

Abstract
Endophilins participate in membrane scission events that occur during endocytosis and intracellular organelle biogenesis through the combined activity of an N-terminal BAR domain that interacts with membranes and a C-terminal SH3 domain that mediates protein binding. Endophilin B1 (Endo B1) was identified to bind Bax, a Bcl-2 family member that promotes apoptosis, through yeast two-hybrid protein screens. Although Endo B1 does not bind Bax in healthy cells, during apoptosis, Endo B1 interacts transiently with Bax and promotes cytochrome c release from mitochondria. To explore the molecular mechanism of action of Endo B1, we have analyzed its interaction with Bax in cell-free systems. Purified recombinant Endo B1 in solution displays a Stokes radius indicating a tetrameric quarternary structure. However, when incubated with purified Bax, it assembles into oligomers more than 4-fold greater in molecular weight. Although Endo B1 oligomerization is induced by Bax, Bax does not stably associate with the high molecular weight Endo B1 complex. Endo B1 oligomerization requires its C-terminal Src homology 3 domain and is not induced by Bcl-xL. Endo B1 combined with Bax reduces the size and changes the morphology of giant unilamellar vesicles by inducing massive vesiculation of liposomes. This activity of purified Bax protein to induce cell-free assembly of Endo B1 may reflect its activity in cells that regulates apoptosis and/or mitochondrial fusion.
Introduction
Endo B12 (1), also called Bif-1 (2) and SH3GLB1 (3), is involved in apoptosis (4), mitochondrial morphogenesis (5), and autophagosome formation (6). Discovered based on yeast two-hybrid screens for proteins that bind the proapoptotic Bcl-2 family member, Bax, Endo B1 has been found to also interact transiently with Bax in mammalian cells upon apoptosis induction (4). RNA interference and genetic knock-out studies show that cells deficient in Endo B1 have a decreased sensitivity to apoptosis induction, although how Endo B1 participates in apoptosis and the significance of its interaction with Bax remains unknown. Whether Bax binding is involved in Endo B1 induction of autophagosomes or in maintenance of outer mitochondrial membrane morphology in healthy cells also remains unclear, although Bcl-2 family members appear to be involved in both processes (7–9).
Endo B1 contains an N-BAR domain toward its N terminus, a domain found in other endophilin family proteins such as endophilin A1 and amphiphysin, to form crescent-shaped dimers and to mediate membrane tubulation (10, 11). Consistent with other BAR domain-containing proteins, Endo B1 induces liposome tubulation in vitro (1). It contains an SH3 domain toward its C terminus that would be predicted to interact with proline-rich domains or PXXP motifs on other proteins. The endophilin A1 SH3 domain, for example, binds to a proline-rich region of dynamin and synaptojanin and mediates colocalization of Endophilin A1 with dynamin around lipid tubules (1).
We have examined the effect of Endo B1 on GUVs. In contrast to results previously obtained with small liposomes (1), purified recombinant Endo B1 alone has no detectable effect on GUVs. However, the addition of Bax induces Endo B1 to assemble into larger molecular weight homo-oligomers and to bud GUVs into smaller vesicles. Importantly, Bcl-xL, a close structural homologue of Bax that lacks proapoptotic activity (12), fails to activate Endo B1 oligomerization. These results demonstrate that Bax triggers formation of Endo B1 oligomers and activates Endo B1 to change lipid membrane curvature. This work also reveals a previously unknown cell-free activity of purified monomeric Bax and suggests how Bax may be activated to promote outer mitochondrial membrane permeabilization (13) and/or how Bcl-2 family members may intersect mitochondrial membrane fusion machineries in healthy cells (14).
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The cDNA of the human Endo B1 composed of 386 residues was cloned into a pET-21 vector (Novagen), and the cDNA of the C-terminally truncated human Endo B1 (Endo B1ΔC) comprising 290 residues was cloned into a pET-41a vector (Novagen). A His6 tag was at the C terminus of both constructs. Escherichia coli BL21 (DE3) pLys was used to express the proteins. Recombinant bacteria were grown in LB in the presence of ampicillin (Endo B1) or kanamycin (Endo B1ΔC). Protein expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside (Sigma), and after 3 h of incubation, cells were harvested. The cell pellet was washed in 25 mm Tris-HCl buffer, pH 8.0, and 100 mm NaCl followed by centrifugation at 10,000 × g for 20 min and resuspended in 50 ml of homogenization buffer (25 mm Tris-HCl pH 8.0, 100 mm NaCl, EDTA-free protease inhibitor mixture (Roche Applied Science), 10% glycerol, and 1 mm 2-mercaptoethanol. The lysate was centrifuged at 20,000 × g for 45 min at 4 °C, and Endo B1, present in the supernatant, was purified by His tag affinity chromatography.
Our preliminary purification efforts indicated that the Endo B1 C-terminal His6 tag associated poorly with the Ni(II) of the column matrix because a significant amount of the protein was not retained. However, in the presence of 6 m guanidine hydrochloride, the binding affinity was restored. Therefore, all of the steps involving affinity chromatography were performed under denaturing conditions. Unfolding was initiated by the addition of 6 m guanidine hydrochloride at 25 °C for 1 h. The Ni2+-nitrilotriacetic acid resin was pre-equilibrated with the binding buffer, which was same as the homogenization buffer used in the previous step except for the addition of 6 m guanidine hydrochloride and 10 mm imidazole. The unfolded Endo B1 was then incubated with the nickel resin overnight at 4 °C on a rotary shaker.
Analysis for protein expression of Endo B1ΔC indicated that the protein was insoluble in the form of inclusion bodies. The pellet was successively washed three times with 30 ml of 0.1% Triton X-100 in a buffer containing 25 mm Tris-HCl buffer (pH 8.0) and 100 mm NaCl. This was followed by three wash steps in the absence of Triton X-100 with the identical buffer. The inclusion bodies were finally solubilized in a buffer containing 25 ml of 6 m guanidine hydrochloride, 25 mm Tris-HCl buffer, pH 8.0, 100 mm NaCl, 10% glycerol, and 1 mm 2-mercaptoethanol and centrifuged at 20,000 × g for 40 min. The supernatant was then added in batch mode to 10 ml of Ni2+-nitrilotriacetic acid resin, which was previously equilibrated with the buffer that had been used for solubilizing the inclusion bodies. For optimal binding, it was left overnight at 4 °C on a rotary shaker.
