

Abstract
The genetic disease ataxia telangiectasia (AT) results from mutations in the ataxia telangiectasia mutated (ATM) gene. AT patients develop a progressive degeneration of cerebellar Purkinje neurons. Surprisingly, while ATM plays a criticial role in the cellular reponse to DNA damage, previous studies have localized ATM to the cytoplasm of rodent and human Purkinje neurons. Here we show that ATM is primarily localized to the nucleus in cerebellar Purkinje neurons in postmortem human brain tissue samples, although some light cytoplasmic ATM staining was also observed. No ATM staining was observed in brain tissue samples from AT patients, verifying the specificity of the antibody. We also found that antibodies against components of the Mre11/Rad50/Nbs1 (MRN) complex showed strong staining in Purkinje cell nuclei. However, while ATM is present in both the nucleoplasm and nucleolus, MRN proteins are excluded from the nucleolus. We also observed very high levels of topoisomerase 1 (TOP1) in the nucleus, and specifically the nucleolus, of human Purkinje neurons. Our results have direct implications for understanding the mechanisms of neurodegeneration in AT and AT-like disorder.
1. Introduction
The genetic disease ataxia telangiectasia (AT) results from mutations in the ataxia-telangiectasia mutated (ATM) gene [1], which encodes a protein kinase that is activated in response to DNA damage [2]. Patients with AT develop a progressive ataxia as a result of degeneration of cerebellar Purkinje neurons, though other brain regions may be affected as well [3,4].
There is no doubt that ATM plays a crucial role in response to DNA damage in eukaryotic cells [2,5-8]. However, the question of whether the neurodegeneration observed in AT patients results from a loss of this DNA damage response function of ATM has been controversial, since several authors have found that ATM protein is located in the cytoplasm of Purkinje neurons in the human [9] and rodent [10,11] brain. Recently however, Shiloh and co-workers [12,13] found that the ATM protein is present in the nucleus of neuronal derived human cells maintained in tissue culture. Also, Barzilai and co-workers [14] showed that nuclear ATM protein is phosphorylated in Purkinje neurons of mice exposed to ionizing radiation.
In light of these findings, we have reinvestigated the intracellular localization of the ATM protein in Purkinje neurons of the human brain. Here, using tissue from both normal and AT patients to verify antibody specificity, we show that the ATM protein is in fact predominantly localized to the nucleus in human Purkinje neurons. Components of the Mre11/Rad50/Nbs1 (MRN) complex are also concentrated in the nucleus of Purkinje neurons, though the distribution of MRN proteins was not identical to that of ATM. Specifically, ATM was detected in both the nucleolus and nucleoplasm, while the MRN proteins were excluded from the nucleolus. We also found that topoisomerase 1 (TOP1) was concentrated in Purkinje cell nuclei, particularly in the nucleolus. We discuss the implications of these results for neurodegeneration in AT patients.
2. Materials and Methods
Tissue samples
The formalin-fixed postmortem brain tissue samples were obtained from the University of Maryland Brain Bank for Developmental Disorders. Information on the donors is listed in Table 1.
Table 1.
| Case # | Age | Postmortem interval (hours) |
|---|---|---|
| 1465 Normal | 17 years 189 days | 4 |
| 917 Normal | 14 years 227 days | 10 |
| 1459 AT | 19 years 342 days | 2 |
| 836 AT | 28 years 271 days | 4 |
Antibodies
The following antibodies were used in this work, at the indicated dilutions: ATM rabbit monoclonal Y170 (Epitomics) 1:100; Histone H1 mouse monoclonal (Santa Cruz) 1:100; Mre11 rabbit polyclonal PC388 (Oncogene/Calbiochem) 1:500; Rad50 mouse monoclonal NB 100-147 (Novus) 1:500; Calbindin mouse monoclonal (Sigma) 1:100 and Calbindin rabbit polyclonal (Sigma) 1:100; TOP1 mouse monoclonal (Topogen) 1:50 and mouse monoclonal antibody against TOP1 (kindly provided by Dr. Igor Bronstein; see [15]) 1:25; S100β rabbit polyclonal (DakoCytomation) 1:50.
Immunofluoresence
Formalin-fixed tissue blocks were cryoprotected in phosphate buffered saline containing 25% sucrose for > 24 hours prior to sectioning. Cryoprotected tissues were carefully frozen and then sectioned at 30 micron thickness. Sections were then processed through a heat-induced antigen retrieval protocol (autoclaving in 10 mM Sodium citrate buffer, pH 6.0, at 121° C for 15 minutes), cooled to room temperature, then processed as free-floating sections.
