

Abstract
Recently, we designed a series of novel HIV-1 protease inhibitors incorporating a stereochemically defined bicyclic fused cyclopentyl (Cp-THF) urethane as the high affinity P2-ligand. Inhibitor 1 with this P2-ligand has shown very impressive potency against multi-drug-resistant clinical isolates. Based upon the 1-bound HIV-1 protease X-ray structure, we have now designed and synthesized a number of meso-bicyclic ligands which can conceivably interact similarly to the Cp-THF ligand. The design of meso-ligands is quite attractive as they do not contain any stereocenters. Inhibitors incorporating urethanes of bicyclic-1,3-dioxolane and bicyclic-1,4-dioxane have shown potent enzyme inhibitory and antiviral activities. Inhibitor 2 (K i = 0.11 nM; IC50 = 3.8 nM) displayed very potent antiviral activity in this series. While inhibitor 3 showed comparable enzyme inhibitory activity (K i = 0.18 nM) its antiviral activity (IC50 = 170 nM) was significantly weaker than inhibitor 2. Inhibitor 2 maintained an antiviral potency against a series of multi-drug resistant clinical isolates comparable to amprenavir. A protein–ligand X-ray structure of 3-bound HIV-1 protease revealed a number of key hydrogen bonding interactions at the S2-subsite. We have created an active model of inhibitor 2 based upon this X-ray structure.
Introduction
The proteolytic enzyme HIV-1 protease is essential for viral assembly and maturation.1 As a consequence, the design of specific inhibitors for HIV-1 protease has become the subject of immense interest. In 1996, protease inhibitors (PIs) were introduced in combination with reverse transcriptase inhibitors to become a highly active antiretroviral therapy (HAART).2 This treatment regimen significantly increased life expectancy, improved quality of life and decreased mortality and morbidity among HIV/AIDS patients. Despite these notable advances, the emergence of drug-resistant HIV-1 variants is severely limiting the efficacy of HAART treatment regimens. Therefore, the development of new broad-spectrum antiretroviral drugs that produce minimal adverse effects remains an important therapeutic objective for the treatment of HIV/AIDS.3 We have recently reported our structure-based design and development of a series of novel HIV-1 protease inhibitors including darunavir,4,5 TMC-126,6 and GRL-06579A (1, Fig. 1).7 These inhibitors were designed with specific features to help combat drug resistance. They have exhibited marked potency in enzyme inhibitory and cell-culture assays. Furthermore, these inhibitors have shown impressive activity against a broad-spectrum of HIV isolates including a variety of multi-PI-resistant clinical strains. Darunavir has been recently approved for the therapy of HIV/AIDS patients who are harboring drug-resistant HIV and do not respond to other antiretroviral drugs.
Fig. 1.

Structure of inhibitors 1–3.
One of our design principles to combat drug resistance is to maximize the ligand-binding interactions in the active site and particularly to promote extensive hydrogen bonding with the active site protein backbone. Indeed, inhibitor 1 incorporates a stereochemically defined bicyclic cyclopentanyltetrahydrofuran (Cp-THF) as the P2-ligand in the hydroxylethylsulfonamide isostere. The protein–ligand X-ray structure of inhibitor 1 revealed extensive hydrogen bonding interactions with the backbone atoms throughout the enzyme active site.8 The cyclic ether oxygen is involved in hydrogen bonding with the backbone NH of Asp29. The presence of this oxygen is critical for its superb antiviral properties, especially against drug resistant HIV strains. Based upon further examination of the protein–ligand X-ray structure of 1-bound HIV-1 protease, we subsequently speculated that a simplified meso-hexahydrocyclopenta-1,3-dioxolane ligand could conceivably maintain similar interactions with respect to the Cp-THF ligand in inhibitor 1. Particularly, it appears that one of the oxygens of this meso ligand can hydrogen bond with the Asp29 NH. Since the Cp-THF ligand in inhibitor 1 contains three chiral centers, incorporation of a meso ligand as shown in inhibitor 2 would remarkably simplify the synthesis compared to the bicyclic Cp-THF ligand. Furthermore, we speculated that the second oxygen atom in the meso-P2-ligand could conceivably engage in further interactions at the S2-subsite. Herein, we report the design, synthesis and biological investigation of a series of protease inhibitors that incorporate structure-based designed symmetrical meso-bicyclic 1,3-dioxolane and 1,3-dioxane derivatives as the P2-ligands. Inhibitors (2 and 3) incorporating these ligands have shown exceedingly potent enzyme inhibitory potency as well as antiviral activity. Furthermore, we evaluated the drug-resistance profile of inhibitor 2 against multi-drug-resistant clinical isolates and it was shown to maintain tremendous potency. The protein–ligand X-ray structure of 3-bound HIV-1 protease has been determined and this structure has provided molecular insight into the ligand-binding site interactions.
