

Abstract
In Escherichia coli, the γ complex clamp loader loads the β-sliding clamp onto DNA. The β clamp tethers DNA polymerase III to DNA and enhances the efficiency of replication by increasing the processivity of DNA synthesis. In the presence of ATP, γ complex binds β and DNA to form a ternary complex. Binding to primed template DNA triggers γ complex to hydrolyze ATP and release the clamp onto DNA. Here, we investigated the kinetics of forming a ternary complex by measuring rates of γ complex binding β and DNA. A fluorescence intensity-based β binding assay was developed in which the fluorescence of pyrene covalently attached to β increases when bound by γ complex. Using this assay, an association rate constant of 2.3 × 107 m−1 s−1 for γ complex binding β was determined. The rate of β binding was the same in experiments in which γ complex was preincubated with ATP before adding β or added directly to β and ATP. In contrast, when γ complex is preincubated with ATP, DNA binding is faster than when γ complex is added to DNA and ATP at the same time. Slow DNA binding in the absence of ATP preincubation is the result of a rate-limiting ATP-induced conformational change. Our results strongly suggest that the ATP-induced conformational changes that promote β binding and DNA binding differ. The slow ATP-induced conformational change that precedes DNA binding may provide a kinetic preference for γ complex to bind β before DNA during the clamp loading reaction cycle.
Introduction
Two accessory factors, a sliding clamp and a clamp loader, enhance the efficiency of DNA replication by increasing the processivity of DNA synthesis. The clamp loader assembles clamps onto DNA. The clamp binds the DNA polymerase to increase the processivity of DNA synthesis from tens to thousands of nucleotides in a single binding event. These processivity factors are conserved from bacteria to humans (reviewed in Refs. 1 and 2). The Escherichia coli β clamp is composed of two identical protein monomers assembled into a ring-shaped dimer, which encircles duplex DNA (3, 4). The clamp slides freely along the DNA while tethering DNA polymerase III to the DNA template. The E. coli clamp loader is composed of seven subunits. At the replication fork, a complete clamp loader includes three copies of the dnaX gene product and one copy each of δ, δ′, χ, and ψ (5–7). The dnaX gene produces two proteins, a full-length protein (τ) and a truncated protein (γ), which is created by a translational frameshift (8–10). Clamp loaders containing either three copies of the τ subunit (τ complex, τ3δδ′χψ) or three copies of the γ subunit (γ complex, γ3δδ′χψ) are fully active in clamp loading (6). The τ subunits have an additional C-terminal extension that coordinates the activities of the replisome (reviewed in Refs. 11 and 12). The clamp loader used in all experiments in this study is the γ complex.
In the presence of ATP, γ complex binds with high affinity to β and DNA to form a ternary complex. Binding to primed template DNA triggers the clamp loader to hydrolyze all three molecules of ATP (13–15). Upon ATP hydrolysis, β is released onto DNA and the clamp loader dissociates from the β·DNA complex. Thus the clamp loading reaction can be divided into two stages based on ATP requirements: 1) formation of a ternary complex requiring ATP binding, and 2) dissociation of the ternary complex to load β on DNA requiring ATP hydrolysis. This article focuses on steps in the first phase involving the formation of the ternary complex.
High affinity binding of the clamp loader to the β clamp and to DNA requires ATP binding by the clamp loader (16–18). Presumably, ATP binding promotes conformational changes within the clamp loader that expose surfaces/residues that interact with the clamp and DNA. When equilibrated with ATP, the γ complex can bind either the clamp or DNA; however, a productive clamp loading reaction most likely requires the clamp loader to bind the clamp before binding DNA. When the γ complex alone binds p/t-DNA,2 the interaction with DNA triggers rapid ATP hydrolysis and dissociation of the clamp loader (19, 20). Therefore, clamp loading would be more efficient if there were some mechanism to favor clamp binding before DNA binding and productive clamp loading. One such mechanism would be a kinetic preference for the clamp loader to bind the clamp before DNA. This kinetic preference could be established simply by the clamp loader binding the clamp at a faster rate than DNA. Alternatively, given that the clamp loader has three ATP binding sites, sequential filling of ATP sites and incremental conformational changes could allow the clamp loader to bind the clamp before DNA. In other words, a subset of sites bind ATP and induce conformational changes that promote β binding faster than the subset of sites that bind ATP and make conformational changes to induce DNA binding. In this article, the questions of whether there is a kinetic preference for γ complex to bind β prior to DNA and how the overall rates of ATP-induced conformational changes contribute to the rates of clamp and DNA binding are addressed.
EXPERIMENTAL PROCEDURES
Nucleotides and Oligonucleotides
Concentrations of ATP (Amersham Biosciences/GE Healthcare) diluted with 20 mm Tris-HCl (pH 7.5) and ATPγS (Roche Diagnostics) diluted with water (Roche Diagnostics) were determined by measuring the absorbance at 259 nm and using an extinction coefficient of 15,400 m−1 cm−1. Synthetic oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA) and purified by 12% denaturing polyacrylamide gel electrophoresis. The sequences of the 60-nucleotide template and the complementary 30-nucleotide primer are as follows: 5′-TTC AGG TCA GAA GGG TTC TAT CTC TGT TGG CCA GAA TGT CCC TTT TAT TAC TGG TCG TGT-3′ and 5′-ACA CGA CCA GTA ATA AAA GGG ACA TTC (C6dT) GG-3′, where C6dT is a T with a C6 amino linker that was covalently labeled with 7-diethylaminocoumarin-3-carboxylic acid succinimidyl ester (Invitrogen) as described (16, 21). Primed templates were annealed by incubating the 30-nucleotide primer with the 60-nucleotide template at 85 °C for 5 min and then allowing the solution to slowly cool to room temperature. For all assays, the molar ratios of primer to template were 1:1.2 in annealing reactions.
Buffers
Assay buffer contained 20 mm Tris-HCl (pH 7.5), 50 mm NaCl, 8 mm MgCl2, and the addition of 4% glycerol where indicated. Proteins were stored in 20 mm Tris-HCl (pH 7.5), 0.5 mm EDTA, and 10% glycerol and γ complex storage buffer also contained 50 mm NaCl.