Sections were washed with buffer containing PBST ( PBS containing 0.1% Tween-20 or 0.1% Triton X-100), then blocked with 10% normal goat serum (Jackson Immunoresearch lab) prepared in PBST for 2 hours at room temperature and incubated with primary antibodies overnight at 4° C. The next day, the sections were washed 3 times for 10 min each with PBST, then incubated for 2 hours at room temperature with secondary antibodies (goat anti-mouse or anti-rabbit IgG labeled with Alexa Fluor 488, or 594, Invitrogen) diluted 1:200. For RNA and DNA staining, some sections were incubated in propidium iodide (PI) diluted 1:2000 in PBS.
For confocal microscopy, following primary and secondary antibody incubation and washings, sections were incubated for 10 minutes in 0.3% Sudan Black in 70% ethanol to reduce endogenous autofluorescence in the tissues, then washed with PBS, mounted in PBS, and coverslipped with Prolong Gold Antifade reagent (Invitrogen). Sections were examined and images prepared using a Zeiss LSM 5 Pascal confocal microscope.
3. Results
Biton et al. [12] found that the antibody Y170 could be used to specifically localize the ATM protein in the nucleus of cultured human neuronal cells. Therefore we utilized this antibody to detect the ATM protein in the cerebellum of human postmortem brain samples from two normal donors. For a negative control, we also used postmortem cerebellar tissue from AT patients (see Table 1). As shown in Figure 1(A and B), the Y170 antibody strongly stained Purkinje neurons in postmortem cerebellar samples from the brains of normal donors. In contrast, no specific staining was observed when the antibody was used on brain sections from two AT patients (Figure 1A and B) demonstrating the specificity of staining. The absence of ATM staining in the AT patient samples cannot be explained by postmortem delay, since the time between death and tissue collection was either equivalent or shorter in the AT patient samples than in controls (see Table 1). The pattern of ATM staining in the cerebellum was consistent in samples from different normal donors (Figure 1C and data not shown). We therefore conclude that the Y170 antibody specifically detects the ATM protein in formalin-fixed human postmortem brain tissue samples.
Figure 1.

The ATM Y170 antibody specifically stains ATM in human Purkinje neurons. A) Low-magnification (25×) images of sections of the postmortem cerebellar tissue from a normal donor #1465 (CON) and AT patient #1459 (AT) stained with ATM antibody Y170 (green) and calbindin (CB), a specific marker for Purkinje neurons (red). Purkinje neurons are identified by arrows. Scale bar = 200 microns. B) Purkinje neurons from normal donor #1465 (CON) and AT patient # 836 (AT) stained with ATM antibody Y170 (green) and CB (red). C) ATM staining (green) with PI counterstain (red) in cerebellar Purkinje neurons from two different control donors #1465 (top) and #917 (bottom) is predominantly nuclear. Scale bars in B and C = 10 microns.
ATM staining in Purkinje neurons appeared to be predominantly nuclear (Figure 1A-C), but to verify this double-labeling studies were carried out using an antibody against histone H1. As shown in Figure 2A, the strongest ATM staining in Purkinje neurons colocalized with histone H1 staining, verifying nuclear localization. However, in addition to the intense ATM staining in Purkinje cell nuclei, it is important to stress that we also observed a light but detectable ATM staining in the cytoplasm of Purkinje neurons (Figure 2B).
Figure 2.

ATM staining is predominantly, but not exclusively, nuclear in human Purkinje neurons. A) Low (top panels) and high (bottom panels) magnification of cerebellar sections from normal donor #1465 showing double-labeling of ATM (red) and the nuclear marker histone H1 (green) in cerebellar tissue. The strongest ATM staining colocalizes with histone H1. Scale bar = 30 microns (top panels) and 10 microns (bottom panels). B) Double-labeling of ATM (green) and CB (red) in Purkinje neuron. ATM staining is predominantly nuclear, but some cytoplasmic staining is also visible. Scale bar = 10 microns.
Localization of ATM in Other Cerebellar Neurons
In addition to the Purkinje neurons, we also examined the localization of ATM in other populations of cerebellar neurons. As shown in Figure 3A-C, a very light signal was detectable in granule neurons, but was barely above background levels. However, scattered strongly staining interneurons [16] were observed within the granule cell layer (Figure 3A-E). Neurons in the molecular layer also showed ATM staining (Figure 3F).
Figure 3.

