

Abstract
Background
Image contrast in clinical MRI is often determined by differences in tissue water proton relaxation behavior. However, many aspects of water proton relaxation in complex biological media, such as protein solutions and tissue are not well understood, perhaps due to the limited empirical data.
Principal Findings
Water proton T1, T2, and T1ρ of protein solutions and tissue were measured systematically under multiple conditions. Crosslinking or aggregation of protein decreased T2 and T1ρ, but did not change high-field T1. T1ρ dispersion profiles were similar for crosslinked protein solutions, myocardial tissue, and cartilage, and exhibited power law behavior with T1ρ(0) values that closely approximated T2. The T1ρ dispersion of mobile protein solutions was flat above 5 kHz, but showed a steep curve below 5 kHz that was sensitive to changes in pH. The T1ρ dispersion of crosslinked BSA and cartilage in DMSO solvent closely resembled that of water solvent above 5 kHz but showed decreased dispersion below 5 kHz.
Conclusions
Proton exchange is a minor pathway for tissue T1 and T1ρ relaxation above 5 kHz. Potential models for relaxation are discussed, however the same molecular mechanism appears to be responsible across 5 decades of frequencies from T1ρ to T1.
Introduction
Image contrast in clinical MRI is often determined by differences in tissue water relaxation behavior. Although the observed properties of proton relaxation in homogeneous liquids such as pure water, ethanol, and glycerol have been successfully explained by the theory of Bloembergen, Purcell, and Pound (BPP) [1], the mechanism of water relaxation in more complex environments such as tissues is still highly speculative. In part to gain insight into tissue relaxation, many studies have evaluated the relaxation characteristics of protein solutions, since for most tissue, relaxation behavior is dominated by the water-macromolecule interaction [2]. However, few studies have attempted to systematically investigate the relationship between the physico-chemical properties of macromolecules and bulk water relaxation, and there are diverse hypotheses concerning the mechanism of water proton relaxation in protein systems—perhaps due to the limited empirical data.
Of particular interest has been the character of the magnetic field dependence (dispersion) of relaxation in these protein systems. Prior investigations have shown that solutions of immobile proteins have spin-lattice relaxation dispersion characteristics similar to that of various soft tissues [3], [4]. Most of these studies measured T1 at low field (<20 MHz) or T1.ρ (spin-lattice relaxation time in the rotating frame, which is measured at B1 field strength [5], [6]) since it is known that water proton T1 at high field is insensitive to significant protein structural changes such as the addition of crosslinks [7]. Rationale for the improved sensitivity of low-field dispersion to detect protein or tissue structural changes includes arguments concerning the long correlation times of motion (τc) in systems containing large macromolecules [8]. For instance, T1ρ will presumably be sensitive to motion with τc on the order of tens of µsecs to msecs, depending on the achievable RF power and proton solvent linewidth, respectively. However, the determination of motional correlation times—whether single, multiple, or even a continuous distribution—requires assumptions about the characteristic shape of the spectral density function. More recently, several investigators have suggested that conventional, BPP-type relaxation theory is inadequate to explain the low field dispersion behavior of solutions of immobilized proteins or tissues, the implications obviously relating to the validity of previous analyses of molecular motion in these systems [6], [9], [10]. For example, Brown and Koenig proposed that the observed low-field dispersion of T1 and T1ρ of tissue water protons is unrelated to a specific correlation time but rather is due to a field dependence of magnetization transfer between water protons and solid-state broadened protein protons [6]. In any case, further data relating specific structural and/or chemical properties of various tissues and protein solutions with properties of water relaxation will be essential to clarify the contributing processes that lead to tissue water relaxation.
In this study, the T1, T2, and T1ρ dispersion of solvent protons in solutions of Bovine Serum Albumin (BSA) were evaluated in detail under conditions of varying crosslink density of proteins, pH, solvents, methylation of proteins, and B0 field strength. The results were compared with a similar evaluation of myocardial tissue and cartilage. In addition, the T1ρ dispersion profiles of both BSA solutions and tissue were analyzed for simple power law or BPP model characteristics. There were two aims: first, to provide data relating water relaxation in protein solutions and tissue to variations in macromolecular environment and structure, and second, to evaluate molecular models of tissue water relaxation using T1ρ dispersion analysis.
Materials and Methods
Experimental Preparation: BSA
Relaxation characteristics were studied using fraction V albumin, which is a mixture of different molecular weight BSA (Sigma Chemical), chromatographically purified monomer BSA (98% pure, Sigma), and dimer BSA (95% pure, Sigma).
Crosslinking
Variations in BSA crosslink density were produced by reacting 10% (1.5 mM) or 20% (3.0 mM) solutions of BSA with different amounts of glutaraldehyde (from 10 to 200 mM GA). In order to control for changes in small solute (i.e. various forms of unreacted GA), a series of BSA samples were reacted with GA, at concentrations of 10 mM to 60 mM, and then dialyzed (3.5 kDa cutoff) in excess distilled and deionized H2O. Grade I GA (50% aqueous solution of pure monomeric GA, stored at −20° C, Sigma) was used for all experiments at 4.7 Tesla and for experiments at 2 Tesla with BSA monomers and dialyzed samples. Grade II GA (25% aqueous solution of monomeric and small quantities of polymeric GA, 25° C, Sigma) was used for fraction V BSA experiments at 2 Tesla. Care was taken to maintain consistent reaction times (≥8 hrs for all experiments) before NMR measurements were obtained.
Validation
Samples of BSA reacted with varying quantities of GA were analyzed with polyacrylamide gel electrophoresis (PAGE) to document progressive increases in BSA molecular weight with increases in [GA]. A standard SDS (Sodium Dodecylsulfate) reducing buffer (Fig. 1a) or a non-denaturing buffer without SDS (Fig. 1b and 1c) was used. Note in lane 1 of figure 1a that purified monomer BSA migrates to a single band near 70 kDa. Lane 2 of figure 1a shows that BSA dimers can exhibit multiple bands. Lanes 4,5, and 7–10 clearly demonstrate that reactions with increasing quantities of GA resulted in the increase not only in the molecular weight of the largest species detected but also in the relative amount of larger to smaller species of BSA. Specifically, as [GA] increases from lane 4 to 10, the monomer band becomes fainter while higher molecular weight species (first BSA in the dimer range, then BSA between 250 and 300 kDa, then BSA polymers that cannot migrate past the 2% stacking gel) become stronger. Figure 1b demonstrates that non-crosslinked, fraction V BSA (lane 2) is composed of a mixture of albumin with different molecular weights unlike purified monomer BSA. The strongest band, however, migrated to a molecular weight of around 70 kDa, similar to monomer purified BSA. In contrast, the strongest bands in the crosslinked, fraction V BSA samples were above 200 kDa. Lanes containing methylated BSA will be described below in Results.
Figure 1. Analysis of bovine serum albumin (BSA) samples by polyacrylamide gel electrophoresis (PAGE).

