

Abstract
The transporter associated with antigen processing (TAP) is an essential machine of the adaptive immune system that translocates antigenic peptides from the cytosol into the endoplasmic reticulum lumen for loading of major histocompatibility class I molecules. To examine this ABC transport complex in mechanistic detail, we have established, after extensive screening and optimization, the solubilization, purification, and reconstitution for TAP to preserve its function in each step. This allowed us to determine the substrate-binding stoichiometry of the TAP complex by fluorescence cross-correlation spectroscopy. In addition, the TAP complex shows strict coupling between peptide binding and ATP hydrolysis, revealing no basal ATPase activity in the absence of peptides. These results represent an optimal starting point for detailed mechanistic studies of the transport cycle of TAP by single molecule experiments to analyze single steps of peptide translocation and the stoichiometry between peptide transport and ATP hydrolysis.
Introduction
The transporter associated with antigen processing (TAP)5 has a key role in the adaptive immune system of higher vertebrates. TAP translocates proteasomal degradation products from the cytosol into the endoplasmic reticulum lumen, where these peptides can bind to major histocompatibility complex (MHC) class I molecules. Kinetically stable peptide-MHC complexes traffic to the cell surface, where they are scanned by cytotoxic T-lymphocytes. Recognition of an antigenic peptide in complex with MHC class I by a specific T-cell receptor induces the elimination of the infected or malignantly transformed cell. Viruses and tumor cells have therefore evolved strategies to shut down antigen presentation, for example by down-regulation or inhibition of TAP (for review see Refs. 1, 2).
TAP belongs to the family of ABC transporters, which translocate a very large range of solutes across membranes under consumption of ATP (3–5). TAP forms a heterodimer of the half-transporters TAP1 and TAP2. Each of the subunits is composed of a transmembrane domain (TMD) and a C-terminal cytosolic nucleotide-binding domain (NBD). Both TMDs together form the translocation pathway, with the peptide-binding site located at cytosolic loops (6). TAP shows a preference for protein fragments of 8–16 residues (7) with a specificity restricted to the three N-terminal and the C-terminal residues (8–10). Peptide binding is ATP-independent and follows a Langmuir (1:1) isotherm (7, 11). However, it is still an open issue whether only one or several peptides bind to TAP with high affinity at the same time. Peptide binding is a two-step process with a fast association followed by a slow structural rearrangement comprising 25% of TAP residues (11, 12).
Each NBD is divided into a helical and catalytic domain and contains the Walker A/B motifs and C-loop (ABC signature) sequence, all of which are involved in the binding and hydrolysis of ATP (13–16). From biochemical and structural studies with soluble NBDs as well as fully assembled ABC transporter complexes, it has become clear that ATP binding induces a rigid body movement of the helical domain toward the catalytic domain, which then allows dimerization of the NBDs in a head-to-tail orientation (17–19). Although ATP binding to TAP is peptide-independent (20), ATP hydrolysis is tightly coupled to peptide binding and translocation (21, 22). However, it is not solved yet whether TAP has a basal ATPase activity as found by other ABC transporters. A short stretch of the cytosolic loop 1 (CL1) of TAP1 is involved in the communication between the NBD and TMD as identified by a chemical protease (23). Interestingly, CL1 and CL2 of TAP1 interact with the X-loop of TAP2 as shown by cysteine cross-linking (24).
For advanced biochemical and biophysical analyses of the molecular mechanism of TAP, it is essential to work with purified protein. Therefore, we evolved in a detailed screen a solubilization, purification, and reconstitution procedure for heterologously expressed TAP. With this we addressed the peptide-binding stoichiometry and basal ATP hydrolysis of TAP.
EXPERIMENTAL PROCEDURES
Cloning and Expression of TAP
For expression of tap, Sf9 insect cells (Spodoptera frugiperda) were infected with baculoviruses containing Cys-less tap1 and wt tap2 (23) or de novo synthesized (GenScript, Piscataway, NJ) wt tap1 and wt tap2 with optimized codon usage for heterologous expression. For the de novo synthesized tap, a sequence coding for an XbaI site followed by a flexible linker (GGGSGGGS), a tobacco etch virus cleavage site (ENLYFQG), and an His10 tag was added to the 3′-end of the tap1 gene. On the 3′-end of the tap2 gene, a sequence coding for SphI site followed by a GGGSGGGS linker, a tobacco etch virus cleavage site, and an anti-PPE peptide (PRGPDRPEGIEE) was introduced (25). Codon-optimized tap1 and tap2 were cloned into EcoRI/HindIII sites and XhoI/KpnI sites of pFastBacDual vector (Invitrogen), respectively. tap1 was under the control of the polyhedrin promoter, and tap2 was under the control of the p10 promoter. To create a mutant unable to hydrolyze ATP, mutations in the Walker A sequence of TAP1 (K544A) and TAP2 (K509A) as well as a mutation in the H-loop of TAP2 (H661A) were introduced. These mutations were generated by ligase chain reaction using codon-optimized tap genes as templates. The following primers were used: tap1 (K544A), 5′-CCTAATGGTTCAGGAGCGTCTACAGTCGCCG-3′; tap2 (K509A), 5′-CCAAACGGGTCAGGAGCATCAACGGTAGCG-3′; and tap2 (H661A), 5′-GTTAGTCATTGCTGCTAGACTGCAGACCG-3′. Correct sequences were confirmed by DNA sequencing. Sf9 cells were infected with an multiplicity of infection of three for single infections and an multiplicity of infection of five for double infections at a cell density of 2.0 × 106 cells/ml. Expression and crude membrane and microsome preparations were performed as described previously (16, 23).
Purification of TAP
Crude membranes were thawed on ice and diluted to 5 mg/ml of total protein with solubilization buffer (20 mm HEPES, 140 mm NaCl, 15% glycerol, 5 mm histidine, 2.5 mm benzamidine, 1 mm phenylmethylsulfonyl fluoride, pH 7.4) containing the indicated detergent (TX-100, Sigma; FC-14 and DDM, Anatrace, Maumee, OH; and digitonin, Calbiochem, Darmstadt, Germany). Importantly, digitonin was prepared as a 20% stock solution in water by boiling for 10 min. After storage for 1 week at 4 °C, the digitonin solution was centrifuged for 10 min at 20,000 × g, and the supernatant was used for solubilization. After 90-min incubation on ice, insoluble material was removed by centrifugation at 100,000 × g for 30 min at 4 °C. The supernatant was applied with a flow rate of 0.5 ml/min to a HiTrap Zn2+-iminodiacetate-Sepharose column (Amersham Biosciences) equilibrated in washing buffer (20 mm HEPES, 140 mm NaCl, 15% glycerol, pH 7.4) containing detergent and 5 mm histidine. The column was washed subsequently with 10 column volumes of washing buffer containing the corresponding detergent and 20 mm or 40 mm histidine. TAP was eluted with washing buffer containing detergent and 200 mm histidine. Buffer exchange was performed by gel filtration (PD-10, Amersham Biosciences), if necessary.
