

Abstract
Voltage-gated potassium (Kv) channels are transmembrane tetramers of individual α-subunits. Eight different Shaker-related Kv subfamilies have been identified in which the tetramerization domain T1, located on the intracellular N terminus, facilitates and controls the assembly of both homo- and heterotetrameric channels. Only the Kv2 α-subunits are able to form heterotetramers with members of the silent Kv subfamilies (Kv5, Kv6, Kv8, and Kv9). The T1 domain contains two subdomains, A and B box, which presumably determine subfamily specificity by preventing incompatible subunits to assemble. In contrast, little is known about the involvement of the A/B linker sequence. Both Kv2 and silent Kv subfamilies contain a fully conserved and negatively charged sequence (CDD) in this linker that is lacking in the other subfamilies. Neutralizing these aspartates in Kv2.1 by mutating them to alanines did not affect the gating properties, but reduced the current density moderately. However, charge reversal arginine substitutions strongly reduced the current density of these homotetrameric mutant Kv2.1 channels and immunocytochemistry confirmed the reduced expression at the plasma membrane. Förster resonance energy transfer measurements using confocal microscopy showed that the latter was not due to impaired trafficking, but to a failure to assemble the tetramer. This was further confirmed with co-immunoprecipitation experiments. The corresponding arginine substitution in Kv6.4 prevented its heterotetrameric interaction with Kv2.1. These results indicate that these aspartates (especially the first one) in the A/B box linker of the T1 domain are required for efficient assembly of both homotetrameric Kv2.1 and heterotetrameric Kv2.1/silent Kv6.4 channels.
Introduction
Eight different voltage-gated K+ (Kv)3 Shaker-related channel subfamilies (Kv1–Kv6 and Kv8–Kv9) have been identified based on the degree of sequence homology (1). Fully assembled Kv channels are composed of four α-subunits arranged around a central pore. Each α-subunit consists of six transmembrane segments S1–S6 with a cytoplasmic N and C terminus. The N terminus contains the T1 domain, a tetramerization domain that facilitates the assembly of α-subunits into functional channels. The presence of a T1 domain is not absolutely required for channel assembly because subunits without a T1 domain could also assemble into a functional tetramer, although less efficiently (2–4). However, the T1 domain not only promotes but also restricts the formation of possible homo- and heterotetramers by preventing incompatible subunits from assembling (5–7). When four compatible T1 domains assemble, they are arranged with the same 4-fold symmetry as the transmembrane segments, forming a hanging gondola structure (8).
The T1 domain contains two subdomains, designated A and B box, which are both highly conserved within the different Kv subfamilies (9). Several residues in both the A and B boxes form the contact interface between neighboring subunits and are commonly assumed to be the key determinants for (subfamily specific) channel tetramerization (6, 10, 11). On the other hand, the amino acid sequence between both subdomains varies in length and sequence conservation and has therefore been assumed to serve no specific role. However, a recent study reported that this linker sequence in Kv2.1 channels is involved in cell surface expression of the channels, voltage-dependent channel activation and phosphorylation-dependent gating modulation (12).
Members of the Kv1–Kv4 subfamilies assemble into functional K+ channels with a homotetrameric configuration. The functional diversity within these subfamilies is increased by forming heterotetramers and by interacting with auxiliary β-subunits. Members of the Kv2 subfamily increase their diversity further by forming heterotetramers with α-subunits of the Kv5, Kv6, Kv8, and Kv9 subfamilies. These latter Kv subfamilies are designated silent α-subunits because they fail to express functional channels in a homotetrameric configuration (13–17) and are retained in the endoplasmic reticulum (ER). This retention is rescued by the selective co-assembly with members of the Kv2 subfamily in which the silent Kv subunits modulate the Kv2 currents by reducing the current amplitude, altering the inactivation and deactivation kinetics and shifting the voltage dependence of inactivation toward more hyperpolarized potentials (13, 15–27).
An alignment of the Kv1–Kv9 subunits revealed a negatively charged sequence (CDD) in the region between the A and B box that is fully conserved in both Kv2 and silent Kv subunits, but absent in the Kv1, Kv3, and Kv4 subfamilies (Fig. 1A). This pattern of amino acid conservation drew our attention regarding the subfamily-specific channel assembly. Therefore, we studied the contribution of this CDD sequence in the assembly of homotetrameric Kv2.1 and heterotetrameric Kv2.1/Kv6.4 channels using biophysical, biochemical, and Förster resonance energy transfer (FRET) analysis of mutant Kv2.1 and Kv6.4 channel subunits.
FIGURE 1.

Location of the CDD sequence. A, sequence alignment of the region between the A and B box of the T1 domain from different Kv channels generated with the program MEGALIGN. For clarity only one member of each subfamily is shown. The CDD sequence (shaded in gray) is absolutely conserved between Kv2 and all known silent Kv subunits but absent in the other subfamilies. B, side view of a structural model of the Kv2.1 T1 domain based on the rat Kv4.2 crystal structure and generated with SWISS-MODEL. Each subunit is represented in a different color. For clarity only three of the four subunits are shown. The Cys (C73) and Asp (D74 and D75) residues are shown in space-fill. C, bottom view of panel B.
EXPERIMENTAL PROCEDURES
Molecular Biology
Human Kv2.1 and Kv6.4 (previously named Kv6.3) were both cloned in the mammalian expression vector pEGFP-N1 (Clontech, Palo Alto, CA) as described previously (13). Mutations were introduced during a PCR amplification using the QuikChange Site-directed mutagenesis kit (Stratagene, La Jolla, CA) and mutant primers. Hemagglutinin (HA) epitope-tagged subunits were obtained by introducing a HA tag in the extracellular S1–S2 linker. The presence of the desired modification and the absence of unwanted mutations were confirmed by DNA sequencing. N-terminal-tagged cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) constructs were obtained by subcloning the channel subunits in the pECFP-C1 (Clontech) and pEYFP-C1 (Clontech) vectors, respectively. The YFP-CFP fusion construct (dimer) was obtained by removing the STOP codon and simultaneously introducing an EcoRI restriction site in the pEYFP-C1 vector. Subsequently, the entire coding sequence for YFP was cut from the vector with EcoRI and ligated in an EcoRI-digested pECFP-N1 vector. The final construct, named YFP-CFP dimer, contains an in-frame sequence with a 17-amino acid long linker between the YFP and CFP.
Electrophysiology
Mouse Ltk− cells (ATCC CCL 1.3) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% horse serum and 1% penicillin-streptomycin under a 5% CO2 atmosphere. The Ltk− cells were transiently transfected with the appropriate channel cDNA, in combination with 0.5 μg of GFP as a transfection marker. Transfections were performed on cultures at 70% confluency with the Lipofectamine 2000 reagent according to the manufacturer's instructions (Invitrogen). The amount of cDNA transfected and overnight incubation conditions (37 or 25 °C) are indicated in the figures and legends. The Kv6.4 subunits were co-transfected with Kv2.1 in a 10:1 ratio to ensure that the presence of homotetrameric Kv2.1 channels was less than 0.01%. Cells were trypsinized 16 to 24 h after transfection and used for electrophysiological analysis within 5 h.