Gels are 7.5% polyacrylamide (PA) with stacking gel of 2% PA and stained with Coomassie Blue. (a) SDS-PAGE: Samples were incubated at 95° C in denaturing sample buffer (containing β-mercaptoethanol and SDS), and each lane was loaded with 10 mg of protein and run with SDS in the buffer. Lane 1: BSA monomer; lane 2: BSA dimer; lane 3: methylated BSA (Sigma Chemicals); lanes 4, 5, 7, 8, 9, 10 contain 10% BSA monomers crosslinked with increasing [GA]: 0.1%, 0.2%, 0.3%, 0.4%, 0.5%, 0.6% GA respectively; lane 6: molecular weight marker (229,126,80,48 kDa). (b) Non-denaturing PAGE. Lane 1: molecular weight marker (85, 50, 35 kDa); lane 2,3,4 are fraction V BSA with 0.0%, 0.4%, and 0.5% GA respectively. (c) Non-denaturing PAGE: Lanes 1–3: methylated BSA (Sigma Chemicals) with decreasing amounts (5, 2.5 and 1.25 ug) of loaded protein; lane 4: molecular weight marker (200, 116, 97, 66, 55 kDa); lanes 5–7: 10% fraction V BSA reacted with 0.4% GA in decreasing amounts (5, 2.5, and 1.25 ug) of protein loaded. See text for details.
pH dependence
Since pH can significantly affect proton chemical exchange rates [11], a subset of BSA relaxation measurements were performed at both pH 5.5 and 7.0. After hydrochloric acid (1 N) was added to the BSA solutions, pH was measured at room temperature using a Mettler pH meter.
DMSO solvent
The importance of chemical exchange effects on relaxation may be studied by substituting dimethyl sulfoxide (DMSO) solvent for water since DMSO does not have exchangeable protons. A 10% solution of crosslinked BSA (60 mM GA) was dialyzed (3.5 kDa cutoff) twice against excess DMSO (ACS Reagent, Sigma) at room temperature for 24 hours each. Both the resultant dialysate as well as the original BSA in water solvent were analyzed. The presence of the methyl proton of DMSO and the absence of observable water resonance was confirmed on 1H NMR spectra of the dialysate.
Methylation
The methylation of BSA in this study refers to the methyl esterification of the carboxyl groups on the BSA molecule (CH3-O-BSA). The lyophilized form of methylated BSA (Sigma Chemicals) was dissolved in H2O (10% w/v) resulting in a clear solution with neutral pH. Deuterated methyl ester of BSA was synthesized by reacting fraction V BSA with deuterated methanol (Aldrich) (CD3OD, 99.8 atom % D) following the protocol of Fraenkel-Conrat [12]. As a control for the synthesis, unlabeled methyl ester of BSA (CH3-O-BSA) was synthesized in the same manner except unlabeled methanol (CH3OH) was used and then compared to the purchased form of methylated BSA.
Experimental Preparation: Tissues
Myocardium
A 3.5 kg New Zealand White rabbit and a 400 g Sprague-Dawley rat were anesthetized with intravenous sodium pentobarbital (c.a. 50 mg/kg) or diethyl ether respectively. The hearts were rapidly excised and then arrested in cold (4°C) cardioplegic solution containing in mM: NaCl 110, NaHCO3 10, KCl 16, MgCl2 16, and CaCl2 1.2. The posterior papillary muscle of the rabbit left ventricle was then quickly excised keeping the majority of the covering intimal layer intact. It was then dabbed dry and placed in a parafilm-sealed glass tube. After the left ventricular free wall of the rat heart was equilibrated in excess saline (4°C), it was also dabbed dry and placed in a sealed glass tube.
Cartilage
Five cubes (4 mm) were cut from a disk of calf patella cartilage which was stored in saline at −20°C. The cubes were thawed, padded dry and equilibrated overnight at room temperature with an excess of one of three different solvents: normal saline (0.9% NaCl in H2O), phosphate buffered solutions (100 mM KH2PO4/K2HPO4 at pH 9.2, 7.0 and 4.4), and DMSO. After equilibration, the cubes were padded dry and sealed in a glass tube for NMR measurements.
NMR Measurements
Relaxation measurements at 2 T or 4.7 T were obtained at room temperature with the sample inside a parafilm-sealed 5 mm diameter spherical glass vial using a 4-turn (6 mm diameter) solenoid RF coil. The size and shape of the samples and coils were designed to minimize the spectral linewidth, as well as the RF power required so that the largest range of B1 values could be studied. T1ρ was measured with solvent proton (water or DMSO) on resonance and linewidth less than 30 Hz for all protein samples. Solvent proton linewidth was less than 110 Hz for the tissue samples. The T1ρ pulse sequence consisted of a hard 90° pulse (15 to 50 µs), a 10 µs delay followed by a spinlock pulse on resonance with a 90° phase offset, and then a 100 µs delay and acquisition. The sequence was repeated with step changes in spinlock pulse duration (from 5 msec to approximately 4-fold T2). T1 was measured using inversion recovery, and T2 was measured using single Hahn spin echoes. At least 11 step changes were used for all T2 and T1ρ measurements, and at least 21 step changes for all T1 measurements. As T2 values varied widely, the upper range of TE also varied and was individually adjusted depending on the signal received (TE ranged from 2 msec to approximately 1–2 fold T2). For T1, T2, and T1ρ measurements, the repetition time (TR) was always at least 5×T1.
Data Analysis
T2 and T1ρ relaxation curves were first plotted in semi-log scale to determine the presence of non-single exponential behavior. Relaxation data of all samples except for rabbit papillary muscle appeared single exponential within the time resolution of the NMR experiment. T1, T2, and T1ρ relaxation times were then obtained by fitting magnitudes of spectral peaks to two-parameter single exponential functions. All relaxation time values were calculated from a one-time measurement. The T1ρ dispersion data of both BSA and tissue samples were analyzed for simple power law or BPP model characteristics using equations of the form:
| (1) |
| (2) |
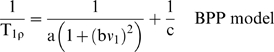 |
(3) |
where ν1 = γB1/2π, and a, b, and c are dispersion parameters whose values are determined by the fitting algorithm. The relaxation times and dispersion parameters were obtained via non-linear least squares fit of the data using the Marquardt-Levenberg algorithm (IDL, Research Systems, Inc.). Convergence occurred when the relative decrease in chi-square between iterations was smaller than 0.01%. Up to 100 iterations were performed before determining a failure to converge.
Results
Table 1 summarizes the relaxation times and T1ρ dispersion characteristics for all samples. The standard error of the estimate for the relaxation time curvefits were on average less than 1.0% of the calculated relaxation time values for all protein samples and less than 2.2% for tissue samples. Unless specifically reported, pH was not measured.
Table 1. Solvent Relaxation Parameters in Various Protein Solutions and Tissues.