Immunoprecipitation of TAP
150 μl of sheep anti-mouse IgG-conjugated magnetic beads (Dynabeads, Invitrogen, Karlsruhe, Germany) were coated with monoclonal α-TAP2 (435.3) or monoclonal HC10 α-MHC class I antibody for 2 h at 4 °C in 20 mm Tris, pH 7.4, 150 mm NaCl, 5 mm MgCl2, 0.1% bovine serum albumin. After washing the beads twice with 1 ml of IP buffer (20 mm HEPES, pH 7.4, 140 mm NaCl, 15% glycerol, 0.1% digitonin), 25 μg of purified TAP were incubated with the beads in a total volume of 500 μl for 3 h at 4 °C. After washing, beads were isolated by a magnet, and bound proteins were eluted with 30 μl of 3× SDS loading buffer. Eluates were separated by SDS-PAGE (10%), transferred to a nitrocellulose membrane, and visualized by immunodetection using monoclonal α-TAP1 (148.3) and α-TAP2 (435.3) antibodies. Band intensities were quantified by Lumi-Imager F1 (Roche Applied Science). TAP1 to TAP2 ratios were normalized to the subunit ratio in crude membranes.
Peptide Binding
Peptides were labeled by fluorophores or by iodine I125 (11, 22). For filter assays, crude membranes (25 μg of protein) and solubilized, purified, or reconstituted TAP were incubated with radiolabeled peptide RRYQKSTEL in 50 μl of solubilization buffer (with or without the corresponding detergent) for 15 min at 4 °C. Samples were transferred to micro filter plates preincubated with 0.3% polyethyleneimine (MultiScreen plates, Durapore membrane, 0.65-μm pore size for crude membranes and reconstituted TAP; glass fiber type B, 1-μm pore size for solubilized TAP, Millipore). Filters were washed twice with 100 μl of ice-cold solubilization buffer with or without the corresponding detergent. The amount of radiolabeled peptide was quantified by a γ-counter (Beckman Coulter). Background binding was determined in the presence of 200-fold excess of unlabeled peptide (RRYQKSTEL). The dissociation constant (KD) was derived from fitting a Langmuir (1:1) isotherm to the data,
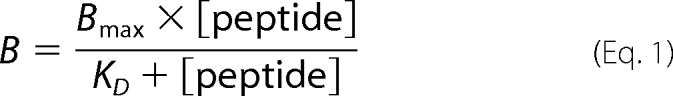 |
where B represents the fraction of peptide bound to TAP. Bmax is the amount of peptide bound under saturating concentrations.
For determination of peptide affinity by a homogenous assay, purified TAP (16 nm) in 200 μl of solubilization buffer supplemented by 0.1% digitonin was added to increasing concentrations of fluorescein-labeled peptide RRYC(F)KSTEL, and the decrease in fluorescence (λex/em = 470/515 nm) was monitored in real-time at 10 °C using a Fluorolog-3 spectrometer (Horiba Jobin Yvon, Munich, Germany). The peptide dissociation kinetics were followed after addition of a 400-fold molar excess of unlabeled RRYQKSTEL peptide. Time courses for association were fitted by a mono-exponential equation (Equation 2),
where Fmax represents the maximal change of fluorescence after reaching equilibrium, and kon is the association rate constant. To derive the peptide dissociation constant, the maximal change of fluorescence (Fmax) was plotted against the peptide concentration and fitted by a Langmuir (1:1) isotherm.
Fluorescence Correlation Spectroscopy
Fluorescence correlation spectroscopy was performed on a Zeiss ConfoCor 2 (Carl Zeiss, Oberkochen, Germany) equipped with an argon laser (488 nm and 25 milliwatts) and a helium-neon laser (633 nm and 5 milliwatts) using a C-Apochromat 40×, 1.2 water immersion objective. Auto- and cross-correlation curves were analyzed with the ConfoCor 2 software. The pinholes in channel 1 (633 nm and 90-μm diameter) and 2 (488 nm and 70-μm diameter) were adjusted with Cy5 and Alexa-Fluor-488-C5-maleimide, respectively. Auto- and cross-correlation curves were fitted with a one- or two-component model,
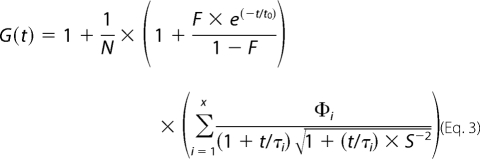 |
where x denotes the number of components (i = 1 or 2), N is the total number of particles in the confocal volume, t is the correlation time, τi is the diffusion time of component i in the confocal volume, F is the triplet fraction, t0 is the triplet time, Φi is the weighting coefficient for the ith component, and S is the structural parameter. The axial structural parameter S is introduced for convenience as S = ωz/ωxy, where ωxy is the half short axis (waist radius of the laser beam) and ωz is the half long axis of the ellipsoidal confocal volume. The structural parameter was determined as 5.8 and 9.0 for the 488 nm laser line and for the 633 nm laser line, respectively. The particle diffusion time τ through the confocal volume can be described as,
 |
where D is the diffusion coefficient. The confocal volumes V were calculated from one-component fits of the respective correlation curves using a diffusion correlation coefficient of 430 μm2/s for Alexa-488-C5 maleimide (26) and 370 μm2/s for Cy5 (27) in water applying Equation 5.
Although the refractive index changes upon addition of glycerol, the confocal volume remains constant (28). Therefore, no calibration methods were applied. The viscosity of the digitonin buffer (washing buffer with 0.1% digitonin) was calculated from the change of diffusion coefficient of free dyes in water and digitonin buffer with Equation 6,
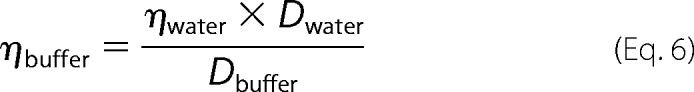 |
where the viscosity, η, of water at 20 °C is 1.003 × 103 Pa s. The hydrodynamic radii RH were determined from the Stokes-Einstein relation, in which RH is inversely proportional to the diffusion coefficient,
 |
where kB is the Boltzmann constant and T the absolute temperature. The translational frictional ratio f represents the ratio of the experimental determined (RH) and the theoretical hydrodynamic (RS) radii,
 |
with,
 |
where M is the molecular mass, NA is the Avogadro constant, and the partial specific volume υ is taken as 0.73 ml/g.
