syk (Fig. 1). Because anti-3BP2 antibodies could not precipitate endogenous chicken 3BP2, HA-tagged mouse 3BP2 was transiently transfected into wild-type, Lyn-deficient (Lyn−), or Syk-deficient (Syk−) DT40 cells and utilized for the experiments. Patterns of protein tyrosine phosphorylation by the engagement of BCR in these cells are shown in Fig. 1A. A BCR-mediated increase in protein-tyrosine phosphorylation was partially reduced in Lyn− cells and almost completely abrogated in Syk− cells compared to that in wild type DT40 cells. Tyrosine phosphorylation of 3BP2 was analyzed by immunoprecipitation experiments. BCR stimulation caused tyrosine phosphorylation of 3BP2 in DT40 wild type and Lyn− but not in Syk− cells (Fig. 1B). Therefore, this result demonstrated that Syk is critical for BCR-mediated tyrosine phosphorylation of 3BP2. Lyn is the upstream PTK that phosphorylates immunoreceptor tyrosine-based activation motif of Igα and Igβ to recruit and activate Syk. BCR-mediated tyrosine phosphorylation of 3BP2 was reduced in Lyn− cells (Fig. 1B). Thus, Lyn might be required for the optimum activation of Syk and, therefore, tyrosine phosphorylation of 3BP2 after BCR stimulation. Tyrosine phosphorylation of 3BP2 in Lyn− cells suggested that some other Src-family PTKs bypasses the activation of Syk. However, those Src-family PTKs could not phosphorylate 3BP2 in Syk− cells (Fig. 1B).
FIGURE 1.
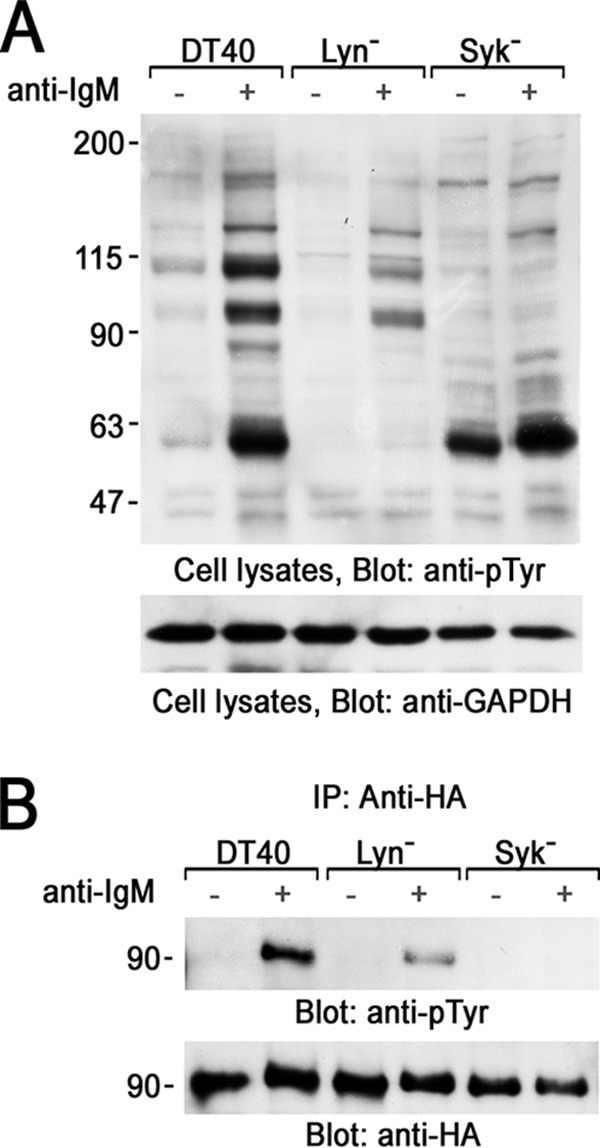
Syk phosphorylates 3BP2 in B cells. A and B, HA-tagged 3BP2 was transiently expressed in DT40 wild type (DT40), Lyn-deficient (Lyn−), or Syk-deficient (Syk) cells. 24 h after transfection cells were unstimulated (−) or stimulated with anti-IgM mAb for 3 min (+). A, detergent-soluble lysates were separated by SDS-PAGE and analyzed by immunoblotting with anti-phosphotyrosine (pTyr) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mAbs. B, anti-HA immunoprecipitates (IP) were separated by SDS-PAGE and analyzed by immunoblotting with anti-Tyr(P) mAb (top panel). The membrane was stripped and reprobed with anti-HA mAb (bottom panel). Molecular size markers are indicated at the left in kilodaltons. The results are representative of three independent experiments.
Pretreatment of DT40 cells with a pan-Src family PTKs pharmacological inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) (40) suppressed BCR-mediated tyrosine phosphorylation of 3BP2 but not completely (data not shown). Therefore, we could not exclude the possible involvement of some other PTKs on 3BP2 tyrosine phosphorylation in B cells.