DNA Polymerase III Proteins
Clamp loader subunits, γ (22), δ (23), δ′ (23), and χψ (24) and the β clamp (25) were purified, and the γ complex (γ3δδ′χψ) was reconstituted (6) as previously described with minor modifications (26). Site-directed mutagenesis was performed to change the glutamine 299 to cysteine in a β construct in which surface cysteines 260 and 333 were replaced with serine so that Cys-299 could be selectively labeled. The QuikChange mutagenesis kit (Stratagene) was used for site-directed mutagenesis with the following primers: Q299C, 5′-CAC CGC CAA CAA CCC GGA ATG CGA AGA AGC GGA AGA GAT C; C260S, 5′-CAT CTG GAA GCT GGC TCA GAT CTG CTC AAG CAG GCG; and C333S, 5′-CTG AAC GCG CTG AAA CTG AGA ACG TCC GCA TGA TGC. Concentrations of γ complex were determined by measuring the absorbance at 280 nm in 6 m guanidine hydrochloride and using the calculated extinction coefficient (220,050 m−1 cm−1). The β clamp was covalently labeled on Cys-299 with N-(1-pyrene)maleimide (Molecular Probes) and purified as described previously (27, 28). The protein concentration of βPY was determined using the modified Lowry protein assay (Pierce). Based on the concentration of protein and the concentration of pyrene determined by its absorbance, the typical labeling efficiency was 60%.
Steady State Fluorescence Assays
Fluorescence emission spectra were measured using a QuantaMaster QM-1 fluorometer (Photon Technology International) or an Edinburgh Analytical Instruments FS900 fluorometer. 7-Diethylaminocoumarin-3-carboxylic acid (DCC)-labeled DNA was excited at 440 nm and emission scanned from 450 to 550 nm using a 3.6-nm bandpass on the Edinburgh SF900 fluorometer. PY-labeled β was excited at 345 nm and emission scanned from 355 to 455 nm using a 5-nm bandpass on the Photon Technology International fluorometer. Equilibrium binding of γ complex to βPY was done by adding reagents sequentially to the cuvette starting with 72 μl of assay buffer with ATP, followed by 4 μl of βPY, and finally 4 μl of γ complex for a total reaction volume of 80 μl. Stoichiometric binding of γ complex to βPY or DNA-DCC was done by adding reagents sequentially to the cuvette starting with 68 μl of assay buffer, followed by 4 μl of βPY or p/t-DNA-DCC, 4 μl of γ complex, and finally 4 μl of ATP (or ATPγS for DNA) for a total reaction volume of 80 μl. For both equilibrium and stoichiometric βPY binding assays, emission spectra were measured following each addition, and relative intensities at 375 nm were plotted as a function of γ complex concentration. The buffer (+ATP) background was subtracted from all spectra and made relative to the free PY or DCC signal during analysis. The dissociation constant (KD) of the βPY·γ complex was calculated by fitting the observed intensity data (Iobs) to Equation 1 in which γc is the concentration of γ complex, β is the concentration of βPY, Imin is the intensity of free βPY, and Imax is the intensity of γ complex·βPY. The intensity of γ complex·βPY (Imax) and KD were fit as adjustable parameters by nonlinear regression using KaleidaGraph.
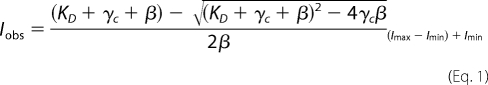 |
Equation 1 was also used to fit the quench in DCC fluorescence as a function of γ complex in the DNA binding assay to calculate the relative intensity of a clamp loader·DNA complex and a KD value. In this case, the concentration of DNA was substituted for β, Imax represents the intensity of free DNA, and Imin represents the intensity of bound DNA.
Pre-Steady State Fluorescence Assays
Assays were done using an Applied Photophysics SX20MV stopped-flow apparatus at 20 °C. Single-mix reactions were performed by mixing equal volumes (50 μl) of reagents immediately before they entered the cuvette. Data were collected for a total of 5 s at intervals of 1 ms. Final concentrations for each experiment in assay buffer with 4% glycerol are indicated in the figure legends and/or under “Results.” Measurements of DCC fluorescence were made using a 455-nm cut-on filter to collect emission while exciting at 430 nm with a 3.72-nm bandpass. A 365-nm cut-on filter was used to collect PY emission when exciting at 345 nm using a 3.72-nm bandpass for βPY binding experiments. Time courses for βPY binding were empirically fit to a single exponential rise, y = a(1 − e−kobs·t) + c, using KaleidaGraph to determine values for observed rate constants, kobs, as a function of γ complex concentration.
Kinetic Modeling
DNA binding data in Figs. 4C, 6, and 7 were globally fit to the model illustrated in Fig. 8 using DynaFit to calculate the rate constants given in the figure. All rate constants were treated as adjustable parameters; none were set at fixed values. The relative intensity for free DNA and for the initial clamp loader·DNA complex prior to the DNA-induced conformational change were set to 1, and the relative intensities for all other clamp loader·DNA complexes were set to the value of 0.58 derived from the titration in Fig. 4B. Concentrations of protein and DNA were fixed based on the concentrations determined experimentally from absorbances at 280 and 260 nm, respectively, as described above. In experiments in which γ complex was preincubated with ATP, the “incubate” and “dilute” functions in DynaFit were used to generate the equilibrium concentrations of the two conformational states of γ complex that exist in the presence of ATP (15). These functions were used to: 1) mix a solution of γ complex, twice as concentrated as in the final reaction, with ATP; 2) dilute the γ complex in half to the final concentration; and 3) incubate the solution for 1 s in silico to generate the equilibrium populations of conformational states that are added to DNA to initiate the binding reactions. In experiments in which the γ complex was not preincubated with ATP, reactions were initiated in silico by adding free γ complex to ATP and DNA.
FIGURE 4.

Binding of γ complex to DNA. A, emission spectra of DCC were measured using an excitation wavelength of 455 nm. The black trace is a scan of free p/t-DNA-DCC, the dashed dark gray trace is a scan repeated after the addition of γ complex, and the light gray trace is a scan repeated after the addition of ATPγS. Final concentrations were: 100 nm p/t-DNA-DCC, 2000 nm γ complex, and 0.5 mm ATPγS in assay buffer. B, the relative intensity of DCC in a clamp loader·DNA complex was determined by measuring DCC fluorescence at 480 nm as a function of γ complex concentration. The maximal quench in DCC fluorescence was calculated by fitting these data to a quadratic equation (see “Experimental Procedures”). The solid line through the data is the result of the fit, which gave a value of 0.58 for the relative intensity of DCC in a clamp loader·DNA complex and a KD value of 375 ± 7 nm for clamp loader·DNA dissociation. Final concentrations were: 100 nm p/t-DNA-DCC and 0.5 mm ATPγS in assay buffer. C, the rate of γ complex binding to DNA was measured in reactions in which a solution of γ complex and ATP was added to a solution of p/t-DNA-DCC and ATP. The relative fluorescence of DCC is plotted as a function of time. Final concentrations were: 125, 250, and 500 nm γ complex and p/t-DNA-DCC and 0.5 mm ATP in assay buffer with 4% glycerol. The concentrations of γ complex and DNA are indicated by the shade of gray; darker indicates a higher concentration. Solid lines through the reaction time course were generated from the kinetic model illustrated in Fig. 8.