Whole cell current recordings were obtained at room temperature (20–25 °C) using an Axopatch-200B amplifier (Axon Instruments, Union City, CA). The current recordings were low-pass filtered and sampled at 1–10 kHz with a Digidata 1200A data acquisition system (Axon Instruments). The pClamp8 software (Axon Instruments) was used to control command voltages and data storage. Patch pipettes, with a resistance of 1.5–2.5 mΩ in the standard extracellular solution, were pulled with a laser puller P2000 (Sutter Instruments, Novato, CA) from 1.2-mm borosilicate glass (World Precision Instruments, Sarasota, FL) and then heat polished. The cells were superfused continuously with an extracellular solution containing (in mm): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 Hepes, 10 glucose and adjusted to pH 7.35 with NaOH. The pipettes were filled with an intracellular solution containing (in mm): 110 KCl, 5 K4-1,2-bis(2-aminophenoxyl)ethane-N,N,N′,N′-tetraacetic acid, 5 K2ATP, 1 MgCl2, and 10 Hepes with the pH adjusted to 7.2 using KOH. Junction potentials were zeroed with the filled pipette in the bath solution. The access resistance varied from 2 to 10 mΩ and cells were excluded from analysis if the series resistance exceeded 3 mΩ after compensation to ensure that voltage errors did not exceed 5 mV.
Pulse Protocols and Data Analysis
Voltage protocols (as shown in the figures and legends) were adjusted to characterize the biophysical properties of mutant channel subunits adequately. The interpulse interval was at least 15 s but was increased to 30 s for some protocols to prevent channels from accumulating in the inactivated state. The voltage dependence of channel activation or inactivation was fitted with a Boltzmann equation according to: y = 1/{1 + exp(− (V − V½)/k)}, in which V represents the voltage applied, V½ the voltage at which 50% of the channels are activated or inactivated, and k the slope factor. Kinetics of activation and deactivation were fitted with a single or double exponential function. Off-line leak corrections were performed for all current density data shown. Results are presented as mean ± S.E. Statistical analysis was done using the Student's t test or a Mann Whitney U Rank Sum test in case unequal variances between the data sets were present. p values < 0.05 were considered to be significantly different.
Immunofluorescence Staining
HEK293 cells were cultured on coverslips in modified Eagle's medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% non-essential amino acids under a 5% CO2 atmosphere. The HEK293 cells were transiently transfected with 1 μg of the N-terminal CFP-tagged construct according to the lipofection method using Lipofectamine (Invitrogen). Cells were fixated 24 to 48 h after transfection with 4% paraformaldehyde. After fixation, cells were incubated overnight with the K39/25 antibody (1:50), a mouse monoclonal antibody generated against the extracellular S1–S2 loop of Kv2.1 (28), dissolved in a 0.1 m phosphate-buffered saline solution containing 10% horse serum and 0.1% bovine serum albumin (BSA-c, Aurion, Wageningen, The Netherlands). R-Phycoerythrin (PE)-labeled anti-mouse (Invitrogen) was used as secondary antibody (1:100) and was dissolved in a 0.1 m phosphate-buffered saline solution supplemented with 1% horse serum. After a 1-h incubation with the secondary antibody, confocal images were obtained on a Zeiss CLSM 510 microscope equipped with an argon laser (excitation 458 nm) and a helium-neon laser (excitation 543 nm) for visualization of CFP-tagged channels (emission signal recorded in the 480–520-nm bandwidth) and PE-labeled antibodies (emission signal recorded beyond the 560-nm bandwidth), respectively. The cell surface expression level of the different channel constructs was determined on three independent experiments.
FRET Analysis
HEK293 cells were cultured on coverslips and transfected with 1 μg of channel cDNA as described above. YFP-tagged Kv2.1 subunits were co-transfected with CFP-tagged Kv2.1 or CFP-tagged Kv6.4 constructs in a 2:1 cDNA ratio to ensure that all FRET donor molecules were paired with the FRET acceptor molecules. Cells were used 24–48 h after transfection for FRET analysis.
The fluorescent emission light of CFP (donor dye) and YFP (acceptor dye) molecules was determined using the Zeiss CLSM 510 microscope as detailed above. The confocal images were recorded with a lower stack size to minimize nonspecific bleaching of the fluorophores. FRET efficiencies were determined using Equation 1.
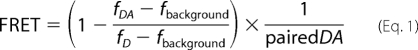 |
The CFP emission signal was recorded in the 464–490-nm bandwidth (after excitation at 458 nm) in both the presence of YFP (fDA, fluorescence signal of donor in the presence of the acceptor) and in the absence of YFP (fD, fluorescence of donor only). To determine fD, the YFP acceptor molecule was bleached by a 30-s full power excitation at 514-nm laser light (quality check for this bleach was a drop in the emission light in the 520–580-nm bandwidth). Both fDA and fD were corrected for background signal (fbackground) by determining the emission light in the 464–490-nm bandwidth after additional bleaching of CFP with a 30-s full power 458-nm laser light exposure. The cDNA ratio of the YFP- and CFP-tagged constructs used was chosen to minimize the fraction of unpaired donor and the paired DA fraction was therefore assumed to be 1 in the situation of WT channel subunits. FRET efficiencies were determined from three or more independent transfections and only calculated for cells in which the YFP:CFP ratio was higher than 2.5. Given that for a YFP-CFP fluorophore pair the Förster distance (Ro) is 49.2 Å (29), the distance between both chromophores (R) can be calculated from the obtained FRET efficiencies using the following equation.
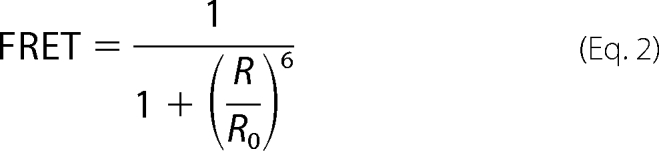 |
Co-immunoprecipitation (Co-IP)
HA-tagged Kv2.1 subunits were co-transfected with CFP-tagged Kv2.1 subunits into HEK293 cells in a 1:1 cDNA ratio. Cells were solubilized in phosphate-buffered saline supplemented with 5 mm EDTA, 1% Triton X-100, and a complete protease inhibitor mixture (Roche Diagnostics). For the immunoprecipitation, 1.7 μg/μl of the total soluble fraction was incubated with 2 μl of anti-GFP (Abcam, Cambridge, UK) followed by incubation with 20 μl of Protein G-agarose beads (Roche Diagnostics), which were preblocked with 2% milk. After extensive washing of the beads with solubilization buffer, proteins were eluted by boiling the beads in NuPAGE LDS sample buffer (Invitrogen) for 15 min. Subsequently, the eluted protein complexes were separated on a 4–12% BisTris SDS-PAGE gel (Invitrogen). The separated protein complexes were transferred to a polyvinylidine difluoride membrane (GE Healthcare) and the blot was blocked with 5% milk. Immunoprecipitated proteins were detected by incubation of the blot with anti-HA (Roche Diagnostics) followed by incubation with anti-rat IgG (GE Healthcare) and subsequently ECL detection (GE Healthcare).