After elution, the two proteins were refolded by flash-dilution with constant stirring into 100 ml of a cold refolding buffer (25 mm Tris-HCl, pH 8.0, 100 mm NaCl, 0.5 m arginine-HCl, 10% glycerol, and 1 mm 2-mercaptoethanol) and dialyzed overnight against the refolding buffer followed by ultracentrifugation at 100,000 × g at 4 °C for 40 min. A second step of Ni(II) metal ion affinity chromatography in the absence of guanidine was performed. The proteins were further purified by gel filtration with a Superdex-200 column on ÅKTA Prime Plus FPLC (Amersham Biosciences) using as running buffer (25 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10% glycerol, and 1 mm dithiothreitol).
The purity of the recombinant proteins was assessed by Western blot, Coomassie-stained native, and SDS-polyacrylamide gels. Furthermore, the Endo B1 protein was analyzed by CD spectroscopy (data not shown) in order to validate the efficacy of the protein refolding method adopted in the purification protocol.
E. coli BL21 DE3 pLys (Invitrogen) was used to express human Bcl-xL. Recombinant bacteria (transformed with the expression plasmid pET16b containing the cDNA for Bcl-xL) were grown in 1 liter of Super Broth (3.2% Tryptone, 2.0% yeast extract, 0.5% NaCl, pH 7.5, (KD Medical)) containing 50 μg/ml ampicillin (Sigma) in 2-liter flasks at 37 °C. Protein expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside (Sigma) when the A600 reached 0.8–1. After an overnight incubation, cells were harvested by centrifugation at 5,000 × g, and, after resuspension in His tag binding buffer (5 mm imidazole, 20 mm Tris-HCl, pH 7.9, 0.5 m NaCl), pellets were lysed by homogenization. The supernatant containing the soluble Bcl-xL protein was centrifuged at 20,000 × g for 30 min in order to remove any traces of cell debris, and it was filtered through a 0.45-μm membrane prior to performing His tag affinity chromatography. The column was loaded with the prepared extract and washed with 5 volumes of binding buffer and 6 volumes of washing buffer (60 mm imidazole, 20 mm Tris-HCl, pH 7.9, 0.5 m NaCl). The bound protein was eluted with 4 volumes of elute buffer (1 m imidazole, 20 mm Tris-HCl, pH 7.9, 0.5 m NaCl). The eluted protein was dialyzed in 20 mm Tris-HCl, pH 8, overnight at 4 °C and further purified by ion exchange chromatography on a MonoQ column (Amersham Biosciences) connected to an AKTA FPLC (Amersham Biosciences). Final yield of Bcl-xL was 1 mg/liter of culture. Purification of recombinant human Bax was performed as described previously (15).
Gel Filtration
The apparent molecular mass of the different proteins and complex was determined by gel filtration on Superdex 200 10/300 GL equilibrated with 50 mm Tris-HCl, pH 7.4, and 0.1 m NaCl and calibrated with thyroglobulin (669 kDa), apoferritin (443 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), carbonic anhydrase (29 kDa), and cytochrome c (12.4 kDa). The proteins were loaded onto the column and eluted at a flow rate of 0.3 ml/min. The elution volume (Ve) of each fraction was determined from the absorbance at 280 nm. The void volume (V0) was determined with blue dextran. The molecular mass was calculated by plotting log molecular mass versus the Ve/V0 ratio. The fractions were analyzed by Western blot using Endo B1 and Bax antibodies from Imgenex and Upstate, respectively.
Protein Labeling
The Endo B1 proteins were labeled with Alexa Fluor-488 carboxylic acid 2,3,5,6-tetrafluorophenyl ester (Invitrogen) in a ratio of protein to dye of 3:1 at 25 °C as per the protocol provided by the manufacturer. The labeled protein was dialyzed against 50 mm borate buffer, pH 8.5, overnight followed by dialysis against 25 mm Tris-HCl, pH 8.0.
For FCS measurements, Alexa-488-labeled Endo B1, which was prepared in Millipore water or in 25 mm Tris-HCl, pH 8, was diluted to a 200 nm concentration and then centrifuged (Beckman Optima Ultracentrifuge, rotor TLA 55) at 100,000 × g for 20 min at 22 °C. Only the top 80% of supernatant of the sample was used. The 1:1 (mol/mol) mixtures of the supernatant Endo B1 and Bax or Bcl-xL were preincubated for ∼1 h on ice prior to the FCS measurements.
FCS Experimental Setup
The basic principles of FCS have been described elsewhere (16, 17). This technique has been successfully used to determine changes in the diffusion of various proteins following their oligomerization or aggregation (17–19). The FCS measurements were carried out with a Hamamatsu FCS instrument (model C9413) equipped with a 473-nm LD-pumped solid-state laser, a high sensitivity photomultiplier tube with low afterpulsing, a 25-μm diameter pinhole for confocal detection, a water immersion objective (Olympus UApo ×40 W/340; numerical aperture 1.15), and a built-in numerical code for correlating the time sequence of the photocounts. In most measurements, the 1-milliwatt input laser beam was attenuated to 3 microwatts, and the cut-off wavelength of the high band emission filter was set to 495 nm. Fitting of the measured correlation functions and calculation of photocounting histograms were performed using the built in software package provided by Hamamatsu.