FIGURE 6.

γ complex binding DNA with and without preincubation of γ complex with ATP. The change in DCC fluorescence due to γ complex binding DNA was measured as a function of time. In one assay (black trace, gray line), a solution of γ complex and ATP was added to a solution of p/t-DNA-DCC and ATP. In the second (gray trace, black line), a solution of γ complex that did not contain ATP was added to a solution of p/t-DNA-DCC and ATP. The relative intensity of DCC is plotted as a function of time. Final concentrations were: 200 nm p/t-DNA-DCC and γ complex, and 0.5 mm ATP in assay buffer with 4% glycerol. Solid lines through the time courses are fits to the model in Fig. 8.
FIGURE 7.

Effect of ATP concentration on γ complex·DNA binding kinetics. Rates of DNA binding were measured as a function of ATP concentration in a reaction in which γ complex was not preincubated with ATP. A solution of γ complex and p/t-DNA-DCC was mixed with a solution of ATP, and the DCC fluorescence was measured as a function of time. Final concentrations were: 200 nm p/t-DNA-DCC and γ complex; and A, 25 μm (red trace); B, 50 μm (green trace); C, 250 μm (orange trace); and D, 500 μm (blue trace) ATP relative to the same reaction at 100 μm ATP (black trace) in assay buffer with 4% glycerol.
FIGURE 8.

Kinetic model fit to p/t-DNA binding data. The model starts on the left-hand side with free γ complex. (Note: the γ complex used in our studies contains seven polypeptides, γ3δδ′χψ, but only five, γ3δδ′, are illustrated in the diagram.) The γ complex binds 3 molecules of ATP in a single step. ATP binding promotes a conformational change that allows the clamp loader to bind DNA. DNA binding induces a second conformational change that activates the ATP sites for hydrolysis and three molecules of ATP are hydrolyzed sequentially at the same rate. DNA and then ADP are released to allow the γ complex to recycle. The relative intensity of free DNA was set at 1 and the relative intensities for all bound DNA species set at 0.58. The data in Figs. 4C, 6, and 7 were fit to this model using DynaFit (34).
RESULTS
Equilibrium Binding of γ Complex to β
The clamp loader when charged with ATP has a high affinity for the clamp. The equilibrium dissociation constant (KD) of γ complex·β is in the low nanomolar range (18). In previous studies, an anisotropy assay was used to measure γ complex·β binding (21, 28), but a limitation of this assay is that it is not sensitive enough to work at low nanomolar protein concentrations, where KD values are accurately determined. Therefore, a more sensitive intensity-based fluorescence assay was developed to measure clamp loader·clamp binding. In this assay, the Gln-299 in β was mutated to Cys, and Cys-299 was covalently labeled with PY. Based on available structural data and similarities between the E. coli and yeast clamps and clamp loaders (5, 29, 30), residue 299 in β is likely to be near a site where a γ subunit contacts the surface of the β clamp in a clamp loader-clamp complex (Fig. 1). Given the head-to-tail symmetry of the β dimer and the anticipated contacts between γ complex and β, the fluorescence of PY is likely to be affected by γ complex binding only at the position near the middle γ subunit (Fig. 1). When γ complex binds to βPY, PY fluorescence increases (Fig. 2A). At saturating concentrations of γ complex, the intensity of PY is just over two and a half times greater than for free βPY.
FIGURE 1.

Fluorescence intensity-based β clamp binding assay. Upper panel, ribbon diagram of the β clamp is shown with one monomer in cyan and the other in magenta. Glutamine 299 (spheres), located on the surface of β to which the γ complex binds, was converted to cysteine. Two surface cysteines, Cys-260 and Cys-333, were converted to serine, so that Cys-299 could be selectively labeled with PY. Lower panel, the β clamp has a C2 axis of symmetry through the center of the ring such that two PY fluorophores (yellow starbursts) covalently attached to Cys-299 are located on opposite sides of the ring. When the γ complex binds βPY, one γ subunit (dark blue) likely binds at or near a PY to alter its environment and increase PY fluorescence (larger yellow starburst). The second PY molecule is unlikely to interact with the γ complex so that only a single PY molecule is reporting on the binding interaction.
FIGURE 2.

Equilibrium binding of γ complex to β. A, emission spectra of βPY were taken at excitation wavelength of 345 nm. The light gray trace is a scan of free βPY and the black trace is after the addition of γ complex and ATP. Final concentrations were: 80 nm βPY, 240 nm γ complex, and 0.5 mm ATP. B, equilibrium binding of γ complex to β was determined by measuring the intensity of PY as a function of γ complex concentration, where γ complex was titrated into βPY solutions in assay buffer containing ATP. Final concentrations were: 10 nm βPY and 0.5 mm ATP in assay buffer. C, stoichiometric binding of γ complex to β was measured in the absence (circles) and presence (squares) of ATP. The γ complex and ATP were added sequentially to a solution of βPY in assay buffer. The relative intensity of PY at 375 nm is plotted as a function of γ complex concentration. Final concentrations after the addition of ATP were 80 nm βPY and 0.5 mm ATP in assay buffer.
This increase in PY fluorescence was used to measure equilibrium binding of γ complex to βPY and calculate the dissociation constant for the interaction. A KD value of 5.0 ± 2.4 nm was calculated from three independent experiments at 10 nm βPY (average values are shown in Fig. 2B). This KD value is in agreement with the value of 3.2 nm previously reported (18). Binding was measured at a higher concentration of βPY (80 nm) to determine the dissociation constant for the weaker ATP-independent binding reaction (Fig. 2C, filled circles) and to determine the stoichiometry of binding in the presence of ATP (Fig. 2C, filled squares). The KD value calculated for ATP-independent binding was 135 ± 32 nm, which is in agreement with the value of 151 nm reported previously (28). In the presence of ATP, γ complex binds β with the expected 1:1 stoichiometry as indicated by achieving saturation at a concentration of around 80 nm γ complex, and these data also give a calculated KD value of 2.9 nm for ATP-dependent binding. Interestingly, the maximum increase in PY fluorescence in assays without ATP is less than 2-fold, whereas it is greater than 2.5-fold in the presence of ATP. This difference could reflect differences in conformational states of γ complex and/or β in the presence and absence of ATP. ATP is shown to cause a conformational change in γ complex that gives it a high affinity for the clamp (18). The clamp is likely to be open in clamp loader-clamp complexes in assays with ATP, whereas it is likely to be closed in clamp loader-clamp complexes in assays without ATP (31).