3BP2 Promotes BCR-mediated Transcriptional Activation of NFAT through Tyr183 and the SH2 Domain
In the TCR-signaling pathway, a point mutation of Tyr183 or Tyr446 or in the SH2 domain of 3BP2 results in the suppression of 3BP2-mediated activation of NFAT (31). Therefore, next we examined whether these candidate tyrosine phosphorylation sites and the SH2 domain of 3BP2 could regulate the activation of NFAT in BCR signaling pathway. The schematic diagram of 3BP2 wild type and its mutant forms used in this experiment is shown in Fig. 2A. BCR-mediated activation of NFAT was increased by the transient expression of wild type 3BP2 (Fig. 2B). Among three tyrosine phosphorylation sites, BCR-mediated activation of NFAT was suppressed by the point mutation of Tyr183 only but not Tyr174 or Tyr446. Point mutation of Arg486, a critical residue for the function of SH2 domain, also suppressed the activation of NFAT. Phorbol 12-myristate 13-acetate plus ionomycin-mediated activation of NFAT was not affected by the expression of various 3BP2 (data not shown). Fig. 2B, bottom panel, shows the level of 3BP2 protein expression. Thus, this result demonstrated that Tyr183 and the SH2 domain of 3BP2 positively regulate 3BP2-mediated activation of NFAT in BCR signaling. Similar results were obtained when human B cell line Daudi cells were analyzed by the same experiment (data not shown).
FIGURE 2.

Structural and functional analysis of 3BP2-mediated activation of NFAT in B cells. A, shown is a schematic diagram of 3BP2 wild type (WT) and mutants used in this study. Tyr174 was substituted for phenylalanine (Y174F), Tyr183 was substituted for phenylalanine (Y183F), and Tyr446 was substituted for phenylalanine (Y446F), respectively. Arg486 in the SH2 domain of 3BP2 was substituted for lysine (R486K) (30). The HA tag (HA), N-terminal pleckstrin homology domain (PH), and three proline-rich regions (Pro-rich) were represented. B and C, shown is a luciferase assay. NFAT-luc reporter, Renilla-luc reporter, and various pMT3-HA-3BP2 cDNAs were transiently cotransfected into wild type DT40 cells (B) or Lyn− and Syk− cells (C). 48 h after the transfection, cells were unstimulated (−) or stimulated with anti-IgM mAb for 6 h (+). The normalized luciferase activities were expressed as -fold of increase compared with that in unstimulated pMT3 vector (Vec)-transfected cells. The results are the mean values ± S.D. from three independent experiments. Cell lysates were separated by SDS-PAGE and analyzed by immunoblotting with anti-HA and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mAbs (bottom panels). Molecular size markers are indicated at the left in kilodaltons. The results are representative of three independent experiments.
Previously, it was reported that Syk-dependent activation of NFAT is elevated in Lyn− DT40 B cells (41). As reported, BCR-induced activation of NFAT was elevated in Lyn− cells compared to that in wild type DT40 cells. Overexpression of 3BP2 moderately increased NFAT activity in the resting cells but not in BCR-stimulated Lyn− cells (Fig. 2C). Thus, the effect of overexpression of 3BP2 was limited in Lyn− cells. More importantly, BCR-induced NFAT activation was completely abrogated in Syk− cells, and overexpression of 3BP2 could not mimic the activation of NFAT (Fig. 2C). This clearly demonstrated that Syk-dependent tyrosine phosphorylation of 3BP2 is required for BCR-induced activation of NFAT.
Both Tyr183 and the SH2 Domain of 3BP2 Are Required for BCR-mediated Tyrosine Phosphorylation of 3BP2
To address the role of Tyr183 and SH2 domain of 3BP2, we examined BCR-mediated tyrosine phosphorylation of 3BP2 by immunoprecipitation experiments using human Ramos B cells expressing SV40 virus T antigen (Ramos-T) for transient expression experiments (Fig. 3). Point mutation of Tyr183 to Phe (Y183F) in 3BP2 resulted in the suppression of BCR-mediated tyrosine phosphorylation of 3BP2 (Fig. 3A). It suggested that Tyr183 is the major phosphorylation site of 3BP2 in BCR signaling. The SH2 domain of 3BP2 was shown to interact with Syk (24). Syk is necessary for tyrosine phosphorylation of 3BP2 (Fig. 1). A point mutation of Arg486 to Lys (R486K) also suppressed BCR-mediated tyrosine phosphorylation of 3BP2 (Fig. 3B). Therefore, Tyr183 and the SH2 domain of 3BP2 are necessary for BCR-mediated tyrosine phosphorylation of 3BP2. Unlike chicken pro-B DT40 cells, there was some phosphorylation of 3BP2 before the stimulation in human Ramos-T cells. This could be due to the difference of animal (chicken or human) or the stage of the differentiation of cultured B cells.
FIGURE 3.