| Sample Group | Sample Description | T1 (ms) | T2 (ms) | T2/T1 | % change in T1ρ (1–30 kHz) | ||
| 1–5 kHz | 5–10 kHz | 10–30 kHz | |||||
| BSA (Fraction V) in H2O (2T) | 10% BSA, 0mM GA, pH 5.5 | 1650 | 473.2 | 0.287 | 83.29 | 6.72 | 9.99 |
| 10% BSA, 0mM GA, pH 7.0 | 1222 | 338.0 | 0.277 | 85.78 | 6.06 | 8.16 | |
| 10% BSA, 20mM, GA | 1535 | 392.0 | 0.255 | 69.70 | 8.95 | 21.36 | |
| 10% BSA, 40mM GA | 1632 | 347.0 | 0.213 | 52.94 | 10.80 | 36.25 | |
| 10% BSA, 80mM GA | 1520 | 167.6 | 0.110 | 17.23 | 22.91 | 59.86 | |
| 10% BSA, 100mM GA | 1557 | 136.5 | 0.088 | 25.49 | 21.22 | 53.29 | |
| 20% BSA, 200mM GA | 766 | 58.7 | 0.077 | 17.96 | 23.78 | 58.26 | |
| BSA (Monomers) in H2O (2T) | 10% BSA, 0mM GA | 1873 | 256.8 | 0.137 | 64.32 | 13.11 | 22.57 |
| 10% BSA, 20mM GA | 1793 | 249.0 | 0.139 | 81.74 | 10.10 | 8.16 | |
| 10% BSA, 40mM GA | 1746 | 229.0 | 0.131 | 44.65 | 17.20 | 38.16 | |
| 10% BSA, 60mM GA | 1656 | 206.7 | 0.125 | 14.54 | 18.02 | 67.44 | |
| 10% BSA, 80mM GA | 1559 | 155.4 | 0.100 | 13.32 | 22.53 | 64.15 | |
| 10% BSA, 100mM GA | 1617 | 136.8 | 0.085 | 20.07 | 22.02 | 57.90 | |
| 10% BSA dimer | 1674 | 275.5 | 0.165 | 74.05 | 16.99 | 8.96 | |
| BSA (Fraction V) in H2O (4.7T) | 10% BSA, 0mM GA | 1767 | 233.1 | 0.132 | 63.63 | 21.89 | 14.48 |
| 10% BSA, 20mM GA | 1715 | 257.0 | 0.150 | 56.84 | 18.78 | 24.38 | |
| 10% BSA, 40mM GA | 1695 | 204.0 | 0.120 | 25.93 | 18.47 | 55.60 | |
| 10% BSA, 60mM GA | 1703 | 168.0 | 0.099 | 21.49 | 18.78 | 59.73 | |
| 10% BSA, 80mM GA | 1833 | 109.3 | 0.060 | 21.44 | 20.89 | 57.67 | |
| 10% BSA, 200mM GA | 1852 | 82.6 | 0.045 | 20.55 | 19.03 | 60.42 | |
| 10% BSA (Fraction V), 60mM GA (2T) | undialyzed | 1453 | 146.3 | 0.101 | 19.95 | 20.89 | 59.16 |
| dialyzed in DMSO | 1121 | 54.0 | 0.048 | 12.42 | 21.43 | 66.15 | |
| dialyzed in H2O | 1542 | 52.4 | 0.034 | 23.43 | 22.65 | 53.92 | |
| 10% BSA (methylated) (2T) | methylated BSA (Sigma) | 1999 | 190.3 | 0.095 | 42.38 | 17.59 | 40.04 |
| methylated BSA (synthesized) | 2080 | 275.3 | 0.132 | 47.13 | 14.27 | 38.60 | |
| 2H-methylated BSA (synthesized) | 2313 | 272.8 | 0.118 | 41.84 | 11.91 | 46.24 | |
| 4.7T | rabbit myocardium | 1396 | 44.0 | 0.032 | 26.80 | 20.83 | 52.37 |
| 2T | rat myocardium | 1131 | 50.5 | 0.045 | 19.84 | 20.37 | 59.79 |
| Cartilage (2T) | in saline | 847 | 64.2 | 0.076 | 26.10 | 24.61 | 49.29 |
| in DMSO | 351 | 6.4 | 0.018 | 11.19 | 20.56 | 68.25 | |
| in phosphate buffer pH 9.2 | 1047 | 27.7 | 0.026 | 18.48 | 21.98 | 59.54 | |
| in phosphate buffer pH 7.0 | 987 | 48.1 | 0.049 | 26.25 | 24.40 | 49.35 | |
| in phosphate buffer pH 4.35 | 953 | 40.3 | 0.042 | 23.33 | 26.11 | 50.56 | |
BSA = Bovine serum albumin, GA = Glutaraldehyde, DMSO = Dimethyl sulfoxide.
BSA Samples
Figure 2a demonstrates two distinct patterns of T1ρ dispersion for fraction V BSA samples at 2 Tesla. In the absence of crosslinking (0% GA), T1ρ sharply increased for γB1 from 1 to 5 kHz (21% and 34% increase in T1ρ values for acidic and neutral samples respectively) and then quickly plateaued beyond 10 kHz (<3% increase up to 60 kHz for both acidic and neutral samples). High concentrations of GA (≥80 mM), in contrast, led to smooth and monotonically increasing T1ρ values from 1 to 60 kHz. For example, T1ρ increased 25% from 1 to 5 kHz and 53% from 10 to 60 kHz for the sample reacted with 80 mM GA, and the dispersion curves no longer displayed an acute transition zone near 5 kHz. Since BSA reacted with 80 mM GA was a homogeneous liquid and BSA reacted with 100 mM GA was a gel, data in Fig. 2a also demonstrate that gelation, by itself, has little effect on T1ρ dispersion from 1 to 60 kHz.
Figure 2. T1ρ dispersion characteristics of various protein solutions.

(a) T1ρ of water protons in solutions of 10% BSA (fraction V) versus B1 field strength at various glutaraldehyde (GA) concentrations. Samples were evaluated at 2T. Uncrosslinked BSA samples were studied at pH 5.5 and 7.0. The data points for 80 and 100 mM GA were fitted to the relaxation-time power law of Eq. [1]. (b) T2 measurements were incorporated into the data of panel a to show T2/T1ρ ratios as a function of B1 field strength. See text for details.
Figure 2b shows the T2/T1ρ ratio as a function of γB1 for the same samples as in Fig. 2a. Note that this ratio approaches unity at 1 kHz γB1 as [GA] increases, indicating that highly crosslinked BSA has minimal dispersion (i.e. T1ρ changes little with B1) below 1 kHz. Uncrosslinked samples, on the other hand, had ratios significantly less than 1, pointing to significant dispersion below 1 kHz. The maximum difference in T1ρ between the acidic and neutral uncrosslinked samples occurred at zero field (measured as T2) where the T2 of the acidic sample was 40% above that of the neutral sample.
Figure 3 shows T1ρ dispersion plots of purified monomers (or dimer) of BSA rather than fraction V BSA. T1ρ values for native (0% GA) purified BSA dimers nearly coincided with those of monomers reacted with a low concentration of GA (20 mM) and showed only subtle changes compared to uncrosslinked BSA monomers (see also Table 1). Similar to the dispersion plots of fraction V BSA, purified monomers of BSA treated with increasing [GA] up to 60 mM showed increasing T1ρ dispersion for γB1 beyond 5 kHz. Increasing [GA] above 60 mM did not significantly change the dispersion characteristics of crosslinked monomers of BSA.
Figure 3. T1ρ dispersion characteristics of BSA solutions derived from purified monomers.

T1ρ of water protons in solutions of 10% BSA (purified monomers) versus B1 field strength at various glutaraldehyde (GA) concentrations evaluated at 2T. Data from uncrosslinked purified BSA dimers are also shown. The solid lines represent the fit to the relaxation-time power law of Eq. [1].
Figure 4 plots the T2/T1 ratio of BSA samples reacted with varying concentration of GA at both 2.0 T and 4.7 T and for dialyzed samples. Although T2/T1 ratios were found to generally decrease with increasing [GA], there was a transition zone between 40 and 60 mM GA where most of the changes in the T2/T1 ratios occurred. This range of [GA] was also the transition zone for changes in the shape of T1ρ dispersion seen in figures 2 and 3. T2/T1 ratios for crosslinked BSA samples dialyzed in excess H2O to remove the possible effects of unreacted GA showed a similar dependence on [GA] as undialyzed samples although the transition zone was narrower. T2/T1 ratios and T1ρ dispersion of BSA samples obtained at 4.7 T demonstrated a similar dependence on [GA] as those obtained at 2 T (see Table 1).
Figure 4. T2/T1 ratio of BSA solutions reacted with various concentrations of glutaraldehyde.

All samples contain 10% BSA (fraction V), except for one sample (*) with 20% BSA. Measurements were performed at both 2 and 4.7 T. See text for details.