Kinetics of γ Complex Binding β
To determine the rate of γ complex binding β, the increase in PY intensity was measured in real time when a solution of γ complex and ATP was mixed with a solution of βPY and ATP. Reactions contained 20 nm βPY and γ complex concentrations of 10, 20, 40, and 80 nm (Fig. 3A). These time courses were empirically fit to exponential rises to calculate apparent rate constants, kobs. These kobs values were plotted as a function of γ complex concentration and fit to line to calculate an apparent on-rate constant, kon(app), of 2.3 × 107 m−1 s−1 and an apparent dissociation rate constant, koff(app), of 0.14 s−1 from the slope and y intercept, respectively (32) (Fig. 3B). The apparent KD value calculated from these kinetic constants was 6 nm, which is in agreement with the value determined from equilibrium measurements.
FIGURE 3.

Kinetics of γ complex binding to βPY. A, the increase in PY intensity was measured as a function of time when a solution of γ complex and ATP (0.5 mm) was added directly to a solution of βPY and ATP (0.5 mm). Reactions contained 20 nm βPY and 10 nm (red), 20 nm (blue), 40 nm (green), or 80 nm (orange) γ complex in assay buffer with 4% glycerol. Solid black lines through time courses are the result of an empirical fit of the data to an exponential rise. B, observed rate constants, kobs, calculated from exponential fits of reaction time courses in A are plotted against the concentration of γ complex. A linear fit of these data gave a slope of 22.7 μm−1 s−1 and a y intercept of 0.14 s−1.
Binding of γ Complex to DNA
A fluorescence intensity-based assay was used in which the primer strand of p/t-DNA is labeled with DCC 3 nucleotides from the 3′ end, p/t-DNA-DCC, was used to measure the kinetics of γ complex binding to DNA (21). When the clamp loader binds DNA at the primer/template junction, the environment of DCC is altered and the fluorescence decreases. The γ complex requires ATP binding for high affinity binding to DNA, but DNA binding triggers hydrolysis of ATP and dissociation of the clamp loader so that binding is transient (19, 33). Therefore, γ complex·DNA binding under equilibrium conditions was measured in assays with the non-hydrolyzable ATP analog, ATPγS. When γ complex binds p/t-DNA-DCC, the fluorescence is quenched and the emission maximum of DCC is blue-shifted by 5–6 nm (Fig. 4A). To determine the magnitude of the fluorescence quench and relative quantum yield of a clamp loader·DNA complex, p/t-DNA-DCC was titrated with γ complex in the presence of ATPγS (Fig. 4B). These titration data showed that the fluorescence of the protein·DNA complex is 58% of the fluorescence of free p/t-DNA-DCC.
Transient binding of γ complex to DNA in assays with ATP was measured in real time stopped-flow fluorescence experiments. A solution of γ complex and ATP was added to a solution of p/t-DNA-DCC and ATP, and the intensity of DCC was measured as a function of time (Fig. 4C). Binding rates were measured with increasing concentrations of γ complex and DNA at 125, 250, and 500 nm. In each case, a rapid decrease in DCC fluorescence due to γ complex binding p/t-DNA-DCC was followed by a rapid increase in fluorescence due to dissociation of γ complex from DNA following ATP hydrolysis. As the concentrations of γ complex and DNA were increased, the binding reaction became faster and the magnitude of the fluorescence quench increased. However, the magnitude of the decrease in fluorescence at the lowest point was not as large as measured in assays with ATPγS (Fig. 4B). Moreover, these reactions are not approaching a limiting value for the minimum fluorescence, but continue to decrease with increasing concentrations. Assuming that the magnitude of the quench in fluorescence is the same for clamp loader·DNA complexes in assays with ATP and ATPγS, this shows that the ATP hydrolysis-induced γ complex dissociation is fast relative to the DNA binding reaction so that only a fraction of p/t-DNA-DCC is bound when the fluorescence reaches a minimal value. Given a relative fluorescence of 58% for the clamp loader·DNA complex, about 14% of the p/t-DNA-DCC is bound when the intensity reaches a minimum value of about 0.94 in the reaction containing 500 nm γ complex and p/t-DNA-DCC.
Rates of γ Complex Binding β with and without Preincubation of γ Complex with ATP
ATP binding promotes conformational changes in γ complex that give it a high affinity for the clamp and DNA. It is possible that the rates of these conformational changes govern the rates of clamp and DNA binding to the clamp loader. To test this possibility, the rate of γ complex binding βPY was measured in assays in which γ complex was preincubated with ATP (Fig. 5A, gray trace) and in which γ complex is added to ATP at the same time as βPY (Fig. 5A, black trace). After mixing, both reactions contained identical concentrations of βPY (200 nm), γ complex (200 nm), and ATP (0.5 mm). There was no difference between the binding rates when γ complex was preincubated or not with ATP before adding β. This suggests that ATP binding and ATP-induced conformational changes that give γ complex a high affinity for β are relatively rapid. When βPY binding was measured in the absence of ATP (Fig. 5B, gray trace), binding was relatively rapid but a smaller fraction of β was bound than in a reaction with ATP (Fig. 5B, black trace). The smaller fraction bound is consistent with the weaker ATP-independent binding reaction (Fig. 2C). This result provides additional support that ATP binding and ATP-induced conformational changes that give γ complex a high affinity for β are relatively rapid.
FIGURE 5.

Rates of γ complex binding β with and without preincubation of γ complex with ATP. A, the rate of γ complex binding βPY was measured in assays with (gray trace) and without (black trace) preincubation of γ complex and ATP. The relative intensity of PY is plotted as a function of time for a reaction in which a solution of γ complex and ATP was added to a solution of βPY and ATP (gray trace) and a reaction in which a solution of γ complex was added to a solution of βPY and ATP (black trace). The inset is the first 0.2 s of the reaction time courses. B, rates of γ complex binding βPY were measured in the absence of ATP (gray trace) and in the presence of ATP but without preincubation of γ complex with ATP (black trace). Final concentrations were: 200 nm βPY and γ complex and 0.5 mm ATP in assay buffer with 4% glycerol.