Theoretical Background of FCS
The basic principles of FCS have been described elsewhere (16, 19, 20). Briefly, FCS is an optical technique in which fluctuations in detected fluorescence are exploited, typically, to measure the movement of nanoparticles within a medium such as an aqueous buffer. A focused laser beam is used to excite the fluorescence of the nanoparticles, and the signal emitted from a small volume (∼1 fl) is measured as a function of time. Due to movement of the nanoparticles in and out of the volume or changes in the photodynamics of the nanoparticles, the emitted fluorescence signal fluctuates. The fluctuating signal is time-correlated using the following function,
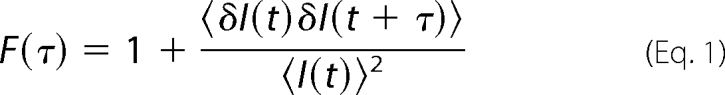 |
where δI(t) = I(t) − 〈I(t)〉 denotes the deviation of the emitted intensity, I(t), at time t from the average intensity, 〈I(t)〉. Analysis of this correlation function can reveal the underlying mechanisms behind the fluctuations. For example, in the case of monodisperse nanoparticles diffusing freely in a solution, one can derive the expression,
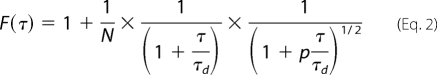 |
where r0 and z0 characterize the ideal Gaussian profile of the beam,
N denotes the average number of nanoparticles in the solution; p = (r0/z0)2 is a constant; and τd = r02/4D is the diffusion time, D being the translational diffusion coefficient. Moreover, for dilute solutions, the Stokes-Einstein relation, D = kBT/3πηdH, expresses the diffusion coefficient in terms of the viscosity, η, of the solvent (η = 0.1 centipoise for water); the hydrodynamic diameter of the nanoparticles, dH; the temperature, T (295 K, room temperature), in Kelvin; and the Boltzmann constant, kB = 1.38 × 10−23 J/K. In this latter expression, dH describes an overall size of the nanoparticle, and, for spherical nanoparticles, it is just their diameter.
For two independent diffusing species, the correlation function correspondingly becomes the following,
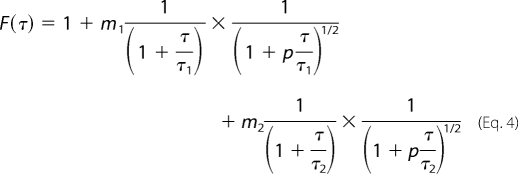 |
where m1 and m2 depend on the quantum yield and average numbers of each of the diffusing species in the sampling volume, and τ1 and τ2 are the diffusion times of the two species. Often with a probe that is exogenously labeled with a fluorophore, it is difficult to remove all of the free fluorophores from the solution after labeling. Even if all of the free fluorophores are removed by slow dialysis or appropriate rapid column centrifugation, a thermodynamic equilibrium state between free fluorophores and fluorophore-protein species may emerge after a while. For fitting the correlation data, we use the expression in Equation 4, where one diffusing species is the free fluorophores (i.e. Alexa-488) and the other is the labeled probe (i.e. labeled Endo B1).
Preparation of GUVs
GUVs were prepared by the modified electroswelling method (21). The optical chamber for GUVs preparation consisted of a 2.5-mm-thick double adhesive rubber spacer with four 9-mm diameter holes (Grace Bio-Labs) sandwiched between two conductive indium tin oxide-coated glass coverslips (Bioptechs Inc.). GUVs were prepared from dioleoylphosphatidylcholine mixed with 0.5% fluorescent rhodamine-dioleoylphosphatidylethanolamine lipid. Lipids were from Avanti Polar Lipids. 5 μl of 0.1 mg/ml lipid solution in chloroform was deposited on four prearranged 9-mm diameter circles on the conductive face of one indium tin oxide-coated glass coverslip and dried under vacuum overnight or lyophilized for 2 h to remove any residual organic solvent. A sandwich of two inward-facing indium tin oxide-covered coverslips and the rubber spacer was gently pressed and sealed, forming four identical 150-μl wells with dried lipid on the bottom of each well. Degassed Millipore water was injected into each well with a 100-μl syringe. Each conducting coverslip was connected to a function waveform generator (Agilent 33220A), and a 3-Hz square wave of 2-V amplitude was applied for ∼2 h, which resulted in the formation of multiple GUVs in all four wells. Based on previous observations of GUVs made by the electroformation method (21), we assumed that the majority of vesicles were unilamellar. After GUVs were formed, the proteins were injected into one of the experimental wells using a 10-μl Hamilton syringe. The added volumes of the protein solutions were kept less than 3 μl so that the final concentration of Tris-HCl buffer in the well never exceeded 0.5 mm. In order to take into account the minor effect of Tris-induced osmotic gradients on GUV shape, control experiments with identical aliquots of Tris were always run in parallel in one of the four wells. To determine the size distribution of a population of GUVs, several liposome images from a well were taken by focusing the microscope at different locations and different planar levels. The set of GUV pictures taken from the same well was considered as one experiment.
Confocal images were taken using an IX70 inverted microscope (Olympus), a confocal spinning disk analyzer (PerkinElmer Life Sciences) attached to a cooled 512X512 CCD camera (model C9100, Hamamatsu), an air-cooled ion-argon laser (Melles-Griot) with two basic excitation lines (488 and 568 nm), a piezo-electric driven device (Physik Instrumente) for z-positioning of the objective, and a two-dimensional XY-Proscan stage (Prior, Rockland). The system was set on a low vibration insulation table (Newport). Images were collected with a ×60, numerical aperture 1.2 water Olympus objective. IPLab software (BD Biosciences) was used to control various devices and to acquire and analyze the images.
RESULTS
FCS and Gel Filtration
Endo B1 was expressed in bacteria and purified to more than 90% homogeneity. Analysis of gel filtration data shows the molecular mass of Endo B1 (∼180,000 Da) to be about 4 times larger than the molecular mass of the monomer (46,400 Da), indicating that Endo B1 in solution forms a tetramer (data not shown) in contrast to the dimeric nature of recombinant purified endophilin A1 and A2 (22). In cell extracts endogenous Endo B1 migrates on blue native PAGE at apparent dimeric and tetrameric molecular masses of ∼90,000 and ∼180,000 Da.