First, the diffusion times of Alexa-488-C5-maleimide, Cy5, Alexa-488-labeled (RRYCA488KSTEL), and Alexa-633-labeled peptide (RRYCA633KSTEL) were determined in water as well as in digitonin buffer by fitting the auto-correlation curves with a one-component model. To determine peptide affinity by fluorescence auto-correlation spectroscopy, increasing TAP concentrations were incubated with Alexa-488-labeled or Alexa-633-labeled peptide (2.5 nm) for 30 min on ice to allow equilibration. After 5 min at room temperature, fluorescence fluctuations were recorded for 10 s and auto-correlation curves were calculated, averaged over 10 repeats, and fitted by a two-component model with the fixed fast diffusion time of free peptide determined separately.
For cross-correlation experiments, increasing TAP concentrations (up to 100 nm) were incubated with Alexa-488-labeled peptide (2.5 nm) and Alexa-633-labeled peptide (2.5 nm). The red laser was adjusted to a higher intensity than the green one, thereby minimizing the crosstalk of the green emission into the red channel. The cross-correlation curve was fitted with a one-component model (Equation 3). The auto-correlation data obtained in the green and red channels were fitted with a two-component model assuming fractions of free and bound substrates. The diffusion times of the free substrates were derived from one-component fits of an analogous experiment, where the Alexa-488 and the Alexa-633-labeled peptide (2.5 nm each) were monitored in both channels without TAP. The higher detectable concentration of the free green substrate in comparison to the free red substrate is due to the higher quantum yield of the green dye.
ATPase Assay
To determine the ATPase activity of solubilized TAP, a colorimetric assay, based on malachite green, was used (29). TAP (1.9 μg) was incubated for 1 h at 32 °C in 25 μl of ATPase buffer (digitonin buffer supplemented with 2 mm MgCl2, 1 mm ouabain, 5 mm NaN3, and 50 μm EGTA) containing 1 mm ATP, in the absence or presence of 2 μm high affinity peptide RRYQKSTEL or non-binder peptide EPGNTWDED (10). To inhibit ATPase activity of TAP, ortho-vanadate (2 mm) was added. The reaction was stopped by the addition of 20 mm H2SO4 (175 μl) and incubated with 50 μl of malachite green solution (3.5 mm malachite green, 0.2% Tween 20, 0.8% ammonium molybdate in 20% H2SO4) for 15 min at room temperature. The amount of inorganic phosphate was determined by the absorbance at 620 nm compared with a standard curve using sodium phosphate. To determine ATP hydrolysis kinetics in the dependence of the ATP or peptide concentration, 0.6 μg of TAP purified or reconstituted in proteoliposomes was incubated for 10 min at 37 °C in 25 μl of ATPase buffer or ATPase buffer without digitonin, respectively, containing ATP in the presence or absence of high affinity peptide RRYQKSTEL peptide as indicated. Peptide-stimulated phosphate released was determined and fitted by using the Michaelis-Menten equation,
 |
with L representing ATP or peptide for determination of the Michaelis-Menten constant Km,ATP or Km,peptide, respectively.
Reconstitution of the TAP Complex
For reconstitution of TAP, a polar lipid extract of Escherichia coli (Avanti Polar Lipids, Alabaster, AL, 20 mg/ml) was dissolved in chloroform and evaporated. The lipid film was rehydrated in reconstitution buffer (20 mm HEPES, pH 7.4, 140 mm NaCl, 5% (w/w) glycerol) by bath sonification. Liposomes (10 mg/ml) were frozen in liquid N2 and thawed at room temperature for three times, and, subsequently, they were extruded 11 times through a polycarbonate filter of a pore size of 400 nm (Avestin, Mannheim, Germany). Liposomes were diluted into reconstitution buffer to a final concentration of 2.5 mg/ml and then destabilized by adding TX-100 to maximal light scattering at 540 nm. For reconstitution, purified TAP complex was added to destabilized liposomes in a lipid-to-protein ratio of 20:1 (w/w) and incubated for 30 min at 4 °C. To remove detergent, aliquots of 40 mg/ml (wet weight) of polystyrene beads (Bio-Beads SM-2, Bio-Rad), washed with methanol and water and then equilibrated in reconstitution buffer, were added sequentially four times and incubated each time for at least 1 h at 4 °C by gentle agitation. Bio-Beads were removed by using a Bio-spin column (Bio-Rad). The proteoliposomes were pelleted by centrifugation at 300,000 × g for 45 min at 4 °C. Finally, the pellet was resuspended in reconstitution buffer to a lipid concentration of 5–10 mg/ml.
Peptide Transport Assay with Proteoliposomes
TAP (0.75 μg) containing proteoliposomes (46 μg of phospholipids) was preincubated at 4 °C in 50 μl of phosphate-buffered saline containing 5 mm MgCl2 and additives as mentioned. Transport was started by adding 1 μm of RRYC(F)KSTEL peptide. After incubation for 15 min at 32 °C, transport was stopped by the addition of 200 μl of ice-cold stopping buffer (10 mm EDTA in phosphate-buffered saline) containing 100 μm unlabeled peptide RRYQKSTEL to compete for peptides bound to TAP. Proteoliposomes were transferred to micro filter plates preincubated with 0.3% polyethyleneimine (MultiScreen plates, Durapore membrane, 0.65-μm pore size, Millipore). Samples were washed twice with 250 μl of ice-cold stopping buffer. Subsequently, filters were incubated with 250 μl of elution buffer (1% SDS in phosphate-buffered saline) for 5 min at room temperature. The transported peptide was quantified by a fluorescence plate reader (λex/em = 485/520 nm, Polarstar Galaxy, BMG, Offenburg, Germany).
RESULTS
Functional Purification of TAP
A prerequisite for a detailed mechanistic analysis of membrane proteins is their functional isolation. Although it is often laborious to establish a mild purification method for soluble proteins, this process is still a major challenge for membrane proteins and in particular for heteromeric membrane protein complexes, such as TAP. Here the choice of the optimal detergent is very critical. In an attempt to identify optimal solubilization conditions for TAP in terms of yield and long term stability, we performed a solubilization screen of 80 different detergents with TAP-containing microsomes from insect cells (supplemental Table S1). In initial screens, a glycerol concentration of 15% and a microsomal lipid concentration of 3 mg/ml, assuming a 1:1 lipid-to-protein ratio (w/w), were determined to be optimal. For all detergents, a ρ-value of two was taken. To determine solubilization efficiency and long term stability over 3 days at 4 °C, solubilized TAP was centrifuged at 100,000 × g for 30 min, and the supernatant was analyzed by immunoblotting using TAP1- and TAP2-specific antibodies. From these studies, FC-14, TX-100, DDM, and digitonin showed appropriate efficiencies in TAP solubilization and kept the peptide transporter stable in solution over 3 days (supplemental Table S1).