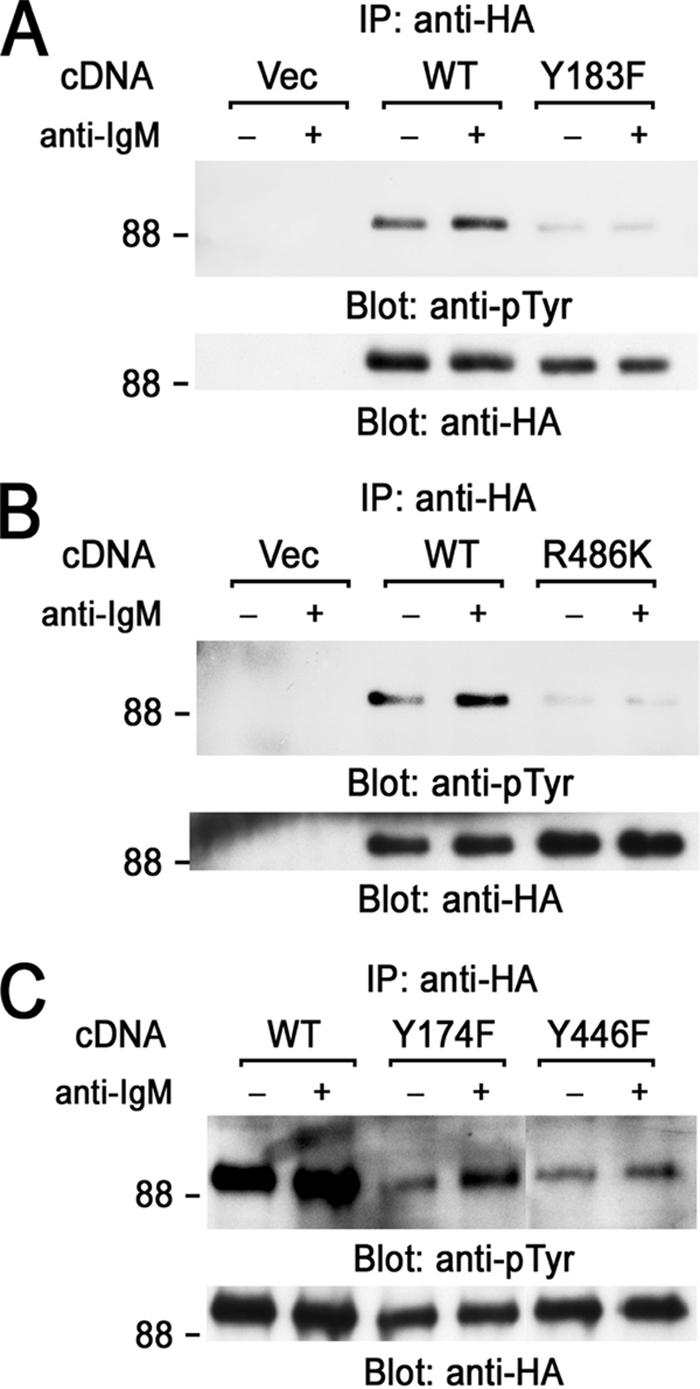
Analysis of BCR-mediated tyrosine phosphorylation of 3BP2. Ramos-T cells were transiently transfected with pMT3 vector (Vec), HA-3BP2 wild type (WT), and HA-3BP2-Y183F (Y183F) (A), HA-3BP2-R486K (R486K) (B), HA-3BP2-Y174F (Y174F), and HA-3BP2-Y446F (Y446F) (C). After 24 h cells were unstimulated (−) or stimulated with anti-IgM mAb for 3 min (+). Anti-HA immunoprecipitates (IP) were separated by SDS-PAGE and analyzed by immunoblotting with anti-Tyr(P) (upper panels) and anti-HA mAbs (bottom panels). Molecular size markers are indicated at the left in kilodaltons. The results are representative of three independent experiments.
Analysis of the residual tyrosine phosphorylation sites in 3BP2 showed that BCR-stimulation induces tyrosine phosphorylation of 3BP2-Y174F and 3BP2-Y446 mutants, although the degrees of their tyrosine phosphorylation were lower than that of wild type (Fig. 3C). The inducible tyrosine phosphorylation of 3BP2-Y174F and 3BP2-Y446F may be sufficient for BCR-induced activation of NFAT (Fig. 2B).
BCR Stimulation Induces the Complex Formation of Signaling Molecules with 3BP2
To address the biological function of 3BP2, we examined the proteins complexed with endogenous 3BP2 in B cells. Ramos B cells were solubilized with digitonin lysis buffer and immunoprecipitated with polyclonal anti-3BP2 antibody, which was capable for the immunoprecipitation of 3BP2 (32). Endogenous 3BP2 was tyrosine-phosphorylated by BCR stimulation (arrows) and appeared to be complexed with other tyrosine-phosphorylated proteins in Ramos B cells (Fig. 4A). Immunoblotting experiments showed that 3BP2 associated with PLC-γ2, CD19, and Vav1 in unstimulated cells, and these associations were slightly increased after BCR stimulation. Because 3BP2 was shown to associate with the linker for activation of T cells (LAT) in T cells and in mast cells, we next examine whether 3BP2 could interact with BLNK in B cells (24, 29). Immunoblotting experiments demonstrated that 3BP2 associated with BLNK (Fig. 4A, bottom panel). This association was reconfirmed by immunoblotting BLNK immunoprecipitates with anti-3BP2 antibody. 3BP2 inducibly associates with BLNK in BCR-stimulated B cells (Fig. 4B).
FIGURE 4.