ATM localization in other cerebellar neurons. Section through the granular layer (GL) of the cerebellar cortex from normal donor stained for ATM (A) and PI (B). Panel C) is merge of A and B. Note the very light staining in granule cell neurons compared to the Purkinje neuron in the upper right corner. Two interneurons within the granule cell layer showing strong ATM staining are indicated with arrows. D) High magnification merged view of cells indicated with arrows in C. E) Merged image of the granular layer showing ATM staining (green) and PI (red) in a large Golgi neuron [16] (indicated by arrow). F) Merged view of ATM staining (green) and PI (red) in the molecular layer (ML) of the cerebellum. A neuron in the molecular layer with strong ATM staining is indicated by the arrow. A Purkinje neuron (lower right), an interneuron showing ATM staining (left), and a few granule neurons (lower left) are also visible. Scale bars = 20 microns for A-C, 10 microns for D-F.
High Levels of MRN Proteins in Purkinje Neurons and Bergmann Glial Cells
The MRN complex acts upstream of ATM in a DNA damage response pathway [17]. Mutations in the MRE11 gene result in a neurological disease, ataxia telangiectasia like-disorder (ATLD), that is similar but not identical to AT [18]. Jacobsen et al. [19] noted that within the human cerebellar cortex of elderly donors, Nbs1 staining was particularly strong in the nucleus of Purkinje neurons. We also observed strong staining for Mre11 and Nbs1 (Figure 4A) and Rad50 (Figure 4B) and in the nucleus of Purkinje neurons from juvenile donors. In addition to Purkinje neurons, however, we also detected a population of smaller cells located within the Purkinje cell layer which showed strong staining for MRN proteins (Figure 4A and B). Based on their size and location, we suspected that these cells might be Bergmann glial cells, a specialized type of glial cell that is intimately associated, both structurally and functionally, with Purkinje neurons [20]. Using an antibody against the S100β protein as a marker, we confirmed that these cells are in fact Bergman glial cells (Figure 4B).
Figure 4.
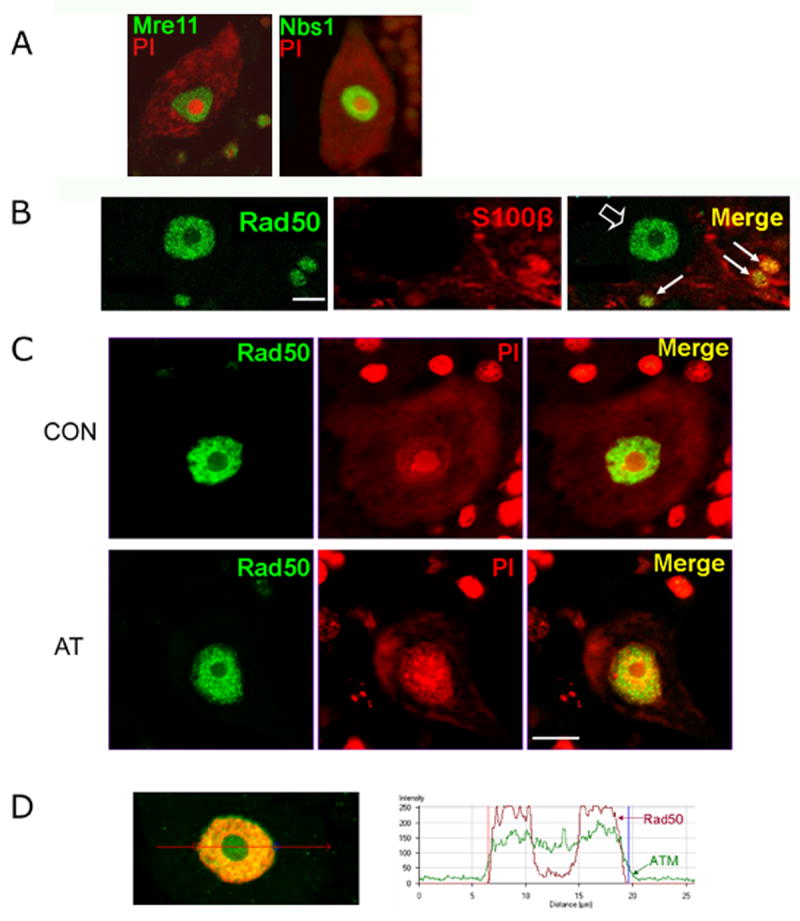
Localization of MRN proteins and ATM in human Purkinje neurons. A) Single Purkinje neurons stained for Mre11 (green, left panel) or Nbs1 (green, right panel), with PI counterstaining in (red, both panels). Three Bergmann glial cells are also visible in the left panel. B) Cerebellar section stained with antibodies against Rad50 (green), S100β (a marker for Bergmann glial cells; red) and merged image. Bergmann glial cells labeled with antibody against S100β showing elevated levels of Rad50 staining are identified by arrows. Rad50 staining in a Purkinje cell nucleus is indicated by open arrowhead. C) Rad50 protein is present in a punctate pattern throughout the nucleoplasm but largely excluded from the nucleolus in Purkinje neurons of control donors (CON), as well in an AT patient (AT). D) Double-labeling of the nucleus of a Purkinje neuron from control donor with antibodies against ATM (green) and Rad50 (red), showing that ATM is present throughout nucleoplasm and nucleolus, while Rad50 is excluded from the nucleolus. A graph showing the intensity of ATM and Rad50 staining on the line drawn through the nucleus of the Purkinje neuron shown at left. The graph was created using the Profile function of the Zeiss LSM Image Browser software. Scale bars = 10 microns.