Sequence Alignment and Homology Modeling
Sequence alignment of the different Kv channel subfamilies (shown in Fig. 1) was build using MEGALIGN (DNAstar, Madison, WI). The Kv2.1 homology model used was generated with the web based interface of the SWISS-MODEL Repository data base (30, 31). Illustrations were produced with the program USCF Chimera (32).
RESULTS
Charge Reversal of the CDD Sequence in Kv2.1 Primarily Affects the Current Density with Limited Biophysical Effects
An alignment of the T1 domain of all members of the Kv1–Kv9 subfamilies revealed an interesting conservation pattern of the net negatively charged CDD sequence, localized in the region between the A and B box; this CDD sequence is fully conserved in both Kv2 and silent Kv subunits but absent in other subfamilies (Fig. 1A). To determine the localization of this conserved CDD sequence, a homology model of the Kv2.1 T1 domain was generated with SWISS-MODEL (30, 31) based on the Kv4.2 T1 domain crystal structure (Fig. 1, B and C). Within this structural model the CDD sequence is situated on a discrete loop at the bottom of the Kv2.1 T1 domain. Because this loop was the only striking difference between the Kv2.1 model and the available Kv1.2 (33), Kv3.1 (10), and Kv4.2 (34) crystal structures, we wondered whether this CDD sequence would play an important role in the subfamily-specific interaction within the Kv2 subfamily and between the Kv2 and silent Kv subfamilies.
To investigate the potential role of the CDD sequence in the homotetrameric Kv2.1 channel assembly, we initially replaced the cysteine (at position 73) and the two aspartates (at positions 74 and 75) individually or together with alanine residues (C73A, D74A, D75A, and D74A,D75A, respectively). These mutations resulted in functional channels with biophysical properties similar to WT Kv2.1. However, the current density of the double D74A,D75A mutant was significantly reduced compared with WT (supplemental Fig. S1). For the single D74A mutant a similar trend was observed, albeit not statistically significant. Next we reversed the charge of the aspartates by replacing these amino acids alone or together with arginine residues (D74R, D75R, and D74R,D75R). Typical current recordings of wild type (WT) Kv2.1 and the charge-reversal mutants Kv2.1(D74R), Kv2.1(D75R), and Kv2.1(D74R,D75R) are shown in Fig. 2A. Both the voltage dependence of activation (Fig. 2B, Table 1) and inactivation (see Fig. 2C and Table 1) as well as the kinetics of activation and deactivation (see Fig. 2D and Table 1) were quite similar to those from WT Kv2.1. However, the translocation of the channel subunits to the plasma membrane, as measured by the overall current density, was severely decreased (Fig. 3, A and B). In contrast to WT Kv2.1, no macroscopic time-dependent K+ current could be detected after transfection of 50 ng of cDNA and overnight incubation at 37 °C for the different Kv2.1 charge-reversal (Asp > Arg) mutants, whereas with these conditions WT Kv2.1 yielded 2.7 ± 0.6 nA current (n = 12, Fig. 3B). By increasing the amount of cDNA transfected to 0.5 μg, both charge-reversal mutants displayed time-dependent Kv2.1-type currents but Kv2.1(D74R) still displayed significantly smaller current densities compared with Kv2.1(D75R). This indicated that substituting Asp-74 had a more severe effect than substituting Asp-75. Transfection of 5 μg of cDNA was required to observe measurable currents for the double mutant (D74R,D75R). Interestingly, overnight incubation at 25 °C largely abolished the effect of the different Asp > Arg mutants (Fig. 3B).
FIGURE 2.

Biophysical properties of WT Kv2.1 and charge-reversal mutants. A, whole cell current recordings of WT Kv2.1 (○), Kv2.1(D74R) (▲), Kv2.1(D75R) (●), and Kv2.1(D74R,D75R) (■). The voltage protocol is given on top with the corresponding currents below. The same scale bars were used for all recordings. B, voltage dependence of activation derived from plotting the normalized tail current amplitudes at −25 mV as a function of the prepulse potential (ranging from −50 to +70 mV with 10-mV increments) and fitted with a Boltzmann function (solid line). C, voltage dependence of inactivation obtained from the normalized peak current amplitude at +60 mV after a 5-s prepulse plotted as a function of the prepulse potential. D, time constants of activation and deactivation derived from a single or double exponential fit of the raw current traces (panel A). Note that the biophysical properties of the different charge-reversal Kv2.1 mutants were similar to those of WT Kv2.1.
TABLE 1.
Biophysical properties of WT and mutant Kv2.1 channel subunits
Values are given as mean ± S.E. and n the number of cells analyzed. The midpoints of activation and inactivation (V1/2) and both slope factors (k) were obtained from a single Boltzmann fit (“Experimental Procedures”). Time constants (tau) were obtained from fitting current activation and deactivation at the indicated voltage with a single or double exponential function, respectively.
| Kv2.1 | Kv2.1 |
|||
|---|---|---|---|---|
| D74R | D75R | D74R,D75R | ||
| Activation | ||||
| V½ | 6.1 ± 2.2 | 1.7 ± 2.1 | −2.2 ± 1.2 | 12.0 ± 2.9 |
| Slope | 9.0 ± 0.4 | 8.1 ± 0.6 | 7.9 ± 0.3 | 12.4 ± 0.7 |
| Tau (60 mV) | 23.7 ± 1.8 | 22.4 ± 2.9 | 23.0 ± 2.5 | 26.1 ± 4.4 |
| n | 7 | 6 | 5 | 5 |
| Inactivation | ||||
| V½ | −18.2 ± 2.3 | −21.4 ± 3.0 | −22.0 ± 1.6 | −12.9 ± 0.7 |
| Slope | 6.4 ± 0.5 | 4.7 ± 0.4 | 4.8 ± 0.4 | 5.2 ± 0.4 |
| n | 7 | 5 | 5 | 4 |
| Deactivation | ||||
| Tau 1 (−20 mV) | 26.9 ± 1.8 | 23.1 ± 3.4 | 23.9 ± 3.5 | 25.6 ± 4.7 |
| Tau 2 (−20 mV) | 126.8 ± 10.8 | 154.5 ± 22.5 | 217.6 ± 22.7 | 247.6 ± 39.2 |
| n | 6 | 5 | 4 | 5 |
FIGURE 3.

Current density of WT and charge-reversal Kv2.1 channels. A, whole cell current recordings of WT Kv2.1 and Kv2.1(D74R) elicited by the same voltage protocol as in Fig. 2A with the incubation conditions and amount of cDNA transfected indicated. The same scale bars were used for all recordings. B, current density of WT and mutant channels determined at the end of a 500-ms step to +30 mV. Note that 10–100 times more cDNA of the charge-reversal Kv2.1 mutants needed to be transfected to approach the WT current density. The expression levels improved after overnight incubation at 25 °C instead of the usual 37 °C, consistent with temperature-sensitive folding problems. The mutant Kv2.1 channels with a current density that were statistically different from WT Kv2.1 are indicated with an asterisk (p < 0.05). The numbers above every bar plot indicate the number of cells analyzed. N.D., not determined (already current at lower DNA concentrations).