3 We also examined the molecular mass of purified Endo B1 by FCS (Fig. 1). Using a two-component expression (see Equation 4), we fit the correlation function in Fig. 1 where the components are the free Alexa-488 fluorophore and Alexa-488-labeled Endo B1 protein. The fit yields a diffusion coefficient of Endo B1: D(Endo B1) = 40 ± 4 μm2/s, which can be compared with that of EGFP (a naturally-fluorescent, globular protein with molecular mass of 27 kDa) in water, D(EGFP) = 78 μm2/s (20). Therefore, we estimate the size of Endo B1 to be about 9.7 nm by using the known diameter of EGFP (∼5 nm). Further, assuming Endo B1 to be compact and spherical, we estimate its molecular mass to be ∼200,000 Da, which is consistent with a tetrameric form of the protein, although this value is slightly larger than that calculated from the protein sequence (185,700 Da) or gel filtration. This difference may be attributed to the assumptions of sphericity and compactness of the Endo B1 tetramers used in the FCS calculations.
FIGURE 1.

Oligomerization of Endo B1 in the presence of Bax. Normalized FCS correlation functions show a time shift of the correlation of Endo B1 plus Bax solution with respect to that of Endo B1 or Endo B1 plus Bcl-xL, indicating oligomerization of Endo B1 in the presence of Bax. The solid lines are fits of the data, where a two-component model is used (Equation 4). Each curve is labeled with the components in the solutions.
We analyzed the effects of purified recombinant Bax and Bcl-xL on the diffusion of Endo B1. Comparison of the correlation functions obtained from Endo B1 and a mixture of Endo B1 and Bax shows a time shift when Bax is mixed with the solution of Endo B1, indicating that Bax induces oligomerization of Endo B1 (Fig. 1). In contrast, there is no significant shift in Endo B1 diffusion when exposed to Bcl-xL (Fig. 1). To estimate the size of the oligomers found in the Endo B1 and Bax mixture, we fit the measured correlation functions with a two-component model (Fig. 1). However, this fit is relatively poor, as can be seen in Fig. 1, especially at longer delay times (τ > 1 ms), suggesting that Bax-induced oligomers of Endo B1 are not monodisperse in size and probably exhibit a range of different molecular weight Endo B1 oligomers.
These analyses by FCS do not reveal whether or not Bax, a protein that binds Endo B1 under certain conditions, is incorporated into the larger oligomeric Endo B1 complexes. We therefore examined Endo B1 migration by gel filtration in the absence and presence of purified Bax. Consistent with the FCS experiments, Bax induced a decrease in the tetrameric form of Endo B1 and a large increase in higher molecular weight protein species appearing in the exclusion volume of the column (Fig. 2). Interestingly, there was no apparent decrease in the Bax peak, suggesting that it is not incorporated into the exclusion volume protein complex (Fig. 2A). To directly examine if Bax binds the oligomeric form of Endo B1, we performed Western blots for Endo B1 and Bax across the gel filtration fractions. Although Endo B1 identified by Western blot shifts location to higher molecular weight fractions upon incubation with Bax, Bax does not change elution pattern in the process of Endo B1 oligomerization and is not detectable in the column exclusion volume fractions with Endo B1 (Fig. 2C). Thus, we see no evidence of Bax-Endo B1 complex formation but rather an apparent chaperone activity of Bax that changes Endo B1 conformation to induce oligomerization. To test the chaperone model, we incubated Endo B1 with a substoichiometric amount of Bax (Fig. 3). Even a 10-fold lower concentration of Bax relative to Endo B1 resulted in Endo B1 migrating to higher molecular weight forms after 4 h (Fig. 3A), although the process was slower, with less Endo B1 assembling into oligomers by 1 h, relative to equimolar levels of Bax and Endo B1 (Fig. 3, A and B).
FIGURE 2.

Bax induces oligomerization of Endo B1. Gel filtration profiles of Endo B1 or Endo B1ΔC before and after incubation for 1 h with Bax (A) or Bcl-xL (B). Gel filtration of Bax and Bcl-xL alone was performed for comparison of their elution times (A and B). Fractions 8–18 from the Endo B1, Bax, and Endo B1 plus Bax gel filtration columns were analyzed by Western blot using anti-Bax and anti-Endo B1 antibodies (C).
FIGURE 3.

Substoichiometric levels of Bax oligomerize Endo B1. A, gel filtration profiles of Endo B1 (2 nmol) incubated with Bax (2 nmol) or with Bax (0.2 nmol) for 1 and 4 h as well as Bax and Endo B1 alone. Fractions 8–18 from the Endo B1 alone, Bax alone, and Endo B1 (2 nmol) incubated with Bax (2 nmol or 0.2 nmol) gel filtration columns were analyzed by Western blot using anti-Bax and anti-Endo B1 antibodies (B).
Although Bcl-xL is a close structural homologue of Bax, it has the opposite biological activity and inhibits apoptosis (12). We therefore examined if Bcl-xL affects Endo B1 tetrameric structure by gel filtration. As seen in FCS experiments (Fig. 1), Bcl-xL had no detectable effect of Endo B1 quaternary structure measured by gel filtration after incubation at room temperature and neutral pH (Fig. 2B).
To test if the SH3 domain of Endo B1 is responsible for homo-oligomerization, we produced a variant Endo B1 lacking this C-terminal region (Endo B1ΔC-(310–400)) and examined if Bax was able to induce Endo B1ΔC oligomerization. As seen in gel filtration experiments (Fig. 2A), Endo B1ΔC, like full-length Endo B1, migrates with a predicted tetrameric molecular mass of ∼150,000 Da relative to the calculated 33,873 Da molecular mass of the monomeric protein. However, in contrast to full-length Endo B1, incubation with Bax did not induce it to oligomerize or otherwise change its migration on gel filtration (Fig. 2A). Endophilin A1 and A2 form primarily dimers in solution mediated by the central coiled coil domain (22). The tetramerization, or dimerization of dimers of Endo B1 appears to be mediated by either the BAR domain and/or central coiled coil region, whereas the Bax-induced higher order assembly requires the SH3 domain.
