

Abstract
A growing body of evidence indicates that peroxisome proliferator-activated receptor α (PPARα) not merely serves as a transcriptional regulator of fatty acid catabolism but also exerts a much broader role in hepatic lipid metabolism. We determined adaptations in hepatic lipid metabolism and related aspects of carbohydrate metabolism upon treatment of C57Bl/6 mice with the PPARα agonist fenofibrate. Stable isotope procedures were applied to assess hepatic fatty acid synthesis, fatty acid elongation, and carbohydrate metabolism. Fenofibrate treatment strongly induced hepatic de novo lipogenesis and chain elongation (±300, 150, and 600% for C16:0, C18:0, and C18:1 synthesis, respectively) in parallel with an increased expression of lipogenic genes. The lipogenic induction in fenofibrate-treated mice was found to depend on sterol regulatory element-binding protein 1c (SREBP-1c) but not carbohydrate response element-binding protein (ChREBP). Fenofibrate treatment resulted in a reduced contribution of glycolysis to acetyl-CoA production, whereas the cycling of glucose 6-phosphate through the pentose phosphate pathway presumably was enhanced. Altogether, our data indicate that β-oxidation and lipogenesis are induced simultaneously upon fenofibrate treatment. These observations may reflect a physiological mechanism by which PPARα and SREBP-1c collectively ensure proper handling of fatty acids to protect the liver against cytotoxic damage.
Introduction
Fatty acids are cytotoxic molecules. Both their oxidation and storage as triglycerides (TGs)2 may be important in protecting the liver against lipotoxicity. Recently it was postulated that an increased conversion of saturated fatty acids into monounsaturated fatty acids (MUFAs) stimulates storage as TG and prevents non-esterified fatty acid (NEFA)-induced hepatocellular apoptosis (1). However, the mechanisms underlying this lipogenic response have remained enigmatic. Peroxisome proliferator-activated receptors (PPARs) represent likely candidates to mediate this response because these nuclear receptors act as cellular fatty acid sensors.
PPARα induces the remodeling of hepatic lipid metabolism under conditions of increased fatty acid influx such as fasting and high fat feeding (2–4). Upon activation, PPARα induces the expression of a multitude of genes encoding proteins involved in peripheral lipid mobilization and fatty acid oxidation (2, 3, 5–7). In addition PPARα plays a role in hepatic lipid droplet formation (8, 9) and mediates adaptive responses to prevent oxidative stress and the accumulation of cytotoxic NEFAs (10). For example, PPARα promotes the degradation of lipid-derived inflammatory mediators (11) and induces mitochondrial uncoupling as well as antioxidant systems to protect against oxidative damage associated with (incomplete) β-oxidation (12–18). As a consequence, PPARα activity protects against hepatic inflammation in mice (19–22).
Fibrates are pharmacological PPARα agonists that are used clinically to treat dyslipidemia (23). Interestingly, PPARα agonist treatment has also been shown to promote 3H2O incorporation into hepatic lipids in wild-type mice but not in Pparα−/− mice (24). This strongly suggests that in response to PPARα activation, both hepatic fatty acid oxidation and fatty acid synthesis are increased. How this observation relates to hepatic MUFA synthesis and TG storage in the liver (1) remains to be elucidated. In this respect it is interesting to note that stearoyl-CoA desaturase 1 (Scd1), the lipogenic enzyme controlling MUFA synthesis, has been reported to be a direct PPARα target gene (25).
Considering the regulatory role of PPARα under conditions of increased fatty acid influx, specific changes in the hepatic processing of fatty acids are to be expected upon PPARα activation. To gain insight into these changes, we used sophisticated stable isotope techniques to quantify de novo lipogenesis and fatty acid elongation in vivo in mice that were treated with the PPARα agonist fenofibrate. To evaluate the interactions between hepatic glucose and lipid metabolism, we also determined relevant hepatic carbohydrate fluxes.
EXPERIMENTAL PROCEDURES
Animals and Experimental Design
To assess the effects of PPARα activation on metabolite concentrations and metabolite fluxes in vivo, male C57Bl/6 mice (Charles River, L'Arbresle Cedex, France) were housed in a light- and temperature-controlled facility (lights on 6:30 a.m. to 6:30 p.m., 21 °C). They were fed a standard laboratory chow diet (diet no. A03, UAR, Villemoisson-sur-Orge, France) with or without fenofibrate (0.2% w/w) for 2 weeks and had free access to drinking water. Experimental procedures were approved by the Ethics Committees for Animal Experiments of the University of Groningen. To determine transcriptional regulation of lipogenic gene expression, female Srebp-1c−/− and Chrebp−/− mice and their wild-type littermates (26, 27) were housed in a light- and temperature-controlled facility (lights on 6:00 a.m. to 6:00 p.m., 21 °C). They were fed a standard laboratory chow diet (diet no. 7002, Harlan Teklad Premier Laboratory Diets, Madison, WI) with or without fenofibrate (0.2% w/w) for 2 weeks and had free access to drinking water. The experiments involving the Srebp-1c−/− and Chrebp−/− mice were approved by the Institutional Animal Care and Research Advisory Committee at the University of Texas Southwestern Medical Center (Dallas, TX).
Metabolite and Gene Expression Analysis
The C57Bl/6 mice were fasted from 6:00 a.m. to 1:00 p.m. with drinking water available and were subsequently sacrificed by cardiac puncture under isoflurane anesthesia. Srebp-1c−/− and Chrebp−/− mice and their wild-type littermates were fasted from 7:00 to 11:00 a.m. with drinking water available and were subsequently sacrificed by isoflurane overdose. Livers were removed quickly, freeze-clamped, and stored at −80 °C. Blood was centrifuged (4000 × g for 10 min at 4 °C), and plasma was stored at −20 °C. Plasma TG and β-hydroxybutyrate concentrations were determined using commercially available kits (Roche Diagnostics). Plasma fibroblast growth factor 21 (FGF-21) concentrations were determined using a mouse radioimmunoassay (Phoenix Pharmaceuticals, Burlingame, CA). Frozen liver was homogenized in ice-cold phosphate-buffered saline. Hepatic protein contents were determined according to Lowry et al. (28). Hepatic TG and total cholesterol contents were assessed using commercially available kits (Roche Diagnostics and Wako Chemicals GmbH, Neuss, Germany) after lipid extraction (29). Hepatic fatty acid composition was analyzed by gas chromatography (30). Δ9-Desaturation indices were calculated from the ratios between C16:1(n-7) and C16:0 and C18:1(n-7/n-9) and C18:0, respectively. Hepatic Glc-6-P and glycogen content were determined as described previously (31, 32).