The Decrease in Current Density of the Charge-reversal Kv2.1 Mutants Reflects a Reduced Plasma Membrane Expression
The decreased current density of the Asp > Arg mutants suggested that less channels reached the plasma membrane. We investigated this by analyzing the surface expression of CFP-tagged WT or mutant Kv2.1 channels with immunofluorescence. Transfected HEK293 cells were stained with the K39/25 antibody followed by a PE-labeled secondary antibody (see “Experimental Procedures”), without permeabilizing the cells. Thus, only the channels that were able to translocate from the ER to the plasma membrane were stained because the K39/25 antibody epitope is situated in the extracellular S1–S2 loop. We tested the specificity of this approach and the K39/25 antibody by staining cells expressing WT CFP-Kv2.1 (positive control) and WT CFP-Kv4.2 (negative control) in which the CFP tag served as a transfection marker. Immunofluorescent staining of WT CFP-Kv2.1 generated a clear plasma membrane (PM) staining (red fluorescence) indicating a PM expression of WT CFP-Kv2.1 (Fig. 4A). This was verified by the observed blue PM fluorescence originating from the genetically encoded CFP tag (Fig. 4A). In contrast, immunofluorescent staining of WT CFP-Kv4.2 caused no noticeable PM staining (red fluorescence) although the channels were robustly expressed at the PM as indicated by the peripheral blue fluorescent signal from the genetically encoded CFP molecule (Fig. 4B). Next, we analyzed the surface expression of the charge-reversal Kv2.1 mutants. Immunofluorescent staining of cells expressing CFP-Kv2.1(D74R) and CFP-Kv2.1(D74R,D75R) exhibited scarce (for the D74R mutant) or virtually no (for the D74R,D75R mutant) PM staining. Furthermore, the localization of the CFP fluorescence of these two mutants, no blue PM staining but a more diffuse intracellular staining, indicated that the mutant channel subunits were synthesized but were unable to translocate from the ER to the PM. In contrast, CFP-Kv2.1(D75R) displayed a pattern of immunofluorescent staining similar to that of WT Kv2.1. Thus, the results on the surface expression level for the different Asp > Arg mutants, obtained from the immunocytochemistry, corresponded well with the electrophysiological observations.
FIGURE 4.

Immunofluorescent analysis of plasma membrane expression of WT and charge-reversal mutant Kv2.1 channels. Immunofluorescence detection of WT CFP-Kv2.1 (A), WT CFP-Kv4.2 (B), CFP-Kv2.1(D74R) (C), CFP-Kv2.1(D75R) (D), and CFP-Kv2.1(D74R,D75R) (E) by incubation with a K2.1-specific antibody (K39/25) followed by a PE-labeled secondary antibody. The left, middle, and right columns represent CFP fluorescence, PE fluorescence, and the overlay of both, respectively. The CFP fluorescence (originating from the genetically encoded CFP molecule) gave an indication of the localization of all the Kv subunits in the whole cell, whereas PE fluorescence (originating from the antibody against the extracellular S1-S2 loop) represented only the Kv subunits located at the PM. For the Kv subunits that have a PM localization, a maximal overlap between the CFP and PE fluorescence could be observed. Scale bars = 10 μm.
The Reduced Plasma Membrane Expression of the Charge-reversal Kv2.1 Mutants Is Due to a Deficiency in Assembly
The reduced density of the mutant channels in the plasma membrane could be caused by either a deficiency in their assembly in the ER or a problem in trafficking from the ER to the PM. To discriminate between these two possibilities we determined FRET between genetically encoded CFP and YFP fluorophores that were added at the N-terminal end of WT Kv2.1 and mutant subunits. N-terminal labeled channel subunits were used because we and others (35) observed no FRET signal when the CFP and YFP molecules were attached to the C-terminal end of the channel subunits, probably because in this configuration the distance between both fluorophores in a functional channel was too large (above 100 Å) for FRET. This is strengthened by the observation that both the N- and C-terminal-tagged Kv2.1 channel subunits with CFP or YFP yielded functional channels that displayed biophysical properties and a current density level similar to WT Kv2.1 (data not shown). With the N-terminal-tagged Kv2.1 constructs a FRET efficiency of 11.1 ± 0.9% (calculated with Equation 1 under “Experimental Procedures”, n = 21) was obtained, corresponding to an YFP/CFP fluorophore separation of 71.2 ± 1.3 Å (calculated with Equation 2 under “Experimental Procedures”). As a positive control, we created an YFP-CFP dimer that displayed FRET efficiency of 38.5 ± 1.5% (n = 18) reflecting an YFP/CFP fluorophore separation of 52.9 ± 0.8 Å. This distance is within the expected range based on their molecular size and a structural model for this dimer (supplemental Fig. S2). As a negative control, the FRET efficiency was determined between N-terminal-tagged Kv2.1 and Kv4.2 subunits because these subunits fail to form heterotetramers. As expected, no FRET was observed for both combinations YFP-Kv2.1 + CFP-Kv4.2 (n = 11, Fig. 5C) and CFP-Kv2.1 + YFP-Kv4.2 (data not shown).
FIGURE 5.

Channel assembly determined with FRET using CFP- and YFP-tagged channel constructs. A, a representative cell expressing both CFP and YFP N-terminal-tagged Kv2.1 channel subunits in a 1:2 ratio (0.33 μg of CFP-Kv2.1 and 0.66 μg of YFP-Kv2.1, respectively). The panels on the left and right show the emission observed in the CFP and YFP bandwidths after excitation with a 458-nm laser line, respectively. The panels on top represent CFP and YFP emissions before bleaching YFP in the whole cell. If FRET occurs the CFP emission is underestimated by transfer of the CFP emission light onto the nearby YFP, which is relieved after bleaching YFP (bottom panels): note the increase in CFP intensity and the marked decrease in YFP emission after YFP bleach. B, emission spectra of a selected PM region (indicated in A as a red box) before (black line) and after (green line) bleaching YFP. Note the increase in light intensity in the CFP bandwidth (464–490 nm, red circle) after YFP is bleached in the whole cell. Background signal (yellow line) was determined after fully bleaching CFP also. C, average FRET efficiencies for WT and the mutant Kv2.1 constructs determined in the PM and cell region. A dimeric YFP-CFP construct was used as positive control and a CFP-Kv2.1 + YFP-Kv4.2 combination as negative. Note that the FRET efficiencies for WT Kv2.1 determined in the cell interior and PM region were similar and that the D74R and D74R,D75R mutations displayed significantly decreased FRET efficiencies compared with WT Kv2.1 (indicated with asterisk, p < 0.05). The numbers above every bar indicate the number of cells analyzed.
The above controls indicate that fully functional Kv2.1 tetramers, residing in the PM, yield a significant FRET efficiency of ∼11%. To use FRET for addressing whether channel mutants failed to assemble or not, we first tested whether assembled Kv2.1 could be detected in the ER using FRET. Therefore, FRET efficiencies between CFP- and YFP-tagged Kv2.1 subunits were compared between a PM region and an intracellular region that should be representative for the ER (Fig. 5, A and C). Because only fully assembled Kv2.1 channels reside in the PM, the FRET efficiency obtained in the PM region served as a positive control for the FRET signal obtained from the cell interior. Fig. 5C shows that a similar FRET efficiency was obtained for both regions indicating that channel tetramers (or at least dimers) still residing in the cell interior (ER) can be detected with the FRET approach. This makes FRET between genetically encoded CFP and YFP fluorophores an adequate technique to determine whether channel assembly in living cells is impaired or not.
