

Abstract
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)2 is a classic glycolytic enzyme that also mediates cell death by its nuclear translocation under oxidative stress. Meanwhile, we previously presented that oxidative stress induced disulfide-bonded GAPDH aggregation in vitro. Here, we propose that GAPDH aggregate formation might participate in oxidative stress-induced cell death both in vitro and in vivo. We show that human GAPDH amyloid-like aggregate formation depends on the active site cysteine-152 (Cys-152) in vitro. In SH-SY5Y neuroblastoma, treatment with dopamine decreases the cell viability concentration-dependently (IC50 = 202 μm). Low concentrations of dopamine (50–100 μm) mainly cause nuclear translocation of GAPDH, whereas the levels of GAPDH aggregates correlate with high concentrations of dopamine (200–300 μm)-induced cell death. Doxycycline-inducible overexpression of wild-type GAPDH in SH-SY5Y, but not the Cys-152-substituted mutant (C152A-GAPDH), accelerates cell death accompanying both endogenous and exogenous GAPDH aggregate formation in response to high concentrations of dopamine. Deprenyl, a blocker of GAPDH nuclear translocation, fails to inhibit the aggregation both in vitro and in cells but reduced cell death in SH-SY5Y treated with only a low concentration of dopamine (100 μm). These results suggest that GAPDH participates in oxidative stress-induced cell death via an alternative mechanism in which aggregation but not nuclear translocation of GAPDH plays a role. Moreover, we observe endogenous GAPDH aggregate formation in nigra-striatum dopaminergic neurons after methamphetamine treatment in mice. In transgenic mice overexpressing wild-type GAPDH, increased dopaminergic neuron loss and GAPDH aggregate formation are observed. These data suggest a critical role of GAPDH aggregates in oxidative stress-induced brain damage.
Introduction
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a classic glycolytic enzyme that is also involved in cell death and neuropsychiatric conditions (1, 2). GAPDH mediates cell death under oxidative stress conditions at least in part through nuclear translocation together with Siah (3). In the nucleus, GAPDH activates p300/CBP and regulates gene transcription (4). The pathway can be blocked by deprenyl (Selegiline), a neuroprotective compound (5). Although nuclear translocation of GAPDH is known to cause cell death, other mechanisms of GAPDH-associated cell death may also exist.
Several neurodegenerative diseases are characterized by the accumulation of misfolded proteins, resulting in intracellular and extracellular protein aggregates (6, 7). For instance, conformational changes in β-amyloid (Aβ) in Alzheimer disease and α-synuclein in Parkinson disease lead to the formation of abnormal oligomers and amyloid fibrils (8). Similar to Aβ and α-synuclein, GAPDH is also amyloidogenic (9–14). We previously reported the molecular mechanism underlying oxidative stress-induced amyloid-like aggregation of GAPDH using the purified rabbit GAPDH and demonstrated the critical role of the active site cysteine in its aggregation (15). The active site cysteine also plays a key role in nuclear translocation of GAPDH during oxidative stress-mediated cell damage (3). Thus, this specific cysteine residue has two roles in oxidative stress-related events.
Methamphetamine (METH) is a psychostimulant that increases extracellular dopamine levels, which frequently results in oxidative stress and its associated cellular damage (16, 17). The pathway by which METH causes dopaminergic neurotoxicity shares similarities with other neurodegenerative pathways, including the GAPDH-Siah pathway (3–5), which is activated in both animals and cells by the Parkinson disease-inducing agent, 1-methyl-4-phelyl-1,2,3,6,-tetrahydropyridine (MPTP) (18). Together with the observation that METH causes an increase in striatal GAPDH protein levels (19), these findings led us to the hypothesis that GAPDH might participate in the METH neurotoxicity pathway.
Here, we show that human GAPDH aggregation is triggered by oxidation of the specific cysteine (Cys-152) and participates in dopamine-induced cell death of dopaminergic neuroblastoma SH-SY5Y. Treatment of mice with METH causes GAPDH aggregate formation in vivo. Moreover, in transgenic mice overexpressing wild-type GAPDH, METH-induced dopamine neuron loss and GAPDH aggregate formation are increased.
EXPERIMENTAL PROCEDURES
Chemicals, Plasmids, and Antibodies
Unless otherwise noted, chemicals were of analytical grade. Deprenyl (Selegiline) is kindly provided from Fujimoto Pharmaceutical (Osaka, Japan).
The cloning of human wild-type (WT) GAPDH cDNA was performed as reported previously (15). For bacterial expression, cDNA was cloned into pBAD-HisA (Invitrogen) using the SacI-KpnI sites. Briefly, 1 μg of total RNA, isolated using an RNeasyTM mini kit (Qiagen) from human neuroblastoma cells SH-SY5Y, was reverse-transcribed at 37 °C for 1 h using OmniscriptTM reverse transcriptase (Qiagen) with oligo(dT) primers to produce each of the cDNAs. The sequences of the primers for amplification of the full-length wild-type GAPDH cDNA were 5′-GGCCGCGAGCTCATGGGGAAGGTGAA-3′ (forward) and 5′-CCTCTAGAGGTACCTTACTCCTTGGAG-3′ (reverse). The bold letters in the sequences were designed for SacI and KpnI restriction sites for the forward and reverse primers respectively. The PCR products were digested, purified, and cloned into the pBAD-HisA bacterial expression vector with 5′-His6 according to the manufacturer's protocol (Invitrogen). For mammalian cell line expression, cDNA was cloned into pcDNA4- TO-Myc/HisA (Invitrogen) using the EcoRI-EcoRV sites. Alternatively, GAPDH cDNAs were re-amplified using the pcDNA3.1/ Myc-His-human GAPDH (generously provided from Drs. Yamaji and Harada at Osaka Prefecture University) as a template according to the manufacturer's protocol. The sequences of the primers were 5′- CAAGCTTGGAATTCCGTTATGGGGAAGGTG-3′ (forward) and 5′-TCCCATATGAGATATCCCTCCTTGGA-3′ (reverse). The bold letters in the sequences were designed as EcoRI and EcoRV restriction sites for the forward and reverse primers, respectively. All constructs were transformed into Escherichia coli DH5α (TOYOBO, Tokyo, Japan), and then several colonies were grown in LB broth plus 100 μg/ml ampicillin. The plasmids were purified by a QIAfilterTM plamid midi kit, and the complete sequences were confirmed using a 373A DNA sequencer (PerkinElmer Life Sciences). The sequence of the cloned cDNA of human GAPDH was completely identical to that reported (accession no. M33197).