Methylation
Figure 5 compares the T1ρ dispersion of methylated BSA with crosslinked (60 mM GA) and uncrosslinked BSA. Dispersion curves were normalized to the respective T1ρ values at the maximum frequency studied (23 kHz) in order to allow direct comparison of dispersion shapes. Figure 5 shows that above 5 kHz γB1 methylated BSA had nearly the same T1ρ dispersion as crosslinked BSA. Below 5 kHz, the dispersion of methylated BSA resembled uncrosslinked BSA. These results were consistent, independent of whether the methyl groups were protonated or 99% deuterated. Deuteron labeling of the methyl groups of methylated BSA led to a minor increase in T1 (2313 vs. 2080 ms) and no change in T2 values (273 vs. 275 ms) compared with 1H-methylated BSA (Table 1). The T1ρ dispersion of methylated BSA synthesized in the same manner as 2H-methylated BSA (see Methods) is shown as a control. The similarity of T1ρ dispersion of methylated BSA and crosslinked BSA should be interpreted in light of our results in Fig. 1c, which shows that methylated BSA tends to form large aggregates in aqueous solutions (non-denaturing PAGE analysis). Absence of covalent bonding in these aggregates is evidenced by the monomeric appearance of the methylated BSA in denaturing PAGE (Fig. 1a, lane 3).
Figure 5. Normalized T1ρ dispersion plots of methylated BSA solutions.

Samples were evaluated at 2T. Plots of native and crosslinked BSA (fraction V) are also shown for comparison. Note the similarity of T1ρ dispersion of methylated BSA with crosslinked BSA above 5 kHz, and the similarity with native BSA below 5 kHz.
Tissues
Figure 6a shows the T2/T1ρ ratio as a function of γB1 for cartilage samples as well as for rat myocardial tissue. Note the similarity of these curves with crosslinked BSA in Fig. 2b. Tissue samples uniformly showed smooth monotonically increasing T1ρ over the entire frequency range studied. For instance, of the total T1ρ dispersion seen from 1 to 30 kHz for cartilage (pH 7.0, 2 T), 26% occurred between 1 and 5 kHz, 24% between 5 and 10 kHz, and 49% between 10 and 30 kHz. The frequency breakdown in T1ρ dispersion for crosslinked BSA (10% solution, 100 mM GA) was remarkably similar (25%, 21%, and 53% respectively) as compared to uncrosslinked BSA where generally over 80% of the T1ρ dispersion occurring from 1 to 30 kHz occurred below 5 kHz (see Table 1). Changes in solvent pH had minor effects on cartilage T1ρ dispersion, although the T2/T1ρ ratio at 1 kHz γB1 for the acidic and basic samples were closer to unity than the neutral sample, indicating less dispersion below 1 kHz for these samples.
Figure 6. T2/T1ρ ratio as a function of B1 field strength (a) and normalized T1ρ dispersion plots (b) of various tissue samples.

Calf patella cartilage at various pH and rat myocardium samples were evaluated at 2T. Plots of cartilage and crosslinked BSA (fraction V) in DMSO solvent are also shown for comparison in panel b. See text for details.
To compare the T1ρ dispersion characteristics of cartilage in water solvent to cartilage in DMSO solvent, normalized dispersion curves are demonstrated in Fig. 6b. For γB1 above 5 kHz, the methyl protons of DMSO showed a remarkably similar dispersion curve to that of water protons. Below 5 kHz, cartilage in DMSO displayed minimal T1ρ dispersion reaching nearly zero slope below 2 kHz. The dispersion curve of crosslinked BSA (10% solution, 60 mM GA) in DMSO nearly coincided with the dispersion curve of cartilage in DMSO from 1 to 33 kHz.
Dispersion Modeling
As a preliminary test for power law behavior, T1ρ 2 was plotted against γB1 for various BSA and tissue samples. Figure 7 demonstrates a near linear relationship between T1ρ 2 and γB1 for crosslinked BSA, which did not exist for uncrosslinked BSA. Near linear relationships were also observed for the water protons of rabbit myocardial tissue and the methyl protons of DMSO equilibrated in cartilage. Although some non-linear behavior was present for crosslinked BSA and tissues, especially at lower frequencies, perfect linearity was not expected since it would require the power law exponent to be exactly ½ and the frequency independent component to be negligible.
Figure 7. Values of T1ρ 2 are plotted against B1 field strength for various samples.

(a) Crosslinked BSA (fraction V) in H2O. (b) Native BSA in H2O. (c) Rabbit myocardial tissue. (d) Cartilage in DMSO solvent. See text for details.
Figure 8 demonstrates the fits of Eq. [1–3] to representative crosslinked BSA and tissue samples. Table 2 shows that the standard error of the estimate of the fit expressed as a percentage of the T1ρ value (% SEE) for crosslinked BSA (GA≥60 mM) and tissue samples was 2.2±1.1% for the relaxation-time power law Eq. [1] compared to 10.5±4.8% for the relaxation-rate power law Eq. [2] and 4.0±2.3% for the BPP model Eq. [3]. The improved fit of T1ρ dispersion using the relaxation-time power law was found to be statistically significant (P<0.005 from analysis of variance with Bonferroni correction for both comparisons [13]). Table 2 lists the fitted values of the parameters (a, b, c) for all samples which were successfully fitted to Eq. [1].
Figure 8. Plots of T1ρ vs. B1 field strength are shown for two tissue and two crosslinked BSA samples (fraction V).

The lines drawn were fitted using Eqs. [1–3]. Note that relaxation-time power law (Eq. [1]) appears to best fit the data. See text for details.
Table 2. T1ρ Dispersion Analysis of Crosslinked BSA and Tissues.
| γB1 Values (kHz) | Model Curve Fit (% Standard Error of the Estimate) | Relaxation-Time Power Law (Eq. [1]) Parameters of Fit | ||||||||
| Sample Groups | Samples | Number of values | Minimum | Maximum | Relaxation-Time Power Law Eq. [1] | Relaxation-Rate Power Law Eq. [2] | BPP Model Eq. [3] | “a” (ms) | “b” (10−6×sec1+C) | “c” (exponent) |
| BSA (Fraction V) in H2O (2T) | 10% BSA, 80mM GA | 13 | 0.97 | 60.0 | 2.4 | 6.8 | 2.9 | 138 | 764 | 0.515 |
| 10% BSA, 100mM GA | 13 | 1.01 | 59.2 | 2.1 | 5.3 | 5.0 | 93.8 | 2247 | 0.425 | |
| 20% BSA, 200mM GA | 22 | 0.14 | 33.1 | 2.3 | 12 | 3.3 | 45.7 | 31.1 | 0.731 | |
| BSA (Monomers) in H2O (2T) | 10% BSA, 60mM GA | 13 | 1.50 | 33.1 | 0.4 | 9.7 | 0.9 | 293 | 3.10 | 0.993 |
| 10% BSA, 80mM GA | 13 | 1.50 | 33.1 | 0.8 | 17 | 1.5 | 187 | 21.7 | 0.833 | |
| 10% BSA, 100mM GA | 13 | 1.50 | 33.1 | 0.9 | 4.0 | 1.9 | 133 | 298 | 0.596 | |
| BSA (Fraction V) in H2O (4.7T) | 10% BSA, 60mM GA | 23 | 1.05 | 154 | 1.2 | 10 | 7.3 | 155 | 334 | 0.591 |
| 10% BSA, 80mM GA | 19 | 1.50 | 100 | 2.0 | 4.7 | 4.5 | 81.8 | 2464 | 0.411 | |
| 10% BSA, 200mM GA | 23 | 1.00 | 154 | 3.0 | 9.4 | 9.5 | 64.2 | 785 | 0.501 | |
| 10% BSA (Fraction V), 60mM GA (2T) | undialyzed | 23 | 0.10 | 33.1 | 1.2 | 8.2 | 1.9 | 156 | 42.7 | 0.757 |
| dialyzed in excess DMSO | 23 | 0.10 | 33.1 | 2.7 | 18 | 1.8 | 53.2 | 3.98 | 0.964 | |
| dialyzed in H2O | 15 | 0.79 | 22.8 | 1.4 | 4.8 | 2.8 | 91.9 | 466 | 0.577 | |
| 4.7T | rabbit myocardial tissue | 19 | 0.59 | 47.6 | 2.0 | 7.2 | 8.4 | 74.5 | 398 | 0.569 |
| 2T | rat myocardial tissue | 22 | 0.10 | 28.5 | 1.2 | 12 | 3.4 | 68.4 | 26.9 | 0.786 |
| Cartilage (2T) | in saline | 23 | 0.21 | 46.0 | 1.7 | 6.0 | 2.9 | 87.4 | 509 | 0.484 |
| in DMSO | 24 | 0.21 | 56.0 | 3.6 | 17 | 3.9 | 9.06 | 5.87 | 0.756 | |
| in phosphate buffer pH 9.2 | 21 | 0.20 | 33.1 | 3.9 | 17 | 4.2 | 26.9 | 25.8 | 0.769 | |
| in phosphate buffer pH 7.0 | 23 | 0.10 | 33.1 | 3.3 | 13 | 4.1 | 51.0 | 224 | 0.591 | |
| in phosphate buffer pH 4.35 | 23 | 0.10 | 33.1 | 4.1 | 16 | 4.7 | 39.6 | 133 | 0.635 | |
BSA = Bovine serum albumin, GA = Glutaraldehyde, DMSO = Dimethyl sulfoxide.