Chemistry
The hexahydrocyclopenta-1,3-dioxolan-5-ol (11), required for the synthesis of 2, was prepared as described in Scheme 1. Commercially available 1,6-heptadien-4-ol 4 was protected as the corresponding t-butyldiphenylsilyl ether using sodium hydride as the base in THF. The resulting diene was subjected to a ring closing metathesis reaction using second generation Grubbs’ catalyst to afford the protected cyclopenten-1-ol 5 in 94% overall yield. Osmium tetroxide-promoted dihydroxylation of olefin 5 was accomplished using a catalytic amount of osmium tetroxide and NMO and pyridine to afford diol 6 as a 6 : 1 mixture of anti- and syn-isomers which were easily separated by column chromatography. The anti- isomer 6 was subsequently treated with paraformaldehyde, preliminarily cracked with aqueous hydrochloric acid in chloroform under reflux,9 affording the cyclic acetal 7 in good yield. Along with the desired compound 7, the trioxepane 8 was also isolated from the reaction mixture in a 1 : 1 ratio. We therefore decided to incorporate the tetrahydro-5aH-cyclopenta[f ][1,3,5]trioxepan-7-yl-moiety as a P2-ligand (resulting in inhibitors 27–28, Table 1) because the higher flexibility of the trioxepane ring could allow an improved adaptability to enzyme amino acid mutations, leading to better activity against HIV–resistant strains. Accordingly, both intermediates 7 and 8 were deprotected using tetrabutyl-ammonium fluoride (TBAF) in THF to provide the anti-alcohols 9 and 10. Compounds 9 and 10 were subsequently subjected to Mitsunobu inversion to afford the corresponding syn-alcohols 11 and 12.
Scheme 1.

Synthesis of alcohols 9–12.
Table 1.
Enzymatic inhibitory activity of compounds 2,3, and 26–30 and antiviral activity of selected inhibitors against HIV-1LAI
| Entry | Inhibitor | Ki/nMa | IC50/μMb |
|---|---|---|---|
| 1 |
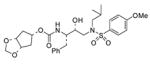 2 |
0.11 ± 0.01 | 0.0038 ± 0.0001 |
| 2 |
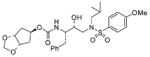 26 |
0.40 ± 0.04 | nd |
| 3 |
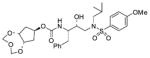 27 |
5.4 ± 0.22 | >1 |
| 4 |
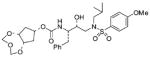 28 |
0.51 ± 0.01 | 0.38 ± 0.02 |
| 5 |
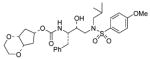 3 |
0.18 ± 0.03 | 0.21 ± 0.04 |
| 6 |
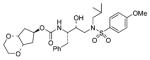 29 |
0.50 ± 0.04 | nd |
| 7 |
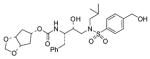 30 |
0.34 ± 0.07 | 0.0077 ± 0.003 |
Values are means of at least two experiments.
MT-2 human T-lymphoid cells exposed to HIV-1LAI ; antiviral activity of amprenavir (APV), saquinavir (SQV) and indinavir (IDV) were 0.03 μM, 0.02 μM and 0.03 μM respectively in this assay. nd: not determined.