RNA was extracted from livers using Tri reagent (Sigma-Aldrich), and cDNA obtained by reverse transcription was amplified using the appropriate primers and probes. Primer and probe sequences for 18S, acetyl-coenzyme A carboxylase 1 (Acc1), ATP-binding cassette a1/g1/g5 (Abca1/g1/g5), fatty acid transporter (Cd36), carnitine palmitoyl transferase 1a (Cpt1a), carbohydrate response element-binding protein (Chrebp), diacylglycerol acyltransferases 1 and 2 (Dgat1 and -2), fatty-acid synthase (Fas), glucokinase (Gk), glucose-6-phosphate catalytic subunit (G6pc), glucose-6-phosphate translocase (G6pt), glucose transporter 2 (Glut2), glycerol-3-phosphate acyltransferase (Gpat), 3-hydroxy-3-methylglutaryl-coenzyme A synthase 2 (Hmgcs2), liver X receptor α (Lxrα), phosphoenolpyruvate carboxykinase (Pepck), peroxisome proliferator-activated receptor γ co-activator 1α/β (Pgc-1α/β), pyruvate dehydrogenase kinase 4 (Pdk4), pyruvate kinase (Pk), Scd1, Srebp-1c, and uncoupling proteins 2 and 3 (Ucp2 and -3) have been published. Source will be provided upon request. The sequences for all other primers and probes are given in supplemental Table 1. All mRNA levels were calculated relative to the expression of 18S and normalized for the expression levels of control mice.
Determination of de Novo Lipogenesis and Chain Elongation in Vivo in C57Bl/6 Mice
Mice were equipped with a permanent jugular vein catheter (33) and allowed a recovery period of at least 3 days. On the day of the experiment, the mice were individually housed and fasted from 6:00 to 10:00 a.m. All infusion experiments were performed in conscious, unrestrained mice. A 0.3 m sodium [1-13C]acetate (99 atom %, Isotec/Sigma- Aldrich) solution was infused via the jugular vein catheter at an infusion rate of 0.6 ml/h. After 6 h of infusion, animals were sacrificed by cardiac puncture under isoflurane anesthesia. Livers were removed quickly, freeze-clamped, and stored at −80 °C. Liver homogenates were prepared in ice-cold phosphate-buffered saline, and TG fractions were obtained using thin layer chromatography as described previously (34). TGs were hydrolyzed in HCl/acetonitrile (1:22 v/v) for 45 min at 100 °C. Fatty acids were extracted in hexane and derivatized for 15 min at room temperature using Br-2,3,4,5,6-pentafluorobenzyl/acetonitrile/triethanolamine (1:6:2 v/v). Derivatization was stopped by adding HCl, and the fatty acid-pentafluorobenzyl derivatives were extracted in hexane.
The fatty acid-pentafluorobenzyl derivatives mass isotopomer distributions were measured using an Agilent 5975 series GC/MSD (Agilent Technologies, Santa Clara, CA). Gas chromatography was performed using a ZB-1 column (Phenomenex, Torrance, CA). Mass spectrometry analysis was performed by electron capture negative ionization using methane as the moderating gas.
The normalized mass isotopomer distributions measured by GC-MS (m0 − mx) were corrected for natural abundance of 13C by multiple linear regression (35) to obtain the excess fractional distribution of mass isotopomers (M0 − Mx) due to the incorporation of [1-13C]acetate. This distribution was used in mass isotopomer distribution analysis (MIDA) algorithms to calculate acetyl-CoA precursor pool enrichment (pacetate), fractional palmitate synthesis rates (fC16:0), and the fraction of palmitate and oleate generated by elongation of de novo synthesized palmitate (fC18:0/1(C16DNL)) or by elongation of pre-existing palmitate (fC18:0/1(C16PE)) as described (36).
In Vivo Hepatic Carbohydrate Flux Measurements in C57Bl/6 Mice
Mice were equipped with a permanent jugular vein catheter as described above. After recovery, the mice were fasted from 6:00 to 10:00 a.m. Conscious, unrestrained mice were infused with a solution containing [U-13C]glucose (7 mm), [2-13C]glycerol (82 mm), [1-2H]galactose (17 mm), and paracetamol (1 mg/ml) for 6 h at an infusion rate of 0.6 ml/h as described previously (37). Blood glucose concentrations were measured every 30 min. Blood and urine spots were collected every 60 min. Analytical procedures for extraction of glucose from blood spots, derivatization of the extracted compounds, and GC-MS measurements of derivatives were performed according to van Dijk et al. (38) and corrected for natural abundance of 13C (35). Hepatic carbohydrate fluxes were calculated using MIDA as described previously (39). supplemental Fig. 1 depicts the isotopic model used.
Scrambling of [U-13C]Glucose in Vivo
During stable isotope infusion, both glycolysis and pentose phosphate pathway (PPP) cycling contribute to triple labeled triose phosphate (M3) generated from the infused [U-13C]glucose (M6). Reconversion of triple labeled triose phosphate into blood and UDP glucose via the gluconeogenic pathway in turn results in M3 abundance in these pools.
Statistics
All data are presented as mean values ± S.E. Statistical analysis was performed using the Mann-Whitney U test and SPSS for Windows software (SPSS 12.02; SPSS Inc., Chicago). The null hypothesis was rejected at the 0.05 level of probability.
RESULTS
The Catabolic Phenotype of Fenofibrate-treated Mice Is Accompanied by Induction of Hepatic Lipogenic Gene Expression and Accumulation of Triglycerides in the Liver
Fenofibrate treatment induced a catabolic phenotype, characterized by weight loss without affecting food intake (Table 1). Hepatic Fgf-21 mRNA expression (Table 2) was induced, and FGF-21 plasma concentrations (Table 1) were increased. Plasma NEFA concentrations did not differ between fenofibrate-treated animals and controls (Table 1), whereas plasma TG concentrations were decreased by fenofibrate treatment (Table 1). Fenofibrate treatment induced hepatic peroxisome proliferation, which resulted in increased liver weight and hepatic protein content (Table 1). Hepatic fatty acid oxidation was promoted by fenofibrate treatment, as indicated by the anticipated increase in the expression of genes involved in fatty acid transport, ketogenesis, and peroxisomal and mitochondrial β-oxidation (Table 2) as well as the elevated plasma β-hydroxybutyrate concentrations (Table 1). The expression of genes encoding proteins involved in uncoupling of oxidative phosphorylation was also increased (Table 2).
TABLE 1.
General characteristics, plasma, and hepatic metabolite levels
Values are given as means ± S.E. for n = 6; *, p < 0.05 fenofibrate vs. control (Mann-Whitney U test).
| Parameter | Control | Fenofibrate |
|---|---|---|
| Body weight change (%) | 7.8 ± 1.0 | −6.4 ± 2.0* |
| Food intake (g/day) | 4.5 ± 0.1 | 4.8 ± 0.4 |
| Plasma FGF-21 (ng/ml) | 1.1 ± 0.1 | 4.7 ± 0.6* |
| Plasma NEFA (mmol/liter) | 0.27 ± 0.04 | 0.20 ± 0.01 |
| Plasma β-hydroxybutyrate (mmol/liter) | 0.15 ± 0.03 | 1.00 ± 0.12* |
| Plasma triglycerides (mmol/liter) | 0.53 ± 0.04 | 0.10 ± 0.01* |
| Liver weight (% body weight) | 4.8 ± 0.1 | 13.2 ± 0.5* |
| Hepatic protein (mg/g) | 160 ± 3 | 171 ± 2* |
| Hepatic C16 desaturation index | 0.07 ± 0.01 | 0.13 ± 0.02* |
| Hepatic C18 desaturation index | 1.20 ± 0.10 | 4.06 ± 0.20* |
| Hepatic triglycerides (μmol/g) | 15.4 ± 2.6 | 24.6 ± 2.0* |
| Hepatic cholesterol (μmol/g) | 7.7 ± 0.4 | 8.2 ± 0.3 |
TABLE 2.