GUV Transformation
We explored if Bax-induced oligomerization affects the ability of Endo B1 to alter membrane curvature, a characteristic of BAR domain-containing proteins (11). Analysis of shape changes in GUVs is a highly sensitive method for detecting the interaction of proteins with lipid membranes (23–25). However, the shape of spherical GUVs is difficult to change without interaction with other GUVs. Therefore, we used GUVs prepared by a modified electroswelling method (21), where liposomes form an expanded network of spherical liposomes interconnected with each other by lipid tethers (21, 26). This method allows one to create homogenous populations of unilamellar GUVs, view shape deformations in real time by confocal microscopy, and analyze GUV shape and size in the presence and absence of different proteins. These GUVs display a rather uniform appearance (Fig. 4A) with an average diameter of around 20 μm. The GUV size distribution is shown in Fig. 4B.
FIGURE 4.

Endo B1 with Bax induces massive vesiculation and liposome size reduction. A, control image before protein addition (upper left). Endo B1 or Bax does not induce liposome shape deformation (upper right and lower left, respectively). The sphericity of these GUVs indicates a lack of osmotic stress. Confocal images were taken 30 min after the addition of 490 nm Endo B1 or Bax. Endo B1 preincubated with Bax affects shape and size of GUVs (lower right). The image was taken 20 min after the Endo B1 plus Bax (1:1 mol/mol) addition. Scale bars here and elsewhere, 20 μm. B, size distribution of GUVs obtained before and after the addition of Endo B1 plus Bax, Endo B1, or Bax. Normalized number of GUVs is plotted versus their size. Data are the summary of the analysis of at least three independent experiments for each condition with multiple images taken in each experiment. C, a representative image of large GUVs with entrapped multiple smaller vesicles. D, number of GUVs with smaller vesicle inclusions increases after the addition of Endo B1 plus Bax. A summary of the statistical analysis of the percentage of GUVs with inclusions before and after the addition of Endo B1 plus Bax, Endo B1, Bax, or corresponding aliquots of Tris buffer as a control. The number of liposomes analyzed per experiment was ∼150. Statistical analysis was done using a two-tailed t test (a = 0.05) as a comparison with control after the addition of 0.2 mm Tris buffer (**, p < 0.04) and as a comparison with control before protein injection (*, p < 0.1).
Endo B1 preincubated with an equimolar amount of Bax was injected into the GUV chamber to yield a final concentration of 490 nm Endo B1 and Bax. This resulted in extensive shape transitions of GUVs, as shown in the representative confocal images (Fig. 4, A (lower right) and C). First, there was an apparent reduction in the overall size of the GUVs induced by Endo B1 plus Bax (Fig. 4A). Second, Endo B1 plus Bax induced massive liposome vesiculation, which was seen through the formation of multiple small vesicles inside larger ones apparently due to a form of “inward budding” (Fig. 4C). Third, there appears to be an activation of liposome “fission” and “fusion” by the Endo B1 plus Bax observed in real time (data not shown). In contrast, Endo B1 or Bax alone did not affect the shape or size of GUVs (Fig. 4A). These results were confirmed by the analysis of the size distributions of the GUVs, which show a reduction in average liposome diameter to <5 μm after the addition of Endo B1 plus Bax, whereas the same amount of Endo B1 or Bax alone did not change the size distribution of GUVs (Fig. 4B). The relative number of GUVs containing inclusions of smaller liposomes increased 4-fold following the addition of Endo B1 plus Bax into the GUV chamber (Fig. 4D), whereas Endo B1 or Bax alone only slightly increased the number of GUVs with inclusions. Furthermore, this small effect of Endo B1 or Bax individually most likely is a result of the osmotic gradient generated by the protein buffer, ∼0.2 mm Tris, because buffer alone caused a similar minor effect (Fig. 4D; also see supplemental Fig. S1). The effect of the osmotic gradients on GUV shape deformation is a well described phenomenon (21, 27, 28). When Bax was preincubated with C-terminal truncated Endo B1ΔC, there was no noticeable affect on liposome vesiculation (supplemental Fig. S2).
As the vesiculation of GUVs is accompanied by a decrease in average size, small liposomes probably derive from the membrane of the initial large GUVs, in a kind of internal (and external) budding process. To test this hypothesis, the soluble fluorescent dye Alexa-488 was added to preformed GUVs prior to the addition of Endo B1 plus Bax, allowing visualization of dark (empty) GUV lumens within the fluorescent solution (Fig. 5B, left image). Upon the addition of Endo B1 plus Bax, small vesicles filled with Alexa-488 appear within the larger, Alexa-488-excluding vesicles (Fig. 5A). GUV membranes otherwise are impermeable to free Alexa-488 added to the buffer, as seen in the control experiment (Fig. 5B). This indicates that the outside medium with free dye becomes encapsulated inside small vesicles during their formation, probably resulting from inward budding of vesicles from the outer membrane of GUVs. Endo B1 covalently attached to Alexa-488 and preincubated with Bax showed the same luminal labeling of encapsulated GUVs as soluble Alexa-488 displayed (supplemental Fig. S3). These experiments support the model that Endo B1 combined with Bax induces an inward (and outward) budding of liposomal membranes.
FIGURE 5.

Internal small vesicles appear to bud off from the GUV membrane. A, images of GUVs generated after sequential addition of Alexa-488 and unlabeled Endo B1 preincubated with Bax. Free dye fills some of the small vesicles (green circles) inside the larger one without dye (empty dark green circles). B, confocal images of GUVs in the presence of free Alexa-488 (green) show that dye does not penetrate through the membrane of GUV (up to 2 h). Images at the left were taken at the 488-nm excitation wave and show Alexa-488. The same images at the right were taken at 568 nm and show liposome membrane labeled with rhodamine-dioleoylphosphatidylethanolamine (red).