For the preparation of inhibitors 3 and 29, alcohols 15 and 16 were synthesized as described in Scheme 2. Diol 6 was heated under reflux in toluene in the presence of dibutyltin oxide with azeotropic removal of water. The resulting stannylene acetal intermediate was treated with chloroethanol to obtain the monoalkylated derivative 13 in 68% overall yield.10 Subsequently, the primary alcohol was selectively tosylated with p-toluenesulfonyl chloride in the presence of pyridine. Exposure of the resulting compound to sodium hydride resulted in an intramolecular substitution reaction leading to the corresponding cyclization compound 14. TBAF-mediated deprotection furnished the target anti-alcohol 15 in good overall yield. The syn-alcohol 16 was then obtained after Mitsunobu inversion of 15 as described above.
Scheme 2.

Synthesis of alcohols 15 and 16.
The synthesis of the active carbonates required for the synthesis of the various inhibitors is shown in Scheme 3. Alcohol 9 was converted to the succinimidyl-derivative 17 by treatment with N,N′-succinimidylcarbonate in the presence of Et3N as described previously.11 Alcohols 10–12, 15 and 16 were activated by conversion to the corresponding p-nitrophenylcarbamates 18–22 (81–95% yield) by using p-nitrophenylchloroformate and N-methylmorpholine in THF. The general procedure for the synthesis of inhibitors 2, 3, and 26–30 is outlined in Scheme 4. Epoxide 2312 was converted into intermediate 24 following our previously reported procedure.8 Deprotection of 24 by using trifluoroacetic acid followed by reaction with the activated alcohols 17–22 furnished inhibitors 2,3 and 26–29 in 43–85% yields.
Scheme 3.

Synthesis of activated alcohols 17–22.
Scheme 4.

Synthesis of inhibitors 2,3 and 26–30.
Finally, inhibitor 30 was synthesized from the known8 amine 25. This amine was reacted with the activated carbonate 17 in the presence of diisopropylethylamine in THF at 23 °C to provide 30. Inhibitor 30 was obtained in 63% yield.
Results and discussion
The inhibitory potencies of the synthetic inhibitors were evaluated using the assay protocol of Toth and Marshall,13 and the results are shown in Table 1. As can be seen, inhibitor 2 has shown an enzyme inhibitory potency of 0.11 nM. It appears that the bicyclic 1,3-dioxolane ring can be accommodated by the S2-subsite of HIV-1 protease. Inhibitor 26 with a meso ligand containing a trans-bicyclic-1,3-dioxolane ring is 2.5-fold less potent than the syn-isomer 2. We have examined the effect of both syn and anti-trioxepane rings as P2-ligands in inhibitors 27 and 28. The syn-isomer 28 is significantly more potent (K i = 0.51 nM) than the anti-isomer 27. Considering the acid sensitivity of 1,3-dioxolane rings, we not only speculated that the stable 1,4-dioxane ring may fill the hydrophobic S2-site, but also that the oxygens on the dioxane ring may interact with backbone atoms or residues in the active site. As shown, the meso ligand in inhibitor 3 with a syn-bicyclic-1,4-dioxane ring has shown an enzyme inhibitory potency of 0.18 nM (Ki value). Consistent with previous results, the corresponding anti-isomer 29 is significantly less potent. As reported previously, the P2-ligand Cp-THF with a P2′-hydroxymethyl sulfonamide (inhibitor 1) is significantly more potent than the corresponding P2′-methoxybenzene sulfonamide derivative. We have, therefore, compared the inhibitory potency of inhibitor 30, containing a P2′-hydroxymethyl benzene sulfonamide derivative, with inhibitor 2. However, inhibitor 30 did not exhibit this potency enhancing effect.