We next compared these four detergents according to their ability to preserve activity during purification. TAP-containing membranes were solubilized at a ρ-value of one in the case of FC-14, DDM, and digitonin or a ρ-value of two for TX-100. The solubilization efficiencies ranged between 64 and 96% compared with SDS-solubilized TAP (Table 1). Solubilized TAP was purified by metal affinity chromatography via a His10 tag at the C terminus of TAP1. Instead of imidazole, which disrupted the heterodimeric complex during extensive washing steps, histidine was used in all washing and elution buffers. In addition, the detergent concentration during chromatography was reduced to a concentration slightly above the critical micelle concentration (1.5- to 6.0-fold above the critical micelle concentration). The purity of TAP ranged between 80 and 95% (Fig. 1A).
TABLE 1.
Efficiency of detergents in solubilization und purification
| TAP1/TAP2 solubilization efficiencya | TAP1/TAP2 recovery after purificationa | TAP2/TAP1 ratio after purificationb | |
|---|---|---|---|
| % | % | ||
| FC-14 | 75/95 | 30/1 | 0.1 |
| DDM | 88/94 | 68/69 | 1.0 |
| TX-100 | 69/74 | 69/75 | 1.0 |
| Digitonin | 77/96 | 36/31 | 1.0 |
a To determine the efficiency in solubilization and recovery of TAP, the amount of TAP in crude membranes was compared with TAP after solubilization and purification by densitometry of immunoblots using monoclonal α-TAP1 (148.3) and α-TAP2 (435.3) antibodies.
b The molar ratio of TAP1 and TAP2 in crude membranes was taken as 1.
FIGURE 1.

Purification of TAP. A, purification of TAP with different detergents. TAP-containing membranes (5 mg/ml of protein) were solubilized by different detergents and purified by metal affinity chromatography. Aliquots of membranes (M), solubilization (S), flow through (FT), wash with 20 mm (W1), wash with 40 mm histidine (W2), and elution with 200 mm of histidine (E) were analyzed by SDS-PAGE (9%, Coomassie-stained) and immunoblotting using monoclonal α-TAP1 (148.3) and α-TAP2 (435.3) antibodies. B, stoichiometry of TAP. TAP-containing membranes (10 mg/ml protein) were solubilized by 2% digitonin, purified by the His10 tag at the C terminus of TAP1 via metal affinity chromatography, and subsequently immunoprecipitated via monoclonal α-TAP2 (435.3) antibody to isolate exclusively heterodimeric TAP complexes or α-MHC I (HC10) antibody as isotope control. Aliquots of membranes (M), solubilization (S), metal affinity-purified TAP (P), and immunoprecipitated TAP (IP) were analyzed by SDS-PAGE (10%) and immunoblotting using monoclonal α-TAP1 (148.3) and α-TAP2 (435.3) antibodies. The ratio of the TAP1 and TAP2 intensities in each fraction was normalized to the membrane level. The ratios depict the mean of three independent experiments.
To determine the subunit stoichiometry after metal affinity chromatography via the His10 tag at the C terminus of TAP1, the eluate of digitonin-solubilized TAP was immunoprecipitated by a monoclonal α-TAP2 antibody. The relative amount of TAP1 and TAP2 during expression, solubilization, purification, and orthogonal immunoprecipitation was compared by quantitative immunoblotting using monoclonal α-TAP1 and α-TAP2 antibodies. Because the TAP1 and TAP2 ratios of the metal affinity purification (0.79 ± 0.05) and the orthogonal immunoprecipitation (0.82 ± 0.06) were identical, all purified TAP complexes exist as heterodimers, and no monomers or homodimers are present after purification (Fig. 1B). The same ratio of both subunits was detected also in the solubilized state (0.80 ± 0.06). However, in crude membranes TAP1 is in a surplus compared with TAP2. Therefore, the single TAP1 subunit seems not to be solubilized under these conditions.
Apart from digitonin, TAP1 and TAP2 were co-purified in TX-100 and DDM in 1:1 stoichiometry. For DDM and TX-100, the TAP recovery was 2-fold higher than for digitonin (Table 1). Importantly, FC-14 dissociated the TAP complex, as only TAP1 was eluted from the column while TAP2 was predominantly found in the flow-through.
Because detergents can inactivate membrane proteins without affecting their solubility, the activity of TAP during purification with different detergents was analyzed by peptide binding studies using a filter assay. TAP function was determined in the presence of the radiolabeled peptide RRYQKSTEL (300 nm). This concentration is slightly below the dissociation constant determined in membranes (KD = 0.6 μm). For all four detergents, TAP activity was reduced during solubilization and purification (Fig. 2A). Solubilization by FC-14 and TX-100 reduced TAP activity almost to 0%, although ∼75% of TAP was solubilized. For DDM or digitonin, 65 and 40% of TAP-binding activity was preserved, respectively, whereas 77 to 88% of TAP was solubilized. After purification in DDM, the TAP activity dropped down to 1%, although 70% of TAP was isolated as determined by immunoblotting. After purification in digitonin, 10% of TAP activity was detected while 30% of TAP was isolated. Notably, the recovery of functional purified TAP is underestimated, because solubilized TAP has a slightly decreased peptide affinity and retention by the filter assay of TAP in crude membranes was higher than that of TAP in detergent solution.
FIGURE 2.

Activity of solubilized TAP. A, stability of TAP during purification. To determine the stability of the TAP complex during purification, crude membranes derived from 1 liter of insect cell culture were solubilized by different detergents and purified by metal affinity chromatography. Subsequently, fractions of crude membranes (black bars), after solubilization (dark gray bars) and purified TAP (light gray bars) containing nominal equal amounts of TAP were incubated with radiolabeled peptide RRYQKSTEL (300 nm) in the presence or absence of 200-fold excess of unlabeled peptide. After 15-min incubation at 4 °C, bound peptides were separated by filter assay and quantified by γ-counting. Specific binding to TAP in crude membranes was normalized to 100%. B, long term stability of TAP. To compare the stability and TAP activity over time, TAP purified in DDM (filled circles) or digitonin (open circles) was stored at 4 °C. Peptide binding was analyzed as described in A. Specific peptide binding to TAP at day zero was normalized to 100%. In digitonin, TAP was active with a half-life longer than 16 days, whereas in DDM the half-life was only 10.5 h. Each data point is derived from triplicate measurements. Error bars show the standard deviation.