In view of the evidence that the MRN complex acts upstream of ATM in the DNA damage response [17], it was of interest to examine the pattern of MRN staining in Purkinje neurons from AT patients. As shown in Figure 4C, the qualitative pattern of MRN staining in Purkinje neurons from AT patients was not noticeably different from the pattern in Purkinje neurons from normal control brains.
To directly compare the distribution of ATM with that of the MRN proteins in the same cell, we carried out double staining using antibodies against ATM and Rad50. As shown in Figure 4D, while ATM is found throughout the nucleoplasm and nucleolus, Rad50 is excluded from the nucleolus. Thus the localization of ATM and the MRN complex are not identical in Purkinje neurons.
Topoisomerase I is Also Concentrated in the Nucleus of Purkinje Neurons
Topoisomerase I (TOP1) plays an essential role in maintaining the appropriate level of supercoiling in genomic DNA [21]. Interestingly, in an earlier survey of TOP1 staining patterns in normal human tissues and cancers, Holden et al. [15] noted that TOP1 levels were particularly high in the nucleus of human Purkinje neurons of a normal donor, although the age of donor was not given. In view of the potential significance of this observation, not only for AT but also for other neurological diseases resulting from defective DNA repair [22], we sought to confirm the observation of Holden et al. in our juvenile samples, and also to compare the pattern of TOP1 staining with that of ATM and the MRN proteins in Purkinje neurons.
As shown in Figure 5, consistent with the observations of Holden et al. [15], we found very strong TOP1 staining in human Purkinje neurons. Our experiments were done using a commercially available antibody, but we also observed an identical pattern of TOP1 staining in Purkinje neurons using a sample of the same monoclonal anti-TOP1 antibody originally used by Holden et al. (kindly provided by Dr. Igor Bronstein; results not shown). As in the case of the MRN proteins, we did not have access to human cerebellar tissue lacking TOP1, which would be the ideal negative control. However, the fact that two independently generated monoclonal antibodies against TOP1 give an identical pattern of staining provides strong evidence that the staining is specific.
Figure 5.
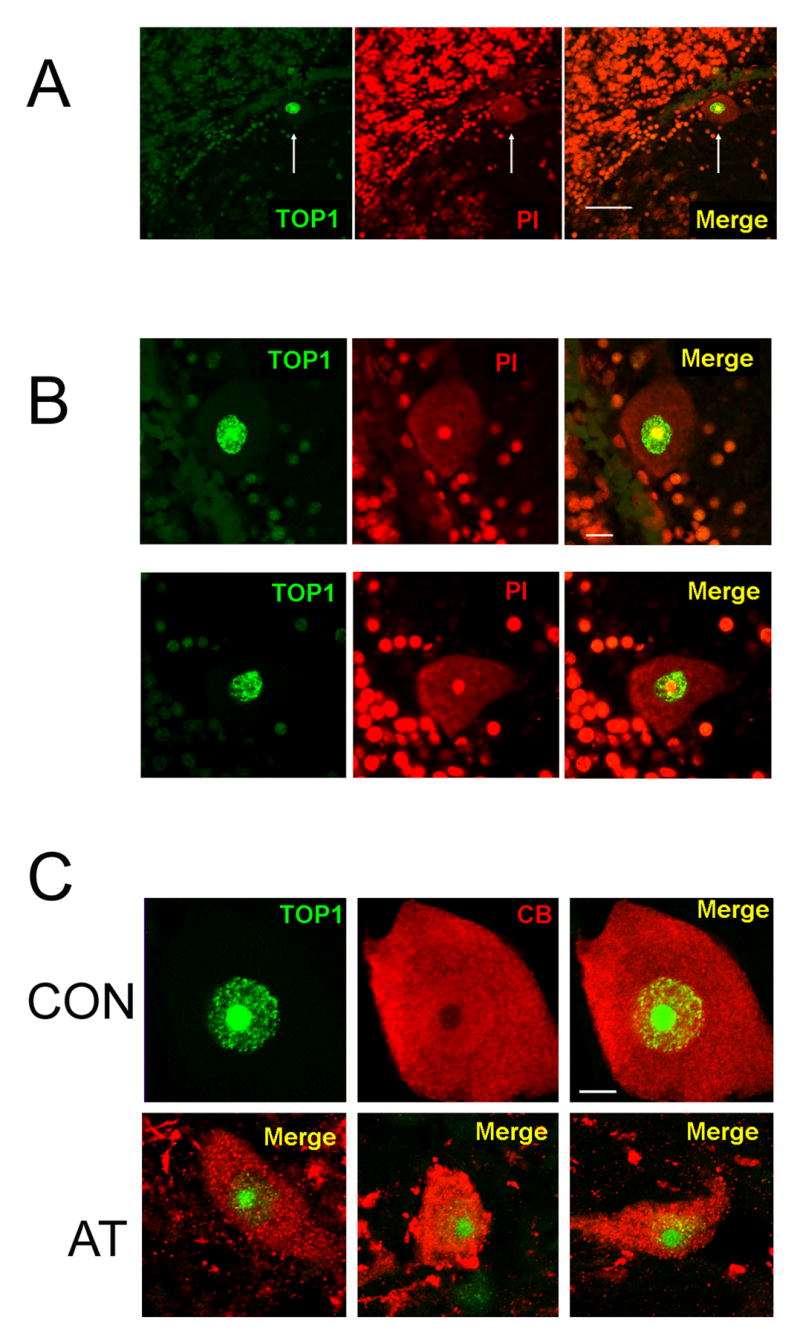
TOP1 is concentrated in Purkinje neurons of normal control and AT patients. A) Section of the cerebellar cortex from a normal donor (#1465) stained for TOP1 (green) and PI (red). Note the very strong TOP1 staining in the nucleus of a Purkinje neuron (arrow), compared to other cells in the section. Scale bar = 50 microns. B) Double staining for TOP1 (green) and PI (red) demonstrating the very strong TOP1 staining in the nucleus of Purkinje neurons. In the cell on the top, TOP1 staining is strongest in the nucleolus, which is the typical pattern. In the cell on the bottom, nucleolar TOP1 staining is less prominent compared to the nucleoplasm. These two cells were chosen to illustrate the diversity of TOP1 staining patterns in different Purkinje neurons in a normal individual. C) Top panels show double staining for TOP1 (green) and CB (red) in a Purkinje neuron from a normal donor (CON). The bottom panels show merged images of TOP1 and CB staining in three different Purkinje neurons from an AT patient (AT). Scale bars on B and C = 10 microns.