Using the WT GAPDH as a template, the alanine- or serine- substituted mutants C152A, C156S, and C247A-GAPDH were generated using the QuikChange site-directed mutagenesis kit according to the manufacturer's protocol (Stratagene). The complementary primer pairs were designed as follows: C152A, 5′-ATCAGCAATGCCTCCGCCACCACCAAC-3′; C156S, 5′-TCCTGCACCACCAACTCCTTAGCACCCCTG-3; C247A, 5′-GTGGTGGACCTGACCGCCCGTCTAGAAAAA-3′. The bold letters in the sequences represent the site of the point mutation. The sequences of all mutants were also confirmed as described above.
Antibodies were purchased from the following companies; anti-GAPDH monoclonal antibody from Chemicon (MAB374); anti-GAPDH polyclonal antibody from Abcam (ab9485); anti-Myc monoclonal antibody from Nacalai tesque (#04362-34, Kyoto, Japan); anti-tyrosine hydroxylase (ΤΗ) monoclonal antibody from Roche Applied Science (1017-381); anti-dinitrophenol monoclonal antibody from Monosan (MONX10960); anti-histone H2B monoclonal antibody from Upstate Cell Signaling solutions (07-371).
Expression and Purification of Recombinant GAPDH
The pBAD-HisA vector carrying WT- or C152A-GAPDH cDNA was transformed into gap(−) E. coli. Strain W3CG (20). These recombinant GAPDH proteins were expressed and then purified as previously described (15). Briefly, the transformants were cultured for 2 h at 37 °C in M63 minimal medium containing both 50 μg/ml ampicillin and 15 μg/ml tetracycline, and then 0.2% (w/v) l-(+)arabinose was added to the medium. After 24 h the cells expressing recombinant proteins were collected by centrifugation (3000 × g, 15 min at 4 °C) and resuspended in lysis buffer containing 50 mm sodium phosphate (pH 8.0), 300 mm NaCl, 30 mm imidazole, 10% glycerol, and 2 mm 2-mercaptoethanol. The suspensions were sonicated on ice and centrifuged at 15,000 × g for 30 min (4 °C). The supernatants were incubated with nickel-nitrilotriacetic acid-agarose resin (50% slurry) for 2 h at room temperature with rocking. The resin was washed with 50 mm phosphate buffer (pH 8.0) containing 300 mm NaCl, 50 mm imidazole, 10% glycerol, and 2 mm 2-mercaptoethanol. The proteins bound to the resin were eluted with 50 mm phosphate buffer (pH 8.0) containing 300 mm NaCl, 300 mm imidazole, 10% glycerol, and 2 mm 2-mercaptoethanol, and the eluates were immediately mixed with 1 mm NAD+, 1 mm dithiothreitol, and 1 mm EDTA followed by incubation at 4 °C overnight. The reduced proteins were directly loaded onto a PD-10 column (GE Healthcare) equilibrated with buffer G2′ containing 50 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA, and 5% glycerol. The fractions containing GAPDH were pooled and concentrated using Amicon Ultra-15 (Millipore Japan, Tokyo, Japan). The protein concentrations were determined spectrophotometrically assuming a ϵ0.1% at 280 nm = 1.0.
GAPDH Enzymatic Assay
GAPDH enzymatic assay for purified recombinant proteins were performed as previously described (21) with some modifications. Briefly, the reaction mixture (100 μl) was 100 mm triethanolamine hydrochloride buffer (pH 8.9) containing 1 mm EDTA, 0.1 m KCl, 10 mm K2HPO4, 0.2 mm NAD+, and 0.8 mm d-glyceraldehyde-3-phosphate. The reaction was started by the addition of purified GAPDH (0.5–1 μg) to the reaction mixture at room temperature. The initial velocity of an increase in absorbance at 340 nm due to the formation of NADH was measured for 1 min using a VERSA Max microplate reader (Molecular Devices, Sunnyvale, CA).
In Vitro Aggregation Assay
In vitro GAPDH aggregation assays for purified recombinant proteins were performed as previously described (15). The recombinant GAPDH (0.6 mg/ml) was treated with various concentrations of NOR3 (1-(±)-(E)-4-ethyl-2-[(E)-hydroxyimino]-5-nitro-3-hexenamide (300 μm)), a nitric oxide donor, and the period of incubation with NOR3 was varied (1–48 h). To measure the turbidity of solutions derived from aggregated GAPDH, the absorbance at 405 nm was recorded using a VERSA Max microplate reader (Molecular Devices). Alternatively, the reaction mixtures were centrifuged (22,000 × g, 30 min), and the supernatants (soluble GAPDH) were collected. The pellets (insoluble GAPDH) were washed twice, resuspended in an equal volume of buffer G2′, and sonicated on ice for 10 s. The samples were subjected to either reducing or non-reducing SDS-PAGE.
Structural Analysis of Aggregated GAPDH
A subset of structural analyses of GAPDH for purified recombinant proteins was performed according to published methods (15).
Circular Dichroism
After GAPDH (0.6 mg/ml) was treated with a control or 100 μm NOR3 for 1 h at 37 °C, the reaction mixture was desalted with a NAP-5 column (GE Healthcare), and the eluate was centrifuged at 22,000 × g for 30 min, providing cleared supernatant. The circular dichroism (CD) spectrum of GAPDH was measured with a spectropolarimeter model J-820 (Jasco, Tokyo, Japan). The temperature of the solutions in the cuvette was controlled at 37 °C by circulating water. The path length of the optical quartz cuvette was 1.0 mm for far-UV CD measurements at 200–250 nm and 10 mm for near-UV CD measurements at 250–300 nm. GAPDH was dissolved in buffer G2′ at a concentration of 0.1 to 0.2 mg/ml. Spectra were obtained as the average of 10 successive scans with a bandwidth of 2.0 nm. The data were expressed as molar residue ellipticity (θ).