For reconstitution or crystallization, long term stability of the purified protein is essential. We therefore followed the activity of TAP, which was purified in DDM or digitonin, over 9 days (Fig. 2B). Purified TAP was stored in the corresponding elution buffer at 4 °C, and peptide binding was analyzed over time. TAP activity was fully preserved over the first 3 days in digitonin and after 9 days still 70% of TAP activity was detected. In DDM, however, the activity of TAP dropped to <20% within the first 24 h. No peptide binding was detectable after 9 days. Importantly, the inactivation did not correlate with protein aggregation, because TAP stayed in solution over the entire period (data not shown). Thus, caution must be taken to probe the stability of membrane proteins solely by aggregation screens, because an inactivation of the protein may not be detected. In conclusion, digitonin was the only detergent under the conditions followed that resulted in sufficient amounts of functional TAP for further biochemical and biophysical analysis.
Peptide Binding to Isolated TAP
Peptide binding to TAP is a prerequisite for transport. We therefore analyzed peptide binding of purified TAP by different techniques. We compared these results to membrane-bound TAP to ensure that solubilization and purification did not alter TAP activity in subtle ways. First, peptide affinity of TAP was determined by a filter binding assay using the radiolabeled epitope RRYQKSTEL. Membrane-embedded TAP had a 2-fold higher peptide affinity (KD = 0.6 μm) in comparison to solubilized TAP or solubilized and purified TAP (KD = 1.3 μm) (Table 2). Because filter binding assays can suffer from dissimilar recovery of membrane-bound proteins versus solubilized proteins, as well as non-equilibrium conditions during the washing steps, TAP affinity was also determined under equilibrium conditions using a homogeneous binding assay, where the association of the fluorescein-labeled peptide RRYC(F)KSTEL was followed by fluorescence quenching (supplemental Fig. 1). Peptide binding resulted in a monoexponential fluorescence decay, which was recovered by the addition of a 400-fold excess of unlabeled competitor peptide RRYQKSTEL. Fitting the concentration-dependent maximal change in fluorescence by a Langmuir (1:1) isotherm resulted in a dissociation constant of 30 nm, which is again 2-fold higher than the KD value of membrane-bound TAP determined by this method (KD = 12 nm (11)). The different affinities derived from the filter binding and fluorescence quench assays were not dependent on the differently labeled peptides, because the radiolabeled and fluorescein-labeled peptides have the same affinity in the filter binding assay (data not shown). Therefore, the differing affinities most likely result from the different assays.
TABLE 2.
Peptide affinity of TAP
| TAP complex | Filter binding KD | Fluorescence quenching KD |
|---|---|---|
| μm | nm | |
| Membranes | 0.6 ± 0.2 | 12.0 ± 1.0a |
| Solubilized | 1.4 ± 0.4 | NDb |
| Purified | 1.3 ± 0.3 | 30.0 ± 5.0 |
| Reconstituted | 0.3 ± 0.1 | ND |
a Taken from Neumann and Tampé (11).
b ND, not determined.
Because P-glycoprotein (MDR1, ABCB1) possesses multiple solute-binding sites, the peptide-TAP stoichiometry represents an important unresolved issue. Equilibrium dialysis or isothermal titration calorimetry would be the method of choice to analyze the stoichiometry. However, these methods require large amounts of stable protein and are critically dependent on an accurate determination of the protein concentration, which is difficult to achieve for membrane proteins. We therefore applied fluorescence cross-correlation spectroscopy, which requires very low amounts of protein. We first analyzed the association of the photostable Alexa-488- or Alexa-633-labeled peptide RRYCKSTEL by fluorescence auto-correlation spectroscopy. The diffusion coefficient of both peptides alone and with increasing concentrations of TAP was determined. In the absence of TAP, only one species of fast diffusing particles was detected with a diffusion coefficient of 104 and 103 μm2/s for Alexa-488- and Alexa-633-labeled peptide, respectively (Fig. 3A). Due to the higher viscosity of the glycerol-containing buffer, the diffusion coefficients of both peptides were 2.2-fold lower than in water. By adding TAP (320 nm final concentration), a second population with a 10-fold slower diffusion coefficient (10 μm2/s) became apparent, reflecting fluorescent peptides bound to TAP (Fig. 3B). A 200-fold excess of unlabeled peptide RRYQKSTEL was efficient in competition, demonstrating that the slowly diffusing population is TAP-specific. The slow component increased hyperbolically with raising TAP concentrations and followed a Langmuir (1:1) isotherm with dissociation constants of 20 ± 9 nm and 30 ± 9 nm for the Alexa-488- and Alexa-633-peptide, respectively. These affinity constants are close to those determined by fluorescence quenching (Fig. 3C). At TAP concentrations 10-fold higher than the dissociation constant, only 50% of peptide was bound to TAP, although saturation was reached. The reason for this effect could not be determined. However, uncoupled fluorophores or aberrant by-products do not cause this effect, because the labeled peptides were identified as a single population by reversed-phase high-performance liquid chromatography and by electrospray ionization-mass spectrometry.
FIGURE 3.

Peptide-binding affinity determined by fluorescence correlation spectroscopy. A, fluorescence auto-correlation of free peptide. Auto-correlation data (gray line) obtained for Alexa-633-peptide RRYCKSTEL (2.5 nm) were fitted by a one-component fit (Equation 3, black line) taking a fraction of the triplet state into account (τtriplet = 4.4 μs). A diffusion time (τfree) of 200 μs resulted from the fit. B, fluorescence auto-correlation of TAP-bound peptides. 2.5 nm of the Alexa-633-peptide RRYCKSTEL was incubated with 320 nm of solubilized and purified TAP for 30 min at 4 °C. The fluorescence correlation spectroscopy measurements were performed at 22 °C. Auto-correlation data (gray line) were fitted by a two-component model assuming fractions of free and bound peptides (Equation 3, black line) taking a fraction of the triplet state into account (τtriplet = 5.7 μs). The diffusion time for free peptide (τfree) was fixed to 200 μs, resulting in a diffusion time of bound peptide (τbound) of 1953 μs. The lower graph displays the residuals of the fit. C, Alexa-488- or Alexa-633-peptides RRYCKSTEL (2.5 nm of each) were incubated with increasing concentrations of TAP. Auto-correlation curves were fitted by a two-component model assuming fractions of free and bound peptides. The fraction of bound peptide (%) was plotted against the TAP concentration and fitted by a Langmuir (1:1) isotherm. For the Alexa-488 peptide, and the Alexa-633 peptide a KD of 20 ± 9 nm (open circles) and 30 ± 6 nm (closed circles) was deduced, respectively.
From the diffusion coefficient and the viscosity of the buffer, a hydrodynamic radius (RH) of 81 Å was determined for the TAP-peptide complex (Table 3). Based on the size of TAP, a theoretical hydrodynamic radius (RS) of 35.1 Å was calculated assuming a spherical molecule. A translational frictional ratio of 2.3 implies that TAP has a non-spherical structure or possesses an abnormally large water and probably detergent shell.
TABLE 3.