In most Purkinje neurons from normal donors, TOP1 staining was strongest in the nucleolus, but was also detectable throughout the nucleoplasm (Figure 4B, top row). This pattern is consistent with other data showing that TOP1 is primarily localized to the nucleolus [23], but shuttles between the nucleolus and the nucleoplasm [24,25], and therefore provides further evidence of the specificity of staining. In some Purkinje neurons however, this pattern was reversed, with the amount of TOP1 being lower in the nucleolus than in the nucleoplasm (Figure 5B, bottom row). Since cells with these different TOP1 staining patterns could be observed within the same tissue section, these different patterns could not be due methodological factors. Instead, the differences most likely reflect the steady-state level of the enzyme in the nucleolus versus nucleoplasm in different Purkinje neurons.
As we did with the MRN proteins, we also examined the expression of TOP1 in Purkinje neurons from AT patients. As shown in Figure 5C, the qualitative pattern of TOP1 staining in the nucleus of Purkinje neurons from AT patients was within the range observed in normal donors, even in cells from the AT patient that appear clearly abnormal based on calbindin staining. However, given the variation in TOP1 staining patterns in the nucleus of Purkinje neurons from normal donors (Figure 5B), the possibility of subtle quantitative abnormalities in TOP1 distribution in Purkinje neurons of AT patients cannot be ruled out.
4. Discussion
Determining the intracellular localization of a protein is essential to understanding function, and therefore to the development of rational therapies for genetic diseases in which specific proteins are lost as a result of mutations. As described in the Introduction, a previous study [9] using postmortem human brain tissue concluded that the ATM protein is located in the cytoplasm of human Purkinje neurons. Cytoplasmic localization implied that the function of ATM in Purkinje neurons is something separate from the established role of the ATM protein, which is the detection and repair of DNA damage. However, in the earlier work [9], control studies using brain tissue from AT patients as a negative control were not employed, thus raising questions about the specificity of the ATM staining. Here, using an antibody previously demonstrated to specifically stain ATM in human neuronal-like cells in culture [12], we show that ATM is predominantly localized in the nucleus of Purkinje neurons in postmortem tissue from the juvenile human brain. Importantly, our conclusion is supported by control experiments demonstrating the absence of staining in Purkinje neurons from AT patients, which represents the most conclusive evidence for the specificity of the staining. As such, this localization is consistent with the hypothesis that a major function of ATM in fully differentiated human Purkinje neurons is to detect nuclear DNA damage, as it is in all other cell types examined.