If the Kv2.1 charge-reversal mutations (D74R, D75R, and D74R,D75R) only impair channel trafficking to the plasma membrane, the channel subunits would still be able to assemble and a positive FRET signal should be observed in the cell interior. On the other hand, if the observed decrease in current density is caused by an underlying assembly deficiency, then limited or no FRET should be observed. When the FRET efficiencies of the different charge-reversal Kv2.1 mutants were determined in the cell interior (Fig. 5C), a significant decrease in FRET efficiency was indeed observed for the Kv2.1(D74R) mutant compared with WT Kv2.1. The FRET efficiency for the double mutant Kv2.1(D74R,D75R) was even further reduced and was only marginally above the background fluorescence (Fig. 5C). On the other hand, the substitution D75R that had a markedly lower impact on channel assembly than the other two mutants, based on the electrophysiological and immunocytochemical data, displayed FRET efficiency similar to WT Kv2.1.
To confirm the results obtained from the FRET approach, we performed co-IP experiments. For this purpose, CFP- and HA-tagged Kv2.1 subunits were heterologously expressed, cells were solubilized, and the CFP-Kv2.1/HA-Kv2.1 complexes were precipitated from the soluble fraction with GFP antibodies. Immunoprecipitated Kv subunits were examined by Western blotting. Incubation of the blots with HA antibodies demonstrated that the assembly of the Kv2.1(D75R) mutant was similar to WT Kv2.1 (Fig. 6A). However, assembly of Kv2.1(D74R) was strongly diminished, whereas assembly of Kv2.1(D74R,D75R) was not detectable (Fig. 6A). These co-IP results thus corroborated the FRET results indicating that the inability of the charge-reversal Kv2.1 mutants to translocate from the ER to the PM was due to a deficiency in assembly and not to impaired trafficking of fully assembled channels.
FIGURE 6.

Co-IP of CFP- and HA-tagged Kv2.1 subunits. After co-expression of CFP- and HA-tagged Kv2.1 subunits, CFP-Kv2.1/HA-Kv2.1 channel complexes were precipitated from the soluble cell fraction with GFP antibodies. Immunoprecipitated Kv subunits were analyzed with Western blotting using HA antibodies. Note that HA-tagged WT Kv2.1 and Kv2.1(D75R) subunits could readily be detected after precipitation of CFP-tagged subunits indicating that these subunits assembled into tetramers. Kv2.1(D74R) was also detectable but much more weakly, whereas HA-tagged Kv2.1(D74R,D75R) could not be detected.
Reversal of the CDD Charge in Kv6.4 Influences the Formation of Kv2.1/Kv6.4 Heterotetramers
Because Kv2.1 interacts with members of the silent Kv subfamilies and because this CDD sequence is also conserved in these silent Kv subunits (Fig. 1A), we investigated the potential role of this sequence in the heterotetrameric Kv2.1/silent Kv subunit assembly. The interaction between WT Kv2.1 and mutant Kv6.4 subunits was examined for this purpose. Because the charge reversal mutations affected assembly of Kv2.1 most profoundly, we investigated in Kv6.4 only the effect of replacing aspartate residues Asp-102 and Asp-103 (the equivalent residues of Asp-74 and Asp-75 in Kv2.1) one-by-one or together by arginine residues. Thus, the following channel mutants were created: Kv6.4(D102R), Kv6.4(D103R), and Kv6.4(D102R,D103R). Fig. 7A shows typical current recordings of WT Kv2.1 alone (left) and upon co-expression with WT Kv6.4 (right). The modulating effects of Kv6.4 subunits on the voltage dependence of activation were limited and it was therefore difficult to discriminate between homo- or heterotetrameric channels based on the activation properties. Consequently, we focused on the voltage dependence of inactivation. Wild type Kv6.4 shifted the voltage dependence of inactivation of Kv2.1 by approximately −40 mV in the hyperpolarizing direction (see Fig. 7B and Table 2). In contrast, no shift of the voltage dependence of inactivation could be observed after co-expression of Kv6.4(D102R) or Kv6.4(D102R,D103R) with WT Kv2.1. The voltage dependence of inactivation of the Kv6.4(D103R) mutant displayed two components suggesting the presence of both homotetrameric Kv2.1 and heterotetrameric Kv2.1/Kv6.4(D103R) channels that are characterized by a V½ of −18 and −59 mV, respectively (Table 2).
FIGURE 7.

Electrophysiological and FRET analysis of Kv2.1 heterotetramers with WT and mutant Kv6.4 channel subunits. A, current recordings of 0.5 μg of WT Kv2.1 alone (left) and upon co-transfection with 5 μg of WT Kv6.4 (right). B, voltage dependence of inactivation. The inactivation curves were obtained as described in the legend to Fig. 2C. WT Kv6.4 (●) shifts the V½ with approximately −40 mV in the hyperpolarized direction compared with WT Kv2.1. This shift was not observed upon co-transfection of the Kv6.4(D102R) and Kv6.4(D102R,D103R) mutants suggesting that the mutant subunits did not interact with WT Kv2.1. The data of the Kv6.4(D103R) mutant was fitted with a sum of 2 Boltzmann functions (solid lines), suggestive for the presence of two populations: homotetrameric Kv2.1 and heterotetrameric Kv2.1/Kv6.4 channels. C, current density of WT Kv2.1 (0.5 μg) alone and upon co-transfection with WT KCNE1, WT Kv6.4, or mutant Kv6.4 subunits (0.5 + 5 μg). The current densities obtained from WT Kv2.1 co-expressed with WT Kv6.4 or mutant Kv6.4 subunits that were significantly different from the WT Kv2.1/WT KCNE1 co-expression are indicated with an asterisk (p < 0.05). D, average FRET efficiencies for the co-expression of YFP-tagged Kv2.1 subunits (0.66 μg) with WT or mutant CFP-tagged Kv6.4 constructs (0.33 μg) determined in the PM and/or cell interior (ER) region. Note that FRET efficiencies of the Kv2.1/WT Kv6.4 co-expression determined in the cell interior and PM region were similar showing that heterotetrameric Kv2.1/WT Kv6.4 channel complexes could be detected in the cell interior. The FRET efficiencies in the cell interior for the Kv2.1/Kv6.4(D102R) and Kv2.1/Kv6.4(D102R,D103R) co-expressions were significantly decreased compared with the Kv2.1/WT Kv6.4 combination (specified with asterisk, p < 0.05). The numbers above every bar plot indicate the number of cells analyzed. Note that the same Kv6.4(D102R) and Kv6.4(D102R,D103R) mutations that eliminated the WT Kv6.4 suppression of the Kv2.1 current (panel C) did not display FRET (panel D), consistent with a lack of interaction and heterotetrameric channel assembly.
TABLE 2.