Parameters derived from fluorescence auto-correlation spectroscopy
| Excitation wavelength | 488 nm | 633 nm |
| Dye | Alexa-488 | Alexa-633 |
| τdye water/buffer (μs) | 35/86 | 53/109 |
| Ddye water/buffer (μm2/s) | 430a/175 | 370b/180 |
| Confocal volume (fl) | 0.5 | 1.1 |
| Viscosity of buffer 20 °C (*103 N*s/m2) | 2.5 | 2.2 |
| Structure parameter, S | 5.8 | 9 |
| τpeptide water/buffer (μs) | 50/120 | 94/190 |
| Dpeptide water/buffer (μm2/s) | 250/104 | 209/103 |
| Confocal volume (fl) | 0.5 | 1.1 |
| Particle number | 6.3 ± 0.5 | 7.4 ± 3.6 |
| Peptide concentration (nm) | 21.8 | 11.2 |
| RH peptide (Å) | 8.4 | 7.0 |
| τpeptide TAP (μs) | 1500 | 2000 |
| Dpeptide TAP (μm2/s) | 10 | 10 |
| RH (Å) | 87.6 | 73.7 |
| RS (Å) | 35.1 | 35.1 |
| RH/RS | 2.5 | 2.1 |
We next examined the association of two peptide molecules to TAP by fluorescence cross-correlation spectroscopy. Notably, Alexa-488 and Alexa-633 are optimal fluorophores for cross-correlation studies with high quantum yield and good photo-stability. These fluorophores are also insensitive to environmental changes, and their emission spectra are well separated to exclude any fluorescence resonance energy transfer and cross-talk. To follow fluorescence cross-correlation, we incubated a mixture of Alexa-488- and Alexa-633-peptide (2.5 nm of each) with different concentrations of TAP (up to 100 nm). Under all conditions tested, a cross-correlation could not be detected. So it is unlikely that TAP is loaded with two peptides under conditions, in which TAP is in 10-fold excess. Therefore, we preincubated TAP with a 200-fold excess of labeled peptides. TAP-bound peptide was separated from free peptide by gel filtration using mini-spin columns. However, even after this rapid separation (<1 min), no cross-correlation signal could be detected. Furthermore, we could not detect any fluorescence resonance energy transfer signal in studies using peptides labeled with fluorescence resonance energy transfer pairs (data not shown). In conclusion, TAP binds only one peptide with high affinity at a time.
Peptide Binding Induces ATP Hydrolysis of the Isolated TAP Complex
In their solubilized state, ABC transporters often show an uncoupling between solute binding and ATP hydrolysis, most probably due to a lack of an optimal lipid environment. We therefore examined the ATPase activity of solubilized and purified TAP. In the presence of a saturating concentration of the peptide RRYQKSTEL (2 μm), TAP showed a 6-fold higher ATPase activity than in its absence (Fig. 4). Notably, the non-binder peptide EPGNTWDED did not stimulate the ATP hydrolysis of TAP, whereas ortho-vanadate inhibited the peptide-stimulated ATPase activity to background levels. To prove whether the observed peptide-dependent ATPase activity is TAP-specific, we created an ATPase-inactive mutant of TAP by replacing the conserved lysine in the P-loop of TAP1 and TAP2 as well as the invariant histidine of the H-loop in TAP2 by alanine. This triple mutant was inactive in peptide transport and showed no peptide-stimulated ATPase activity, although it has the same peptide-binding affinity as wild-type TAP (data not shown). Therefore, the observed peptide-stimulated ATPase activity is TAP-dependent. Strikingly, TAP does not hydrolyze ATP in the absence of peptide. The very low background ATPase activity originates from a co-purified ATP-hydrolyzing enzyme, because a similar background activity was found for the inactive TAP mutant. In conclusion, the solubilized and purified TAP complex shows an ATPase activity that is strictly coupled to peptide binding.
FIGURE 4.

Peptide binding induces ATP hydrolysis of the solubilized TAP complex. 500 nm of purified wt (filled bars) or inactive TAP variant (TAP1 K544A/TAP2 K509A, H661A) (open bars) were incubated with 1 mm MgATP in the presence or absence of high affinity peptide (2 μm RRYQKSTEL), low affinity peptide (2 μm EPGNTWDED), or ortho-vanadate (2 mm) in combination with high peptide (2 μm) for 1 h at 32 °C. Release of inorganic phosphate was quantified by the malachite green assay and normalized to the TAP-dependent ATPase activity in the presence of RRYQKSTEL. Data are derived from triplicate measurements. Error bars show the standard deviation.
We next determined the kinetic parameters of purified TAP by measuring ATP hydrolysis in dependence of peptide or ATP (Table 4). In the presence of a saturating concentration of RRYQKSTEL peptide (1 μm), ATP hydrolysis showed a hyperbolic dependence on the ATP concentration with a Michaelis-Menten constant (Km,ATP) of 152 ± 30 μm (supplemental Fig. 2). Under a saturating ATP concentration (3 mm), the ATPase activity was hyperbolically dependent on the peptide concentration with a Michaelis-Menten constant (Km,peptide) of 131 ± 62 nm. This good agreement of Km,peptide with the peptide affinity determined under equilibrium conditions suggests a tight coupling between peptide binding and ATP hydrolysis and that each peptide, bound to TAP, induces ATP hydrolysis. In summary, the Michaelis-Menten parameter Km,peptide directly reflects the affinity for peptide binding.
TABLE 4.
Kinetic parameters of TAP
| TAP complex | Km,ATP | Km,peptide | Vmax |
|---|---|---|---|
| μm | nm | μmol/min*mg TAP | |
| Purified | 152 ± 30 | 131 ± 62 | 1.40 ± 0.03 |
| Reconstituted | 94 ± 20 | 63 ± 33 | 1.92 ± 0.02 |
Reconstitution of TAP
Up to now, detailed mechanistic studies have been hampered by the lack of functionally reconstituted, highly purified TAP complexes. With isolated TAP that is functional and stable in detergent over days, we have established an optimal starting point for the functional reconstitution. TX-100-destabilized liposomes of polar E. coli lipids were mixed with TAP in a lipid-to-protein ratio of 20:1 (w/w). Subsequently, the detergent was slowly removed by adding polystyrene beads. As a result, reconstituted TAP was fully functional in respect of peptide binding, because the binding affinity was identical to TAP in microsomes (Table 2). Strikingly, for the first time the transport activity of purified TAP was recovered in proteoliposomes (Fig. 5). Peptide transport was not detected in the absence of ATP or if the ATPase-deficient TAP mutant was analyzed. In addition, transport was blocked by ortho-vanadate. Therefore, peptide transport is ATP-specific. Moreover, transport of fluorescence peptide is blocked to background by an excess of unlabeled peptide or the viral inhibitor ICP47, demonstrating that transport is both peptide- and TAP-specific.