DNA Binding with and without ATP Preincubation
To determine whether the rate of ATP binding and ATP-induced conformational changes could limit the rate at which γ complex binds DNA, DNA binding was measured in real time assays in which γ complex was preincubated with ATP or in which γ complex was added to ATP at the same time as p/t-DNA-DCC. When a solution of γ complex and ATP was added to a solution of p/t-DNA-DCC and ATP (ATP preincubation), a rapid quench in fluorescence occurred that reached a minimum value in about 60–70 ms and was followed by an increase in fluorescence that reached a steady state level within about 500 ms (Fig. 6, black trace, gray line). In contrast, when a solution of γ complex without ATP was added to a solution of p/t-DNA-DCC and ATP (no ATP preincubation), the decrease in DCC fluorescence is slower overall (Fig. 6, gray trace, black line). There is a short lag of at least 25 ms before fluorescence begins to decrease and the rate of decrease is slower, such that the maximal quench occurs between 150 and 200 ms. Final concentrations of γ complex (200 nm) and pt/-DNA-DCC (200 nm) were the same in both reactions. Because the overall binding rate is slower, but the subsequent rates of ATP hydrolysis and γ complex·DNA dissociation are likely to be the same, the amplitude of the change in fluorescence is smaller when the γ complex was not preincubated with ATP. This experiment was repeated with ATPγS instead of ATP to measure DNA binding in the absence of DNA-triggered ATP hydrolysis and subsequent clamp loader dissociation (supplemental Fig. S1). Although the reaction in which γ complex was preincubated with ATPγS was slightly faster than the non-preincubated case, overall both reactions were much slower and took 6–8 s to reach completion. It is possible that DNA binding reactions are slower in assays with ATPγS because ATPγS does not have the efficacy of ATP in promoting conformational changes that allow the clamp loader to bind DNA. We have also observed a decrease in the affinity of γ complex for β in assays with ATPγS compared with ATP (21). For this reason, we chose to focus on assays with ATP even though the kinetics are complicated by hydrolysis. In any case, a slow ATP-dependent step is bypassed when γ complex is preincubated with ATP.
Effect of ATP Concentration on γ Complex·DNA Binding Kinetics
Either a slow ATP binding step or a slow ATP-induced conformational change could limit the rate at which γ complex binds DNA. To determine which step is slow, rates of γ complex·DNA binding were measured as a function of ATP concentration in reactions in which γ complex was not preincubated with ATP. A solution of γ complex and p/t-DNA-DCC was mixed with solutions of ATP, and DCC fluorescence was measured as a function of time. In Fig. 7, reactions containing 25 (A, red), 50 (B, green), 250 (C, orange), and 500 μm (D, blue) ATP are plotted in the same graph with a plot of a reaction containing 100 μm ATP (black trace). At the lowest concentration of ATP used, the reactions approached a maximal rate, becoming slightly faster with increasing concentrations of ATP, and reaching a maximum rate at 100 μm ATP. In all previous experiments, reactions contained 500 μm ATP, therefore, ATP binding was unlikely to be rate-limiting. The slower rates of DNA binding in assays in which γ complex was not preincubated with ATP must be due to a slow ATP-induced conformational change.
Kinetic Modeling of DNA Binding Reactions
DNA binding data were modeled to get an estimate of the rate of the ATP-induced conformational change that precedes DNA binding to determine whether there could be a kinetic preference for binding β prior to DNA. A kinetic model from previous studies was adapted to fit these DNA binding data (15). The kinetic model was developed based on experiments measuring rates of ATP hydrolysis in reactions in which the γ complex was preincubated with ATP for varying periods of time before adding p/t-DNA. The earlier studies revealed that the rate of ATP hydrolysis was limited by an ATP-dependent conformational change and suggested that a slow ATP-induced conformational change activates the clamp loader for DNA binding. Experiments in Figs. 6 and 7 confirm this by establishing that a slow ATP-induced conformational change precedes DNA binding. The complete kinetic mechanism is quite complex and contains too many forward and reverse rate constants to define in a single set of experiments. Therefore, the kinetic model was simplified by including only forward rates for most of the steps and allowing three molecules of ATP to bind and three molecules of ADP to dissociate as a unit in a single step. All of the rate constants shown in Fig. 8 were derived by globally fitting the data in Figs. 4C, 6, and 7 to the model shown using DynaFit (34). The forward and reverse rate constants for the ATP-induced conformational change obtained from DNA binding data were 3.3 and 1.7 s−1 compared with values of 6.5 and 3.9 s−1, respectively, obtained previously from fits of ATP hydrolysis data (15). After the conformational change, p/t-DNA binding occurred at a rate of 4.0 × 107 m−1 s−1. The rate of DNA binding is similar to the rate of clamp binding (2.3 × 107 m−1 s−1) and on the order of what would be expected for a diffusion-limited rate for two macromolecules interacting. However, in reactions in which the γ complex is not preincubated with ATP, the rate of DNA binding is limited by the slow, 3.3 s−1 conformational change, whereas the rate of β binding is not.
The interaction with p/t-DNA triggers a change in the clamp loader that makes the ATP sites competent for hydrolysis, and in the model used here, three molecules of ATP are hydrolyzed sequentially before DNA is released. ADP release allows the clamp loader to go through the cycle again. The DNA binding data do not provide a direct measure of ATP hydrolysis, and in terms of these data, the ATP hydrolysis steps in the kinetic model provide a time lag between observed DNA binding and release. The model used to fit DNA binding data here differs from that used to fit ATP hydrolysis data in that two molecules of ATP were hydrolyzed rapidly and the third slowly in the ATP hydrolysis model. The DNA binding data could be fit equally well by a model in which ATP hydrolysis occurred in two phases (data not shown), hydrolysis of two molecules of ATP rapidly before DNA release, and hydrolysis of one molecule of ATP slowly after DNA release as proposed previously based on ATP hydrolysis data (15). When DNA binding data were fit to this two-phase model for ATP hydrolysis, similar rates for ATP binding (kon(ATP) = 12.6 μm−1 s−1), the ATP-induced conformational change (kconf = 3.3 s−1, krev(conf) = 1.7 s−1), DNA binding (3.9 × 107 m−1 s−1), and DNA release (88 s−1) were obtained. Therefore, for the purposes of this study, which focuses on the overall rate of DNA binding, we used the simpler model in which all three molecules were hydrolyzed at the same rate.