DISCUSSION
Bax, a key proapoptotic protein in the Bcl-2 family, translocates from the cytosol to mitochondria during apoptosis, where it inserts into the outer mitochondrial membrane, forms oligomeric complexes, and mediates the release of cytochrome c (12). Recapitulating some of these steps in vitro, Bax can interact with tBid in artificial membranes to trigger Bax insertion into the membrane and oligomerization (29). Despite intense study, only one cell-free activity of Bax, the ability to permeabilize artificial lipid membranes and isolated mitochondria, has yielded insight into its mechanism of action in cells (13). We report here a new cell-free activity of monomeric Bax that occurs in the absence of tBid, the ability to catalyze Endo B1 oligomerization and activate membrane budding. Strikingly, Endo B1 oligomers exclude Bax from the complex, consistent with a chaperone-like activity of monomeric Bax to catalyze this conversion. Furthermore, Bax can act slowly over time in substoichiometric amounts to oligomerize Endo B1. In contrast, the antiapoptotic Bcl-2 family member Bcl-xL lacks the ability to oligomerize Endo B1. Bax does not induce oligomerization of Endo B1ΔC, indicating that the SH3 domain is responsible for either Endo B1 interaction with Bax or Endo B1 homo-oligomerization. Because the only known Bax interaction region of Endo B1 exists at the extreme N terminus (3, 30), the SH3 domain most likely mediates Endo B1 oligomerization.
Correlating with conditions of Endo B1 oligomerization, Bax activates Endo B1 to mediate membrane budding of GUVs. Endo B1 combined with Bax appears to mediate budding of vesicles out of GUVs as well as into GUVs, consistent with the induction of both positive and negative membrane curvature changes. Although it is difficult to track and quantify vesicles budding off GUVs, the internally budded vesicles become trapped within GUVs, allowing their detection and analysis. That these vesicles bud inward is shown by the derivation of their luminal contents from outside GUVs and distinct from the internal milieu of the parent GUVs. Such concave membrane deformation has been previously demonstrated for I-BAR (31) but not N-BAR or F-BAR proteins. Outward as well as inward vesiculation of GUVs could be induced by very different external forces, such as low osmotic stress (21, 27, 28), mild detergents (32), lysophospholipids (33), DNA (34), and local pH gradients (35). A variety of membrane surface-active agents, such as a mild detergent, a lysophospholipid, or an amphiphile, which tends to modify the surface ratio of the two bilayer leaflets and therefore the spontaneous curvature, is expected to promote budding and fission (32). Such shape changes in GUVs are reasonably explained by the “bilayer coupled hypothesis” also known as the “area difference-elasticity” model (25, 32, 33). The rationale of this theory is that the insertion of an amphiphile into the outer leaflet of the lipid bilayer induces a lateral tension of this leaflet that cannot be relieved by lateral expansion because the two leaflets must remain together. This promotes a tendency to increase the outside/inside surface ratio of the two leaflets by budding (32). Assuming that N-BAR domains of Endo B1 and endophilin A1 share structural similarities, we can apply this model to our system. Two distinct amphipathic helixes in the N-BAR domain of endophilin A1 that penetrate into the external leaflet of the bilayer (10, 11) could promote convex curvature of a region of a GUV membrane to induce outward budding. The concave budding of GUVs we observed with Endo B1 plus Bax may be a consequence of the displacement of lipid from the inner leaflet of an Endo B1-mediated convex bulge forcing excess lipid in the inner leaflet out of the bulge that may be predicted to induce a reciprocal concave bulge outside the BAR domain occupied region in compensation. However, the lipid continuities of electroswollen GUVs may negate such a requirement for stoichiometric lipid curvature compensations, and Endo B1 may actively mediate inward as well as outward GUV budding. Thus, at this point, we cannot ascertain if Endo B1 plus Bax induces both concave and convex budding of vesicles from the GUVs or if they induce only one directional budding that forces reverse budding to compensate for excessive lipid mass in the opposite leaflet. Importantly, the tetrameric form of Endo B1 alone is not sufficient to induce GUV budding. GUV budding induced by Endo B1 occurred upon preincubation with Bax, which also induces Endo B1 to oligomerize, suggesting that it is the high molecular weight homo-oligomeric complexes of Endo B1 that initiate GUV shape deformation. Further linking the oligomerization of Endo B1 to GUV vesiculation is the failure of SH3 domain-truncated Endo B1, which does not change gel filtration pattern upon incubation with Bax, to vesiculate GUVs in the presence of Bax. Thus, the mechanism of GUV budding by Endo B1 may resemble the mechanism proposed recently for oligomeric forms of F-BAR domains (36). F-BAR domains, sharing overall shape similarities with N-BAR domains (11), assemble in parallel arrays on flat membrane surfaces that can rotate in curved convex ensembles, thereby bending the membrane lipid bilayers into tubules (37). High molecular weight Endo B1 complexes could adsorb on the essentially flat surface of GUVs (considering the vesicle size relative to the Endo B1 complexes) and squeeze membrane vesicles off of the flat surface or invert them into the GUV. We could visualize this model as oligomeric Endo B1 forming a “collar-like” structure outside of convex membrane buds or inside of the neck of inward concave buds as they emerge off or into GUVs, respectively. Here we suggest a dual curvature-generating mechanism previously proposed for BAR proteins (10, 11), where Endo B1 ensembles anchored into the external leaflet of the bilayer promote local bilayer uncoupling and membrane budding.