FIGURE 5.

Functional reconstitution of TAP in proteoliposomes. Proteoliposomes (46 μg of E. coli lipids) containing wt (filled bars) or inactive TAP (open bars; 0.75 μg of each) were incubated in 50 μl with 1 μm RRYC(F)KSTEL in the presence of MgATP or MgADP (3 mm of each). The assay was performed in the presence of unlabeled competitor peptide (200 μm RRYQKSTEL), the viral TAP inhibitor ICP47 (30 μm), or ortho-vanadate (3 mm) as indicated. Data are derived from triplicate measurements. Error bars show the standard deviation.
The kinetic parameters of reconstituted TAP were determined for comparison in the same way as for solubilized TAP. The ATP as well as the peptide concentration-dependent ATP hydrolysis showed a hyperbolic behavior with Km,ATP of 94 ± 20 μm and Km,peptide of 63 ± 33 nm, which are slightly decreased in comparison to purified TAP (Table 4 and supplemental Fig. 2). Taken together, this is the first report of the functional reconstitution of highly purified TAP.
DISCUSSION
A detailed mechanistic analysis of any protein requires its functional purification and, in the case of membrane proteins, its reconstitution into lipid bilayers. By carefully evaluating each step of solubilization, purification, and reconstitution, we succeeded in the functional reconstitution of purified TAP. Importantly, the activity of solubilized TAP could be checked in every step and lead to important results: (i) Peptide binding triggers ATP hydrolysis of TAP in the solubilized state. Notably, in contrast to other ABC transporters, TAP does not possess a basal ATPase activity. (ii) By single molecule analyses, we further demonstrated that TAP binds only one peptide at a time.
An essential step in the purification of membrane proteins is their functional solubilization. By testing 80 different detergents for solubilization efficiency and long term stability, four detergents yielded good results. After purification, only digitonin could fully preserve TAP activity. In FC-14, the heterodimeric TAP complex was disrupted as also reported for other oligomeric membrane proteins (30). TAP inactivation by TX-100 and DDM was most probably due to delipidation, whereas digitonin is a very mild detergent often used to preserve protein-protein interactions within membranes. Most likely, digitonin maintains a belt of lipids around the protein, thus retaining the TAP function. Three parameters were critical for the purification: First, using Zn2+ instead of Ni2+ as the central cation strongly improved the purity of TAP. Second, in an initial screen, the presence of 15% glycerol was essential for long term stability. Third, TAP purification was performed in the presence of histidine instead of imidazole, because prolonged incubation with imidazole disrupted the heterodimeric complex as also shown for the bacterial oligopeptide transporter OppA (31).
Solubilized and purified TAP was fully functional with respect to peptide binding and ATP hydrolysis. However, the maximal ATP turnover rate was decreased in the purified state in comparison to reconstituted TAP. This change can be explained by a lower lateral membrane pressure in the solubilized state, affecting the spatial packing of the transmembrane helices (32, 33). Interestingly, the peptide-binding affinity and Km,peptide of purified TAP decreased 2- to 3-fold in comparison to membrane-embedded TAP. This difference may be due to a local enrichment of positively charged peptides at the negatively charged lipid interface. An even stronger effect was observed for the herpesviral inhibitor ICP47, which showed a 50-fold reduced affinity for solubilized as for membrane-embedded TAP (34).
Based on the diffusion coefficient determined by fluorescence correlation spectroscopy, the hydrodynamic radius (RH) of peptide-bound and, therefore, active TAP was calculated to be 81 Å corresponding to a molecular mass of 1.8 MDa assuming a spherical molecule. Based on the calculated molecular mass of TAP (156 kDa), the theoretical Stokes radius (RS) was 38.6 Å taking an additional water shell of 3 Å into account. This discrepancy does not originate from higher oligomeric states, because only one slow diffusing population of TAP-peptide complexes was detected. Furthermore, TAP oligomers should lead to a cross-correlation signal, which was not detected under any of the conditions tested. Therefore, the apparent high molecular mass most likely is caused by bound digitonin and lipids, which has been found to account for 80% of the mass of the yeast mitochondrial ADP/ATP exchanger (35). If this also applies for TAP, the frictional ratio of TAP is 1.22, which is within the range of 1.2–1.3 commonly found for globular species. A similar value (f = 1.18) was determined for the ABC transporter BmrA determined by sedimentation centrifugation and size-exclusion chromatography (36). The frictional ratio of Sav1866 (f = 1.35) determined from the x-ray structure by the program HYDRO is somewhat higher than the one estimated from TAP (37, 38). The more spherical structure of TAP could result from the two extra N-terminal domains (TMD0) linked to the core-TAP complex (39).
We previously succeeded in the reconstitution of enriched TAP derived from the human B-lymphoma cell line Raji (21). The kinetic parameters of reconstituted TAP derived from insect cells (Table 4) and of reconstituted TAP isolated from Raji cells (Km,peptide = 161 ± 15 nm; Km,ATP 0.3 ± 0.06 mm; Vmax = 2.02 ± 0.13 μmol/min × mg of TAP) are in good agreement showing that the different expression hosts, purification procedures, and lipid compositions of the proteoliposomes do not influence the activity of this translocation machinery. However, Raji cells would never provide a sufficient amount of purified TAP for detailed biochemical and biophysical studies. This presses the need for a heterologous expression system. However, the method originally developed for Raji cells did not work for TAP expressed in insect cells. Thus we invested another 5 years of research to succeed in the functional reconstitution of heterologously expressed and highly purified TAP. The most obvious difference between both strategies was the use of different detergents as well as different purification protocols. Combining our observations from extensive screens, we believe that TAP from insect cells is faster deactivated by delipidation during solubilization and purification than TAP from Raji cells, most likely because of the different lipid composition of the endoplasmic reticulum of insect cells and of Raji cells. Therefore, only the mild detergent digitonin can be used for the functional solubilization and reconstitution of heterologously expressed TAP by preserving the lipid environment around the solubilized protein.
The new established purification and reconstitution procedure presented herein gave us for the first time the possibility of deciphering the peptide stoichiometry as well as the basal ATPase activity of TAP. In future, we will dissect the single steps of the transport cycle by single molecule analysis and analyze mechanistic details of peptide translocation in the reconstituted system.
Supplementary Material
Acknowledgments
We thank Gudrun Illig and Eckhard Linker for excellent technical assistance. We thank Drs. Ute Scheffer (Goethe-University Frankfurt), Claus Seidel, and Suren Felekyan (University of Düsseldorf) for helpful advice and discussion in fluorescence correlation spectroscopy. We thank Christine Le Gal, Katrin Schulze, and Dr. David Parcej for helpful discussions and for preparation of the manuscript.