Discussion
Low Field Dispersion Behavior
In this study we have obtained low field relaxation data of protein solutions and tissue under varying conditions. We show that the T1ρ dispersion profiles of native BSA solutions are clearly distinct from that of crosslinked BSA. Above 5 kHz γB1 the T1ρ dispersion of 10% native BSA was essentially flat. This result is similar to that of Zhou and Bryant [5] and Koenig and Brown [7] who found minimal T1 dispersion of native BSA from 10 to 100 kHz. Below 5 kHz, we found a steep dispersion profile (i.e. T1ρ changed rapidly with B1). Our measured ratio of T2 to T1ρ at 1 kHz for native BSA (Fig. 2b) also suggested continued dispersion below 1 kHz. This sharp dispersion from 0 to 5 kHz was sensitive to changes in solution pH and likely is due to chemical exchange of the water protons with the ionizable protons of the protein. As far as we are aware, only Virta et al. [10] and Mäkelä el al. [14] have also measured the T1ρ of native BSA below 10 kHz. Although Virta et al. observed insignificant T1ρ dispersion, only two T1ρ data points were obtained below 5 kHz. In contrast, Mäkelä et al. demonstrated similar findings to the current study in that native BSA solutions showed significant T1ρ dispersion below 5 kHz and were strongly affected by pH.
For our experimental conditions, at least 60 mM GA was required to alter the BSA T1ρ dispersion to match the smooth monotonically increasing profile that was seen for tissues. Although 40 mM GA was sufficient to form ≈300 kDa BSA oligomers, 60 mM GA formed an additional species of BSA polymers that were unable to migrate through the pores of the 2% stacking gel (Fig. 1a) suggesting at least an order of magnitude increase in molecular weight for this band. The formation of these large BSA polymers was associated with significant T1ρ dispersion above 5 kHz as well as an abrupt change in the T2/T1 ratio (fig. 4). This result indicates that a high degree of immobilization is required for protein solutions to accurately model tissue. Not surprisingly, Gore and Brown [15] evaluating proteins with molecular weight range from 1.4 to 483 kDa, and Menon and Allen [16] assessing serum proteins from 69 to 725 kDa found these protein solutions to be poor models for tissue relaxation behavior. Increasing GA above 80 mM, which obviously led to macromolecular structural changes since gelation occurred between 80 and 100 mM GA, did not lead to further changes in dispersion profile (Fig. 2–3) or the T2/T1 ratio even with an increase in [BSA] to 20%. Apparently a plateau is reached whereby further increases in macromolecular crosslinking does not enhance relaxation. Our finding of a plateau for samples with GA/BSA mole ratios greater than 53 (80 mM GA, 10% BSA) is in contrast to the results of Zhou and Bryant [5]. They showed increases in T1 relaxation dispersion with increasing concentrations of GA with no sign of plateau even at a mole ratio of 256 (8.25% BSA). Since BSA polymerization is known to be highly sensitive to concentrations of BSA as well as GA [17], their results may relate to poor production of sufficiently large BSA polymers even at high GA concentrations.
It should be noted that the discussion, so far, assumes only intermolecular crosslinking of BSA is important in the observed changes in water relaxation. It is known that BSA is a rather rigid globular protein in its native state and that the addition of intramolecular crosslinks can warp or stiffen it only slightly [18]. Thus, intramolecular crosslinking of BSA is unlikely to affect protein motion significantly or lead to dipolar interactions that will substantially enhance relaxation.
Surprisingly, methylated BSA showed essentially the same T1ρ dispersion as that of crosslinked BSA above 5 kHz (Fig. 5). The mechanism of this low field relaxation, however, is clearly independent of dipole-dipole interactions of the methyl protons either directly by spin exchange with tightly bound solvent protons or indirectly by spin diffusion with the protein protons. Significant reduction in both the intra- and intermolecular dipolar interactions of methyl protons by the substitution of deuterons for protons had essentially no effect on the T1ρ dispersion profile. Rather, the similarity of T1ρ dispersion of methylated BSA and crosslinked BSA can be explained by Fig. 1c. which shows that methylated BSA tends to form large aggregates in aqueous solutions (non-denaturing PAGE analysis). Presumably, the addition of methyl ester side groups to BSA allows nonspecific intermolecular binding of BSA monomers which, similar to crosslinking BSA, slows macromolecular tumbling below a critical threshold. The absence of covalent bonding in these aggregates is evidenced by the single monomer band of methylated BSA in denaturing PAGE (Fig. 1a). Since the methylated BSA solutions contain mixtures of different sized aggregates (Fig. 1c), the sharp dispersion below 5 kHz can be accounted for by the presence of methylated BSA monomers and small aggregates that behave similar to native BSA, and the continued dispersion above 5 kHz is due to large aggregates which are functionally “immobilized” and behave similarly to crosslinked BSA. Thus, the methylated BSA data provide further evidence that differences in the relaxation properties of native and crosslinked BSA are a consequence of the increase in the polymerization of BSA, rather than other effects of GA, such as its attachment as a chemical side group to BSA. Although motion of proton containing side groups, such as methyl groups have been suggested to provide significant relaxation sinks for large proteins [19], [20], their high mobility [19], [21] implies that no significant enhancement of relaxation can be expected below 100 MHz much less in the kHz regime of T1ρ [20].
Modeling
For tissue and solutions of sufficiently crosslinked BSA, significant T1ρ dispersion was seen for the entire range of γB1 studied (up to 150 kHz for some samples, Table 2). Our plots of T1ρ versus γB1 were remarkably similar to the plots of T1 versus γB0 shown by Bottomley et al. [22] for many tissues. Specifically, both T1ρ and T1 dispersion profiles showed a weak field dependence, which was distinct from the T1∝ν2 relationship expected for magnetic dipolar interactions in simple homogeneous systems (BPP model, see Eq. [3]). Although, the BPP equation can present a concave-down frequency relationship over a local range (Fig. 8), the same relationship cannot occur over an extended range from kHz to hundreds of MHz—5 decades of frequencies from T1ρ to T1—unless multiple or continuously distributed correlation times are assumed [8], [23], [24].