Hepatic gene expression levels
Expression levels were normalized to 18S expression, and values are given as means ± S.E. for n = 6; *, p < 0.05 fenofibrate vs. control (Mann-Whitney U test).
| Gene | Control | Fenofibrate |
|---|---|---|
| Fatty acid mobilization/uptake | ||
| Fgf-21 | 1.0 ± 0.3 | 4.4 ± 0.4* |
| Cd36 | 1.0 ± 0.1 | 7.2 ± 0.7* |
| β-Oxidation and ketogenesis | ||
| Aox | 1.0 ± 0.1 | 4.5 ± 0.5* |
| Cpt-1a | 1.0 ± 0.1 | 1.4 ± 0.1* |
| Lcad | 1.0 ± 0.1 | 2.6 ± 0.2* |
| Hmgcs2 | 1.0 ± 0.1 | 1.9 ± 0.2* |
| Mitochondrial uncoupling | ||
| Ucp2 | 1.0 ± 0.1 | 2.9 ± 0.2* |
| Ucp3 | 1.0 ± 0.2 | 109.8 ± 9.6* |
| Fatty acid synthesis | ||
| Srebp-1c | 1.0 ± 0.1 | 1.3 ± 0.1 |
| Pgc-1ß | 1.0 ± 0.1 | 1.0 ± 0.1 |
| Acc1 | 1.0 ± 0.1 | 1.7 ± 0.1* |
| Fas | 1.0 ± 0.1 | 1.5 ± 0.1* |
| Elovl6 | 1.0 ± 0.2 | 1.3 ± 0.1 |
| Scd1 | 1.0 ± 0.1 | 2.6 ± 0.2* |
| Elovl5 | 1.0 ± 0.1 | 3.2 ± 0.3* |
| Fads1 | 1.0 ± 0.1 | 1.5 ± 0.1* |
| Fads2 | 1.0 ± 0.2 | 2.7 ± 0.3* |
| Triglyceride synthesis | ||
| Gpat | 1.0 ± 0.1 | 1.5 ± 0.1* |
| Dgat1 | 1.0 ± 0.1 | 2.0 ± 0.2* |
| Dgat2 | 1.0 ± 0.1 | 0.7 ± 0.1* |
| Glucose uptake/glycolysis | ||
| Chrebp | 1.0 ± 0.1 | 0.7 ± 0.1* |
| Glut2 | 1.0 ± 0.1 | 0.5 ± 0.1* |
| Gk | 1.0 ± 0.1 | 0.4 ± 0.0* |
| Pk | 1.0 ± 0.1 | 0.2 ± 0.0* |
| Pdk4 | 1.0 ± 0.3 | 27.0 ± 2.8* |
| Gluconeogenesis | ||
| Pgc-1α | 1.0 ± 0.1 | 1.1 ± 0.1 |
| Pepck | 1.0 ± 0.1 | 0.8 ± 0.1 |
| G6pc | 1.0 ± 0.3 | 0.8 ± 0.1 |
| G6pt | 1.0 ± 0.0 | 0.5 ± 0.2* |
| Gyk | 1.0 ± 0.1 | 1.8 ± 0.1* |
| PPP and NADPH synthesis | ||
| G6pdh | 1.0 ± 0.1 | 1.0 ± 0.1 |
| 6Pdgh | 1.0 ± 0.0 | 2.2 ± 0.1* |
| Taldo1 | 1.0 ± 0.1 | 1.8 ± 0.2* |
| Tkt | 1.0 ± 0.1 | 1.2 ± 0.1 |
| Me1 | 1.0 ± 0.1 | 5.9 ± 0.5* |
The catabolic phenotype of fenofibrate-treated mice was associated with an increase in hepatic TG content, whereas cholesterol content remained unaffected (Table 1). The hepatic expression of genes encoding enzymes involved in de novo lipogenesis, e.g. Acc1 and Fas, was higher in these animals, whereas that of Srebp-1c and its co-activator Pgc-1β remained unaltered (Table 2). In addition, the expression of genes encoding enzymes involved in fatty acid elongation and desaturation, e.g. Elovl5, Scd1, Fads1, and Fads2, as well as TG synthesis (Dgat1 and Gpat), was markedly induced upon treatment with fenofibrate (Table 2). Changes in hepatic fatty acid synthesis and the elongation/desaturation gene expression pattern translated into altered hepatic fatty acid composition (Table 3) with a marked increase in the abundance of MUFA, resulting in increased hepatic Δ9-desaturation indices (Table 1).
TABLE 3.
Hepatic fatty acid profiles
Values are given as means ± S.E. for n = 6 and expressed in μmol/g liver; *, p < 0.05 fenofibrate vs. control (Mann-Whitney U test).
| Fatty Acid | Control | Fenofibrate |
|---|---|---|
| C14:0 | 0.24 ± 0.03 | 0.27 ± 0.01 |
| C16:1(n-7) | 1.76 ± 0.22 | 4.55 ± 0.56* |
| C16:0 | 24.04 ± 0.98 | 34.77 ± 0.95* |
| C18:3(n-6) | 0.23 ± 0.02 | 0.26 ± 0.01 |
| C18:2(n-6) | 20.69 ± 1.05 | 16.09 ± 0.67* |
| C18:3(n-3) | 0.62 ± 0.07 | 0.26 ± 0.04* |
| C18:1(n-9) | 12.76 ± 1.28 | 28.57 ± 1.21* |
| C18:1(n-7) | 2.07 ± 0.13 | 5.79 ± 0.20* |
| C18:0 | 12.32 ± 0.34 | 8.50 ± 0.30* |
| C20:4(n-6) | 11.44 ± 0.26 | 10.82 ± 0.36 |
| C20:5(n-3) | 0.70 ± 0.03 | 0.51 ± 0.03* |
| C20:3(n-9) | 0.14 ± 0.01 | 0.61 ± 0.02* |
| C20:3(n-6) | 1.12 ± 0.04 | 5.43 ± 0.22* |
| C20:2(n-6) | 0.33 ± 0.02 | 0.42 ± 0.02* |
| C20:1(n-9) | 0.36 ± 0.03 | 0.55 ± 0.02* |
| C20:0 | 0.20 ± 0.01 | 0.08 ± 0.00* |
| C22:5(n-6) | 0.13 ± 0.01 | 0.14 ± 0.01 |
| C22:6(n-3) | 7.91 ± 0.26 | 6.72 ± 0.16* |
| C22:4(n-6) | 0.21 ± 0.01 | 0.41 ± 0.02* |
| C22:5(n-3) | 0.58 ± 0.03 | 0.85 ± 0.04* |
| C22:0 | 0.44 ± 0.02 | 0.18 ± 0.01* |
| C24:1(n-9) | 0.48 ± 0.01 | 0.39 ± 0.01* |
| C24:0 | 0.37 ± 0.01 | 0.14 ± 0.00* |
Massive Induction of the Lipogenic Flux Contributes to Hepatic TG Accumulation in Fenofibrate-treated Mice
To establish the physiological relevance of the induction in lipogenic gene expression, we determined de novo lipogenesis and fatty acid elongation and their contributions to hepatic TG in vivo. We therefore infused [1-13C]acetate into mice for 6 h and applied MIDA to quantify fatty acid biosynthesis from 2-carbon precursors and to differentiate between de novo lipogenesis and fatty acid elongation as described (36). Fenofibrate treatment resulted in a massive increase in de novo lipogenesis (Fig. 1A). Elongation of both de novo synthesized and pre-existing palmitate was also higher in fenofibrate-treated mice, which resulted in an increase in stearate and oleate synthesis (Fig. 1, B and 1C). These results are consistent with the increased expression of genes encoding enzymes involved in fatty acid synthesis, elongation, and desaturation upon fenofibrate treatment (Table 1). Moreover, the results from the isotope infusion studies explain the changes observed in the hepatic fatty acid profile (Table 3), i.e. the higher desaturation indices and the ∼50% increase in oleate content. Interestingly, the synthesis rates of TG-associated palmitate, oleate, and stearate (Fig. 1, A–C) were very similar to the values observed for the total hepatic synthetic rates of these fatty acids. Hence, the acetyl-CoA precursor pool enrichments in total and TG-associated palmitate were similar. Strikingly, the acetyl-CoA precursor pool enrichment remained unaffected upon fenofibrate treatment (Fig. 1D), indicative of a similar and rapid turnover of the acetyl-CoA precursor pool.