Thioflavin-S Fluorescence
50 μl of GAPDH sample was mixed with 450 μl of thioflavin-S solution (3.2 mg/ml in 20 mm MOPS (pH 6.5)), and the fluorescence intensity was measured at an excitation wavelength of 450 nm and an emission of 482 nm using a Fluorescence Spectrophotometer F-2000 (Hitachi, Tokyo, Japan).
Congo Red Birefringence
Aliquots (100 μl) of GAPDH (0.6 mg/ml) as is or treated with 100 μm NOR3 for 24 h at 37 °C were added to 900 μl of Congo Red solution (25 μg/ml in PBS). This mixture was incubated for 30 min at room temperature and then centrifuged at 15,000 × g for 30 min. The pellet was resuspended in 100 μl of Milli-Q water (Millipore). The drops of this suspension were allowed to dry on a slide glass for 10 min. The birefringence was determined with an Eclipse LV100POL microscope equipped with a polarizing stage (Nikon, Tokyo, Japan). As a positive control, amyloidal aggregates of β-amyloid (1–42) (Peptide Institute, Osaka, Japan) were prepared according to the established procedures (22).
Cell Culture, Subcellular Fractionation, and Western Blotting
Human SH-SY5Y neuroblastoma cells (ATCC) were grown in Dulbecco's modified Eagle's medium/Ham's F-12 medium supplemented with 10% fetal bovine serum, 2 mm glutamine, and antibiotics-antimycotics (Invitrogen) at 37 °C in a 5% CO2 humidified incubator. Human carcinoma HeLa cells (ATCC) were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mm glutamine, and antibiotics-antimycotics in the same environment. Subcellular fractionation and detection of intracellular GAPDH oligomers by Western blotting were performed as previously described (15). After treatment with either a control or oxidative stress (dopamine or NOC18), cells were washed twice with PBS and then incubated for 5 min in ice-cold PBS containing 40 mm iodoacetamide to protect unmodified thiols from oxidation during fractionation. All the subsequent steps were performed at 4 °C. Cells were scraped in 500 μl of buffer A containing 10 mm Tris-HCl (pH 7.5), 10 mm NaCl, 3 mm MgCl2, 0.05% Nonidet P-40, 0.5% Triton-X100, 40 mm iodoacetamide, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture. After 10 min, the suspensions of cells were mixed vigorously for 15 s, and the aliquots were collected as a total cell lysate. The remaining lysates were centrifuged at 800 × g for 5 min, and the pellets were collected. The pellets were then resuspended in 200 μl of buffer B containing 10 mm Hepes-KOH (pH 7.4), 25 mm NaCl, 3 mm MgCl2, 300 mm sucrose, 40 mm iodoacetamide, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture and washed 2 times by centrifugation (3000 × g for 10 min) followed by suspension in buffer. After the addition of 100 μl of buffer B, the pellets were sonicated for 30 s and finally obtained as particulate fractions. All samples were stored at −80 °C until use. Protein concentrations of the samples were determined by the Bradford assay (Bio-Rad). Both total cell lysates and fractionated proteins were mixed with an equal volume of SDS-sample buffer containing 0.25 m Tris-HCl (pH 6.8), 2% SDS, 30% glycerol, 0.01% bromphenol blue in the presence (reduced) or the absence (non-reduced) of 100 mm dithiothreitol and then heated at 100 °C for 5 min. These samples were separated by 5–20% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). The membranes were incubated for 1 h with Blocking One (Nacalai Tesque, Kyoto, Japan) to block nonspecific binding. The membrane was then incubated for 2 h at room temperature with an anti-GAPDH monoclonal antibody (1:300), an anti-Myc monoclonal antibody (1:1000), or an anti-TH monoclonal antibody (1:200) in 10% Blocking One-PBST (0.05% Tween 20, and 0.02% NaN3 in PBS) followed by incubation for 1 h at room temperature with a peroxidase-conjugated affinity-purified secondary antibody (Zymed Laboratories Inc.). The membranes were also reprobed with an anti-histone H2B polyclonal antibody (1:5000). Detection was performed using ECL plus and HyperFilm according to the manufacturer's protocol (GE Healthcare). The intensity of the bands was measured using Scion image software.
Generation of Stable Cell Lines for Inducible Expression of GAPDH
SH-SY5Y cells were co-transfected with both pcDNA6/TR (Invitrogen) and pcDNA4-TO/Myc-HisA vector carrying WT- or C152A-GAPDH (or mock-transfected) using Lipofectamine 2000 (Invitrogen) or HilyMax (Dojindo, Kumamoto, Japan). After 2–3 weeks, colonies resistant to both blasticidin (20 μg/ml) and zeocin (100 μg/ml) were dual-selected for Tet-repressor and recombinant GAPDH. Stable cells resistant to these antibiotics were treated without or with doxycycline (1 μg/ml) for 1–9 days followed by screening for their capacity to induce Myc-tagged GAPDH proteins using Western blotting.
Cell Immunofluorescence
We performed immunofluorescence as described in Nakajima et al. (15). After treatment with either a control or oxidative stress (dopamine or NOC18), cells were washed twice with ice-cold PBS and fixed with 4% paraformaldehyde in PBS (pH 7.4) for 10 min at room temperature. The cells were incubated with Blocking One for 1 h at room temperature to block nonspecific binding and permeabilized with PBS containing 0.1% Triton X-100 for 5 min. The cells were then incubated with an anti-GAPDH polyclonal antibody (1:1000) or anti-Myc monoclonal antibody (1:1000) in 10% Blocking One-PBST overnight at 4 °C. After four washings with PBST, the specific signals were visualized by staining the cells with an Alexa488 or an Alexa568-conjugated secondary antibody (Invitrogen, 1:2000) using a confocal scanning microscope (C1si-TE2000-E; Nikon). For nuclear staining, the cells were labeled with 4′,6-diamidino-2-phenylindole (Dojindo, 1 μg/ml) for 10 min.