Interaction of endogenous 3BP2 with signaling molecules. Ramos B cells were unstimulated (−) or stimulated with anti-IgM mAb for 3 min (+) and then solubilized with 1% digitonin lysis buffer (A) or 1% Triton lysis buffer (B). Detergent-soluble lysates and anti-3BP2 (A) and normal rabbit serum (NRS) and anti-BLNK (B) immunoprecipitates (IP) were separated by SDS-PAGE and analyzed by immunoblotting with anti-Tyr(P), anti-PLC-γ2, anti-CD19, anti-Vav1, anti-3BP2, and anti-BLNK antibodies, as indicated. Molecular size markers are indicated at the left in kilodaltons. The results are representative of three independent experiments.
3BP2-Tyr183 Directly Binds to PLC-γ2 and Vav1
To characterize 3BP2-binding proteins, affinity purification by peptide binding experiments were carried out. A sequence of synthesized biotinylated 3BP2 peptides along with the sequence around Tyr174, Tyr183, or Tyr446 of 3BP2 is shown in Fig. 5A. Either phosphorylated or non-phosphorylated peptide was reacted with Daudi cell lysates, and proteins interacted with these peptides were analyzed by immunoblotting (Fig. 5B). Among them, only the phosphorylated Tyr183 (phospho-Tyr183) peptide of 3BP2 could react with PLC-γ2 and Vav1 in B cell lysates. These signaling molecules could not react with either phospho-Tyr174 or phospho-Tyr446 peptide, suggesting that this interaction was specific. Prediction of SH2 domain binding sequence by degenerated peptide library showed that the sequence after phospho-Tyr183 matched the preference of binding sequence of the SH2 domains of PLC-γ1 (isozyme of PLC-γ2) and Vav1 (42). Therefore, next we tested whether these interactions were direct or not, because we could not exclude the possibility that PLC-γ2 and Vav1 can interact with some other 3BP2-associating proteins and indirectly complex with 3BP2. To address this issue, bacterial GST, GST fusion proteins of PLC-γ2-SH2 domains (N+C), or GST-Vav1-SH2 domain was reacted with each synthesized 3BP2 peptide, and the interaction of these molecules was analyzed by the immunoblotting (Fig. 5C). Precipitation of both SH2 domains was still observed in phospho-Tyr183 peptide. Thus, this result strongly suggests that phospho-Tyr183 in 3BP2 directly binds to the SH2 domains of PLC-γ2 and Vav1. Similar studies were examined by using COS cells. Either 3BP2 wild type or Y183F was cotransfected with Syk, and cell lysates were reacted with GST-PLC-γ2-SH2 domains. As expected, a point mutation of Tyr183 abrogated the association with GST-PLC-γ2-SH2 domains (data not shown). Alternatively, these results reconfirmed that 3BP2-Tyr183 is required for interaction with PLC-γ2-SH2 domains. Further co-immunoprecipitation experiments by using B cells demonstrated that 3BP2-Y183F mutation impairs the interaction of 3BP2 with tyrosine-phosphorylated PLC-γ2 after BCR stimulation (Fig. 5D).
FIGURE 5.

Interaction of 3BP2 with PLC-γ2 and Vav1. A, shown is a schematic diagram of biotinylated synthesized 3BP2 peptides used in this experiment. Phosphorylated or non-phosphorylated 3BP2 peptides Ser166–Ser182, Glu177–Lys193, and Gly438–Ser453 are shown. Positions of phosphorylation sites are indicated with asterisks. Tyr174, Tyr183, or Tyr446 was phosphorylated in each phosphorylated 3BP2 peptide, respectively. These tyrosine residues were not phosphorylated in non-phosphorylated 3BP2 peptides. B, affinity purification by peptide binding experiments is shown. Daudi cell lysates were reacted with either phosphorylated or non-phosphorylated biotinylated synthesized 3BP2 peptides prebound to streptavidin beads. Interactions of these peptides with PLC-γ2 and Vav1 were analyzed by SDS-PAGE and immunoblotting with anti-PLC-γ2 and anti-Vav1 antibodies. C, interaction of 3BP2 peptides with GST-SH2 domain fusion proteins is shown. Bacterial GST, GST fusion proteins of PLC-γ2-SH2 (N+C), or Vav1-SH2 was reacted with each 3BP2 peptide prebound to streptavidin beads. Interactions of these peptides with SH2 domains were analyzed by SDS-PAGE and immunoblotting with anti-GST mAb. The last two lanes show the positive controls of GST and GST-SH2 fusion proteins of PLC-γ2 (N+C) and Vav1. D, shown is a co-immunoprecipitation experiment. Ramos-T cells were transiently transfected with HA-Myc-3BP2 wild type (WT) and HA-myc-3BP2-Y183F (Y183F). After 24 h cells were unstimulated (−) or stimulated with anti-IgM mAb for 3 min (+). Anti-HA immunoprecipitates (IP) were separated by SDS-PAGE and analyzed by immunoblotting with anti-phospho-PLC -γ2 (upper panel) and anti-HA antibodies (bottom panel). Molecular size markers are indicated at the left in kilodaltons. The results are representative of three independent experiments.