We have examined selected compounds for their activity against HIV-1 using a human CD4+ T-cell line (MT-2 cells). The activity of inhibitor 2 against a variety of multi-drug-resistant HIV-1 variants was also examined in detail using human peripheral blood mononuclear cells (PBMCs) as target cells. We employed two endpoints for the activity against HIV-1: (i) the inhibition of the HIV-1-elicited cytopathic effect for MT-2 cells and (ii) the inhibition of HIV-1 p24 production for PBMCs.5
When examined in MT-2 cells as the target cells, inhibitor 2 displayed an impressive antiviral IC50 of 3.8 nM (Table 1). Inhibitor 3 showed an antiviral IC50 value in the high nanomolar range (IC50 = 210 nM, Table 1), while it exhibited a similar K i to inhibitor 2. We subsequently examined inhibitor 2 for its activity against a clinical wild-type X4-HIV-1 isolate (HIV-1ERS104pre) along with various multi-drug-resistant clinical X4- and R5-HIV-1 isolates (Table 2) using PBMCs as the target cells.5 The activity of inhibitor 2 against HIV-1ERS104pre (IC50 = 29 nM) was comparable to those of currently available protease inhibitors, SQV, APV, and IDV, which display IC50 values of 12, 33, and 26 nM, respectively. Of particular note, the IC50 value of inhibitor 2 in PBMCs (IC50= 29 nM) was nearly 8-fold greater than the IC50 value in MT-2 cells (IC50 = 3.8 nM). With regard to this difference, considering that 2 is highly potent as examined in human T-cells (MT-2 cells) but its activity is slightly less in PBMCs, it is possible that relatively higher concentrations of 2 are required to suppress HIV-1 production in chronically infected macrophages.14 IDV was not capable of efficiently suppressing the replication of most of the multi-drug-resistant clinical isolates examined (HIV-1MDR/MM, HIV-1MDR/JSL, HIV-1MDR/C, and HIV-1MDR/A), with IC50 values of >1.0 μM. The potency of inhibitor 2 against most of the multi-drug-resistant variants was generally comparable to that of SQV and APV, although DRV was found to be the most potent among those tested, including inhibitor 2, against HIV-1ERS104pre as well as all the multi-drug-resistant variants.
Table 2.
Antiviral activity of inhibitor 2 against clinical HIV-1 isolates in PBMC cells
| IC50valuesa(nM) |
|||||
|---|---|---|---|---|---|
| Virusb | 2 | DRVc | SQVd | APVe | IDVf |
| HIV-1ERS104pre (wild-type: X4) | 29 | 3.5 | 12 | 33 | 26 |
| HIV-1MDR/MM (R5) | 150 (5) | 17 (5) | 190 (16) | 300 (9) | >1000 (>38) |
| HIV-1MDR/JSL (R5) | 550 (19) | 26 (7) | 330 (28) | 430 (13) | >1000 (>38) |
| HIV-1MDR/C (X4) | 300 (10) | 7 (2) | 36 (3) | 230 (7) | >1000 (>38) |
| HIV-1MDR/G (X4) | 340 (12) | 7 (2) | 29 (2) | 340 (10) | 290 (11) |
| HIV-1MDR/A (X4) | 21 (1) | 3 (1) | 81 (7) | 100 (3) | >1000 (>38) |
Amino acid substitutions identified in the protease-encoding region compared to the consensus type B sequence cited from the Los Alamos database include L63P in HIV-1ERS104Pre; L10I, K14R, L33I, M36I, M46I, F53I, K55R, 162V, L63P, A71V, G73S, V82A, L90M, and I93L in HIV-1MDR/B; L10I, V11I, T12E, I15V, Ll9I, R41K. M46L, L63P, A71T, V82A, and L90M in HIV-1MDR/G; L10I, K14R. R41K, M46L, I54V, L63P, A71V, V82A, L90M, I93L in HIV-1MDR/TM; L10I, L24I, I33F, E35D, M36I, N37S, M46L, I54V, R57K, I62V, L63P, A71V, G73S, and V82A in HIV-1MDR/JSL; and L10I, K43T, M46L, I54V, L63P, A71V, V82A, L90M, and Q92K in HIV-1MDR/MM. HIV-1ERS104Pre; served as a source of wild-type HIV-1. The IC50 values were determined by employing PHA-PBMC (phytohemaglutinin-activated peripheral blood mononuclear cells) as the target cells and the inhibition of p24Gag protein production as the endpoint. All values were determined in triplicate.