Our T1ρ dispersion profiles for crosslinked BSA and tissue from 1–100 kHz displayed continuous, monotonic increases with frequency that did not suggest obvious inflections. Likewise, T1 tissue dispersion curves from 1–100 MHz summarized by Bottomley et al. [22] and crosslinked BSA dispersion curves from 10 kHz to ∼100 MHz shown by Koenig and Brown [4] (notwithstanding small, local changes in dispersion due to 14N-1H quadrupole dips) do not show obvious inflections. Similar to Bottomley et al. [22] who found an excellent fit to T1 dispersion using the relationship T1 = AνB, the T1ρ dispersion data fit well to the simple relaxation-time power law, T1ρ = a+bνc (Eq. [1]), where parameter “a” was added to account for the zero-field offset, T1ρ(0) = T2. Not only did this equation present a significantly improved fit to the data compared to the relaxation-rate power law (Eq. [2]) and the BPP model (Eq. [3]), it was able to provide a calculated T1ρ(0) value that closely approximated T2. Specifically, the T1ρ(0)/T2 ratio was near unity (1.10±0.32) for the relaxation-time power law, whereas it was significantly higher (P<0.001) for the BPP model (1.40±0.40).
The relaxation-rate power law (Eq. [2]) is similar to the Escanye et al. [23] expression 1/T1 = Aν−1/2+B. This expression was found to adequately fit T1 dispersion of mouse muscle from 7–90 MHz and has the advantage that it can be easily interpreted mechanistically as a fast-exchange two-state model. However, this expression cannot account for properties of relaxation at or near zero field where it predicts T1(0) to be zero. The relaxation-rate power law demonstrated a poor fit to our T1ρ dispersion data.
The exponent “c” in the relaxation-time power law was calculated to be 0.66±0.20, 0.68±0.15, and 0.66±0.12 for crosslinked BSA (GA≥60 mM), myocardial tissue, and cartilage, respectively. Neglecting the effects of the T1ρ(0) offset, these values are higher than the exponent reported by Bottomley et al. [22] for water proton T1 dispersion of skeletal muscle (0.42) and heart muscle (0.36). These values, however, are near the exponent reported by Kimmich et al. [20] for 1H T1 dispersion of either lyophilized or minimally D2O-hydrated (16% by weight) proteins and polypeptides (0.74±0.06). Potential relaxation models that account for simple power law field dependence are discussed below.
Relaxation Mechanisms
Protein-associated water
Nearly all models of water relaxation in macromolecular systems consider one or more new groups of protein-associated water with altered motion that contributes to bulk water relaxation. For example, “hydration layers” at the macromolecular interface have been proposed with increased correlation times in order to explain the dispersion data [23]. In the case of T1ρ relaxation data, invariably an additional correlation time is added to the model to account for the low-field regime [8]. However, observations obtained by high-resolution NMR spectroscopy of proteins [25], relaxation dispersion of water 17O [26], and paramagnetic spin labeling [27], strongly suggest that surface hydration water is highly mobile with sub-nanosecond residence times. Thus, it is unlikely that models based on distributions of surface water with restricted motional characteristics or the “exchange diffusion” of water molecules to and from a bound hydration layer can explain the relaxation dispersion of protein solutions or tissue.
Later models have focused on a small number of water molecules buried inside proteins, which are clearly distinguished from surface hydration water by their longer residence times [25]. Denisov and Halle [26] report that the internal water molecules of the globular protein, bovine pancreatic Trypsin inhibitor (BPTI), have residence times (τRES) on the order of 10−8 to 10−6 seconds, whereas the water molecules on the surface of the protein have an average reorientational correlation time of approximately 20 picoseconds. By studying the relaxation behavior of water 17O nuclei, the complicating effects of cross-relaxation and hydrogen exchange were avoided, and they postulate that the origin of the water 17O relaxation dispersion of BPTI solutions can be explained by a small number of interior water molecules exchanging with bulk water on the submicrosecond time-scale. Although a consensus view is still lacking, our experimental data will be examined considering this model of protein hydration.
Dilute globular protein solutions
Similar to our T1ρ data in native and crosslinked BSA solutions, several investigators have shown that the T1 dispersion profiles of dilute globular protein solutions are clearly distinct from those of immobilized protein solutions [5], [7]. At least for mobile protein solutions, the dispersion relation is generally Lorentzian, and the dispersion inflection frequency of water 1H, 2H, and 17O nuclei has been shown to correspond to τR, the rotational correlation time of the protein molecule [26], [28], [29]. Thus, the conventional BPP model along with the condition of motional narrowing (ω1τc<<1) is apparently applicable in these protein solutions as the effective correlation time of motion, τC, is easily identified with τR. As suggested by Venu et al [28], interior water molecules with residence times greater than τR (∼6 ns for BPTI) can sense the Brownian motion of the protein molecule, exchange with bulk water, and thereby contribute to the observed relaxation dispersion. The intrinsic relaxation rate of these buried relaxation sinks was explained quantitatively by intramolecular dipole couplings (∼70%) and many intermolecular dipole couplings with BPTI protons (∼30%). Labile protein protons were also thought to make a significant contribution to the observed water relaxation rate. Contributions from direct nuclear Overhauser effect (NOE) cross-relaxation between protein protons and interior or surface water protons were found to be negligible, which is not surprising given the motional narrowing condition [28].
Irrespective of the actual mechanism, the relation ω1τc<<1 for the spinlock experiment predicts an essentially flat 1H T1ρ dispersion below ∼1 MHz. As suggested by Hills [30], proton exchange then becomes the remaining relaxation mechanism that is operative in the low-field regime. Our results (Fig. 2–3) show that this is indeed the case. Native BSA solutions exhibited a sharp dispersion profile below 5 kHz that was sensitive to changes in pH and showed a flat dispersion above 5 kHz. Furthermore, the active dispersion range was consistent with the intrinsic proton exchange rates (700–10,000 s−1) measured by Liepinsh and Otting from OH and NH groups of several amino acid side chains under physiologic conditions [31]. Our results, therefore, are consistent with the theoretical T1ρ dispersion of dilute globular protein solutions proposed by Hills [30].
Immobilized proteins and tissues
Rotational immobilization of solute protein can be achieved by a chemical [3] or thermal crosslinking reaction [10] or by non-covalent interactions at high (>50% w/w) protein concentrations [9]. For such solutions and biological tissues, it is assumed that the dispersion inflection frequency no longer reflects protein rotation but instead the residence times of long-lived water molecules that are associated with the protein [32]. However, the dispersion curves are not simply scaled, Lorentzian profiles with shifted inflection frequencies, but are distinctly non-Lorentzian [9]. In addition, immobilized protein solutions also exhibit broader temperature T1 minimums which are characterized by lower T2/T1 ratios than expected by conventional BPP-theory unless a distribution of correlation times is assumed [33]. Although many investigators have incorporated various distributions of correlation times to model the non-Lorentzian T1 dispersion data [34], [35], it should be recognized that given enough variables, successful fitting of data can occur and may simply represent a convenient parameterization without physical meaning. Packer [36] noted that the weakest assumption of the approach incorporating distributions of correlation times is that all motional processes modulate the same magnetic dipolar interaction strength. A wider distribution of motional correlation times then will predict higher relaxation rates at high field than those observed [3]. Moreover, our finding that a power law relationship also holds for T1ρ dispersion from 1–100 kHz would imply an even larger distribution of correlation times in this model. The appeal of a BPP-type model is that it corresponds to a well-defined mechanism of relaxation, and thus, physically meaningful parameters such as correlation times of motion can be extracted from the relaxation dispersion data. Nevertheless, for immobilized protein solutions and tissues, calculation of mechanistic parameters using BPP-type models—with their inherent assumptions about the nature of the local interactions causing relaxation and the shape of the spectral density function—is likely erroneous.