Voltage dependence of inactivation for WT Kv2.1 alone and upon co-transfection with WT and mutant Kv6.4 channel subunits
Values are given as mean ± S.E. and n the number of experiments. The midpoints of inactivation (V1/2) and slope factors (k) were obtained from a single or double Boltzmann fit (“Experimental Procedures”).
| Inactivation | Kv2.1 | +Kv6.4 | +Kv6.4 |
||
|---|---|---|---|---|---|
| D102R | D103R | D102R,D103R | |||
| 1st V½ | −18.2 ± 2.3 | NAa | −22.5 ± 0.6 | −23.7 ± 1.6 | −22.5 ± 1.1 |
| Slope | 6.4 ± 0.5 | NA | 6.5 ± 0.8 | 4.9 ± 0.3 | 5.9 ± 0.4 |
| 2nd V½ | NA | −59.2 ± 2.5 | NA | −59.5 ± 3.0 | NA |
| Slope | NA | 7.4 ± 1.2 | NA | 6.5 ± 1.1 | NA |
| n | 7 | 7 | 5 | 6 | 5 |
a NA, not applicable.
A second pronounced effect of WT Kv6.4 is the down-regulation of the Kv2.1 current density. Therefore, we also investigated the impact of the Kv6.4 mutants on the Kv2.1 current suppression. When 0.5 μg of Kv.2.1 cDNA was co-expressed with 5 μg of Kv6.4 cDNA the current density was suppressed by a 20–100-fold (Fig. 7C). Because this down-regulation may in part be caused by dilution of the Kv2.1 cDNA or by an increased load on the transcription/translation machinery, we transfected 0.5 μg of Kv2.1 cDNA with 5 μg of cDNA of the human membrane protein KCNE1 that does not modulate Kv2.1. This Kv2.1/KCNE1 co-transfection resulted in a current density of 404 ± 58 pA/pF (n = 4), which is 3–5-fold less compared with 0.5 μg of WT Kv2.1 alone but still a factor 10 higher than the 37.0 ± 10.1 pA/pF current density of the Kv2.1/Kv6.4 combination. This indicated that co-expressing WT Kv6.4 resulted in at least a 10-fold current suppression (Fig. 7C). Conversely, expression of Kv6.4(D102R) and Kv6.4(D102R,D103R) with WT Kv2.1 resulted in current densities that were not significantly different from the Kv2.1/KCNE1 co-transfection, indicating that less heterotetramers were created or that the modulating effect of K6.4 on the current density level was impaired. The Kv6.4(D103R) mutant had an intermediate effect on the Kv2.1 current density.
To discriminate whether the effects of the Kv6.4 mutants resulted from a deficient assembly or from a loss of the modulatory mechanism, we performed FRET analysis using CFP-tagged Kv6.4 and YFP-tagged Kv2.1 subunits. FRET efficiencies for Kv2.1/Kv6.4 channels determined in the PM and cell interior region were similar indicating that heterotetrameric Kv2.1/Kv6.4 channels could be detected in the PM and ER. After expression of Kv6.4(D102R) or Kv6.4(D102R,D103R) with WT Kv2.1, FRET efficiencies were drastically decreased, whereas expression of Kv6.4(D103R) with Kv2.1 produced intermediate FRET efficiencies (Fig. 7D). These observations indicated that these aspartate residues in Kv6.4 are a requisite for the assembly with WT Kv2.1 subunits.
DISCUSSION
The tetramerization of four Kv α-subunits into a functional channel is coordinated by residues in the T1 domain, which is situated on the cytoplasmic N terminus of each α-subunit (2–7). Presumably, the (subfamily specific) tetramerization is determined by the residues at the contact interface between the individual T1 domains (6, 10, 11). The majority of the residues at the contact interface are polar and situated in the A and B box regions. The linker between these two subdomains has been assumed to serve no specific role in channel assembly because it varies in length and sequence conservation within and between the Kv subfamilies. Nevertheless, a recent study has shown that this linker region (in rat Kv2.1) interacts with parts of the C terminus and that this interaction is involved in surface expression of the channel (12). In this study we investigated further the role of this linker in channel assembly and more precisely the contribution of the negatively charged sequence (CDD) that is absolutely conserved within the Kv2 and silent Kv (Kv5, K6, Kv8, and Kv9) subfamilies. This CDD sequence represents the last three residues of the T1 section that was reported to interact with the C terminus in rat Kv2.1 (note that human Asp-74 corresponds to rat Asp-70) (12).
Neutralizing the negative charges in Kv2.1 by replacing the aspartates individually with alanine residues did not affect the biophysical properties nor the current density significantly (supplemental Fig. S1). However, when both residues were simultaneously replaced by an alanine the current density was significantly reduced. This decrease in current without altering the biophysical properties of the functional channels was even more pronounced with the charge-reversal arginine substitutions (see Figs. 2 and 3). Apparently, substituting the aspartate at position 74 had a larger effect than replacing Asp-75. Immunocytochemical staining of HEK293 cells expressing WT or mutant Kv2.1 channels showed that the reduced current density of the charge reversal mutants was caused by fewer channels reaching the plasma membrane (see Fig. 4). Thus, both the electrophysiological data (see Figs. 2 and 3) as well as immunocytochemical cell staining (see Fig. 4) showed that substituting the negative charges of the CDD sequence reduced the channel density at the plasma membrane. Lowering the temperature incubation condition to 25 °C largely abolished the effect of the different charge-reversal Kv2.1 mutants suggesting that the inability of the mutant channels to translocate from the ER to the PM was due to a temperature-sensitive folding problem as previously described for other ion channels such as the human ether-a-gogo related gene channel (36).
To discriminate more directly whether the inability of the mutant channels to translocate to the plasma membrane was caused by either a trafficking or tetramerization deficiency, we used FRET measurements. A similar approach was recently used to demonstrate that substitutions of histidine residue 105 (which forms part of the Zn2+-coordinating C3H1 motif) impaired both the formation of homotetrameric Kv2.1 and heterotetrameric Kv6/Kv2 channels (37). Thus, FRET analysis between genetically tagged channel constructs allows to investigate protein complex formation in living cells (38–41). We combined FRET measurements with confocal imaging to determine and compare FRET efficiencies in different cell compartments: PM versus cell interior (ER fraction). Fig. 5C shows that channel tetramers (or at least subunit dimers) could be detected in the cell interior for the tagged WT Kv2.1 subunits. Furthermore, the reduced plasma membrane expression of the channel mutants was accompanied by a significant reduction in FRET, indicating that it was caused by an underlying tetramerization failure. These FRET results obtained in living cells were validated biochemically with co-IP experiments that demonstrated that the assembly of the Kv2.1(D75R) mutant was similar to WT Kv2.1 but that the assembly of Kv2.1(D74R) and Kv2.1(D74R,D75R) was severely diminished (see Fig. 6).