X4 denotes CXCR4-tropic HIV-1 while R5 CCR5-tropic HIV-1.
DRV (darunavir).
SQV (saquinavir).
APV (amprenavir).
IDV (indinavir).
X-Ray crystallography
To obtain molecular insight into the ligand-binding site interactions responsible for the impressive enzyme inhibitory potency of compound 3, we determined the X-ray structure of 3-bound HIV-1 protease. The crystal structure was solved and refined to an R factor of 15.2% at a 1.07 Å resolution. The inhibitor binds with extensive interactions from P2 to P2′ with the protease atoms, and most notable are the favorable polar interactions including hydrogen bonds, as shown in Fig. 2. The transition-state hydroxyl group forms hydrogen bonds to the side chain carboxylate oxygen atoms of the catalytic Asp25 and Asp25′. Of particular interest, the meso-bicyclic 1,4-dioxane ligand appears to be involved in hydrogen bonding interactions with the backbone atoms and residues at the S2-site. One of the dioxane oxygens hydrogen bonds with the backbone NH of Asp29. The other oxygen makes a water–mediated hydrogen bond with the carbonyl oxygen of Gly48. These interactions are described in several peptide substrate analogs.15 However, the design of high affinity ligands incorporating this interaction with Gly48 has not been previously demonstrated. The inhibitor also hydrogen bonds with the protease main chain amide carbonyl oxygen of Gly27, and there are water-mediated interactions with the amides of Ile50 and Ile50− that are conserved in the majority of protease complexes with inhibitors16 and substrate analogs.15 The weaker polar interactions such as C–H ··· O and water-π interactions can be analyzed accurately in atomic resolution structures.17,18 Inhibitor 3 also shows a water-mediated interaction of the π system of the P2′ aromatic ring with the amide of Asp29′, which was also observed for darunavir and inhibitor 1.19 Furthermore, the P2′ methoxy group forms a hydrogen bond to the backbone NH of Asp30′. Importantly, the P2 group forms a hydrogen bond interaction with the carbonyl oxygen of Gly48 and a water-mediated interaction with the amide of Gly48, similar to the interactions described for several peptide substrate analogs.15 These interactions of the P2 group confirm the design strategy of incorporating new polar interactions with conserved backbone regions of the protease.
Fig. 2.

Stereoview of the X-ray structure of inhibitor 3 bound to the active site of wild-type HIV-1 protease.
In an effort to understand the binding interactions of the corresponding meso-1,3-dioxolane ligand in the S2-subsite, we have created an active model of inhibitor 2 (Fig. 3) based upon the X-ray structure of 3-bound HIV-1 protease. The model suggests that both dioxolane oxygens may interact with both active site residues Asp29 and Asp30, as well as Gly48 through the structural water molecule. In comparison, it appears that the dioxane oxygens of inhibitor 3 are not within hydrogen bonding distance of the backbone NH of Asp30. This may explain the marked difference in antiviral activity of inhibitor 2 compared with inhibitor 3.
Fig. 3.

A stereoview of an active model of inhibitor 2 (green) with the X-ray structure of inhibitor 3 (magenta)-bound HIV-1 protease.