DISCUSSION
Clamp loaders catalyze the assembly of sliding clamps onto DNA for use by DNA polymerases. To load clamps, the affinity of the clamp loader for the clamp and DNA must be modulated. Initially, the clamp loader must have a high affinity for the clamp and DNA to bring these macromolecules together, but then the affinity must decrease so that the clamp loader can release the clamp onto DNA. This affinity modulation is achieved by ATP binding and hydrolysis. ATP binding activates the clamp loader for binding the clamp and DNA, whereas ATP hydrolysis deactivates the clamp loader for releasing the clamp and DNA. Thus, the clamp loading reaction can be divided into two phases depending on the ATP requirements: 1) ATP binding-dependent formation of a ternary clamp loader·clamp·DNA complex, and 2) ATP hydrolysis-dependent decay of the ternary complex releasing the clamp on DNA. Much of our previous work focused on the second phase of the reaction by adding a preformed clamp loader-clamp complex to DNA to rapidly form the ternary complex via a single pathway. Here, we focused on the first phase of the reaction, formation of a ternary clamp loader-clamp complex. We asked whether γ complex binds the clamp or DNA faster, and how preincubation of the clamp loader with ATP to form the ATP-activated state affects the rates of clamp loader-clamp and clamp loader-DNA binding.
To measure the rate of clamp binding, a sensitive fluorescence intensity-based assay was developed in which the β clamp was covalently labeled with PY. The intensity of PY increases when the clamp loader binds the clamp. An advantage of this assay is that clamp loader-clamp binding can be measured directly in solution and in real time. The assay is sensitive enough to measure the high affinity binding of γ complex to β under equilibrium conditions as well as measuring binding kinetics on a millisecond time scale. Using this binding assay, a bimolecular rate constant of 2.3 × 107 m−1 s−1 was determined for γ complex binding β in the presence of ATP. This rapid rate indicates the binding interaction is limited by the rate of diffusion of the proteins.
Two Sets of ATP-induced Conformational Changes
High affinity binding of the clamp loader to the clamp and to DNA requires the clamp loader to bind ATP first. Presumably, ATP binding promotes conformational changes in the clamp loader that place amino acid residues and protein surfaces in the appropriate conformation to productively interact with the clamp and DNA (18, 19, 31). Interestingly, our results strongly suggest that a different set of ATP-induced conformational changes is required to promote binding of the clamp loader to the clamp versus binding to DNA (Fig. 9). Given that ATP-induced conformational changes must precede clamp or DNA binding, the overall rate of these conformational changes will contribute to observed binding rates in experiments in which γ complex is not preincubated with ATP prior to adding the clamp or DNA. On the other hand, if the γ complex is preincubated with ATP before adding the clamp or DNA, these conformational changes can take place during the preincubation period and binding rates will not be influenced by the rate of the conformational changes. When the γ complex was preincubated with ATP, the rate of p/t-DNA binding was faster than for reactions in which γ complex was not allowed to bind ATP before p/t-DNA (see Fig. 6). DNA binding kinetics measured as a function of ATP concentration (see Fig. 7) demonstrated that the slower DNA binding kinetics were not due to a slow ATP binding reaction, but instead must be the result of slow ATP-induced conformational changes. In contrast, the observed rate for β binding was the same regardless of whether the γ complex was preincubated with ATP or not (see Fig. 5). The rapid β binding in experiments without ATP preincubation is unlikely to be due to an ATP-independent binding reaction because the ATP-independent binding interaction is weaker (see Fig. 2C) and could only account for a fraction of the binding events (see Fig. 5B). ATP binding and subsequent ATP-induced conformational changes are rapid relative to the rate of β binding. Given that only DNA binding rates are limited by a slow ATP-induced conformational change, this would suggest that a different set of conformational changes gives rise to β and DNA binding. It will be interesting to see which conformational changes promote clamp opening. The first “fast” conformational change could promote both clamp binding and clamp opening as illustrated in Fig. 9. Alternatively, the second slower conformational change could promote clamp opening and subsequent DNA binding.
FIGURE 9.

Model for formation of a ternary clamp loader·clamp·DNA complex. ATP binds the clamp loader and rapidly induces a conformational change (yellow star state) that enables the clamp loader to bind the clamp. A second conformational change (green star state) occurs more slowly and enables the clamp loader to bind p/t-DNA. This second conformational change could either occur within a clamp loader-clamp complex when the clamp is present (upper reaction scheme), or within the free clamp loader in the absence of the clamp (lower reaction scheme) to promote p/t-DNA binding. Note that the γ complex can bind three molecules of ATP, but that we have not determined how many molecules bind at each stage of the assembly reaction. Therefore, the stoichiometry of bound ATP molecules at each step is not given. It is possible that the slow conformational change opens an ATP site, or a subset of sites, to allow binding of additional ATP that promotes DNA binding.
There are a number of interesting observations in the literature that can be used to speculate about a mechanism by which two distinct sets of conformational changes could occur sequentially to promote β and then DNA binding. The γ complex has three ATP sites and sequential filling of these sites could promote sequential sets of conformational changes. A subset of ATP sites could fill first to induce conformational changes rapidly that promote β binding. A slow conformational change could then allow the remaining sites to fill with ATP and promote DNA binding. Incubation of the γ complex with ATP prior to adding β or DNA would give all the sites time to fill with ATP and allow the conformational changes to occur so that the γ complex could bind either the clamp or DNA. Studies with arginine finger mutants of the E. coli and yeast clamp loaders, which bind but do not “sense” or respond to bound ATP, show that ATP binding at different sites has differential effects on clamp and DNA binding (28, 35). Similarly, studies of single Walker A mutants that reduce ATP binding to the yeast clamp loader showed that mutations of individual sites had differential effects on DNA binding (36). Both Arg finger (35) and Walker A (36) mutations that affect ATP sensing/binding to the replication factor C (RFC) 2 and RFC3 subunits of the yeast clamp loader gave the greatest decreases in DNA binding activities. Interestingly, Arg finger mutations in the E. coli clamp loader that affect ATP sensing in the γ subunit that occupies the same position as RFC3 (darkest blue or middle γ subunit in Fig. 9), as well as the γ subunit adjacent to the δ subunit, greatly reduced DNA binding. In a crystal structure of the minimal E. coli clamp loader (γ3δδ′), the middle γ subunit does not contain a bound ATPγS molecule, whereas the other two γ subunits are bound to ATPγS (37). Biochemical characterization showed that this minimal clamp loader (γ3δδ′) had greatly reduced DNA binding activity compared with γ complex (γ3δδ′ψχ), which is consistent with the idea that ATP binding to the middle γ subunit is important for DNA binding (21). That is not to say that a single subunit binds DNA, but instead that ATP binding at one (or a subset of sites) is important for inducing conformational changes within the clamp loader that as a whole increase DNA binding activity. If a slow conformational change were to open this site and cause it to fill last, then this could provide a mechanism for sequential binding of the clamp and DNA.