However, we cannot exclude the possibility that Bax may participate with the N-BAR protein, Endo B1, to mediate vesicle budding. A synergistic effect between Endo B1 (Bif-1) and Bax activated by tBid was found to promote Bax oligomerization and liposome permeabilization. This effect was also observed on small unilamellar liposomes formed from mitochondrial outer membrane-mimicking lipid compositions (30). Interestingly, the synergistic effect of Endo B1 with Bax was observed only in the presence of tBID and apparently distinct from our results reported here, where tBID is absent. Consistent with a distinct interaction between Bax and Endo B1 in the absence of tBID, we see no stable binding of Bax to Endo B1 or Bax oligomerization in solution. Thus, although it may be possible for Bax to participate with Endo B1 in GUV vesiculation, because we have no tBid present, we favor the model that oligomeric Endo B1 alone, dependent on Bax primarily for the induction of oligomerization, mediates GUV fission and fusion. A several-step mechanism by which Bax triggers cytochrome c release from mitochondria has been proposed (29, 38). In this model, the first steps are Bax conformational activation and insertion into the mitochondria outer membrane (MOM), and the last step is the formation of large pores and MOM permeabilization. Etxebarria et al. (30) concluded that Endo B1 stimulates the permeabilization function of Bax at the membrane level. Interestingly, Bax inserts selectively into membranes with higher curvature (39), and Endo B1-mediated curvature may activate Bax insertion as previously proposed (30). However, it is important to distinguish the mechanism of membrane activity of Bax proposed here from models of its pore-forming activity (40, 41). Oligomeric Bax, which is known to form pores in lipid membranes (42), induces rupture of the entire GUV (data not shown), in marked contrast to what we see with monomeric Bax or monomeric Bax plus Endo B1, where no vesicle rupture is observed, indicating little or no Bax-mediated pore formation. This is also consistent with the gel filtration data indicating that despite Bax induction of Endo B1 oligomerization, Bax stays monomeric during the process. Because membrane curvature may in turn activate Bax insertion into membranes, our results suggest that Bax could promote its own membrane insertion by activating Endo B1 oligomerization and thereby the induction of mitochondrial membrane curvature, upstream of pore formation. A second step could be BH3-only protein promoting the oligomerization of the membrane-inserted Bax, leading to MOM permeabilization. This model is consistent with the co-localization of Bax and Endo B1 specifically during apoptosis (4).
Alternatively or additionally, because Endo B1 is normally involved in mitochondrial morphogenesis in healthy cells, our results suggest how Bax may activate Endo B1 specifically during apoptosis to mediate the mitochondrial fission that occurs close in time to Bax translocation and cytochrome c release (14). Bax stimulation of EndoB1 membrane activity and alteration of its quarternary structure shown here may also reflect the in vivo activity of Bax on outer mitochondrial membrane dynamics of Endo B1 (5) or the effect of Bax on mitochondrial fusion rates (8) in healthy, non-apoptotic cells. If Bax alters Endo B1 quaternary structure in healthy cells it may be mediated by the monomeric non-apoptotic conformation of Bax, as is used in the cell-free assays reported here and as occurs predominantly in non-apoptotic cells. However, we find that most Endo B1 appears in dimers and tetramers in the cytosol of cells, and only a minor fraction of Endo B1 on mitochondria in healthy cells would be predicted to form oligomers. In contrast, concentrated foci of Endo B1, perhaps representing oligomers identified here, occur on mitochondria during apoptosis (5). Endo B1 lacking the SH3 domain, which fails to oligomerize upon cell-free incubation with Bax and fails to induce GUV vesicle budding, acts as a dominant negative inhibitor of Endo B1 in healthy cells by inhibiting mitochondrial outer membrane fission and inducing long filaments of interconnected outer mitochondrial membrane (5). We suggest that the Endo B1 and Bax may work together to affect mitochondrial outer membrane dynamics, although, because EndoB1 knockdown cells show an inhibition of outer mitochondrial membrane fission and Bax is required for normal rates of mitochondrial fusion, other proteins probably impact their activity. For example, Endophilin B2, a close Endo B1 homologue that is reported not to bind Bax (3) is found in cells bound to Endo B1,4 and Bax may alter the interaction between these two proteins. Last, how Bak, a close structural and functional homologue of Bax, may interact with Endo B1 is unknown and, because Bak can also stimulate mitochondrial fusion in healthy cells, the potential interplay between Endo B1 and Bak may be influenced by Bax. How these healthy cell activities of Bax and Endo B1 on the mitochondrial outer membrane, that may be reflected in the GUV fission processes documented here, may be coupled to the MOM permeabilization during apoptosis remains to be elucidated.
Supplementary Material
Acknowledgments
T. K. R. is grateful to Sergey Bezrukov for fruitful discussions. We thank Jenny Hinshaw, Kristi Norris, and Chunxin Wang for critical reading of the manuscript.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Programs of the Eunice Kennedy Shriver NICHD (to T. K. R., H. B., and B. S.) and NINDS (to A. A., S. B., and R. J. Y.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
C. Wang, unpublished results.
C. Wang and R. J. Youle, unpublished data.
- Endo B1
- endophilin B1
- Endo B1ΔC
- C-terminally truncated human Endo B1
- GUV
- giant unilamellar vesicle
- FCS
- fluorescent correlation spectroscopy
- MOM
- mitochondrial outer membrane
- SH3
- Src homology 3
- EGFP
- enhanced green fluorescent protein.