Conclusions
In summary, a series of novel HIV-1 protease inhibitors were designed and synthesized by incorporating bicyclic meso-1,3-dioxolane and 1,4-dioxane derivatives as the P2-ligands. A number of inhibitors have shown very impressive enzyme inhibitory and antiviral potency, similar to inhibitor 1 with a stereochemically defined Cp-THF ligand. The design of meso-1,3-dioxolane and 1,4-dioxane P2-ligands as exemplified in inhibitors 2 and 3, respectively, has remarkably simplified the stereochemical complexity as well as chemical synthesis over the Cp-THF ligand in inhibitor 1. We have developed efficient synthetic routes to these ligands. Inhibitor 2 has shown potent antiviral activity in both MT-2 cells and PBMCs. Inhibitor 2 was profiled against a series of multi-drug-resistant clinical isolates. While inhibitor 2 is less potent than darunavir, it is significantly more potent than IDV and comparable to APV and SQV in suppressing the replication of multi-drug-resistant isolates MDRMM and MDRJSL. A protein–ligand X-ray structure of 3-bound HIV-1 protease revealed extensive interactions of the inhibitor with the active site of HIV-1 protease. Most notably, both oxygens of the meso-P2-ligand are involved in hydrogen bonding interactions with the protein backbone atoms. In particular, a water-mediated hydrogen bond to the Gly48 carbonyl is very unique. An active model of inhibitor 2 indicates similar ligand binding site interactions. Our design principle of increasing ‘backbone binding’ appears to maintain key interactions in the enzyme active site leading to retained potency against multi-drug-resistant variants. Further design and ligand optimization involving these interactions is in progress.
Experimental
General
All moisture sensitive reactions were carried out under a nitrogen or argon atmosphere. Anhydrous solvents were obtained as follows: THF, diethyl ether, and benzene, distilled from sodium and benzophenone; dichloromethane, pyridine, triethylamine, and diisopropylethylamine, distilled from CaH2. All other solvents were HPLC grade. Column chromatography was performed with Whatman 240–400 mesh silica gel under low pressure (5–10 psi). TLC was carried out with E. Merck silica gel 60 F254 plates. 1H and 13C NMR spectra were recorded on Varian Mercury 300 and Bruker Avance 400 and 500 spectrometers. Optical rotations were measured using a Perkin-Elmer 341 polarimeter. IR spectra were recorded on a Mattason Genesis II FT-IR spectrometer.
4-(tert-Butyldiphenylsilyloxy)-4H-cyclopentene (5)
To a suspension of sodium hydride (60% in mineral oil, 0.92 g, 23 mmol) in THF (10 mL), cooled to 0 °C, 1,6-heptadien-4-ol 4 (1 mL, 7.7 mmol) was added dropwise over 10 min. The resulting suspension was stirred at 0 °C for 30 min and then tert-butyldiphenylchlorosilane (2 mL, 7.9 mmol) was added. The reaction mixture was stirred at 23 °C for 4 h and then quenched with a saturated solution of ammonium chloride. The solvent was removed in vacuo and the aqueous phase was extracted with CH2Cl2. The organic extracts were dried (Na2SO4), the solvent removed and the residue purified by flash-chromatography (1 : 10, EtOAc–Hex) to afford 4-(tert-butyldiphenylsilyloxy)hepta-1,6- diene (2.6 g, 96%) as a colorless oil: IR νmax (NaCl; cm−1) 3066, 2951, 1421, 1103 and 696; δH (300 MHz, CDCl3) 7.70 (4 H, dd, J 1.6, 7.6 Hz, ArH), 7.47–7.38 (6 H, m, ArH), 5.83–5.69 (2 H, m, 2 × CH=CH2), 5.02–4.91 (4 H, m, 2 × CH=CH 2), 3.87–3.80 (1 H, m, CHOSi), 2.31–2.12 (4 H, m, 3-H2, 5-H2) and 1.08 [9 H, s, C(CH3)3; δC (75 MHz, CDCl3) 135.9, 134.7, 134.3, 129.5, 127.5, 117.1, 72.4, 40.5, 27.0 and 19.4; m/z (CI) 351 (M + H, 100); HRMS (M + H)+ calcd for C23H31OSi, 351.2144; found, 351.2146.