Furthermore, unlike for mobile protein solutions, abundant evidence exists for direct NOE cross-relaxation between immobilized protein and solvent protons [37]. Bryant et al. [3] have suggested that the longitudinal relaxation of water protons in solutions of immobilized proteins and tissue is due to magnetic coupling of macromolecular protons with water protons and that the magnetic field dependence of the solid component could be transferred at least partially to the liquid component. The simple power law dispersion profiles found for solid protein protons has been explained by intrinsic motions characteristic of protein backbones by Kimmich and Winter [38]. Independent of the mechanism by which protein protons acquire their relaxation field dependence, Zhou and Bryant [5] have proposed that cross-relaxation could then allow “water spins to report a scaled replica” of the relaxation behavior of the solid system. Efficient coupling is required to allow cross-relaxation, and long-lived water molecules buried inside macromolecules (τRES up to 200 µs for BPTI [39]) could be an important pathway for the magnetization transfer. Long-lived hydration water in junction zones formed by protein crosslinking have also been postulated [40]. In considering this cross-relaxation model, we note that the similarity of our T1ρ dispersion profiles to that of published T1 profiles for immobilized proteins [20] and tissue [22] indicate a common relaxation mechanism is dominant across 5 decades of modulating field strength. The importance of this model is not settled, however, other relaxation models will need to consider dynamic processes that span this large range of frequencies.
An additional mechanism by which magnetization can be transferred from the solid phase to the solvent phase has been suggested by Hills [30]. A three-site model whereby spin diffusion in the solid phase allows spin exchange between non-exchangeable and exchangeable protein protons followed by proton exchange between water and exchangeable protein protons could provide a quantitative interpretation of the relaxation data without the need to invoke special hydration water. Our T1ρ dispersion profiles of the methyl protons of DMSO for cartilage and crosslinked BSA, however, show strong field dependence throughout the studied range (Fig. 6b). In fact, normalized dispersion profiles for the DMSO solvent samples were nearly identical to the dispersion profiles of the corresponding samples with water solvent above 5 kHz. Since the methyl protons of DMSO are not exchangeable, proton exchange is obviously not necessary for the strong field dependence of DMSO protons in immobilized protein solutions or tissue. In addition, the minor effect of solvent pH on water T1ρ dispersion of cartilage as compared to native BSA solutions suggests that protein immobilization attenuates the contribution of proton exchange to water relaxation (Fig. 2b & 6a). Thus, proton exchange appears to have a minor role on T1ρ dispersion in immobilized protein solutions and tissues above 5 kHz. Interestingly, Mäkelä el al. [14] using some similar sample preparations, drew nearly the opposite conclusion. Namely, they conclude that there is “…a crucial role of proton exchange on R1ρ and R1ρ dispersion in immoblilzed protein solution mimicking tissue relaxation properties.” However, we note that Mäkelä el al. assessed T1ρ over a much narrower range of γB1 (1–11 kHz), did not evaluate DMSO solvent samples, and did not study any tissues. The latter is particularly important, since Mäkelä el al. selectively focus on their results from heat-denatured rather than glutaraldehyde cross-linked BSA, postulating that glutaraldehyde treated BSA is a poor model for tissue, albeit without tissue data to support this supposition.
We also note that our T1ρ dispersion profiles of BSA and tissue samples showed no significant ω0 dependence between 86 and 200 MHz (2 T and 4.7 T). Therefore, exchange models that produce ω1 dependence because of a resonance offset, δω, between water and labile protein protons or between long-lived protein associated water and bulk water, cannot account for the T1ρ dispersions measured in this study.
The similarity of our DMSO and water solvent T1ρ dispersions implies similar molecular mechanisms for relaxation. Long-lived DMSO molecules, if present, should also have comparable residence times to that of water molecules. It is consistent then that Denisov and Halle [26] indicate that buried water molecules have long residence times due to the free energy cost of local protein unfolding rather than due to a full complement of strong hydrogen bonds. In addition, we note that DMSO solvent in crosslinked BSA and cartilage show nearly identical 1H-T1ρ dispersion (Fig. 6b), despite obvious differences in macromolecular content. This result suggests that buried solvent molecules, which presumably function as relaxation centers, lack sensitivity to details of macromolecular structure.
Thus, in summary, the data of the current study suggest the following relaxation mechanisms. For dilute globular proteins, the conventional BPP model appears to be applicable with the effective correlation time corresponding to protein rotation. Proton exchange is an important contributor to the observed water relaxation rate, whereas cross-relaxation between protein protons and water protons is negligible given the motional narrowing condition. For immobilized proteins and tissue, proton exchange appears to be a minor pathway for T1ρ and T1 relaxation above 5 kHz. The data are consistent with special water protons, perhaps located internally, that have enhanced relaxation. The relaxation of these special water protons is possibly due to cross-relaxation with immobilized protein protons, although intra- or intermolecular dipole interactions of these special water protons may also contribute. The smooth monotonic relaxation dispersion across 5 decades of frequencies (from T1ρ to T1) may or may not reflect the relaxation behavior of the solid system, but nonetheless, implies a failure of the simple BPP model.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: The authors have no support or funding to report.