Assessment of GAPDH Aggregate Formation and Nuclear Translocation of GAPDH in Cell Staining
Detailed procedures are described in the legend to supplemental Fig. S3. For semiquantification of cells with aggregates, five microscopic fields were selected at random, and the number of cells with aggregates among at least 500 total cells was quantified (%). GAPDH nuclear translocation was evaluated by 4′,6-diamidino-2-phenylindole staining. Semiquantification of cells with GAPDH nuclear translocation was performed using the same procedures.
Cytotoxicity
Cytotoxicity was measured as cell viability using the Cell Titer Glo Luminescent Cell Viability Assay kit (Promega) according to the manufacturer's protocol.
Animal Procedures
All procedures related to animals were performed according to the guidelines set by the Animal Ethical Committee of Osaka Prefecture University. The detailed procedure of preparing GAPDH transgenic mice was described in supplemental Fig. S5. METH ((2S)-N-methyl-1-phenyl-propan-2-amine (Dainippon Sumitomo Pharma Co., Ltd., Osaka, Japan)) treatment of mice (2 × 5 mg/kg, intraperitoneal, 2-h interval) were performed as previously described (23). Double immunofluorescence staining of paraffin-embedded brain sections were performed using both an anti-GAPDH polyclonal antibody (1:1000) and an anti-TH antibody (1:200). Protein carbonyls in the striatum, a hallmark for oxidative stress, were detected by Western blotting using anti-dinitrophenol antibody (24). The dopamine contents in the striatum were measured by electrochemical detection-HPLC (16). Briefly, acidic extracts of striatum were applied to a C18 reversed phase column (Mightysil R18 GP250-4.6, Kanto Chemical, Japan) equilibrated with a mobile phase composed of 70 mm sodium acetate, 78 mm citric acid, 0.014 mm Na2EDTA, 1 mm sodium 1-heptanesulfonic acid with 22% methanol at a flow rate of 1 ml/min. l-Dopamine, 3,4-dihydroxyphenylacetic acid, and homovanillic acid were detected using an analytical electrochemical detection (ESA Coulochem III electrochemical detector; E1 = −100 mV, E2 = 500 mV). Commercial l-dopamine, 3,4-dihydroxyphenylacetic acid, and homovanillic acid were used as the standard.
RESULTS
In a previous study we demonstrated that two of four Cys residues present in rabbit GAPDH play key roles in aggregate formation under oxidative stress conditions (15). Aggregate formation requires oxidation of Cys-149 and is accelerated by further oxidation at Cys-281. In human GAPDH the equivalent to Cys-149 (Cys-152) is conserved, but a serine residue is found at the equivalent to the Cys-281 position. Thus, we hypothesized that Cys-152 in human GAPDH might also play a key role in aggregate formation. To test this idea we purified recombinant human WT GAPDH protein as well as three human GAPDH mutants in which each of the cysteine residues was substituted (Fig. 1A) and tested whether or not aggregate formation was altered by these mutations. Mutation at Cys-152 but not at the other two cysteines altered GAPDH glycolytic activity (Fig. 1B). In addition, aggregate formation of GAPDH induced by the nitric oxide donor NOR3 was almost totally suppressed by mutating Cys-152 (Fig. 1C). Cys-152-specific effects on the formation of disulfide-bonded oligomers were consistently observed by non-reduced SDS-PAGE (Fig. 1, D and E). Other oxidants such as H2O2, S-nitrosoglutathione, peroxynitrite, and dopamine also elicited GAPDH aggregation (supplemental Fig. S1, A and B). To validate these in vitro biochemical observations, HeLa cells were transfected with WT or C152A mutant GAPDH. Similar results were obtained from these cell-based assays (supplemental Fig. S1, C and D).
FIGURE 1.

The active site cysteine (Cys-152) plays an essential role in oxidative stress-induced disulfide-bonded GAPDH aggregate formation. A, shown is a ribbon diagram of human GAPDH monomer (PDB code 1U8F). Side chains of three cysteines (152, 156, and 247) are indicated as yellow bonds. B, enzymatic activities of WT-, C152A-, C156S-, and C247A-GAPDH are indicated as a percentage of that of WT GAPDH. Data are indicated as the means ± S.D. of four samples (t test; **, p < 0.01). C, time course and concentration dependence of the increase in turbidity of GAPDH solutions in the presence of NOR3 are shown. Recombinant GAPDH (0.6 mg/ml) was treated with or without NOR3 (100 μm) at 37 °C for the indicated times (left panel) or treated with or without the indicated concentrations of NOR3 at 37 °C for 72 h (right panel). Data are presented as the means ± S.D. of three samples. D, NOR3-induced disulfide-bonded aggregates of GAPDH are shown. Samples without NOR3 were used as controls (left panel). The reaction mixtures treated with or without NOR3 (100 μm) at 37 °C for 72 h were centrifuged, generating supernatants (S) and pellets (P). The samples were subjected to either reduced or non-reduced 5–20% SDS-PAGE (right panels). E, semiquantification of the insolubility of GAPDH derived from D is shown as a pellet/(pellet + supernatant) ratio. The pellet or supernatant value is expressed as all of intensity of each lane, which was measured by Scion image software. Data are indicated as the means ± S.D. of three samples (t test; **, p < 0.01 versus WT).