FIGURE 1.

Hepatic fatty acid synthesis in control and fenofibrate-treated mice. Conscious, unrestrained C57Bl/6 mice were infused with sodium [1-13C]acetate. Fatty acids from total liver homogenates and TG fractions were derivatized, and isotopomer patterns were determined by GC-MS analysis. Synthesis rates and contribution of de novo lipogenesis and chain elongation were calculated using MIDA. A, fractional synthesis rates of total and TG-derived palmitate from de novo lipogenesis. B, fractional synthesis rates of total and TG-derived stearate from elongation of labeled (de novo synthesized; C16:0 DNL) and unlabeled (pre-existing; C16:0 PE) palmitate. C, fractional synthesis rates of total and TG-derived oleate from elongation of labeled and unlabeled palmitate. D, acetyl-CoA precursor pool enrichments in total and TG-derived palmitate. Open bars, control group; filled bars, fenofibrate-treated group. Values represent means ± S.E. for n = 6–8; *, p < 0.05 fenofibrate versus control (Mann-Whitney U test).
Induction of Lipogenic Genes upon Fenofibrate Treatment Is Regulated Transcriptionally by SREBP-1c
PPARα agonist treatment is reportedly associated with an increased abundance of nuclear SREBP-1c (24), which might be responsible for the observed induction in lipogenic gene expression. On the other hand, lipogenic gene expression might be controlled transcriptionally by ChREBP (40). To assess whether the induction of the lipogenic genes upon fenofibrate treatment depended on SREBP-1c and/or ChREBP, Srebp-1c−/− and Chrebp−/− mice were treated with fenofibrate. Fig. 2 shows the expression profiles of genes encoding enzymes controlling de novo lipogenesis as well as fatty acid elongation and desaturation in the knock-out mice and their wild-type littermates. In Srebp-1c−/−mice, as compared with their wild-type littermates, the induction of fatty acid synthesis genes upon treatment with fenofibrate was clearly blunted. In Chrebp−/−mice, however, the induction of these genes was maintained. Similarly, the induction of genes encoding enzymes controlling fatty acid esterification as well as NADPH synthesis was blunted in Srebp-1c−/−mice only (supplemental Table 2).
FIGURE 2.
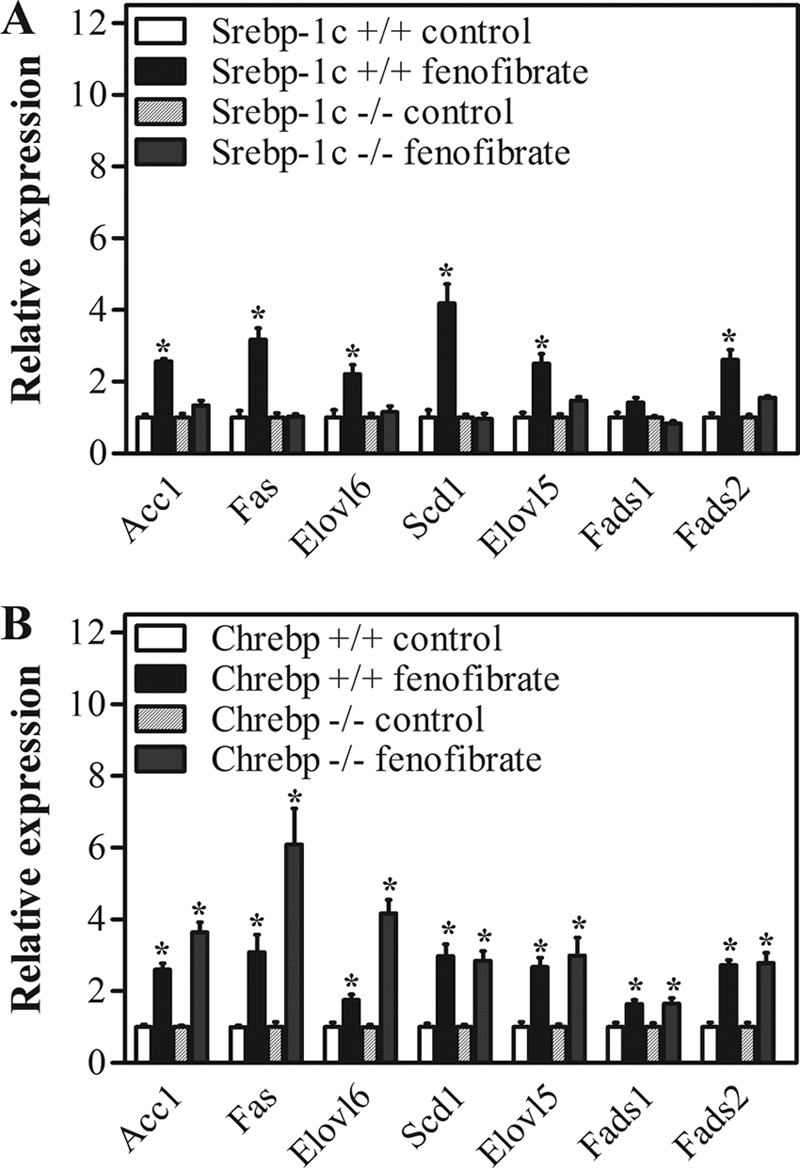
Transcriptional control of lipogenic gene expression in control and fenofibrate-treated mice. Srebp-1c−/− and Chrebp−/− mice and their wild-type littermates were sacrificed by cardiac puncture. Gene expression levels in livers were determined by quantitative PCR and normalized for 18S expression. Expression levels of untreated mice of each genotype were set to 1. A, expression of genes involved in fatty acid synthesis in Srebp-1c−/− mice and wild-type littermates. B, expression of genes involved in fatty acid synthesis in Chrebp−/− mice and wild-type littermates. Open bars, wild-type control group; filled bars, wild-type fenofibrate-treated group; dashed bars, knock-out control group; dotted bars, knock-out fenofibrate-treated group. Values represent means ± S.E. for n = 4; *, p < 0.05 fenofibrate versus control (Mann-Whitney U test).