Phosphorylated Tyr174 could interact with GST-Vav1-SH2 at lower magnitude (Fig. 5C). This peptide (Tyr(P)174) could not react with the intact Vav1 in B cell lysates even in the long exposure (data not shown). However, it reacted with Vav1 in cultured osteoclasts.3 The biological relevance of the interaction of phosphorylated Tyr174 and Vav1-SH2 domain remains unclear.
3BP2-SH2 Domain Directly Binds to BLNK
To characterize the functional requirement of the SH2 domain of 3BP2 in BCR-mediated signaling, pulldown experiments were carried out. Incubation of the GST-3BP2-SH2 domain with unstimulated or BCR-stimulated Ramos B cell lysates resulted in coprecipitation of tyrosine-phosphorylated proteins in a BCR stimulation-dependent manner (Fig. 6A). Immunoblotting experiments demonstrated that the 3BP2-SH2 domain inducibly bound to CD19 and BLNK after BCR stimulation and constitutively bound to PLC-γ2 (Fig. 6A). It is important to note that the point mutation of Arg486 in the SH2 domain completely abrogated the interaction with these signaling molecules (Fig. 6, A, lanes 3 and 4 versus 5 and 6). Recently, Foucault et al. (25) demonstrated that 3BP2-SH2 inducibly interacts with Vav1, PLC-γ2, and Syk in Daudi cells after BCR stimulation. Presumably, the discrepancy of the inducibility of the 3BP2-SH2 domain with PLC-γ2 is due to the difference of cell line used in the experiments (25). In addition to their findings, we identified that 3BP2-SH2 domain associates with BLNK. 3BP2-SH2 domain is predicted to bind to the YEN motif when it is tyrosine-phosphorylated (42). In fact, Tyr403 and Tyr443 in the cytoplasmic tail of CD19, both of which are located within YEN motif, are capable of mediating the interaction with 3BP2 (33). BLNK has 1 copy of the YEN motif (Tyr72). Therefore, next we examined whether the SH2 domain of 3BP2 can directly bind to BLNK or not. Far Western experiments showed that GST-3BP2-SH2 domain directly bound to immunoprecipitated BLNK from BCR-stimulated Ramos B cells (Fig. 6B). The point mutation in the SH2 domain of 3BP2 completely abrogated the direct interaction with BLNK, suggesting that 3BP2-SH2 domain directly binds to tyrosine-phosphorylated BLNK.
FIGURE 6.

Interaction of 3BP2 with BLNK. A, in vitro interaction of 3BP2-SH2 domain with cellular proteins in B cells is shown. Ramos B cells were unstimulated (−) or stimulated with anti-IgM mAb for 3 min (+) and solubilized with the binding buffer. Cell lysates were reacted with GST, GST-3BP2-SH2, or GST-3BP2-SH2-R486K immobilized on glutathione-Sepharose 4B beads. Interacted cellular proteins were analyzed by SDS-PAGE and immunoblotting with anti-Tyr(P) mAb. The membrane was stripped and then reprobed with the indicated antibodies. B, interaction of the SH2 domain of 3BP2 with BLNK is direct. BLNK was immunoprecipitated from unstimulated (−) or anti-IgM stimulated Ramos B cell lysates (+) with anti-BLNK mAb. Anti-BLNK immunoprecipitates (IP) were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane. Membrane was blocked with milk and incubated with GST, GST-3BP2-SH2, or GST-3BP2-SH2 R486K. Membranes were then reacted with anti-GST mAb and horseradish peroxidase-conjugated mAb for visualization. A and B, molecular size markers are indicated at the left in kilodaltons. The results are representative of three independent experiments.
DISCUSSION
In the present study we have demonstrated that the adaptor protein 3BP2 facilitates the activation of transcription factor NFAT through tyrosine phosphorylation, which forms a direct complex with PLC-γ2 and Vav1, and SH2 domain binds to BLNK after BCR stimulation (Fig. 7). Engagement of BCR triggers the activation of Lyn to phosphorylate tyrosine residues in the immunoreceptor tyrosine-based activation motif, which then provides the docking sites for Syk. Activated Syk phosphorylates Tyr183 of 3BP2 to interact with the SH2 domains of PLC-γ2 and Vav1. The SH2 domain of 3BP2 is required for tyrosine phosphorylation of 3BP2 as the 3BP2-SH2 domain could bind to Syk (24). Our preliminary results showed that c-Abl, another member of non-receptor type PTK, could phosphorylate 3BP2 on Tyr446 but not Tyr183 in COS cells.4 Thus, phosphorylation of specific tyrosine residues might be determined by the substrate specificity of PTKs. Simultaneously, the SH2 domain of 3BP2 directly binds with BLNK, which then recruits Btk to phosphorylate PLC-γ2 after BCR stimulation. Genetic analysis has demonstrated that both Btk and PLC-γ2 are required for Ca2+ mobilization and the transcriptional activation of NFAT by BCR engagement (4, 43, 44). These 3BP2-mediated signaling complexes lead to the activation of NFAT. BLNK is a central adaptor protein that connects BCR-stimulating PTKs and effector molecules. Tyrosine phosphorylation of BLNK allows inducible association with a number of signaling molecules and is required for BCR-mediated Ca2+ mobilization and NFAT activation (45). In addition to our findings, 3BP2-SH2 domain was shown to interact with PLC-γ2 and Vav1 (25). Our results demonstrated that 3BP2 facilitates the function of BLNK by connecting Syk and effector molecules such as PLC-γ2 and Vav1.