McKinnon and colleagues [26] have shown that in mice, the ATM protein is required for triggering apoptosis in response to neuronal DNA damage during the period shortly after the stage of terminal differentiation. Importantly, however, this observation does not exclude an ongoing role of the ATM protein in Purkinje neurons at later stages (see [27]). In humans, Purkinje neuron differentiation extends from the late fetal period into the first year of life [28]. Therefore, in addition to a role in triggering apoptosis in response to DNA damage in early brain development, our observation of nuclear ATM localization in human Purkinje neurons in normal individuals 14 and 17 years of age is consistent with the hypothesis that ATM also plays an ongoing role in sensing DNA damage in Purkinje neurons long after the terminal differentiation stage.
It is important to note that while ATM staining intensity was clearly highest in the cell nucleus, we also observed some ATM staining in the cytoplasm of Purkinje neurons as well. This staining may correspond to ATM in cytoplasmic vesicles, as described by others [29,30]. While the intensity of cytoplasmic staining was very light compared to the nuclear staining, given the large volume of cytoplasm in human Purkinje neurons, this light staining could represent a substantial amount of the total ATM protein. However, the functional significance of this cytoplasmic ATM, if any, remains to be determined.
While the primary goal of this work was to address the localization of ATM in Purkinje neurons, we also examined ATM staining in other cerebellar neurons. Notably, very weak staining was observed in granule cells, but strong staining was observed in interneurons within the granule cell layer, as well as in neurons in the molecular layer. It is becoming increasingly clear that the cerebellar cortex contains a variety of morphologically and biochemically distinct types of interneurons [16,31], and additional studies would be necessary to fully categorize the ATM staining in each specific type.
A full description of ATM staining in the rest of human brain is beyond the scope of this work. However, consistent with the widespread expression of ATM mRNA in the human brain [32], we detected ATM protein expression by Western blotting in multiple brain regions (cerebellum, pons, cerebral cortex, hippocampus, and thalamus) and detected predominantly nuclear ATM staining in neurons in the human cerebral cortex (data not shown). Since the neuropathology observed in AT patients primarily affects the cerebellum (although cells in the spinal cord, dorsal root ganglia, and basal ganglia are also affected [3]), our observations provide further evidence that ATM expression is not limited to brain regions or cell types that are affected in AT patients, and emphasize the importance of understanding why Purkinje neurons are so severely affected in AT patients.
The MRN-Complex and Topoisomerase I are also Concentrated in Human Purkinje neurons
In contrast to ATM, we did not have access to human tissues from patients lacking the MRN proteins or TOP1 as negative controls for antibody specificity. With regard to TOP1, such material will never be available, since TOP1 is an essential protein. In lieu of negative control tissues, we relied on the concordant results of multiple independent antibodies to demonstrate staining specificity. This approach is analogous to the use of multiple siRNAs directed against different regions of an mRNA to demonstrate specificity [33]. Also, our localization of the MRN proteins in human Purkinje neurons is completely consistent with the observations of Jacobsen et al. [19]. With regard to TOP1, in addition to obtaining identical results using two independently generated monoclonal antibodies, our observation that TOP1 is localized to the nucleolus and nucleoplasm is consistent with other studies on the distribution of this enzyme within the nucleolus [23], and nucleus, including observations of GFP-tagged TOP1 in living human cells [24,25]. It should be noted that in one study [34], cytoplasmic TOP1 staining was observed in the mouse Purkinje neurons. However, in another study [35] TOP1 staining was observed in the nucleolus of postnatal rat Purkinje neurons, consistent with our findings. Whether the discrepant observations [34] represent a species difference, a methodological difference, or non-specific staining remains to be determined.
The fact that the MRN complex and ATM act together in a double-strand break repair pathway in human cells is well established, although the exact details of how this pathway functions is the subject of ongoing investigations [2,5-8]. Our observations of high levels of ATM and MRN proteins in Purkinje neurons are consistent with the hypothesis that this same pathway functions to repair endogenous double-strand breaks (perhaps arising from closely apposed single-strand breaks) in human Purkinje neurons. However, our observation that ATM is present in the nucleolus of Purkinje neurons, while the MRN proteins are essentially excluded from this compartment, raises the possibility that ATM may have additional functions in the nucleolus of Purkinje neurons which are independent of the MRN complex, although further experiments will be necessary to test this possibility.