This work was supported by German Research Foundation Grant SFB 807 (to R. T.) and Grant AB149/1-1 (to R. A.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Table S1, and Figs. 1 and 2.
- TAP
- transporter associated with antigen processing
- ABC
- ATP-binding cassette
- DDM
- n-dodecyl-β-d-maltoside
- FC-14
- Fos-choline-14
- NBD
- nucleotide-binding domain
- MHC
- major histocompatibility complex
- TMD
- transmembrane domain
- TX-100
- Triton X-100
- wt
- wild type
- CL1,-2
- cytosolic loops 1 and 2.
REFERENCES
- 1.Schölz C., Tampé R. (2005) J. Bioenerg. Biomembr. 37, 509–515 [DOI] [PubMed] [Google Scholar]
- 2.Groothuis T. A., Griekspoor A. C., Neijssen J. J., Herberts C. A., Neefjes J. J. (2005) Immunol. Rev. 207, 60–76 [DOI] [PubMed] [Google Scholar]
- 3.Schmitt L., Tampé R. (2002) Curr. Opin. Struct. Biol. 12, 754–760 [DOI] [PubMed] [Google Scholar]
- 4.Davidson A. L., Dassa E., Orelle C., Chen J. (2008) Microbiol. Mol. Biol. Rev. 72, 317–364, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollenstein K., Dawson R. J., Locher K. P. (2007) Curr. Opin. Struct. Biol. 17, 412–418 [DOI] [PubMed] [Google Scholar]
- 6.Nijenhuis M., Hämmerling G. J. (1996) J. Immunol. 157, 5467–5477 [PubMed] [Google Scholar]
- 7.van Endert P. M., Tampé R., Meyer T. H., Tisch R., Bach J. F., McDevitt H. O. (1994) Immunity 1, 491–500 [DOI] [PubMed] [Google Scholar]
- 8.Heemels M. T., Schumacher T. N., Wonigeit K., Ploegh H. L. (1993) Science 262, 2059–2063 [DOI] [PubMed] [Google Scholar]
- 9.Momburg F., Roelse J., Howard J. C., Butcher G. W., Hämmerling G. J., Neefjes J. J. (1994) Nature 367, 648–651 [DOI] [PubMed] [Google Scholar]
- 10.Uebel S., Kraas W., Kienle S., Wiesmüller K. H., Jung G., Tampé R. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 8976–8981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neumann L., Tampé R. (1999) J. Mol. Biol. 294, 1203–1213 [DOI] [PubMed] [Google Scholar]
- 12.Neumann L., Abele R., Tampé R. (2002) J. Mol. Biol. 324, 965–973 [DOI] [PubMed] [Google Scholar]
- 13.Lapinski P. E., Neubig R. R., Raghavan M. (2001) J. Biol. Chem. 276, 7526–7533 [DOI] [PubMed] [Google Scholar]
- 14.Karttunen J. T., Lehner P. J., Gupta S. S., Hewitt E. W., Cresswell P. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 7431–7436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saveanu L., Daniel S., van Endert P. M. (2001) J. Biol. Chem. 276, 22107–22113 [DOI] [PubMed] [Google Scholar]
- 16.Chen M., Abele R., Tampé R. (2004) J. Biol. Chem. 279, 46073–46081 [DOI] [PubMed] [Google Scholar]
- 17.Smith P. C., Karpowich N., Millen L., Moody J. E., Rosen J., Thomas P. J., Hunt J. F. (2002) Mol. Cell 10, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaitseva J., Jenewein S., Jumpertz T., Holland I. B., Schmitt L. (2005) EMBO J. 24, 1901–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergani P., Lockless S. W., Nairn A. C., Gadsby D. C. (2005) Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russ G., Esquivel F., Yewdell J. W., Cresswell P., Spies T., Bennick J. R. (1995) J. Biol. Chem. 270, 21312–21318 [DOI] [PubMed] [Google Scholar]
- 21.Gorbulev S., Abele R., Tampé R. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3732–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M., Abele R., Tampé R. (2003) J. Biol. Chem. 278, 29686–29692 [DOI] [PubMed] [Google Scholar]
- 23.Herget M., Oancea G., Schrodt S., Karas M., Tampé R., Abele R. (2007) J. Biol. Chem. 282, 3871–3880 [DOI] [PubMed] [Google Scholar]
- 24.Oancea G., O'Mara M. L., Bennett W. F., Tieleman D. P., Abele R., Tampé R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5551–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abacioglu Y. H., Fouts T. R., Laman J. D., Claassen E., Pincus S. H., Moore J. P., Roby C. A., Kamin-Lewis R., Lewis G. K. (1994) AIDS Res. Hum. Retroviruses 10, 371–381 [DOI] [PubMed] [Google Scholar]
- 26.Nitsche J. M., Chang H. C., Weber P. A., Nicholson B. J. (2004) Biophys. J. 86, 2058–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loman A., Dertinger T., Koberling F., Enderlein J. (2008) Chem. Phys. Lett. 459, 18–21 [Google Scholar]
- 28.Sherman E., Itkin A., Kuttner Y. Y., Rhoades E., Amir D., Haas E., Haran G. (2008) Biophys. J. 94, 4819–4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henkel R. D., VandeBerg J. L., Walsh R. A. (1988) Anal. Biochem. 169, 312–318 [DOI] [PubMed] [Google Scholar]
- 30.Zhao C., Haase W., Tampé R., Abele R. (2008) J. Biol. Chem. 283, 17083–17091 [DOI] [PubMed] [Google Scholar]
- 31.Doeven M. K., Abele R., Tampé R., Poolman B. (2004) J. Biol. Chem. 279, 32301–32307 [DOI] [PubMed] [Google Scholar]
- 32.Marsh D. (2007) Biophys J. 93, 3884–3899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cantor R. S. (1997) J. Phys. Chem. B. 101, 1723–1725 [Google Scholar]
- 34.Aisenbrey C., Sizun C., Koch J., Herget M., Abele R., Bechinger B., Tampé R. (2006) J. Biol. Chem. 281, 30365–30372 [DOI] [PubMed] [Google Scholar]
- 35.Bamber L., Harding M., Butler P. J., Kunji E. R. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 16224–16229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ravaud S., Do Cao M. A., Jidenko M., Ebel C., Le Maire M., Jault J. M., Di Pietro A., Haser R., Aghajari N. (2006) Biochem. J. 395, 345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dawson R. J., Locher K. P. (2006) Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 38.García De La Torre J., Huertas M. L., Carrasco B. (2000) Biophys J. 78, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koch J., Guntrum R., Heintke S., Kyritsis C., Tampé R. (2004) J. Biol. Chem. 279, 10142–10147 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.