In addition to SREBP-1c and ChREBP, the liver X receptor (LXR) is another important transcriptional regulator of lipogenic genes. Expression analysis of Lxrα and its direct target genes Abca1, Abcg5, and Abcg1 did not provide evidence for increased LXR activity upon treatment with fenofibrate (supplemental Table 3).
Reduced Hepatic Glucose Consumption upon Fenofibrate Treatment Is Compensated for by an Increased Gluconeogenic Flux and Enhanced PPP Cycling
Glucose provides the acetyl-CoA required for fatty acid synthesis via the glycolytic pathway. The interconversions of glucose, Glc-6-P, and glycogen can be quantified by direct labeling of the blood glucose and UDP-glucose pools, whereas the gluconeogenic flux can be estimated by glucose and UDP-glucose biosynthesis from 3-carbon precursors. To assess the contribution of adaptations in hepatic glucose metabolism to the increased lipogenic flux, we determined carbohydrate fluxes in vivo following isotope infusion by analysis of glucose and galactose cycling, whereas we applied MIDA to quantify the gluconeogenic flux through the triose phosphate precursor pool following infusion of labeled glycerol. Fluxes were derived from isotopomer distributions in the blood glucose and urinary UDP-glucose pools as described (38, 39). The isotopic model used is depicted in supplemental Fig. 1, and the primary isotopic parameters are listed in supplemental Table 4. Blood glucose concentrations were comparable during isotope infusion in both groups (supplemental Table 4). Fenofibrate treatment resulted in a lower hepatic glucose uptake, indicated by a decreased flux through glucokinase (Fig. 3A). This was paralleled by a decreased hepatic mRNA expression of Chrebp, Glut2, and Gk (Table 2). In addition, the decreased expression of hepatic Pk mRNA, encoding a key enzyme in glycolysis, and the massive induction of Pdk4 mRNA, encoding the major inhibitor of glycolysis (Table 2), indicated a reduced glycolysis upon fenofibrate treatment. The reduction in hepatic glucose input from the circulation was compensated for by an increased de novo synthesis of Glc-6-P, i.e. an increased gluconeogenic flux (Fig. 3B). Expression of Gyk, which encodes the enzyme that facilitates the use of glycerol as a gluconeogenic substrate, was also induced upon fenofibrate treatment. On the other hand, expression of other gluconeogenic genes, e.g. Pgc-1α, Pepck, and G6pc, remained unaffected, whereas expression of G6pt was reduced upon fenofibrate treatment (Table 2). The increased gluconeogenic flux in fenofibrate-treated mice did not promote hepatic glucose production; hepatic glucose output (i.e. the glucose-6-phosphatase flux) was hardly affected by fenofibrate treatment. This is explained by the lower compartmentation of the gluconeogenic flux toward blood glucose (86 ± 1% versus 80 ± 1%, control versus fenofibrate, p < 0.05) and the reduced glucose cycling (from blood glucose to Glc-6-P back to blood glucose (Fig. 3C)) in fenofibrate-treated mice. Moreover, the increased gluconeogenic flux did not increase glycogen disposition because of an increased flux through glycogen phosphorylase (Fig. 3, D and E). Hepatic Glc-6-P and glycogen content were reduced even in fenofibrate-treated mice (Fig. 4). The increased expression of genes encoding enzymes that mediate the PPP (6Pdgh and Taldo1, Table 2) strongly suggests an enhanced flux through the PPP upon fenofibrate treatment. This is further supported by the higher abundance of triple labeled glucose molecules in the UDP-glucose and blood glucose pools (Fig. 3F). Upon infusion of uniformly labeled glucose, triple labeling of the triose phosphate pool used for gluconeogenesis results either from glycolysis or from the interconversions of the PPP. Our data strongly suggest that glycolysis was reduced, rather than increased, upon fenofibrate treatment. Therefore, the triple label most likely originates from enhanced cycling through the PPP, which in turn cycles back to Glc-6-P via triose phosphate. Cycling through the PPP generates NADPH, which is needed to maintain the increased energy-consuming lipogenic flux in fenofibrate-treated animals. This is consistent with the induction of hepatic of malic enzyme 1 (Me1) expression upon fenofibrate treatment (Table 2). Me1 encodes another NADPH-generating enzyme, and its expression is reportedly controlled by PPARα (41).
FIGURE 3.

Hepatic glucose metabolism in control and fenofibrate-treated mice. Conscious, unrestrained C57Bl/6 mice were infused with different stable isotopes, and carbohydrate fluxes were calculated from GC-MS analysis of blood and urine spots obtained at regular time intervals under steady-state conditions (t = 180–360 min) using MIDA. A, glucokinase flux. B, gluconeogenic (GNG) flux and partitioning toward glucose (light gray bars) and UDP-glucose (dark gray bars). C, hepatic glucose production rate and contribution of gluconeogenic flux or rate of appearance of endogenous glucose (Ra) (glc; endo) (light gray bars) and glucose cycling (dark gray bars). D, glycogen phosphorylase flux. E and F, glycogen balance (E) and abundance (F) of triple labeled molecules in blood and UDP-glucose. Open bars, control group; filled bars, fenofibrate-treated group. Values represent means ± S.E. for n = 5–6; *, p < 0.05 fenofibrate versus control (analysis of variance for repeated measurements).
FIGURE 4.

Hepatic Glc-6-P and glycogen content in control and fenofibrate-treated mice. C57Bl/6 mice were sacrificed by cardiac puncture. Hepatic Glc-6-P and glycogen content were determined using enzymatic assays. Open bars, control group; filled bars, fenofibrate-treated group. Values represent means ± S.E. for n = 6; *, p < 0.05 fenofibrate versus control (Mann-Whitney U test).
DISCUSSION
The current study is the first to establish the remodeling of hepatic intermediary metabolism that occurs upon chronic PPARα activation in mice. These metabolic changes are depicted and highlighted in Fig. 5. In parallel with the well known increase in hepatic fatty acid oxidation, fenofibrate treatment resulted in a massive induction of the lipogenic flux and a concomitant adjustment of hepatic glucose metabolism to provide the NADPH required for fatty acid synthesis and the subsequent esterification to form TG. In addition, we show that fenofibrate treatment reduced glycolysis and thus the acetyl-CoA supply from glucose. Altogether, these data provide evidence for the existence of an adaptive response to an increased fatty acid influx and catabolism that will protect the liver against the toxic effects of excess intracellular NEFA and their oxidation products.