REFERENCES
- 1.Farsad K., Ringstad N., Takei K., Floyd S. R., Rose K., De Camilli P. (2001) J. Cell Biol. 155, 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuddeback S. M., Yamaguchi H., Komatsu K., Miyashita T., Yamada M., Wu C., Singh S., Wang H. G. (2001) J. Biol. Chem. 276, 20559–20565 [DOI] [PubMed] [Google Scholar]
- 3.Pierrat B., Simonen M., Cueto M., Mestan J., Ferrigno P., Heim J. (2001) Genomics 71, 222–234 [DOI] [PubMed] [Google Scholar]
- 4.Takahashi Y., Karbowski M., Yamaguchi H., Kazi A., Wu J., Sebti S. M., Youle R. J., Wang H. G. (2005) Mol. Cell Biol. 25, 9369–9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karbowski M., Jeong S. Y., Youle R. J. (2004) J. Cell Biol. 166, 1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi Y., Coppola D., Matsushita N., Cualing H. D., Sun M., Sato Y., Liang C., Jung J. U., Cheng J. Q., Mulé J. J., Pledger W. J., Wang H. G. (2007) Nat. Cell Biol. 9, 1142–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha S., Colbert C. L., Becker N., Wei Y., Levine B. (2008) Autophagy 4, 989–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karbowski M., Norris K. L., Cleland M. M., Jeong S. Y., Youle R. J. (2006) Nature 443, 658–662 [DOI] [PubMed] [Google Scholar]
- 9.Delivani P., Adrain C., Taylor R. C., Duriez P. J., Martin S. J. (2006) Mol. Cell 21, 761–773 [DOI] [PubMed] [Google Scholar]
- 10.Masuda M., Takeda S., Sone M., Ohki T., Mori H., Kamioka Y., Mochizuki N. (2006) EMBO J. 25, 2889–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallop J. L., Jao C. C., Kent H. M., Butler P. J., Evans P. R., Langen R., McMahon H. T. (2006) EMBO J. 25, 2898–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youle R. J., Strasser A. (2008) Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 13.Chipuk J. E., Green D. R. (2008) Trends Cell Biol. 18, 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suen D. F., Norris K. L., Youle R. J. (2008) Genes Dev. 22, 1577–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki M., Youle R. J., Tjandra N. (2000) Cell 103, 645–654 [DOI] [PubMed] [Google Scholar]
- 16.Webb W. W. (2001) Appl. Opt. 40, 3969–3983 [DOI] [PubMed] [Google Scholar]
- 17.Chen Y., Müller J. D., Berland K. M., Gratton E. (1999) Methods 19, 234–252 [DOI] [PubMed] [Google Scholar]
- 18.Boukari H., Nossal R., Sackett D. L. (2003) Biochemistry 42, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 19.Boukari H., Sackett D. L. (2008) Methods Cell Biol. 84, 659–678 [DOI] [PubMed] [Google Scholar]
- 20.Chen Y., Müller J. D., Ruan Q., Gratton E. (2002) Biophys. J. 82, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathivet L., Cribier S., Devaux P. F. (1996) Biophys. J. 70, 1112–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ringstad N., Nemoto Y., De Camilli P. (2001) J. Biol. Chem. 276, 40424–40430 [DOI] [PubMed] [Google Scholar]
- 23.Römer W., Berland L., Chambon V., Gaus K., Windschiegl B., Tenza D., Aly M. R., Fraisier V., Florent J. C., Perrais D., Lamaze C., Raposo G., Steinem C., Sens P., Bassereau P., Johannes L. (2007) Nature 450, 670–675 [DOI] [PubMed] [Google Scholar]
- 24.Tareste D., Shen J., Melia T. J., Rothman J. E. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 2380–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamba Y., Ohba S., Kubota M., Yoshioka H., Yoshioka H., Yamazaki M. (2007) Biophys. J. 92, 3178–3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bagatolli L. A., Parasassi T., Gratton E. (2000) Chem. Phys. Lipids 105, 135–147 [DOI] [PubMed] [Google Scholar]
- 27.Boroske E., Elwenspoek M., Helfrich W. (1981) Biophys. J. 34, 95–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernard A. L., Guedeau-Boudeville M. A., Jullien L., di Meglio J. M. (2002) Biochim. Biophys. Acta 1567, 1–5 [DOI] [PubMed] [Google Scholar]
- 29.Lovell J. F., Billen L. P., Bindner S., Shamas-Din A., Fradin C., Leber B., Andrews D. W. (2008) Cell 135, 1074–1084 [DOI] [PubMed] [Google Scholar]
- 30.Etxebarria A., Terrones O., Yamaguchi H., Landajuela A., Landeta O., Antonsson B., Wang H. G., Basañez G. (2009) J. Biol. Chem. 284, 4200–4212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattila P. K., Pykäläinen A., Saarikangas J., Paavilainen V. O., Vihinen H., Jokitalo E., Lappalainen P. (2007) J. Cell Biol. 176, 953–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Staneva G., Seigneuret M., Koumanov K., Trugnan G., Angelova M. I. (2005) Chem. Phys. Lipids 136, 55–66 [DOI] [PubMed] [Google Scholar]
- 33.Tanaka T., Sano R., Yamashita Y., Yamazaki M. (2004) Langmuir 20, 9526–9534 [DOI] [PubMed] [Google Scholar]
- 34.Angelova M. I., Hristova N., Tsoneva I. (1999) Eur. Biophys. J. 28, 142–150 [DOI] [PubMed] [Google Scholar]
- 35.Khalifat N., Puff N., Bonneau S., Fournier J. B., Angelova M. I. (2008) Biophys. J. 95, 4924–4933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimada A., Niwa H., Tsujita K., Suetsugu S., Nitta K., Hanawa-Suetsugu K., Akasaka R., Nishino Y., Toyama M., Chen L., Liu Z. J., Wang B. C., Yamamoto M., Terada T., Miyazawa A., Tanaka A., Sugano S., Shirouzu M., Nagayama K., Takenawa T., Yokoyama S. (2007) Cell 129, 761–772 [DOI] [PubMed] [Google Scholar]
- 37.Frost A., Perera R., Roux A., Spasov K., Destaing O., Egelman E. H., De Camilli P., Unger V. M. (2008) Cell 132, 807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Youle R. J. (2007) Science 315, 776–777 [DOI] [PubMed] [Google Scholar]
- 39.Basañez G., Sharpe J. C., Galanis J., Brandt T. B., Hardwick J. M., Zimmerberg J. (2002) J. Biol. Chem. 277, 49360–49365 [DOI] [PubMed] [Google Scholar]
- 40.Antonsson B., Montessuit S., Sanchez B., Martinou J. C. (2001) J. Biol. Chem. 276, 11615–11623 [DOI] [PubMed] [Google Scholar]
- 41.Basañez G., Nechushtan A., Drozhinin O., Chanturiya A., Choe E., Tutt S., Wood K. A., Hsu Y., Zimmerberg J., Youle R. J. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 5492–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roucou X., Rostovtseva T., Montessuit S., Martinou J. C., Antonsson B. (2002) Biochem. J. 363, 547–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.