The Conformational Change That Gives γ Complex a High Affinity for DNA
The observation that the rate of p/t-DNA binding is limited by the rate of ATP-induced conformational changes is consistent with results from our previous work measuring rates of p/t-DNA-triggered ATP hydrolysis by the γ complex. As with the DNA binding kinetics, we found that rates of DNA-triggered ATP hydrolysis were faster when the γ complex was preincubated with ATP than when there was no ATP preincubation, and that the slower rate was not due to slow ATP binding (15). Based on rates of ATP hydrolysis as a function of preincubation time with ATP, a forward rate constant of 6.5 s−1 and reverse rate constant of 3.9 s−1 was calculated for the ATP-induced conformational changes. These values are in good agreement with the values of 3.3 and 1.7 s−1 for the forward and reverse rate constants calculated from kinetic modeling of the DNA binding reaction (see Fig. 8) given the complexity of this reaction and that both data sets were fit independently. Both sets of experiments predict that after equilibration with ATP a little over 60% of the clamp loaders (63% from ATPase experiments and 66% from DNA binding experiments) exist in the conformational state that has high affinity for p/t-DNA. Interaction with the β clamp increases the affinity of the clamp loader for DNA, and it is interesting to speculate that the β clamp may do this by stabilizing the conformational state with a high affinity for DNA and shifting the equilibrium to favor this species (16). Studies with a minimal form of the clamp loader, γ3δδ′, missing the χ and ψ subunits support this idea. The minimal clamp loader is defective in ATP-dependent DNA binding activity. The β clamp can rescue the ATP-dependent DNA binding activity of the minimal clamp loader most likely by stabilizing or promoting the conformational state with high affinity for DNA (21). It is likely that the β clamp also increases the affinity of the clamp loader for DNA by directly interacting with the DNA duplex. The central cavity of the β ring is lined with positively charged amino acid residues (4) and these residues interact with the sugar-phosphate backbone of the duplex (3).
A Kinetic Preference for the Clamp Loader to Bind β before DNA
When equilibrated with ATP, the E. coli γ complex can bind either β or DNA. The same is true for the eukaryotic clamp loader, RFC (38). Although these clamp loaders can bind either the clamp or DNA, productive clamp loading is likely to require these clamp loaders to bind the clamp first. In contrast, the bacteriophage T4 clamp loader can productively load clamps by binding either DNA or the clamp first (39, 40). This may be due to differences in the solution structures of the clamps. The bacteriophage T4 gp45 clamp exists as an open ring in solution (41, 42), whereas the E. coli β clamp (43) and eukaryotic clamp are likely to exist as closed rings in solution (44). When bound to genomic DNA, the geometry of the clamp loader·DNA complex likely prevents the clamp loader from productively binding a closed clamp. In addition, binding of the bacterial and eukaryotic clamp loaders to p/t-DNA triggers rapid ATP hydrolysis and dissociation of the clamp loader from the DNA, such that the clamp loader·DNA complex is transient and not likely to be long-lived enough to efficiently bind clamps (19, 45). Nonproductive interactions between the clamp loaders and DNA would reduce the overall efficiency of clamp loading by engaging the clamp loader in futile cycles of DNA binding and ATP hydrolysis. A mechanism that favored clamp binding prior to DNA binding would increase the overall efficiency of clamp loading. It is possible that the rate-limiting ATP-induced conformational changes that precede DNA binding but not clamp binding provide a kinetic preference for the clamp loader to bind the clamp before DNA and increase the overall efficiency of clamp loading. The fastest rate at which the clamp loader can bind DNA is limited by rate of the ATP-induced conformational change (3.3 s−1), but binding could be slower if DNA concentrations are limiting. The rate of the conformational change that promotes high affinity β binding is faster so that the rate of β binding may be a function of the concentration of β in the cell, 500 nm assuming 300 copies of β (46) in a 1 × 10−15 liter cell volume, and the bimolecular rate constant for binding, 2.3 × 107 m−1 s−1, to give an effective on-rate of 11 s−1. This effective on-rate assumes that β on the lagging strand is rapidly recycled by excess δ subunit and/or clamp loader in the cell such that all the β is free to be loaded (47). Even at saturating concentrations of DNA, given these rates, the γ complex would bind β first at least 75% of the time. During active replication, the timing of primer synthesis on the lagging strand could also provide a mechanism for regulating the order of β and DNA binding. This could be accomplished by synthesizing primers at a rate that would allow the clamp loader to bind a clamp before a new primed template site is formed.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM055596 (to L. B. B.) and R01 GM38839 (to M. O. D.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- p/t-DNA
- primer/template-DNA
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- βPY
- β covalently labeled on residue Q299C with pyrene
- DCC
- 7-diethylaminocoumarin-3-carboxylic acid, succinimidyl ester
- p/t-DNA-DCC
- primed template DNA with an amino linker covalently labeled with DCC
- PY
- N-(1-pyrene)maleimide
- RFC
- replication factor C.