Our previous report also demonstrated that oxidative stress induced conformational changes in rabbit GAPDH, resulting in amyloid-like aggregates (15). We, therefore, examined whether or not NOR3 led to structural changes in human GAPDH by measuring the CD spectra. The absolute far-UV CD intensity increased between the wavelengths of 205–230 nm as compared with untreated WT GAPDH, suggesting augmentation of the secondary structure of human GAPDH (Fig. 2A, left upper panel). The C152A mutant, however, displayed no NOR3-induced increase of the intensity (Fig. 2A, right upper panel). Additionally, the near-UV CD spectrum of WT GAPDH exhibited cotton effects, with negative maxima at 269, 279, 282, 285, and 291 nm and positive maxima at 259, 264, 270, 272, 280, 284, and 289 nm, indicating the anisotropic environment of the aromatic side chains (Fig. 2A, left lower panel) but not in the C152A mutant (Fig. 2A, right lower panel). Moreover, the GAPDH aggregates showed an affinity to amyloid dyes, such as thioflavin-S and Congo Red (Fig. 2, B and C). Consistently, NOR3 barely increased the thioflavin-S-dependent fluorescence intensity of C152A-GAPDH (Fig. 2B). The birefringence of the GAPDH aggregates labeled by Congo Red somewhat resembled that of amyloid β 1–42 (Aβ1–42), but the GAPDH aggregates were granular, whereas Aβ1–42 aggregates were fibrous (Fig. 2C, High magnification). Together, these results indicate that both the increase in the secondary structure and the conformational changes of human GAPDH are triggered by NOR3-treatment via the Cys-152 residue and that these structural changes cause amyloid-like aggregates of human GAPDH, such as rabbit GAPDH.
FIGURE 2.

Structural analysis of aggregated GAPDH. A, shown are far UV (upper panels) and near UV (lower panels) CD spectra of WT- or C152A-GAPDH-treated without (black lines) or with 100 μm NOR3 (red lines) at 37 °C for 1 h. The data are expressed as molar residue ellipticity (θ). B, thioflavin-S binding-dependent fluorescence of recombinant GAPDH treated with 100 μm NOR3 at 37 °C for 72 h is shown. Data are the means ± S.D. of four samples (t test; **, p < 0.01 versus WT). C, Congo Red birefringence of aggregated proteins is shown. WT GAPDH treated with 100 μm NOR3 for 72 h at 37 °C was shown under polarized lights. Aggregated Aβ1–42 (20 μm) was prepared by established methods (22). The magnification is ×20 (upper panels) or ×100 (lower panels). A.U., arbitrary units.
To address whether or not human GAPDH aggregate formation plays a role in cell death, we investigated the relationship between cell viability and GAPDH aggregate formation in SH-SY5Y human dopaminergic neuroblastoma cell lines. Because GAPDH aggregate formation is triggered by oxidative stress in vitro, we applied dopamine to the cells. Dopamine is thought to cause oxidative stress and leads to neuronal cell damage in neuropsychiatric and neurodegenerative diseases (25, 26). We confirmed that dopamine treatment markedly increased the level of oxidative stress in cells (supplemental Fig. S2). Treatment of SH-SY5Y cells with dopamine decreased the cell viability in a concentration-dependent manner (Fig. 3A). The IC50 value was 202 μm. We observed robust formation of disulfide-bonded GAPDH aggregates in cells 48 h after the addition of dopamine (200–300 μm) (Fig. 3, B and C). Consistently, the significant increases in the numbers of cells with GAPDH aggregates were caused by dopamine treatment (200–300 μm) (Fig. 3E). In contrast, nuclear translocation of GAPDH without aggregate formation was observed with lower doses of dopamine (50–100 μm) (Fig. 3, C and E). The relationship between GAPDH aggregate formation and dopamine-induced loss of cell viability was examined (Fig. 4). The aggregation over 20%, which corresponded to the IC50 value (202 μm), was closely correlated with the decrease of the cell viability caused by dopamine treatment (r2 = 0.96).
FIGURE 3.

Dopamine-induced cell death, GAPDH aggregation, and nuclear translocation in SH-SY5Y cells. A, concentration-dependent effects of dopamine on the viability of SH-SY5Y cells are shown. The cell viabilities were measured at 48 h after dopamine treatment. Data are the means ± S.D. of four samples (Dunnet's test; **, p < 0.01 versus no dopamine). The IC50 value (202 μm) is calculated from the concentration dependence of the inhibition curves using nonlinear regression analyses assisted by a GraphPad Prizm 4 software. B, dopamine-induced disulfide-bonded aggregates of insoluble GAPDH in SH-SY5Y cells are shown. The particulate fractions treated without or with dopamine (50–300 μm) were subjected to non-reduced 5–20% SDS-PAGE. WB, Western blot. C, immunofluorescence of GAPDH (red) in SH-SY5Y cells treated without or with dopamine (50–300 μm) for 48 h is shown. Nuclei were stained by 4′,6-diamidino-2-phenylindole (blue). Arrows or arrowheads indicate cells with GAPDH aggregates or nuclear translocation, respectively. The scale bar = 20 μm. IHC, immunohistochemistry; endo, endogenous. D, semiquantifications of the number of the cells with aggregates are shown. Data are the means ± S.D. of three samples (Dunnet's test; **, p < 0.01 versus no dopamine). E, semiquantification of the number of the cells with nuclear translocation are shown. Data are the means ± S.D. of three samples (Dunnet's test; **, p < 0.01 versus no dopamine).
FIGURE 4.
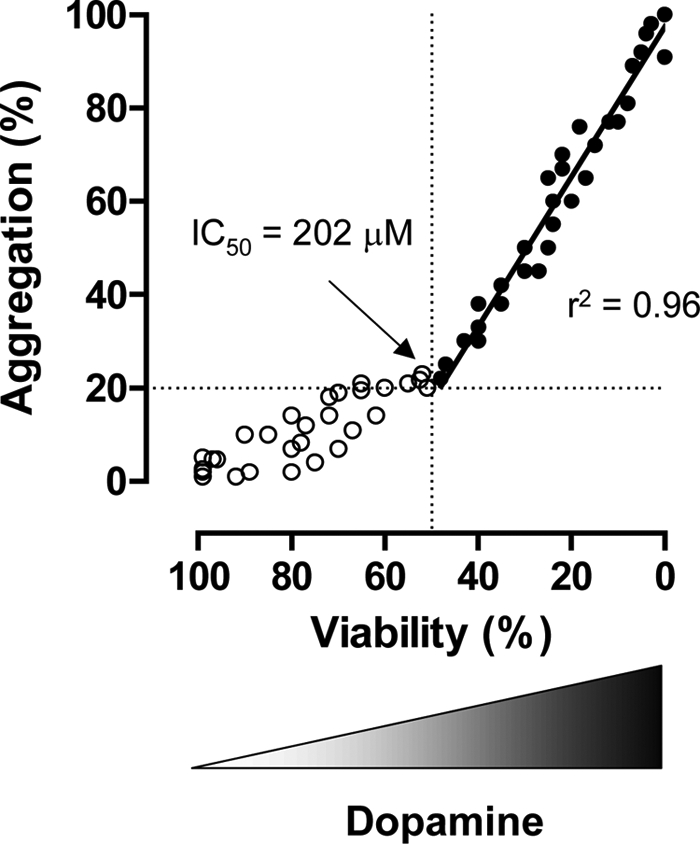
Correlation between GAPDH aggregation and dopamine-induced cell death. These plots indicate all of a datum of both the number of cells with GAPDH aggregates (%) and its viability (%) in single sample. The closed circles indicate as above the IC50 value. The correlation value (r2 = 0.96) is calculated using linear regression analyses assisted by GraphPad Prizm 4 software.