FIGURE 5.

Remodeling of hepatic intermediary metabolism in fenofibrate-treated mice. Fenofibrate treatment promotes adipose tissue lipolysis, thereby enhancing hepatic influx of glycerol and fatty acids. In the liver, fenofibrate promotes fatty acid β-oxidation in peroxisomes and mitochondria. PPARα target genes are indicated in blue (see also Ref. 5). Acetyl-CoA generated by β-oxidation is used for ketogenesis and energy supply but also serves as a substrate for fatty acid synthesis via de novo lipogenesis and fatty acid elongation. Acetyl-CoA transport over the mitochondrial membrane is facilitated by increased pyruvate/malate cycling, which generates NADPH to support the lipogenic flux. In parallel, hepatic glucose uptake and glycolysis are suppressed, and the contribution of acetyl-CoA from hepatic glucose metabolism to the lipogenic flux is consequently reduced. Glycerol is converted into glucose 6-phosphate via the gluconeogenic pathway. Glucose 6-phosphate (G-6-P) cycles through the pentose phosphate pathway to triose phosphate and back to Glc-6-P and PPP, thereby generating NADPH.
PPARα action was originally found to be crucial for the hepatic adaptive response to fasting (2, 3). Increased PPARα activity enhances the flux of fatty acids from the adipose tissue to the liver via the action of FGF-21. Hepatic Fgf-21 is a direct target gene of PPARα, and both fasting and pharmacological PPARα activation result in an increase in circulating concentrations of FGF-21 (6, 7). FGF-21 in turn acts directly on adipose tissue to stimulate lipolysis (6). We observed a 4.5-fold induction of both hepatic Fgf-21 expression and its plasma concentration (Tables 1 and 2) upon fenofibrate treatment. The hepatic expression of Cd36, a major fatty acid transporter, was induced concomitantly (Table 2). Thus, the increased hepatic influx of adipose tissue-derived fatty acids was compensated for by an increased hepatic uptake. As a consequence, circulating NEFA concentrations were maintained.
Interestingly, PPARα agonist treatment has also been shown to promote 3H2O incorporation into hepatic lipids in wild-type but not in Pparα−/− mice (24), strongly suggesting a PPARα-dependent induction of hepatic fatty acid synthesis. However, H2O is used in multiple metabolic pathways; the results with 3H2O therefore do not truly reflect the lipogenic flux, as fatty acid oxidation may also contribute to 3H incorporation into fatty acids. Moreover, the contributions of de novo lipogenesis and fatty acid elongation were not established. Finally, the 3H2O study did not address the relationship between MUFA synthesis and hepatic TG storage, which is of particular interest, as these processes have been reported to protect against lipotoxicity (1). Using [13C]acetate and MIDA, we now show that acetyl-CoA incorporation into the hepatic fatty acids was strongly induced in fenofibrate-treated mice, indicated by an induction of both de novo lipogenesis and fatty acid elongation (Fig. 1). Although quantitative data on the rate of fatty acid catabolism are currently not available, the increased hepatic content of the major TG-derived fatty acids indicates that the rate of fatty acid oxidation was not sufficient to counterbalance the NEFA influx and synthesis.
The induction of lipogenic genes was found to depend on the presence of Srebp-1c−/− rather than that of Chrebp−/−, which supports previous work (24). Expression of Pk, a direct target gene of ChREBP, was actually found to be strongly reduced upon fenofibrate treatment (Table 2). The question arises how an increased PPARα activity enhances SREBP-1c-mediated gene transcription. Although Srebp-1c is a target of LXR and fenofibrate has recently been shown to enhance LXR promoter activity in vivo (42), we did not observe any induction of Abcg5, Abca1, or Abcg1, which argues against the involvement of LXR (supplemental Table 3). Under normal physiological conditions, PPARα and SREBP-1c each acts in an opposite manner. However, the presence of Pparα has been shown to be required for proper Srebp-1c functioning (43), whereas our current observations indicate that Srebp-1c is needed for the induction of Scd1 upon fenofibrate treatment. This was surprising because Scd1 has been identified as a direct PPARα target (25). The relationship between PPARα and SREBP-1c action therefore requires further investigation, particularly because de novo lipogenesis generates endogenous PPARα ligands (44). The increased SREBP-1c activity may be related to changes in the intracellular lipid status secondary to an enhanced fatty acid influx. SCD1 action may be crucial in this process (45). PPARα agonist treatment actually inhibits SREBP-1c activity and TG synthesis in vitro (46). This strongly suggests that a fenofibrate-mediated increase in lipolysis and fatty acid influx into the liver induces hepatic lipogenesis in vivo. Furthermore, pharmacological PPARα agonists fail to induce either Fgf-21 (47) or lipogenic genes (48) in the livers of Pparα−/− mice.
Both de novo lipogenesis and fatty acid elongation require acetyl-CoA and NADPH. In mice, fenofibrate treatment induces both peroxisomal and mitochondrial fatty acid oxidation, processes that generate acetyl-CoA and NADH. Although humans appear to be resistant to the induction of peroxisome proliferation by PPARα agonists, an increased expression of hepatic lipogenic genes is also observed in mice that express human PPARα (48). This lipogenic induction is therefore most likely related to an elevated mitochondrial fatty acid oxidation driven by the increased hepatic influx and uptake of fatty acids upon fenofibrate treatment. Cytosolic acetyl-CoA from peroxisomal β-oxidation has been shown to promote fatty acid synthesis via chain elongation (49), whereas mitochondrial acetyl-CoA is used for ketogenesis and citrate synthesis. Increased NADH levels promote citrate shuttling from the mitochondria into the cytosol, where it is converted into acetyl-CoA and oxaloacetate. The acetyl-CoA is used for de novo lipogenesis and chain elongation (49, 50), whereas oxaloacetate facilitates transport of acetyl-CoA across the mitochondrial membrane via the pyruvate/malate cycle, thereby generating the NADPH required for fatty acid synthesis. Enhanced cycling through this pathway is evident from the ∼6-fold induction of Me1 expression (Table 2). The expression of 6Pdgh, encoding another NADPH-generating enzyme, was also increased upon fenofibrate treatment (Table 2).
We observed comparable acetyl-CoA precursor pool enrichments in treated and untreated mice (Fig. 1D), indicating a similar acetyl-CoA turnover in both groups. Because acetyl-CoA input from peroxisomal and mitochondrial β-oxidation must have been increased in fenofibrate-treated mice, its input from other pathways must have been reduced. Hepatic glucose metabolism provides a major source of hepatic acetyl-CoA, and we therefore determined hepatic carbohydrate fluxes. In fenofibrate-treated mice, the glucokinase flux was reduced by 45%, whereas Pdk4 expression was induced ∼30-fold, indicating reduced hepatic glucose uptake and glycolysis (Fig. 3A and Table 2). This presumably reflects “glucose sparing” (51) and strongly suggests reduced acetyl-CoA supply from glycolysis. Altogether, these observations explain the maintenance of acetyl-CoA precursor pool enrichment in the face of increased β-oxidation upon fenofibrate treatment.