REFERENCES
- 1.Indiani C., O'Donnell M. (2006) Nat. Rev. Mol. Cell Biol. 7, 751–761 [DOI] [PubMed] [Google Scholar]
- 2.Johnson A., O'Donnell M. (2005) Annu. Rev. Biochem. 74, 283–315 [DOI] [PubMed] [Google Scholar]
- 3.Georgescu R. E., Kim S. S., Yurieva O., Kuriyan J., Kong X. P., O'Donnell M. (2008) Cell 132, 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong X. P., Onrust R., O'Donnell M., Kuriyan J. (1992) Cell 69, 425–437 [DOI] [PubMed] [Google Scholar]
- 5.Jeruzalmi D., O'Donnell M., Kuriyan J. (2001) Cell 106, 429–441 [DOI] [PubMed] [Google Scholar]
- 6.Onrust R., Finkelstein J., Naktinis V., Turner J., Fang L., O'Donnell M. (1995) J. Biol. Chem. 270, 13348–13357 [DOI] [PubMed] [Google Scholar]
- 7.Pritchard A. E., Dallmann H. G., Glover B. P., McHenry C. S. (2000) EMBO J. 19, 6536–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blinkowa A. L., Walker J. R. (1990) Nucleic Acids Res. 18, 1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flower A. M., McHenry C. S. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 3713–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsuchihashi Z., Kornberg A. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 2516–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McHenry C. S. (2003) Mol. Microbiol. 49, 1157–1165 [DOI] [PubMed] [Google Scholar]
- 12.O'Donnell M. (2006) J. Biol. Chem. 281, 10653–10656 [DOI] [PubMed] [Google Scholar]
- 13.Bertram J. G., Bloom L. B., Turner J., O'Donnell M., Beechem J. M., Goodman M. F. (1998) J. Biol. Chem. 273, 24564–24574 [DOI] [PubMed] [Google Scholar]
- 14.Hingorani M. M., Bloom L. B., Goodman M. F., O'Donnell M. (1999) EMBO J. 18, 5131–5144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams C. R., Snyder A. K., Kuzmic P., O'Donnell M., Bloom L. B. (2004) J. Biol. Chem. 279, 4376–4385 [DOI] [PubMed] [Google Scholar]
- 16.Bloom L. B., Turner J., Kelman Z., Beechem J. M., O'Donnell M., Goodman M. F. (1996) J. Biol. Chem. 271, 30699–30708 [DOI] [PubMed] [Google Scholar]
- 17.Hingorani M. M., O'Donnell M. (1998) J. Biol. Chem. 273, 24550–24563 [DOI] [PubMed] [Google Scholar]
- 18.Naktinis V., Onrust R., Fang L., O'Donnell M. (1995) J. Biol. Chem. 270, 13358–13365 [PubMed] [Google Scholar]
- 19.Ason B., Bertram J. G., Hingorani M. M., Beechem J. M., O'Donnell M., Goodman M. F., Bloom L. B. (2000) J. Biol. Chem. 275, 3006–3015 [DOI] [PubMed] [Google Scholar]
- 20.Ason B., Handayani R., Williams C. R., Bertram J. G., Hingorani M. M., O'Donnell M., Goodman M. F., Bloom L. B. (2003) J. Biol. Chem. 278, 10033–10040 [DOI] [PubMed] [Google Scholar]
- 21.Anderson S. G., Williams C. R., O'Donnell M., Bloom L. B. (2007) J. Biol. Chem. 282, 7035–7045 [DOI] [PubMed] [Google Scholar]
- 22.Maki S., Kornberg A. (1988) J. Biol. Chem. 263, 6547–6554 [PubMed] [Google Scholar]
- 23.Dong Z., Onrust R., Skangalis M., O'Donnell M. (1993) J. Biol. Chem. 268, 11758–11765 [PubMed] [Google Scholar]
- 24.Olson M. W., Dallmann H. G., McHenry C. S. (1995) J. Biol. Chem. 270, 29570–29577 [PubMed] [Google Scholar]
- 25.Johanson K. O., Haynes T. E., McHenry C. S. (1986) J. Biol. Chem. 261, 11460–11465 [PubMed] [Google Scholar]
- 26.Anderson S. G., Thompson J. A., Paschall C. O., O'Donnell M., Bloom L. B. (2009) Biochemistry 48, 8516–8527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griep M. A., McHenry C. S. (1988) Biochemistry 27, 5210–5215 [DOI] [PubMed] [Google Scholar]
- 28.Snyder A. K., Williams C. R., Johnson A., O'Donnell M., Bloom L. B. (2004) J. Biol. Chem. 279, 4386–4393 [DOI] [PubMed] [Google Scholar]
- 29.Bowman G. D., O'Donnell M., Kuriyan J. (2004) Nature 429, 724–730 [DOI] [PubMed] [Google Scholar]
- 30.Jeruzalmi D., Yurieva O., Zhao Y., Young M., Stewart J., Hingorani M., O'Donnell M., Kuriyan J. (2001) Cell 106, 417–428 [PubMed] [Google Scholar]
- 31.Turner J., Hingorani M. M., Kelman Z., O'Donnell M. (1999) EMBO J. 18, 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson K. A. (1992) in The Enzymes (Sigman D. S. ed) 3rd Ed., Vol. 20, pp. 1–61, Academic Press, Inc., San Diego [Google Scholar]
- 33.Bertram J. G., Bloom L. B., Hingorani M. M., Beechem J. M., O'Donnell M., Goodman M. F. (2000) J. Biol. Chem. 275, 28413–28420 [DOI] [PubMed] [Google Scholar]
- 34.Kuzmic P. (1996) Anal. Biochem. 237, 260–273 [DOI] [PubMed] [Google Scholar]
- 35.Johnson A., Yao N. Y., Bowman G. D., Kuriyan J., O'Donnell M. (2006) J. Biol. Chem. 281, 35531–35543 [DOI] [PubMed] [Google Scholar]
- 36.Schmidt S. L., Gomes X. V., Burgers P. M. (2001) J. Biol. Chem. 276, 34784–34791 [DOI] [PubMed] [Google Scholar]
- 37.Kazmirski S. L., Podobnik M., Weitze T. F., O'Donnell M., Kuriyan J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16750–16755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomes X. V., Schmidt S. L., Burgers P. M. (2001) J. Biol. Chem. 276, 34776–34783 [DOI] [PubMed] [Google Scholar]
- 39.Sexton D. J., Kaboord B. F., Berdis A. J., Carver T. E., Benkovic S. J. (1998) Biochemistry 37, 7749–7756 [DOI] [PubMed] [Google Scholar]
- 40.Zhuang Z., Berdis A. J., Benkovic S. J. (2006) Biochemistry 45, 7976–7989 [DOI] [PubMed] [Google Scholar]
- 41.Alley S. C., Shier V. K., Abel-Santos E., Sexton D. J., Soumillion P., Benkovic S. J. (1999) Biochemistry 38, 7696–7709 [DOI] [PubMed] [Google Scholar]
- 42.Millar D., Trakselis M. A., Benkovic S. J. (2004) Biochemistry 43, 12723–12727 [DOI] [PubMed] [Google Scholar]
- 43.Stukenberg P. T., Studwell-Vaughan P. S., O'Donnell M. (1991) J. Biol. Chem. 266, 11328–11334 [PubMed] [Google Scholar]
- 44.Zhuang Z., Yoder B. L., Burgers P. M., Benkovic S. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2546–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomes X. V., Burgers P. M. (2001) J. Biol. Chem. 276, 34768–34775 [DOI] [PubMed] [Google Scholar]
- 46.Burgers P. M., Kornberg A., Sakakibara Y. (1981) Proc. Natl. Acad. Sci. U.S.A. 78, 5391–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leu F. P., Hingorani M. M., Turner J., O'Donnell M. (2000) J. Biol. Chem. 275, 34609–34618 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.