FIGURE 7.

Model of 3BP2-mediated NFAT activation after BCR stimulation. BCR stimulation induces the sequential activation of non-receptor type of PTKs Lyn, Syk, and tyrosine phosphorylation of Tyr183 in 3BP2. Phosphorylated Tyr183 directly binds to the SH2 domains of Vav1 and PLC-γ2. 3BP2-SH2 domain directly binds to BLNK. In addition, 3BP2-SH2 domain was reported to associate with Syk, Vav1, and PLC-γ2. 3BP2-mediated signaling complex facilitates to the activation of NFAT.
There is a difference in the function of 3BP2 tyrosine phosphorylation between B cells and T cells. The point mutation of Tyr446 results in the suppression of TCR-mediated NFAT activation (31) but not BCR (Fig. 1B). Phosphorylation of Tyr446 allows 3BP2 to act as a positive regulator for Lyn, an upstream PTK of Syk (30). In addition, Tyr446 is required for the interaction with Lck-SH2 domain (31). Proline-rich regions and phosphorylated Tyr446 associate with the SH3 and SH2 domains of Lyn to lead to the possible conformational change of Lyn to activate its kinase activity (30). In T and natural killer cells, another member of Syk family PTK ZAP-70 was expressed. Unlike Syk, the mechanism of ZAP-70 activation is different. Src family PTK Lck activates ZAP-70 by phosphorylating Tyr319 in the linker region and Tyr493 in the activation loop of the kinase domain (46–48). ZAP-70 could not catalyze phosphorylation on these tyrosine residues, whereas Syk could phosphorylate corresponding tyrosine residues by autophosphorylation (49). Therefore, requirement of Tyr446 in TCR-mediated NFAT activation might be explained by the fact that activation of ZAP-70 requires the activation of upstream Src-family kinase (31). Phosphorylation of Tyr446 may trigger the activation of Lck to induce the activation of ZAP-70. Initial tyrosine phosphorylation of 3BP2 could be catalyzed by some other receptor-associating PTKs in T cells. All together, phosphorylation of Tyr446 is required for the activation of NFAT in signaling from TCR but not BCR.
Point mutations in 3BP2 cause human inherited disease cherubism (26). To understand the pathological role of 3BP2 in cherubism, Ueki et al. (50) generated “cherubism” mice in which the most common mutation found in cherubism patients, Pro418 to Arg (Pro416 to Arg in mouse), was inserted into the mouse 3bp2 gene. Analysis of cherubism mice demonstrated that mutant myeloid cells showed increased responses to macrophage-colony-stimulating factor and RANKL (RANK (receptor activation of NF-κB ligand) and enhanced differentiation to macrophages and osteoclasts. These findings suggested that point mutations of 3BP2 in cherubism patients result in the gain of function in macrophage lineage cells. Then, how does cherubism 3BP2 become “active”? 3BP2 does not possess the catalytic activity. Therefore, the substitution of amino acids of 3BP2 may increase the signaling complex formation or decrease the association with the negative regulators. Tyrosine phosphorylation of cherubism 3BP2 might be increased because expression of the mutant form of 3BP2 increased phosphorylation of Tyr346 of Syk (in the mouse protein) in osteoclasts, suggesting that the kinase activity and/or expression amount of Src-family PTK was up-regulated (50). This may enhance the kinase activity of Syk and tyrosine phosphorylation of 3BP2. Alternatively, constitutive association of 3BP2 with 14-3-3 in B cells5 might be decreased as we demonstrated previously in COS cells (38). A lack of the negative regulator may result in the enhancement of the 3BP2-mediated signaling pathway by the increase in the association with signaling molecules or change of the intracellular localization of 3BP2.
The present study demonstrated that Syk-mediated tyrosine phosphorylation and function of SH2 domain in 3BP2 regulate NFAT activation in BCR signaling. 3BP2 is the putative substrate of Syk. Mutational analysis has revealed that both Tyr183 and the SH2 domain are necessary for NFAT activation. These contribute to the formation of an inducible complex of signaling molecules in B cells. Detailed analysis of the role of 3BP2 tyrosine phosphorylation could provide the evidence to develop the immune regulatory drugs.
Acknowledgment
We are grateful to Satomi Nishibata for assistance.
This work was supported in part by research funding from the University of Fukui (to K. S.), by the Takeda Science Foundation (to K. S.), by the Osaka Medical Research Foundation for Incurable Diseases and by grants-in-aid from the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, and Technology, Japan.
K. Nakashima, T. Hatani, and K. Sada unpublished observations.
T. Hatani and K. Sada, unpublished observations.
K. Ogi, T. Hatani, and K. Sada, unpublished observations.
- BCR
- B cell receptor
- SH2
- Src homology 2
- NFAT
- nuclear factor of activated T cells
- PLC
- phospholipase C
- BLNK
- B cell linker protein
- PTK
- protein-tyrosine kinase
- TCR
- T cell receptor
- Tyr(P)
- phosphotyrosine
- HA
- hemagglutinin
- GST
- glutathione S-transferase
- 3BP2
- c-Abl SH3 domain-binding protein-2
- mAb
- monoclonal antibody.