References
- 1.Bloembergen N, Purcell EM, Pound RV. Relaxation effects in nuclear magnetic resonance absorption. Phys Rev. 1948;73:679–712. [Google Scholar]
- 2.Fullerton GD. Physiologic basis of magnetic relaxation. In: Stark DD, Bradley WG , editors. Magnetic Resonance Imaging. Vol 1, 2 edition. St. Louis: Mosby Year Book; 1980. pp. 88–108. [Google Scholar]
- 3.Bryant RG, Mendelson DA, Lester CC. The magnetic field dependence of proton spin relaxation in tissues. Magn Reson Med. 1991;21:117–126. doi: 10.1002/mrm.1910210114. [DOI] [PubMed] [Google Scholar]
- 4.Koenig SH, Brown RD., 3rd A molecular theory of relaxation and magnetization transfer: application to cross-linked BSA, a model for tissue. Magn Reson Med. 1993;30:685–695. doi: 10.1002/mrm.1910300606. [DOI] [PubMed] [Google Scholar]
- 5.Zhou D, Bryant RG. Magnetization transfer, cross-relaxation, and chemical exchange in rotationally immobilized protein gels. Magn Reson Med. 1994;32:725–732. doi: 10.1002/mrm.1910320607. [DOI] [PubMed] [Google Scholar]
- 6.Brown RD, 3rd, Koenig SH. 1/T1 rho and low-field 1/T1 of tissue water protons arise from magnetization transfer to macromolecular solid-state broadened lines. Magn Reson Med. 1992;28:145–152. doi: 10.1002/mrm.1910280115. [DOI] [PubMed] [Google Scholar]
- 7.Koenig SH, Brown RD, 3rd, Ugolini R. Magnetization transfer in cross-linked bovine serum albumin solutions at 200 MHz: a model for tissue. Magn Reson Med. 1993;29:311–316. doi: 10.1002/mrm.1910290306. [DOI] [PubMed] [Google Scholar]
- 8.Knispel RR, Thompson RT, Pintar MM. Dispersion of proton spin-lattice relaxation in tissues. J Magn Reson. 1974;14:44–51. [Google Scholar]
- 9.Lester CC, Bryant RG. Water-proton nuclear magnetic relaxation in heterogeneous systems: hydrated lysozyme results. Magn Reson Med. 1991;22:143–153. doi: 10.1002/mrm.1910220115. [DOI] [PubMed] [Google Scholar]
- 10.Virta A, Komu M, Kormano M. T1rho of protein solutions at very low fields: dependence on molecular weight, concentration, and structure. Magn Reson Med. 1997;37:53–57. doi: 10.1002/mrm.1910370109. [DOI] [PubMed] [Google Scholar]
- 11.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraenkel-Conrat H, Olcott HS. Esterification of proteins with alcohols of low molecular weight. J Biol Chem. 1945;161:259–268. [PubMed] [Google Scholar]
- 13.Glantz SA, Slinker BK. Primer of applied regression and analysis of variance. New York: McGraw-Hill, Health Professions Division; 1990. [Google Scholar]
- 14.Makela HI, Grohn OH, Kettunen MI, Kauppinen RA. Proton exchange as a relaxation mechanism for T1 in the rotating frame in native and immobilized protein solutions. Biochem Biophys Res Commun. 2001;289:813–818. doi: 10.1006/bbrc.2001.6058. [DOI] [PubMed] [Google Scholar]
- 15.Gore JC, Brown MS. Pathophysiological significance of relaxation. In: Partain CL, Price RR, Patton JA, Kulkarni MV, James AEJ, editors. Nuclear Magnetic Resonance Imaging - Physical Principles and Instrumentation. Vol 2. Philadelpia: Saunders; 1988. pp. 1070–1074. [Google Scholar]
- 16.Menon RS, Allen PS. Solvent proton relaxation of aqueous solutions of the serum proteins alpha 2-macroglobulin, fibrinogen, and albumin. Biophys J. 1990;57:389–396. doi: 10.1016/S0006-3495(90)82555-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luft JH. Fixation for biological ultrastructure. I. A viscometric analysis of the interaction between glutaraldehyde and bovine serum albumin. J Microsc. 1992;167:247–258. [PubMed] [Google Scholar]
- 18.Carter DC, He XM, Munson SH, Twigg PD, Gernert KM, et al. Three-dimensional structure of human serum albumin. Science. 1989;244:1195–1198. doi: 10.1126/science.2727704. [DOI] [PubMed] [Google Scholar]
- 19.Kalk A, Berendsen HJC. Proton magnetic relaxation and spin diffusion in proteins. J Magn Reson. 1976;24:343–366. [Google Scholar]
- 20.Kimmich R, Winter F, Nusser W, Spohn KH. Interactions and fluctuations deduced from proton field-cycling relaxation spectroscopy of polypeptides, DNA, muscles, and algae. J Magn Reson. 1986;68:263–282. [Google Scholar]
- 21.Shirley WM, Bryant RG. Proton-nuclear spin relaxation and molecular dynamics in the lysozyme-water system. J Am Chem Soc. 1982;104:2910–2918. [Google Scholar]
- 22.Bottomley PA, Foster TH, Argersinger RE, Pfeifer LM. A review of normal tissue hydrogen NMR relaxation times and relaxation mechanisms from 1–100 MHz: dependence on tissue type, NMR frequency, temperature, species, excision, and age. Med Phy. 1984;11:425–448. doi: 10.1118/1.595535. [DOI] [PubMed] [Google Scholar]
- 23.Escanye JM, Canet D, Robert J. Frequency dependence of water proton longitudinal nuclear magnetic relaxation times in mouse tissues at 20 degrees C. Biochim Biophys Acta. 1982;721:305–311. doi: 10.1016/0167-4889(82)90083-0. [DOI] [PubMed] [Google Scholar]
- 24.Fung BM, McGaughy TW. The state of water in muscle as studied by pulsed NMR. Biochim Biophys Acta. 1974;343:663–673. doi: 10.1016/0304-4165(74)90287-6. [DOI] [PubMed] [Google Scholar]
- 25.Otting G, Liepinsh E, Wuthrich K. Protein hydration in aqueous solution. Science. 1991;254:974–980. doi: 10.1126/science.1948083. [DOI] [PubMed] [Google Scholar]
- 26.Denisov VP, Halle B. Protein hydration dynamics in aqueous solution: a comparison of bovine pancreatic trypsin inhibitor and ubiquitin by oxygen-17 spin relaxation dispersion. J Mol Biol. 1995;245:682–697. doi: 10.1006/jmbi.1994.0055. [DOI] [PubMed] [Google Scholar]
- 27.Polnaszek CF, Bryant RG. Nitroxide radical induced solvent proton relaxation - measurement of localized translational diffusion. J Chem Phys. 1984;81:4038–4045. [Google Scholar]
- 28.Venu K, Denisov VP, Halle B. Water 1H magnetic relaxation dispersion in protein solutions. A quantitative assessment of internal hydration, proton exchange, and cross-relaxation. J Am Chem Soc. 1997;119:3122–3134. [Google Scholar]
- 29.Koenig SH, Brown RD, 3rd, Ugolini R. A unified view of relaxation in protein solutions and tissue, including hydration and magnetization transfer. Magn Reson Med. 1993;29:77–83. doi: 10.1002/mrm.1910290114. [DOI] [PubMed] [Google Scholar]
- 30.Hills BP. The proton exchange cross-relaxation model of water relaxation in biopolymer systems. Mol Phys. 1992;76:489–508. [Google Scholar]
- 31.Liepinsh E, Otting G. Proton exchange rates from amino acid side chains–implications for image contrast. Magn Reson Med. 1996;35:30–42. doi: 10.1002/mrm.1910350106. [DOI] [PubMed] [Google Scholar]
- 32.Halle B, Denisov VP. A new view of water dynamics in immobilized proteins. Biophys J. 1995;69:242–249. doi: 10.1016/S0006-3495(95)79895-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bryant RG, Shirley WM. Water-protein interactions: nuclear magnetic resonance results on hydrated lysozyme. 1980. Presented at 178th Meeting of the American Chemical Society; Washington, D.C.
- 34.Rorschach HE, Hazlewood CF. Protein dynamics and the NMR relaxation time T1 of water in biological systems. J Magn Reson. 1986;70:79–88. [Google Scholar]
- 35.Fung BM. Non-freezable water and spin-lattice relaxation time in muscle containing a growing tumor. Biochim Biophys Acta. 1974;362:209–214. doi: 10.1016/0304-4165(74)90042-7. [DOI] [PubMed] [Google Scholar]
- 36.Packer KJ. The dynamics of water in heterogeneous systems. Philos Trans R Soc Lond B Biol Sci. 1977;278:59–87. doi: 10.1098/rstb.1977.0031. [DOI] [PubMed] [Google Scholar]
- 37.Edzes HT, Samulski ET. Cross relaxation and spin diffusion in the proton NMR or hydrated collagen. Nature. 1977;265:521–523. doi: 10.1038/265521a0. [DOI] [PubMed] [Google Scholar]
- 38.Kimmich R, Winter F. Double-diffusive fluctuation and the v3/4-law of proton spin-lattice relaxation in biopolymers. Prog Colloid Polym Sci. 1985;71:66–70. [Google Scholar]
- 39.Denisov VP, Halle B, Peters J, Horlein HD. Residence times of the buried water molecules in bovine pancreatic trypsin inhibitor and its G36S mutant. Biochemistry. 1995;34:9046–9051. doi: 10.1021/bi00028a013. [DOI] [PubMed] [Google Scholar]
- 40.Hills BP. The proton exchange cross-relaxation model of water relaxation in biopolymer systems .II. The sol and gel states of gelatin. Mol Phys. 1992;76:509–523. [Google Scholar]