To further address that GAPDH aggregate formation might mediate oxidative stress-induced cell death, we generated stable SH-SY5Y neuroblastoma cell lines in which Myc-tagged WT- or C152A-GAPDH was expressed under the control of a doxycycline (DOX)-inducible promoter. The expression level of induced GAPDH treated by DOX was similar to that of endogenous GAPDH (supplemental Fig. S4A). Induced expression of Myc-tagged GAPDH in these cell lines was also confirmed by immunofluorescence (supplemental Fig. S4B). We found that the cellular glycolytic activities were augmented in cells expressing WT GAPDH, but not C152A-GAPDH, consistent with the results obtained from test tube studies (supplemental Fig. S4C).
We next investigated the effects of DOX-inducible overexpression of WT- or C152A-GAPDH on dopamine-induced cell death. Significant augmentation of cell death was observed in cells expressing WT GAPDH (Fig. 5A), but not C152A-GAPDH (Fig. 5B), when 100–300 μm dopamine was added. We observed robust formation of GAPDH aggregates in cells expressing WT GAPDH after the addition of dopamine (200–300 μm) (Fig. 5, C and D). In contrast, more pronounced nuclear translocation without aggregate formation was observed with lower doses of dopamine (50–100 μm) (Fig. 5C). In cells expressing C152A-GAPDH, we did not observe aggregate formation or nuclear translocation even in the presence of dopamine (Fig. 5C). We then examined how much aggregation there is of GAPDH in cells expressing mock, WT GAPDH, and C152A-GAPDH to demonstrate that GAPDH aggregation correlates with the degree of dopamine-induced cell death. Both endogenous and exogenous GAPDH in these cell lines were concomitantly detected by immunofluorescence using anti-GAPDH antibody (Fig. 5D, upper panels). The number of imaged particles was measured (Fig. 5D, lower panels). Significant levels of dopamine-induced increase of aggregation were observed in cells expressing WT GAPDH but not the C152A-GAPDH and mock-transfected (Fig. 5E).
FIGURE 5.

Dopamine-induced GAPDH aggregation, nuclear translocation, and cytotoxicity in DOX-inducible, GAPDH-expressing SH-SY5Y cells. A, effects of dopamine on the viability of WT GAPDH expressing cells with (+) or without (−) DOX treatment are shown. Data are the means ± S.D. of four samples (t test; **, p < 0.01 versus DOX (−) control cells). B, shown are the effects of dopamine on the viability of C152A-GAPDH expressing cells with (+) or without (−) DOX treatment are shown. Data are the means ± S.D. of four samples. C, immunofluorescence of Myc-tagged WT (upper panels) or C152A (lower panels)-GAPDH (green) in DOX-inducible cells treated without or with dopamine (50–300 μm) for 48 h is shown. Nuclei were stained by 4′,6-diamidino-2-phenylindole (red). Arrows or arrowheads indicate cells with GAPDH aggregates or the nuclear translocation, respectively. The scale bar = 20 μm. D, immunofluorescence of total GAPDH (red) including both endogenous and Myc-tagged induced WT- and C152A-GAPDH in DOX-inducible cells treated without or with dopamine (300 μm) for 48 h are shown (upper panels). Mock cells in the presence of DOX indicate a control. The lower panels show the imaged particles, namely total GAPDH aggregates. IHC, immunohistochemistry. E, quantifications of the number of particles per cell are shown. Data are the means ± S.D. of 20 samples (t test; **, p < 0.01 versus mock control cell).
GAPDH mediates cell death under oxidative stress through nuclear translocation (3). The pathway can be blocked by deprenyl (Selegiline), a neuroprotective compound (5). We, therefore, investigated the effect of deprenyl on dopamine-induced aggregation and cell death both in vitro and in cells (Fig. 6). Treatment of purified human WT GAPDH with deprenyl (1–100 nm) failed to block the increase of NOR3-indued turbidities in vitro (Fig. 6A). Consistently, deprenyl did not affect dopamine-induced aggregation of GAPDH in SH-SY5Y cells (Fig. 6B). On the other hand, we found that deprenyl significantly inhibited the dopamine-induced nuclear translocation in SH-SY5Y cells (Fig. 6C) and ameliorated the cell death treated with 100 μm dopamine but not with higher concentrations (200–300 μm) (Fig. 6D).
FIGURE 6.

Effects of deprenyl (DEP) on dopamine-induced GAPDH-aggregation, nuclear translocation, and cytotoxicity. A, no effects of DEP on the NOR3-induced increase of the turbidity of solution of WT GAPDH are shown. Deprenyl at the indicated concentrations were preincubated for 30 min at 4 °C before the NOR3 treatment. Data are the means ± S.D. of four samples. B, no effects of deprenyl on the dopamine-elicited aggregation of endogenous GAPDH in SH-SY5Y cells are shown. Deprenyl at 10 nm was preincubated for 30 min at 37 °C before the dopamine treatment. Data are the means ± S.D. of three samples. C, significant effects of deprenyl on the dopamine-elicited nuclear translocation of endogenous GAPDH in SH-SY5Y cells are shown. Data are the means ± S.D. of three samples (t test; **, p < 0.05 versus control cells). D, partial effectiveness of deprenyl on dopamine-elicited cytotoxity is shown. Deprenyl significantly prevented cytotoxicity only at 100 μm dopamine treatment. Data are the means ± S.D. of four samples (t test; **, p < 0.01 versus control).