In terminally differentiated cells such as neurons, the main role of TOP1 is presumably in transcription, and in particular transcription of ribosomal DNA [23]. The strikingly high level of TOP1 we observed in Purkinje neurons compared to other cerebellar neurons indicates that these cells have an elevated requirement for this enzyme, perhaps due to a high demand for ribosomal RNA synthesis. TOP1 can become covalently attached to the DNA, forming TOP1 cleavage complexes (TOP1ccs) [36], and many types of endogenous DNA modifications can cause TOP1ccs [37]. The observation that spinocerebellar ataxia with axonal neuropathy (SCAN1) results from mutations in the TDP1 gene [22], which encodes a protein involved in the repair of TOP1ccs [38,39] indicates the importance of repair of such complexes in preventing ataxia. In view of the high levels of TOP1 in Purkinje neurons, it follows that these cells would be at elevated risk for TOP1ccs, and thus the loss of ability to repair such complexes would be particularly severe in these cells. If ATM is involved in the repair of TOP1ccs, this would provide a possible explanation of why Purkinje neurons are specifically affected in AT patients. Additional functional studies are necessary to address this point.
Acknowledgments
We thank Cheryl Marietta for advice and helpful comments on the manuscript, and Tracy Gilman for comments on the manuscript and assistance with preparation of the figures. We also thank Dr. Igor Bronstein, NIMR, MRC for his generous gift of his TOP1 monoclonal antibody. Tissue samples were obtained from the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland (NICHD Contract No. N01-HD-4-3368 and NO1-HD-4-3383).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 2.Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–410. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Crawford TO. Ataxia telangiectasia. Semin Pediatr Neurol. 1998;5:287–294. doi: 10.1016/s1071-9091(98)80007-7. [DOI] [PubMed] [Google Scholar]
- 4.Sedgewick R, Boder E. Ataxia-Telangectasia. In: de Jong JMBV, editor. Hereditary Neuropathies and Spinocebellar Atrophies. Elsevier; Amsterdam: 1991. pp. 347–423. [Google Scholar]
- 5.Lavin MF. The Mre11 complex and ATM: a two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair (Amst) 2004;3:1515–1520. doi: 10.1016/j.dnarep.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 6.Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol. 2005;70:99–109. doi: 10.1101/sqb.2005.70.002. [DOI] [PubMed] [Google Scholar]
- 7.Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 8.Pellegrini M, Celeste A, Difilippantonio S, Guo R, Wang W, Feigenbaum L, Nussenzweig A. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature. 2006;443:222–225. doi: 10.1038/nature05112. [DOI] [PubMed] [Google Scholar]
- 9.Oka A, Takashima S. Expression of the ataxia-telangiectasia gene (ATM) product in human cerebellar neurons during development. Neurosci Lett. 1998;252:195–198. doi: 10.1016/s0304-3940(98)00576-x. [DOI] [PubMed] [Google Scholar]
- 10.Kuljis RO, Chen G, Lee EY, Aguila MC, Xu Y. ATM immunolocalization in mouse neuronal endosomes: implications for ataxia-telangiectasia. Brain Res. 1999;842:351–358. doi: 10.1016/s0006-8993(99)01813-2. [DOI] [PubMed] [Google Scholar]
- 11.Barlow C, Ribaut-Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, Larson D, Harrington EA, Haeberle AM, Mariani J, Eckhaus M, Herrup K, Bailly Y, Wynshaw-Boris A. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci U S A. 2000;97:871–876. doi: 10.1073/pnas.97.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biton S, Dar I, Mittelman L, Pereg Y, Barzilai A, Shiloh Y. Nuclear ataxia-telangiectasia mutated (ATM) mediates the cellular response to DNA double strand breaks in human neuron-like cells. J Biol Chem. 2006;281:17482–17491. doi: 10.1074/jbc.M601895200. [DOI] [PubMed] [Google Scholar]
- 13.Biton S, Gropp M, Itsykson P, Pereg Y, Mittelman L, Johe K, Reubinoff B, Shiloh Y. ATM-mediated response to DNA double strand breaks in human neurons derived from stem cells. DNA Repair (Amst) 2007;6:128–134. doi: 10.1016/j.dnarep.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 14.Dar I, Biton S, Shiloh Y, Barzilai A. Analysis of the ataxia telangiectasia mutated-mediated DNA damage response in murine cerebellar neurons. J Neurosci. 2006;26:7767–7774. doi: 10.1523/JNEUROSCI.2055-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holden JA, Rahn MP, Jolles CJ, Vorobyev SV, Bronstein IB. Immunohistochemical detection of DNA topoisomerase I in formalin fixed, paraffin wax embedded normal tissues and in ovarian carcinomas. Mol Pathol. 1997;50:247–253. doi: 10.1136/mp.50.5.