In the study by Mederos y Schnitzler et al. (37) a FRET efficiency of ∼22% was reported for Kv2.1 homotetramers, whereas we obtained a FRET efficiency of ∼11%. Overall, the FRET efficiencies between Kv2.1 homotetramers and Kv2.1/Kv6.4 heterotetramers reported here were ∼10% lower compared with the efficiencies obtained in that study (37). Because our setup for FRET measurements is slightly different from their approach we performed additional control experiments with an YFP-CFP dimer protein. Our dimer construct resulted in a 38.5 ± 1.5% FRET efficiency that falls in the range of expected values based on their molecular structure (supplemental Fig. S2A) (42) and corresponds very well to other reported FRET efficiencies for YFP-CFP dimer constructs that varied from 25 to 42% depending on the size of the linker (35, 38, 40, 41, 43). Łukasiewicz et al. (39) reported an efficiency of 36% for a CFP-YFP dimer with a linker similar to the one we used. Because of laser limitations we excited CFP using a 458-nm laser line. Because YFP could be excited directly by 458-nm wavelength, which might result in YFP bleed-through in the CFP excitation bandwidth, we checked the YFP contribution in the 464–490-nm bandwidth by analysis of the YFP emission upon excitation with 458-nm laser light. This showed that the YFP emission in the 464–490-nm bandwidth was limited to less than 0.1% of its maximal peak emission (supplemental Fig. S2B); taking this into account in our FRET calculations it did not alter the obtained efficiencies. Another difference between our data and those of Mederos y Schnitzler et al. (37) is that our negative control experiments gave no FRET signal, whereas in their case a FRET efficiency of ∼5% was observed. Differences in background correction methodology might thus explain the differences in FRET efficiencies, in addition to the use of different N-terminal labeled channel constructs, with slightly different linkers.
Because the CDD sequence is conserved between Kv2 and the silent Kv subunits we hypothesized that this CDD sequence might also be involved in assembly of heterotetrameric Kv2.1/silent Kv channels and subsequently be a determinant for subfamily-specific tetramerization. Most silent subunits affect the Kv2.1 inactivation and current density markedly, whereas the effects on the Kv2.1 activation are more limited (13, 15–27). This is also the case for Kv6.4: WT Kv6.4 subunits do not affect the activation properties markedly but shift the voltage dependence of inactivation by −40 mV in a hyperpolarized direction and cause a severe down-regulation of the Kv2.1 current density (13) (in the original nomenclature Kv6.3). In contrast to WT Kv6.4 no hyperpolarizing shift in the voltage dependence of inactivation was observed with the charge-reversal Kv6.4 mutants and also the down-regulation of the current density was less. Both observations suggested that fewer heterotetrameric Kv2.1/Kv6.4 channels reached the plasma membrane. Furthermore, FRET measurements showed that the charge-reversal Kv6.4 mutants resulted in markedly reduced FRET signals with Kv2.1 in comparison to the WT Kv6.4 subunits. Similar to the results obtained in Kv2.1, substituting the first aspartate of the CDD sequence in Kv6.4 (Asp-102), which is the equivalent of Asp-74 in Kv2.1, had a greater impact on heterotetrameric channel assembly than mutating the second residue (Asp-103). Indeed, the voltage dependence of inactivation of Kv2.1/Kv6.4(D103R) displayed two components suggesting that a fraction of channels formed were heterotetramers but that most were Kv2.1 homotetramers. Overall, the results show that the aspartates of the CDD sequence are not only involved in the assembly process of homotetrameric Kv2.1 channels but also in that of heterotetrameric Kv2.1/Kv6.4 channels.
Approximately 30% of the amino acids in the entire T1 domain and around 70% of the interface residues are charged, suggesting that specific electrostatic or polar interactions are responsible for subfamily-specific tetramerization (6, 10, 11). The CDD sequence is located at the (cytoplasmic) bottom of the T1 domain in both our homology model (see Fig. 1) and that of others (12, 44) as well as the model based on single particle image reconstruction (45). This area is ∼25% composed of charged residues and the C-terminal region that it has been suggested to interact with, which contains ∼40% charged residues, most of which are positively charged (12). Therefore, it is likely that both aspartates are involved in electrostatic interactions with other residues that may be located within the same subunit (intrasubunit) or in a neighboring subunit (intersubunit). It may also be a combination of both because channel assembly involves both protein folding and subunit tetramerization. The double D74A,D75A mutant displayed a significant reduction in current density level that was corrected by increasing the amount of cDNA transfected to 0.5 μg, which was not the case for the D74R,D75R mutant (Fig. 3B). In this sense the effect of D74A,D75A was intermediate between WT and D74R,D75R. Therefore we suggest that the aspartates of the CDD sequence are part of a bigger electrostatic network that involves multiple interactions and that shapes an energy landscape for channel assembly. Replacing the negative charges by a neutral alanine would prevent the interaction with the normal partner(s) but this loss of interaction is partly compensated by the other contacts within the larger network. However, a charge-reversal arginine substitution would not only prevent the correct interaction(s) from being formed, but would also result in repulsion of the interacting charges. This would have two consequences: (i) the repulsion may be too strong to be compensated by the other interactions and/or (ii) the new charge distribution may result in new electrostatic interactions. Both effects would distort the correct folding of the T1 domain, precluding efficient subunit assembly. Thus the alanine substitutions would increase a specific barrier of the energy landscape for subunit folding and channel assembly to a lesser extent than the charge-reversal mutants. The observation that the charge-reversal mutants can be rescued by lowering the overnight incubation temperature also suggests that it involves small/subtle differences in the energy landscape; once the subunits pass this energy barrier they correctly assemble into functional tetrameric channels with biophysical properties similar to WT Kv2.1 channels.
In conclusion, the two aspartates (Asp-74 and Asp-75 in Kv2.1 and Asp-102 and Asp-103 in Kv6.4), which are located in the linker region between the A and B boxes of the T1 domain, contribute to both homotetrameric Kv2.1 and heterotetrameric Kv2.1/Kv6.4 channel assembly in which the first (Asp-74 and Asp-102) has a greater contribution. Furthermore, we show that subcellular FRET measurements using confocal microscopy with channel subunits labeled with CFP and YFP represent a suitable approach to determine, in living cells, whether a reduced channel expression is the result from either a failure to assemble the tetramer or from an impaired trafficking of fully assembled channel complexes.
Supplementary Material
Acknowledgments
We thank Natacha Ottschytsch and Diane Van Hoorick for helpful discussions. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputting, Visualization, and Informatics at the University of California, San Francisco, supported by National Institutes of Health P41 RR-01081.
This work was supported by a Ph.D. grant of the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen) (to E. B.), Interuniversity Attraction Poles program P6/31 of the Belgian Federal Science Policy Office, Research Foundation Flanders (FWO, Fonds voor Wetenschappelijk Onderzoek Vlaanderen) Grants G.0085.04 and G.0152.06 (to D. J. S.) and 1.5.044.07 (to A. J. L.), and GOA50.4426 from the concerted action fund of the University of Antwerp.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Table S1.