To ascertain whether GAPDH aggregates form in animals, we treated mice with METH, which is known to induce loss of nigra-striatum dopaminergic neurons due to excess release of dopamine and the associated oxidative stress (16, 17). We confirmed that oxidative stress was increased in animal brains after METH treatment (Fig. 7A) and observed a significant reduction in the levels of striatal dopamine (Fig. 7B). In METH-treated animals, we observed GAPDH oligomer and aggregate formation detected by Western blot (Fig. 7C). Consistent with this observation, intracellular, plaque-like foci of GAPDH immunoreactivity were also observed in TH-positive neurons in the substantia nigra (Fig. 7D). To address whether GAPDH mediates METH-induced dopamine cell damage, we examined transgenic mice overexpressing WT GAPDH (supplemental Fig. S5). The level of GAPDH aggregate formation was elevated in the transgenic mice compared with that of their littermate controls (Fig. 7E). Moreover, a marked reduction in striatal dopamine and TH in the midbrain was observed (Fig. 7, F and G).
FIGURE 7.

METH-treated GAPDH transgenic mice show robust GAPDH aggregate formation and a marked reduction in dopaminergic neurons in the nigra-striatum. A, effects of METH treatment on oxidative stress levels in the striatum of WT mice are shown. DNP, dinitrophenol. WB, Western blot. B, effects of METH treatment on dopamine levels in the striatum of WT mice are shown. C, Western blotting analysis of GAPDH aggregate formation in the striatum of METH-treated WT mice are shown. D, double immunofluorescence staining of TH and GAPDH in the substantia nigra of METH-treated WT mice is shown. TH-positive dopaminergic neurons (green) and GAPDH-positive plaques (red) are shown. E, Western blotting analysis of GAPDH aggregate formation in the striatum of METH-treated WT or GAPDH-Tg mice is shown. F, dopamine levels in the striatum of METH-treated GAPDH-Tg mice are shown. G, semiquantification of TH levels in the midbrain of METH-treated GAPDH-Tg mice is shown. All of data in these graphs are the means ± S.E. of WT (littermate controls) (n = 15), WT METH-treated (n = 12), GAPDH-Tg control (n = 8), or GAPDH-Tg METH-treated (n = 8) animals (t test; *, p < 0.05; **, p < 0.01 versus WT).
DISCUSSION
Based on our findings, we propose that disulfide-bonded, amyloid-like aggregate formation of GAPDH in response to oxidative stress might participate in cell death. Under some conditions, the levels of dopamine-induced cell death and GAPDH aggregate formation in dopaminergic SH-SY5Y neuroblastoma cells are correlated. In METH-treated mice, we observed GAPDH aggregate formation in dopaminergic neurons. In transgenic mice overexpressing WT GAPDH, METH treatment elicited more robust formation of GAPDH aggregates and dopaminergic cell loss.
Our data indicate that a specific cysteine residue (Cys-152) of GAPDH might have at least two distinct roles in cell death. Requirement of Cys-152 (Cys-150 in rats) to nuclear translocation of GAPDH has been fully characterized (3). In addition, the present study suggests the role of Cys-152 in GAPDH aggregate formation and its accompanying cell death. Our data provide hints to how these two mechanisms are differentially used in cells. In SH-SY5Y cells, low doses of dopamine elicited nuclear translocation of endogenous GAPDH without robust formation of the aggregates, whereas high doses of dopamine induced endogenous GAPDH aggregate formation with less nuclear translocation (Fig. 3), and the levels of endogenous GAPDH aggregates correlate with a high concentration of dopamine-induced cell death (Fig. 4). These results are also consistent with the data using DOX-inducible SH-SY5Y cells overexpressing WT- or C152A-GAPDH (Fig. 5). It is, therefore, possible that Cys-152-dependent GAPDH aggregate formation has a role in cell death when massive levels of oxidative stress occur. Moreover, deprenyl, a blocker of GAPDH nuclear translocation (5), fails to inhibit the aggregation both in vitro and in cells but reduced cell death in SH-SY5Y treated with only a low concentration of dopamine at 100 μm (Fig. 6). Thus, GAPDH nuclear translocation seems to be an initial event triggered by low to moderate oxidative stress-induced cell death. Then increasing stress promotes GAPDH aggregate formation in cytoplasm, which may lead to disturbance of the nuclear translocation, resulting in more severe cell death.
We also demonstrated that disulfide-bonded endogenous GAPDH aggregation occurs in dopamine neurons of mice treated with METH (Fig. 7). Furthermore, we observed increased dopamine neuron loss accompanied by more robust formation of GAPDH aggregates in METH-treated transgenic mice overexpressing WT GAPDH (Fig. 7). These findings suggest that GAPDH may participate in METH-induced neurotoxicity. This observation may contribute to a better understanding of the brain damage in METH abusers who exhibit various neuropsychiatric manifestations (27).
Supplementary Material
Acknowledgments
We thank Dr. Andreas Pluckthun and Dr. Peter Lindner (Zurich University) for generously providing W3CG, Hideki Yano (Oriental BioService, Inc., Kyoto, Japan) for excellent assistance in generating GAPDH-Tg mice, Fujimoto Pharmaceutical (Osaka, Japan) for kindly providing deprenyl (Selegiline), Tomoaki Ida (Osaka Prefecture University) for excellent assistance in HPLC-electrochemical detection analysis, and Drs. Ryouichi Yamaji, Naoki Harada, and Mitsugu Akagawa (Osaka Prefecture University) for helpful discussions.