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crook J, Hendrickson A, Robinson FR. Co-localization of glycine and gaba immunoreactivity in interneurons in Macaca monkey cerebellar cortex. Neuroscience. 2006;141:1951–1959. doi: 10.1016/j.neuroscience.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 17.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. Embo J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair (Amst) 2004;3:1219–1225. doi: 10.1016/j.dnarep.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Jacobsen E, Beach T, Shen Y, Li R, Chang Y. Deficiency of the Mre11 DNA repair complex in Alzheimer's disease brains. Brain Res Mol Brain Res. 2004;128:1–7. doi: 10.1016/j.molbrainres.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- 21.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 22.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, Stockton DW, Lupski JR. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267–272. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 23.Muller MT, Pfund WP, Mehta VB, Trask DK. Eukaryotic type I topoisomerase is enriched in the nucleolus and catalytically active on ribosomal DNA. Embo J. 1985;4:1237–1243. doi: 10.1002/j.1460-2075.1985.tb03766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christensen MO, Barthelmes HU, Feineis S, Knudsen BR, Andersen AH, Boege F, Mielke C. Changes in mobility account for camptothecin-induced subnuclear relocation of topoisomerase I. J Biol Chem. 2002;277:15661–15665. doi: 10.1074/jbc.C200066200. [DOI] [PubMed] [Google Scholar]
- 25.Christensen MO, Krokowski RM, Barthelmes HU, Hock R, Boege F, Mielke C. Distinct effects of topoisomerase I and RNA polymerase I inhibitors suggest a dual mechanism of nucleolar/nucleoplasmic partitioning of topoisomerase I. J Biol Chem. 2004;279:21873–21882. doi: 10.1074/jbc.M400498200. [DOI] [PubMed] [Google Scholar]
- 26.Lee Y, Chong MJ, McKinnon PJ. Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status. J Neurosci. 2001;21:6687–6693. doi: 10.1523/JNEUROSCI.21-17-06687.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abner CW, McKinnon PJ. The DNA double-strand break response in the nervous system. DNA Repair (Amst) 2004;3:1141–1147. doi: 10.1016/j.dnarep.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Zecevic N, Rakic P. Differentiation of Purkinje cells and their relationship to other components of developing cerebellar cortex in man. J Comp Neurol. 1976;167:27–47. doi: 10.1002/cne.901670103. [DOI] [PubMed] [Google Scholar]
- 29.Lim DS, Kirsch DG, Canman CE, Ahn JH, Ziv Y, Newman LS, Darnell RB, Shiloh Y, Kastan MB. ATM binds to beta-adaptin in cytoplasmic vesicles. Proc Natl Acad Sci U S A. 1998;95:10146–10151. doi: 10.1073/pnas.95.17.10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watters D, Kedar P, Spring K, Bjorkman J, Chen P, Gatei M, Birrell G, Garrone B, Srinivasa P, Crane DI, Lavin MF. Localization of a portion of extranuclear ATM to peroxisomes. J Biol Chem. 1999;274:34277–34282. doi: 10.1074/jbc.274.48.34277. [DOI] [PubMed] [Google Scholar]
- 31.Simat M, Parpan F, Fritschy JM. Heterogeneity of glycinergic and gabaergic interneurons in the granule cell layer of mouse cerebellum. J Comp Neurol. 2007;500:71–83. doi: 10.1002/cne.21142. [DOI] [PubMed] [Google Scholar]
- 32.Soares HD, Morgan JI, McKinnon PJ. Atm expression patterns suggest a contribution from the peripheral nervous system to the phenotype of ataxia-telangiectasia. Neuroscience. 1998;86:1045–1054. doi: 10.1016/s0306-4522(98)00117-1. [DOI] [PubMed] [Google Scholar]
- 33.Echeverri CJ, Perrimon N. High-throughput RNAi screening in cultured cells: a user's guide. Nat Rev Genet. 2006;7:373–384. doi: 10.1038/nrg1836. [DOI] [PubMed] [Google Scholar]
- 34.Plaschkes I, Silverman FW, Priel E. DNA topoisomerase I in the mouse central nervous system: Age and sex dependence. J Comp Neurol. 2005;493:357–369. doi: 10.1002/cne.20793. [DOI] [PubMed] [Google Scholar]
- 35.Tsutsui K, Tsutsui K, Hosoya O, Sano K, Tokunaga A. Immunohistochemical analyses of DNA topoisomerase II isoforms in developing rat cerebellum. J Comp Neurol. 2001;431:228–239. doi: 10.1002/1096-9861(20010305)431:2<228::aid-cne1067>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 36.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 37.Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Adv Cancer Res. 2001;80:189–216. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- 38.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–555. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 39.Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J Biol Chem. 2004;279:55618–55625. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]