The flux through the gluconeogenic pathway was increased upon fenofibrate treatment as indicated by the enhanced glycerol incorporation (Fig. 3B), which supports previous work (52). The gluconeogenic induction did not result in an increased hepatic glucose output (Fig. 3C). Moreover, the increased gluconeogenic flux toward glycogen was balanced by increased glycogen breakdown (Fig. 3, D and E). Therefore, Glc-6-P must have been metabolized via pathways other than those covered by the isotopic model applied, particularly because hepatic Glc-6-P content was reduced by ∼50%. Increased cycling through the PPP upon fenofibrate treatment seems the obvious explanation, because expression of 6Pdgh and Taldo1 was induced in the livers of fenofibrate-treated mice (Table 2). Furthermore, the abundance of triple labeled molecules in the isotopomer patterns of both blood glucose and UDP-glucose was increased (Fig. 3F). We infused [U-13C]glucose, and the M3 abundance can be considered as a measure of the futile cycling of substrates from Glc-6-P through the PPP back to Glc-6-P. Interestingly, the flux through the PPP has been shown to be reduced in Pparα−/− mice (53). PPP activity is also reportedly increased when the flux through glycerol kinase is enhanced (54), as was the case in our experiments. The PPP remodeling of hepatic glucose metabolism upon PPARα activation may not only provide NADPH needed to maintain the lipogenic flux but may also support antioxidant action, because the PPP is coupled to the synthesis of reduced glutathione (55).
In conclusion, we have shown that fatty acid synthesis and esterification are promoted in response to an increased fatty acid influx and catabolism upon pharmacological PPARα activation by fenofibrate. The alterations in hepatic metabolism that occur upon fenofibrate treatment are depicted in Fig. 5. The presence of SREBP-1c appears to be essential for the adaptive processes, in particular for the induction of lipogenic genes. These novel insights add to the growing body of evidence that PPARα does not merely serve as a transcriptional activator of fatty catabolism but exerts a much broader role in lipid metabolism.
Supplementary Material
Acknowledgments
The experiments involving the Srebp-1c−/− and Chrebp−/− mice and their wild-type littermates were performed in the laboratory of Dr. Jay D. Horton at the Department of Molecular Genetics, University of Texas Southwestern Medical Center, Dallas. We thank Drs. Jay D. Horton and Kosuka Uyeda for providing the Srebp-1c−/− and Chrebp −/− mice, respectively. We thank Theo Boer, Trijnie Bos, Tineke Jager, and Frank Perton for excellent technical assistance.
This work was supported by Dutch Diabetes Foundation Grant 2002.00.041 and by EU Grant Hepadip 018734.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1 and Tables 1–4.
- TG
- triglyceride
- ChREBP
- carbohydrate response element-binding protein
- FGF-21
- fibroblast growth factor 21
- Glc-6-P
- glucose 6-phosphate
- GC-MS
- gas chromatography-mass spectrometry
- LXR
- liver X receptor
- MIDA
- mass isotopomer distribution analysis
- MUFA
- monounsaturated fatty acid
- NEFA
- non-esterified fatty acid
- PPAR
- peroxisome proliferator-activated receptor
- PPP
- pentose phosphate pathway
- SCD
- stearoyl-CoA desaturase
- SREBP
- sterol regulatory element-binding protein.
REFERENCES
- 1.Li Z. Z., Berk M., McIntyre T. M., Feldstein A. E. (2009) J. Biol. Chem. 284, 5637–5644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kersten S., Seydoux J., Peters J. M., Gonzalez F. J., Desvergne B., Wahli W. (1999) J. Clin. Investig. 103, 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leone T. C., Weinheimer C. J., Kelly D. P. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 7473–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patsouris D., Reddy J. K., Müller M., Kersten S. (2006) Endocrinology 147, 1508–1516 [DOI] [PubMed] [Google Scholar]
- 5.Mandard S., Müller M., Kersten S. (2004) Cell. Mol. Life Sci. 61, 393–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., Elmquist J. K., Gerard R. D., Burgess S. C., Hammer R. E., Mangelsdorf D. J., Kliewer S. A. (2007) Cell Metab. 5, 415–425 [DOI] [PubMed] [Google Scholar]
- 7.Badman M. K., Pissios P., Kennedy A. R., Koukos G., Flier J. S., Maratos-Flier E. (2007) Cell Metab. 5, 426–437 [DOI] [PubMed] [Google Scholar]
- 8.Dalen K. T., Ulven S. M., Arntsen B. M., Solaas K., Nebb H. I. (2006) J. Lipid Res. 47, 931–943 [DOI] [PubMed] [Google Scholar]
- 9.Martin P. G., Guillou H., Lasserre F., Déjean S., Lan A., Pascussi J. M., Sancristobal M., Legrand P., Besse P., Pineau T. (2007) Hepatology 45, 767–777 [DOI] [PubMed] [Google Scholar]
- 10.Malhi H., Gores G. J. (2008) Semin. Liver Dis. 28, 360–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devchand P. R., Keller H., Peters J. M., Vazquez M., Gonzalez F. J., Wahli W. (1996) Nature 384, 39–43 [DOI] [PubMed] [Google Scholar]
- 12.Kelly L. J., Vicario P. P., Thompson G. M., Candelore M. R., Doebber T. W., Ventre J., Wu M. S., Meurer R., Forrest M. J., Conner M. W., Cascieri M. A., Moller D. E. (1998) Endocrinology 139, 4920–4927 [DOI] [PubMed] [Google Scholar]
- 13.Schrauwen P., Hesselink M. K. (2004) Proc. Nutr. Soc. 63, 287–292 [DOI] [PubMed] [Google Scholar]
- 14.Andrews Z. B., Horvath T. L. (2009) Am. J. Physiol. Endocrinol. Metab. 296, E621–E627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrader M., Fahimi H. D. (2006) Biochim. Biophys. Acta 1763, 1755–1766 [DOI] [PubMed] [Google Scholar]
- 16.Guéraud F., Alary J., Costet P., Debrauwer L., Dolo L., Pineau T., Paris A. (1999) J. Lipid Res. 40, 152–159 [PubMed] [Google Scholar]
- 17.Toyama T., Nakamura H., Harano Y., Yamauchi N., Morita A., Kirishima T., Minami M., Itoh Y., Okanoue T. (2004) Biochem. Biophys. Res. Commun. 324, 697–704 [DOI] [PubMed] [Google Scholar]