This work was supported in part by Grant-in-aid 19580346 for Scientific Research (to H. N.) from the Japan Society for the Promotion of Science and a special research grant (to H. N.) from Osaka Prefecture University.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- NOR3
- (±)-(E)-4-ethyl-2-[(E)-hydroxyimino]-5-nitro-3-hexenamide
- NOC18
- 1-hydroxy-2-oxo-3,3-bis(2-aminoethyl)-1-triazene
- PBS
- phosphate-buffered saline
- WT
- wild type
- Aβ
- amyloid β
- DOX
- doxycycline
- METH
- methamphetamine (2S)-N-methyl-1-phenyl-propan-2-amine
- TH
- tyrosine hydroxylase
- Tg
- transgenic
- HPLC
- high performance liquid chromatography.
REFERENCES
- 1.Hara M. R., Cascio M. B., Sawa A. (2006) Biochim. Biophys. Acta 1762, 502–509 [DOI] [PubMed] [Google Scholar]
- 2.Chuang D. M., Hough C., Senatorov V. V. (2005) Annu. Rev. Pharmacol. Toxicol. 45, 269–290 [DOI] [PubMed] [Google Scholar]
- 3.Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., Sawa A. (2005) Nat. Cell Biol. 7, 665–674 [DOI] [PubMed] [Google Scholar]
- 4.Sen N., Hara M. R., Kornberg M. D., Cascio M. B., Bae B. I., Shahani N., Thomas B., Dawson T. M., Dawson V. L., Snyder S. H., Sawa A. (2008) Nat. Cell Biol. 10, 866–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hara M. R., Thomas B., Cascio M. B., Bae B. I., Hester L. D., Dawson V. L., Dawson T. M., Sawa A., Snyder S. H. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 3887–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross C. A., Poirier M. A. (2004) Nat. Med. 10, S10–S17 [DOI] [PubMed] [Google Scholar]
- 7.Forman M. S., Trojanowski J. Q., Lee V. M. (2004) Nat. Med. 10, 1055–1063 [DOI] [PubMed] [Google Scholar]
- 8.Serpell L. C., Smith J. M. (2000) J. Mol. Biol. 299, 225–231 [DOI] [PubMed] [Google Scholar]
- 9.Burke J. R., Enghild J. J., Martin M. E., Jou Y. S., Myers R. M., Roses A. D., Vance J. M., Strittmatter W. J. (1996) Nat. Med. 2, 347–350 [DOI] [PubMed] [Google Scholar]
- 10.Cumming R. C., Schubert D. (2005) FASEB J. 19, 2060–2062 [DOI] [PubMed] [Google Scholar]
- 11.Lindersson E., Beedholm R., Højrup P., Moos T., Gai W., Hendil K. B., Jensen P. H. (2004) J. Biol. Chem. 279, 12924–12934 [DOI] [PubMed] [Google Scholar]
- 12.Schulze H., Schuler A., Stüber D., Döbeli H., Langen H., Huber G. (1993) J. Neurochem. 60, 1915–1922 [DOI] [PubMed] [Google Scholar]
- 13.Tsuchiya K., Tajima H., Kuwae T., Takeshima T., Nakano T., Tanaka M., Sunaga K., Fukuhara Y., Nakashima K., Ohama E., Mochizuki H., Mizuno Y., Katsube N., Ishitani R. (2005) Eur. J. Neurosci. 21, 317–326 [DOI] [PubMed] [Google Scholar]
- 14.Wang Q., Woltjer R. L., Cimino P. J., Pan C., Montine K. S., Zhang J., Montine T. J. (2005) FASEB J. 19, 869–871 [DOI] [PubMed] [Google Scholar]
- 15.Nakajima H., Amano W., Fujita A., Fukuhara A., Azuma Y. T., Hata F., Inui T., Takeuchi T. (2007) J. Biol. Chem. 282, 26562–26574 [DOI] [PubMed] [Google Scholar]
- 16.Larsen K. E., Fon E. A., Hastings T. G., Edwards R. H., Sulzer D. (2002) J. Neurosci. 22, 8951–8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaVoie M. J., Hastings T. G. (1999) J. Neurosci. 19, 1484–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lotharius J., O'Malley K. L. (2000) J. Biol. Chem. 275, 38581–38588 [DOI] [PubMed] [Google Scholar]
- 19.Mark K. A., Quinton M. S., Russek S. J., Yamamoto B. K. (2007) J. Neurosci. 27, 6823–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganter C., Plückthun A. (1990) Biochemistry 29, 9395–9402 [DOI] [PubMed] [Google Scholar]
- 21.Ishii T., Sunami O., Nakajima H., Nishio H., Takeuchi T., Hata F. (1999) Biochem. Pharmacol. 58, 133–143 [DOI] [PubMed] [Google Scholar]
- 22.Yoshiike Y., Chui D. H., Akagi T., Tanaka N., Takashima A. (2003) J. Biol. Chem. 278, 23648–23655 [DOI] [PubMed] [Google Scholar]
- 23.Iwashita A., Mihara K., Yamazaki S., Matsuura S., Ishida J., Yamamoto H., Hattori K., Matsuoka N., Mutoh S. (2004) J. Pharmacol. Exp. Ther. 310, 1114–1124 [DOI] [PubMed] [Google Scholar]
- 24.Robinson C. E., Keshavarzian A., Pasco D. S., Frommel T. O., Winship D. H., Holmes E. W. (1999) Anal. Biochem. 266, 48–57 [DOI] [PubMed] [Google Scholar]
- 25.Jiang H., Ren Y., Zhao J., Feng J. (2004) Hum. Mol. Genet. 13, 1745–1754 [DOI] [PubMed] [Google Scholar]
- 26.Lee M. K., Kang S. J., Poncz M., Song K. J., Park K. S. (2007) Exp. Mol. Med. 39, 376–384 [DOI] [PubMed] [Google Scholar]
- 27.Uhl G. R., Drgon T., Liu Q. R., Johnson C., Walther D., Komiyama T., Harano M., Sekine Y., Inada T., Ozaki N., Iyo M., Iwata N., Yamada M., Sora I., Chen C. K., Liu H. C., Ujike H., Lin S. K. (2008) Arch. Gen. Psychiatry 65, 345–355 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.