REFERENCES
- 1.Fu C., Turck C. W., Kurosaki T., Chan A. C. (1998) Immunity. 9, 93–103 [DOI] [PubMed] [Google Scholar]
- 2.Goitsuka R., Fujimura Y., Mamada H., Umeda A., Morimura T., Uetsuka K., Doi K., Tsuji S., Kitamura D. (1998) J. Immunol. 161, 5804–5808 [PubMed] [Google Scholar]
- 3.Ishiai M., Kurosaki M., Pappu R., Okawa K., Ronko I., Fu C., Shibata M., Iwamatsu A., Chan A. C., Kurosaki T. (1999) Immunity. 10, 117–125 [DOI] [PubMed] [Google Scholar]
- 4.Takata M., Kurosaki T. (1996) J. Exp. Med. 184, 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taniguchi T., Kobayashi T., Kondo J., Takahashi K., Nakamura H., Suzuki J., Nagai K., Yamada T., Nakamura S., Yamamura H. (1991) J. Biol. Chem. 266, 15790–15796 [PubMed] [Google Scholar]
- 6.Turner M., Schweighoffer E., Colucci F., Di Santo J. P., Tybulewicz V. L. (2000) Immunol. Today 21, 148–154 [DOI] [PubMed] [Google Scholar]
- 7.Sada K., Takano T., Yanagi S., Yamamura H. (2001) J. Biochem. 130, 177–186 [DOI] [PubMed] [Google Scholar]
- 8.Turner M., Mee P. J., Costello P. S., Williams O., Price A. A., Duddy L. P., Furlong M. T., Geahlen R. L., Tybulewicz V. L. (1995) Nature 378, 298–302 [DOI] [PubMed] [Google Scholar]
- 9.Cheng A. M., Rowley B., Pao W., Hayday A., Bolen J. B., Pawson T. (1995) Nature 378, 303–306 [DOI] [PubMed] [Google Scholar]
- 10.Zhang J., Berenstein E. H., Evans R. L., Siraganian R. P. (1996) J. Exp. Med. 184, 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costello P. S., Turner M., Walters A. E., Cunningham C. N., Bauer P. H., Downward J., Tybulewicz V. L. (1996) Oncogene 13, 2595–2605 [PubMed] [Google Scholar]
- 12.Greenberg S., Chang P., Wang D. C., Xavier R., Seed B. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 1103–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mócsai A., Humphrey M. B., Van Ziffle J. A., Hu Y., Burghardt A., Spusta S. C., Majumdar S., Lanier L. L., Lowell C. A., Nakamura M. C. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 6158–6163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole A., Gibbins J. M., Turner M., van Vugt M. J., van de Winkel J. G., Saito T., Tybulewicz V. L., Watson S. P. (1997) EMBO J. 16, 2333–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takata M., Sabe H., Hata A., Inazu T., Homma Y., Nukada T., Yamamura H., Kurosaki T. (1994) EMBO J. 13, 1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurosaki T., Johnson S. A., Pao L., Sada K., Yamamura H., Cambier J. C. (1995) J. Exp. Med. 182, 1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coopman P. J., Do M. T., Barth M., Bowden E. T., Hayes A. J., Basyuk E., Blancato J. K., Vezza P. R., McLeskey S. W., Mangeat P. H., Mueller S. C. (2000) Nature 406, 742–747 [DOI] [PubMed] [Google Scholar]
- 18.Bailet O., Fenouille N., Abbe P., Robert G., Rocchi S., Gonthier N., Denoyelle C., Ticchioni M., Ortonne J. P., Ballotti R., Deckert M., Tartare-Deckert S. (2009) Cancer Res. 69, 2748–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cha H. S., Boyle D. L., Inoue T., Schoot R., Tak P. P., Pine P., Firestein G. S. (2006) J. Pharmacol. Exp. Ther. 317, 571–578 [DOI] [PubMed] [Google Scholar]
- 20.Krishnan S., Juang Y. T., Chowdhury B., Magilavy A., Fisher C. U., Nguyen H., Nambiar M. P., Kyttaris V., Weinstein A., Bahjat R., Pine P., Rus V., Tsokos G. C. (2008) J. Immunol. 181, 8145–8152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawson T., Scott J. D. (1997) Science 278, 2075–2080 [DOI] [PubMed] [Google Scholar]
- 22.Rudd C. E. (1999) Cell 96, 5–8 [DOI] [PubMed] [Google Scholar]
- 23.Ren R., Mayer B. J., Cicchetti P., Baltimore D. (1993) Science 259, 1157–1161 [DOI] [PubMed] [Google Scholar]
- 24.Deckert M., Tartare-Deckert S., Hernandez J., Rottapel R., Altman A. (1998) Immunity 9, 595–605 [DOI] [PubMed] [Google Scholar]
- 25.Foucault I., Le Bras S., Charvet C., Moon C., Altman A., Deckert M. (2005) Blood 105, 1106–1113 [DOI] [PubMed] [Google Scholar]