- Kv
- voltage-gated K+
- PM
- plasma membrane
- ER
- endoplasmic reticulum
- FRET
- Förster resonance energy transfer
- IP
- immunoprecipitation
- HA
- hemagglutinin
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- HEK
- human embryonic kidney
- PE
- R-phycoerythrin
- WT
- wild type
- BisTris
- 2-[bis(2- hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1.Gutman G. A., Chandy K. G., Grissmer S., Lazdunski M., McKinnon D., Pardo L. A., Robertson G. A., Rudy B., Sanguinetti M. C., Stühmer W., Wang X. (2005) Pharmacol. Rev. 57, 473–508 [DOI] [PubMed] [Google Scholar]
- 2.Kobertz W. R., Miller C. (1999) Nat. Struct. Biol. 6, 1122–1125 [DOI] [PubMed] [Google Scholar]
- 3.Tu L., Santarelli V., Sheng Z., Skach W., Pain D., Deutsch C. (1996) J. Biol. Chem. 271, 18904–18911 [DOI] [PubMed] [Google Scholar]
- 4.Zerangue N., Jan Y. N., Jan L. Y. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 3591–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee T. E., Philipson L. H., Kuznetsov A., Nelson D. J. (1994) Biophys. J. 66, 667–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen N. V., Pfaffinger P. J. (1995) Neuron 14, 625–633 [DOI] [PubMed] [Google Scholar]
- 7.Xu J., Yu W., Jan Y. N., Jan L. Y., Li M. (1995) J. Biol. Chem. 270, 24761–24768 [DOI] [PubMed] [Google Scholar]
- 8.Kobertz W. R., Williams C., Miller C. (2000) Biochemistry 39, 10347–10352 [DOI] [PubMed] [Google Scholar]
- 9.Drewe J. A., Verma S., Frech G., Joho R. H. (1992) J. Neurosci. 12, 538–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bixby K. A., Nanao M. H., Shen N. V., Kreusch A., Bellamy H., Pfaffinger P. J., Choe S. (1999) Nat. Struct. Biol. 6, 38–43 [DOI] [PubMed] [Google Scholar]
- 11.Kreusch A., Pfaffinger P. J., Stevens C. F., Choe S. (1998) Nature 392, 945–948 [DOI] [PubMed] [Google Scholar]
- 12.Mohapatra D. P., Siino D. F., Trimmer J. S. (2008) J. Neurosci. 28, 4982–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ottschytsch N., Raes A., Van Hoorick D., Snyders D. J. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 7986–7991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stocker M., Kerschensteiner D. (1998) Biochem. Biophys. Res. Commun. 248, 927–934 [DOI] [PubMed] [Google Scholar]
- 15.Hugnot J. P., Salinas M., Lesage F., Guillemare E., de Weille J., Heurteaux C., Mattéi M. G., Lazdunski M. (1996) EMBO J. 15, 3322–3331 [PMC free article] [PubMed] [Google Scholar]
- 16.Patel A. J., Lazdunski M., Honoré E. (1997) EMBO J. 16, 6615–6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salinas M., Duprat F., Heurteaux C., Hugnot J. P., Lazdunski M. (1997) J. Biol. Chem. 272, 24371–24379 [DOI] [PubMed] [Google Scholar]
- 18.Castellano A., Chiara M. D., Mellström B., Molina A., Monje F., Naranjo J. R., López-Barneo J. (1997) J. Neurosci. 17, 4652–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerschensteiner D., Stocker M. (1999) Biophys. J. 77, 248–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kramer J. W., Post M. A., Brown A. M., Kirsch G. E. (1998) Am. J. Physiol. 274, C1501–C1510 [DOI] [PubMed] [Google Scholar]
- 21.Post M. A., Kirsch G. E., Brown A. M. (1996) FEBS Lett. 399, 177–182 [DOI] [PubMed] [Google Scholar]
- 22.Richardson F. C., Kaczmarek L. K. (2000) Hear. Res. 147, 21–30 [DOI] [PubMed] [Google Scholar]
- 23.Salinas M., de Weille J., Guillemare E., Lazdunski M., Hugnot J. P. (1997) J. Biol. Chem. 272, 8774–8780 [DOI] [PubMed] [Google Scholar]
- 24.Sano Y., Mochizuki S., Miyake A., Kitada C., Inamura K., Yokoi H., Nozawa K., Matsushime H., Furuichi K. (2002) FEBS Lett. 512, 230–234 [DOI] [PubMed] [Google Scholar]
- 25.Shepard A. R., Rae J. L. (1999) Am. J. Physiol. 277, C412–C424 [DOI] [PubMed] [Google Scholar]
- 26.Vega-Saenz de Miera E. C. (2004) Brain Res. Mol. Brain Res. 123, 91–103 [DOI] [PubMed] [Google Scholar]
- 27.Zhu X. R., Netzer R., Böhlke K., Liu Q., Pongs O. (1999) Receptors and Channels 6, 337–350 [PubMed] [Google Scholar]
- 28.Lim S. T., Antonucci D. E., Scannevin R. H., Trimmer J. S. (2000) Neuron 25, 385–397 [DOI] [PubMed] [Google Scholar]
- 29.Patterson G. H., Piston D. W., Barisas B. G. (2000) Anal. Biochem. 284, 438–440 [DOI] [PubMed] [Google Scholar]
- 30.Kopp J., Schwede T. (2004) Nucleic Acids Res. 32, D230–D234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 32.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 33.Minor D. L., Lin Y. F., Mobley B. C., Avelar A., Jan Y. N., Jan L. Y., Berger J. M. (2000) Cell 102, 657–670 [DOI] [PubMed] [Google Scholar]
- 34.Nanao M. H., Zhou W., Pfaffinger P. J., Choe S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8670–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobrinsky E., Stevens L., Kazmi Y., Wray D., Soldatov N. M. (2006) J. Biol. Chem. 281, 19233–19240 [DOI] [PubMed] [Google Scholar]
- 36.Delisle B. P., Anson B. D., Rajamani S., January C. T. (2004) Circ. Res. 94, 1418–1428 [DOI] [PubMed] [Google Scholar]
- 37.Mederos y Schnitzler M., Rinné S., Skrobek L., Renigunta V., Schlichthörl G., Derst C., Gudermann T., Daut J., Preisig-Müller R. (2009) J. Biol. Chem. 284, 4695–4704 [DOI] [PubMed] [Google Scholar]
- 38.Erickson M. G., Alseikhan B. A., Peterson B. Z., Yue D. T. (2001) Neuron 31, 973–985 [DOI] [PubMed] [Google Scholar]
- 39.Łukasiewicz S., Błasiak E., Faron-Górecka A., Polit A., Tworzydło M., Górecki A., Wasylewski Z., Dziedzicka-Wasylewska M. (2007) Pharmacol. Rep. 59, 379–392 [PubMed] [Google Scholar]
- 40.Grailhe R., Merola F., Ridard J., Couvignou S., Le Poupon C., Changeux J. P., Laguitton-Pasquier H. (2006) Chemphyschem. 7, 1442–1454 [DOI] [PubMed] [Google Scholar]
- 41.Hoppe A., Christensen K., Swanson J. A. (2002) Biophys. J. 83, 3652–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ormö M., Cubitt A. B., Kallio K., Gross L. A., Tsien R. Y., Remington S. J. (1996) Science 273, 1392–1395 [DOI] [PubMed] [Google Scholar]
- 43.Pentcheva T., Edidin M. (2001) J. Immunol. 166, 6625–6632 [DOI] [PubMed] [Google Scholar]
- 44.Ju M., Stevens L., Leadbitter E., Wray D. (2003) J. Biol. Chem. 278, 12769–12778 [DOI] [PubMed] [Google Scholar]
- 45.Adair B., Nunn R., Lewis S., Dukes I., Philipson L., Yeager M. (2008) Biophys. J. 94, 2106–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.