- 18.Inoue I., Noji S., Awata T., Takahashi K., Nakajima T., Sonoda M., Komoda T., Katayama S. (1998) Life Sci. 63, 135–144 [DOI] [PubMed] [Google Scholar]
- 19.Stienstra R., Mandard S., Patsouris D., Maass C., Kersten S., Müller M. (2007) Endocrinology 148, 2753–2763 [DOI] [PubMed] [Google Scholar]
- 20.Shiri-Sverdlov R., Wouters K., van Gorp P. J., Gijbels M. J., Noel B., Buffat L., Staels B., Maeda N., van Bilsen M., Hofker M. H. (2006) J. Hepatol. 44, 732–741 [DOI] [PubMed] [Google Scholar]
- 21.Yeon J. E., Choi K. M., Baik S. H., Kim K. O., Lim H. J., Park K. H., Kim J. Y., Park J. J., Kim J. S., Bak Y. T., Byun K. S., Lee C. H. (2004) J. Gastroenterol. Hepatol. 19, 799–804 [DOI] [PubMed] [Google Scholar]
- 22.Seo Y. S., Kim J. H., Jo N. Y., Choi K. M., Baik S. H., Park J. J., Kim J. S., Byun K. S., Bak Y. T., Lee C. H., Kim A., Yeon J. E. (2008) J. Gastroenterol. Hepatol. 23, 102–109 [DOI] [PubMed] [Google Scholar]
- 23.Staels B., Maes M., Zambon A. (2008) Nat. Clin. Pract. Cardiovasc. Med. 5, 542–553 [DOI] [PubMed] [Google Scholar]
- 24.Knight B. L., Hebbachi A., Hauton D., Brown A. M., Wiggins D., Patel D. D., Gibbons G. F. (2005) Biochem. J. 389, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller C. W., Ntambi J. M. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 9443–9448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang G., Yang J., Horton J. D., Hammer R. E., Goldstein J. L., Brown M. S. (2002) J. Biol. Chem. 277, 9520–9528 [DOI] [PubMed] [Google Scholar]
- 27.Iizuka K., Bruick R. K., Liang G., Horton J. D., Uyeda K. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 7281–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 29.Bligh E. G., Dyer W. J. (1959) Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 30.Muskiet F. A., van Doormaal J. J., Martini I. A., Wolthers B. G., van der Slik W. (1983) J. Chromatogr. 278, 231–244 [DOI] [PubMed] [Google Scholar]
- 31.Hohorst H. J. (1970) in Methoden der Enzymatischen Analyse (Bergmeyer H. U. ed) pp. 1200–1204, Verlag Chemie, Weinheim, Germany [Google Scholar]
- 32.Keppler D., Decker K. (1970) in Methoden der Enzymatischen Analyse (Bergmeyer H. U. ed) pp. 1089–1094, Verlag Chemie, Weinheim, Germany [Google Scholar]
- 33.Kuipers F., Havinga R., Bosschieter H., Toorop G. P., Hindriks F. R., Vonk R. J. (1985) Gastroenterology 88, 403–411 [DOI] [PubMed] [Google Scholar]
- 34.van der Veen J. N., Havinga R., Bloks V. W., Groen A. K., Kuipers F. (2007) J. Lipid Res. 48, 337–347 [DOI] [PubMed] [Google Scholar]
- 35.Lee W. N., Byerley L. O., Bergner E. A., Edmond J. (1991) Biol. Mass Spectrom. 20, 451–458 [DOI] [PubMed] [Google Scholar]
- 36.Oosterveer M. H., Van Dijk T. H., Tietge U. J., Boer T., Havinga R., Stellaard F., Groen A. K., Kuipers F., Reijngoud D. J. (2009) PLoS One 4, e6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derks T. G., Van Dijk T. H., Grefhorst A., Rake J. P., Smit G. P., Kuipers F., Reijngoud D. J. (2008) Hepatology 47, 1032–1042 [DOI] [PubMed] [Google Scholar]
- 38.Van Dijk T. H., Boer T. S., Havinga R., Stellaard F., Kuipers F., Reijngoud D. J. (2003) Anal. Biochem. 322, 1–13 [DOI] [PubMed] [Google Scholar]
- 39.Oosterveer M. H., Van Dijk T. H., Grefhorst A., Bloks V. W., Havinga R., Kuipers F., Reijngoud D. J. (2008) J. Biol. Chem. 283, 25437–25445 [DOI] [PubMed] [Google Scholar]
- 40.Kabashima T., Kawaguchi T., Wadzinski B. E., Uyeda K. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 5107–5112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Castelein H., Gulick T., Declercq P. E., Mannaerts G. P., Moore D. D., Baes M. I. (1994) J. Biol. Chem. 269, 26754–26758 [PubMed] [Google Scholar]
- 42.Ogata M., Tsujita M., Hossain M. A., Akita N., Gonzalez F. J., Staels B., Suzuki S., Fukutomi T., Kimura G., Yokoyama S. (2009) Atherosclerosis 205, 413–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hebbachi A. M., Knight B. L., Wiggins D., Patel D. D., Gibbons G. F. (2008) J. Biol. Chem. 283, 4866–4876 [DOI] [PubMed] [Google Scholar]
- 44.Chakravarthy M. V., Pan Z., Zhu Y., Tordjman K., Schneider J. G., Coleman T., Turk J., Semenkovich C. F. (2005) Cell Metab. 1, 309–322 [DOI] [PubMed] [Google Scholar]
- 45.Sampath H., Miyazaki M., Dobrzyn A., Ntambi J. M. (2007) J. Biol. Chem. 282, 2483–2493 [DOI] [PubMed] [Google Scholar]
- 46.König B., Koch A., Spielmann J., Hilgenfeld C., Hirche F., Stangl G. I., Eder K. (2009) Eur. J. Pharmacol. 605, 23–30 [DOI] [PubMed] [Google Scholar]
- 47.Lundåsen T., Hunt M. C., Nilsson L. M., Sanyal S., Angelin B., Alexson S. E., Rudling M. (2007) Biochem. Biophys. Res. Commun. 360, 437–440 [DOI] [PubMed] [Google Scholar]
- 48.Cheung C., Akiyama T. E., Ward J. M., Nicol C. J., Feigenbaum L., Vinson C., Gonzalez F. J. (2004) Cancer Res. 64, 3849–3854 [DOI] [PubMed] [Google Scholar]
- 49.Wong D. A., Bassilian S., Lim S., Paul Lee W. N. (2004) J. Biol. Chem. 279, 41302–41309 [DOI] [PubMed] [Google Scholar]
- 50.Reszko A. E., Kasumov T., David F., Jobbins K. A., Thomas K. R., Hoppel C. L., Brunengraber H., Des R. C. (2004) J. Biol. Chem. 279, 19574–19579 [DOI] [PubMed] [Google Scholar]
- 51.Sugden M. C., Holness M. J. (2003) Am. J. Physiol. Endocrinol. Metab. 284, E855–E862 [DOI] [PubMed] [Google Scholar]
- 52.Patsouris D., Mandard S., Voshol P. J., Escher P., Tan N. S., Havekes L. M., Koenig W., März W., Tafuri S., Wahli W., Müller M., Kersten S. (2004) J. Clin. Investig. 114, 94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu J., Chang V., Joseph S. B., Trujillo C., Bassilian S., Saad M. F., Lee W. N., Kurland I. J. (2004) Endocrinology 145, 1087–1095 [DOI] [PubMed] [Google Scholar]
- 54.Sriram G., Rahib L., He J. S., Campos A. E., Parr L. S., Liao J. C., Dipple K. M. (2008) Mol. Genet. Metab. 93, 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spolarics Z. (1998) J. Leukoc. Biol. 63, 534–541 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.