- 26.Ueki Y., Tiziani V., Santanna C., Fukai N., Maulik C., Garfinkle J., Ninomiya C., doAmaral C., Peters H., Habal M., Rhee-Morris L., Doss J. B., Kreiborg S., Olsen B. R., Reichenberger E. (2001) Nat. Genet. 28, 125–126 [DOI] [PubMed] [Google Scholar]
- 27.Hatani T., Sada K. (2008) Curr. Med. Chem. 15, 549–554 [DOI] [PubMed] [Google Scholar]
- 28.Deckert M., Rottapel R. (2006) Adv. Exp. Med. Biol. 584, 107–114 [DOI] [PubMed] [Google Scholar]
- 29.Sada K., Miah S. M., Maeno K., Kyo S., Qu X., Yamamura H. (2002) Blood 100, 2138–2144 [DOI] [PubMed] [Google Scholar]
- 30.Maeno K., Sada K., Kyo S., Miah S. M., Kawauchi-Kamata K., Qu X., Shi Y., Yamamura H. (2003) J. Biol. Chem. 278, 24912–24920 [DOI] [PubMed] [Google Scholar]
- 31.Qu X., Kawauchi-Kamata K., Miah S. M., Hatani T., Yamamura H., Sada K. (2005) Biochemistry 44, 3891–3898 [DOI] [PubMed] [Google Scholar]
- 32.de la Fuente M. A., Kumar L., Lu B., Geha R. S. (2006) Mol. Cell. Biol. 26, 5214–5225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen G., Dimitriou I. D., La Rose J., Ilangumaran S., Yeh W. C., Doody G., Turner M., Gommerman J., Rottapel R. (2007) Mol. Cell. Biol. 27, 3109–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao N., Ghosh A. K., Ota S., Zhou P., Reddi A. L., Hakezi K., Druker B. K., Wu J., Band H. (2001) EMBO J. 20, 7085–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miah S. M., Sada K., Tuazon P. T., Ling J., Maeno K., Kyo S., Qu X., Tohyama Y., Traugh J. A., Yamamura H. (2004) Mol. Cell. Biol. 24, 71–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qu X., Sada K., Kyo S., Maeno K., Miah S. M., Yamamura H. (2004) Blood 103, 1779–1786 [DOI] [PubMed] [Google Scholar]
- 37.Yankee T. M., Keshvara L. M., Sawasdikosol S., Harrison M. L., Geahlen R. L. (1999) J. Immunol. 163, 5827–5835 [PubMed] [Google Scholar]
- 38.Miah S. M., Hatani T., Qu X., Yamamura H., Sada K. (2004) Genes Cells 9, 993–1004 [DOI] [PubMed] [Google Scholar]
- 39.Lupher M. L., Jr., Rao N., Lill N. L., Andoniou C. E., Miyake S., Clark E. A., Druker B., Band H. (1998) J. Biol. Chem. 273, 35273–35281 [DOI] [PubMed] [Google Scholar]
- 40.Chung S. C., Limnander A., Kurosaki T., Weiss A., Korenbrot J. I. (2007) J. Cell Biol. 177, 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma H., Yankee T. M., Hu J., Asai D. J., Harrison M. L., Geahlen R. L. (2001) J. Immunol. 166, 1507–1516 [DOI] [PubMed] [Google Scholar]
- 42.Songyang Z., Shoelson S. E., McGlade J., Olivier P., Pawson T., Bustelo X. R., Barbacid M., Sabe H., Hanafusa H., Yi T., Ren R., Baltimore D., Ratnofsky S., Feldman R. A., Cantley L. C. (1994) Mol. Cell. Biol. 14, 2777–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antony P., Petro J. B., Carlesso G., Shinners N. P., Lowe J., Khan W. N. (2003) Exp. Cell Res. 291, 11–24 [DOI] [PubMed] [Google Scholar]
- 44.Hao S., Kurosaki T., August A. (2003) EMBO J. 22, 4166–4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu C. W., Dalton M., Ishiai M., Kurosaki T., Chan A. C. (2002) EMBO J. 21, 6461–6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams B. L., Irvin B. J., Sutor S. L., Chini C. C., Yacyshyn E., Bubeck Wardenburg J., Dalton M., Chan A. C., Abraham R. T. (1999) EMBO J. 18, 1832–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pelosi M., Di Bartolo V., Mounier V., Mège D., Pascussi J. M., Dufour E., Blondel A., Acuto O. (1999) J. Biol. Chem. 274, 14229–14237 [DOI] [PubMed] [Google Scholar]
- 48.Chan A. C., Dalton M., Johnson R., Kong G. H., Wang T., Thoma R., Kurosaki T. (1995) EMBO J. 14, 2499–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furlong M. T., Mahrenholz A. M., Kim K. H., Ashendel C. L., Harrison M. L., Geahlen R. L. (1997) Biochim. Biophys. Acta 1355, 177–190 [DOI] [PubMed] [Google Scholar]
- 50.Ueki Y., Lin C. Y., Senoo M., Ebihara T., Agata N., Onji M., Saheki Y., Kawai T., Mukherjee P. M., Reichenberger E., Olsen B. R. (2007) Cell 128, 71–83 [DOI] [PubMed] [Google Scholar]