To a solution of the above compound (2.0 g, 5.7 mmol) in dry CH2Cl2 (20 mL), second generation Grubbs’ catalyst (48 mg) was added and the resulting mixture was heated under reflux for 2 h. Subsequently, the reaction mixture was cooled to 23 °C, the solvent removed under reduced pressure and the residue purified by flash-chromatography (1 : 10 EtOAc–Hex) to afford 5 (1.8 g, 98%) as a colorless oil: IR νmax (NaCl; cm−1) 3067, 2853, 2736, 1428, 1109 and 702; δH (300 MHz, CDCl3) 7.67 (4 H, dd, J 1.8, 7.8, ArH), 7.45–7.34 (6 H, m, ArH), 5.61 (2 H, s, 1-H, 2-H), 4.57– 4.51 (1 H, m, 4-H), 2.47–2.33 (4 H, m, 3-H2, 5-H2) and 1.05 [9 H, s, C(CH3)3]; δC (100 MHz, CDCl3) 135.7, 134.5, 129.5, 128.3, 127.5, 73.5, 42.4, 26.9 and 19.1; m/z (CI) 323 (M + H, 100); HRMS (M + H)+ calcd for C21H27OSi, 323.1831; found, 323.1834.
(1α,2α,4β)-4-(tert-Butyldiphenylsilyloxy)-1,2-cyclopentanediol (6)
A mixture of 5 (5.1 g, 15.8 mmol), osmium tetroxide (2.5 wt% solution in tert-butanol, 4 mL), N-methylmorpholine-N-oxide (2.6 g, 22.2 mmol), and pyridine (1.3 mL, 15.8 mmol) in a 3 : 2 : 1 mixture of tert-butanol, THF, and water (80 mL) was heated under reflux for 4 h. The reaction mixture was cooled to 23 °C and treated with a 20% aqueous solution of sodium bisulfite (10 mL). The organic solvents were removed under reduced pressure and the aqueous phase was extracted with EtOAc. The organic extracts were washed with 1 N hydrochloric acid, water and brine, and dried (Na2SO4). The solvent was removed in vacuo and the residue was purified by flash-chromatography (1 : 1 EtOAc–Hex) to yield diol 6 (5.3 g, 94%) as a colorless oil: IR νmax (NaCl; cm−1) 3006, 2676, 1427, 1112 and 702; δH (300 MHz, CDCl3) 7.62 (4 H, dd, J 1.8, 7.5, ArH), 7.45–7.33 (6 H, m, ArH), 4.84–4.42 (1 H, m, 4-H), 4.30–4.29 (2 H, m, 1-H, 2-H), 2.22 (2 H, br. s, 2 × OH), 1.99–1.80 (4 H, m, 3-H2, 5-H2) and 1.04 [9 H, s, C(CH3)3]; δC (100 MHz, CDCl3) 135.6, 134.0, 129.6, 127.6, 72.4, 71.2, 41.9, 26.8 and 19.0; m/z (EI) 356 (M, 100); HRMS (M)+ calcd for C21H28O3Si, 356.1808; found, 356.1803.
(1β,2β,4α)-4-(tert-Butyldiphenylsilyloxy)-1,2-(methylenedioxy)-cyclopentane (7) and (5aα,7β,8aα)-7-(tert-butyldiphenylsilyloxy)-tetrahydrocyclopenta[f ]-1,3,5-trioxepane (8)
A mixture of paraformaldehyde (0.77 g, 25.7 mmol) and con- centrated hydrochloric acid (2 mL) in CHCl3 (2 mL) was stirred at 23 °C until a clear solution was formed (6 h) and then a solution of 6 (0.2 g, 0.54 mmol) in CHCl3 (2 mL) was added. The resulting mixture was heated under reflux overnight and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and evaporated under reduced pressure to yield 7 (0.18 g, 86%) after flash-chromatography (1 : 10, EtOAc–Hex): IR νmax (NaCl; cm−1) 2791, 1589, 1471, 1428, 822 and 699; δH (300 MHz, CDCl3) 7.64 (4 H, d, J 6.3, ArH), 7.45–7.35 (6 H, m, ArH), 4.78 (1 H, s), 4.60 (1 H, s), 4.51 (2 H, d, J 5.4, 1-H, 2-H), 4.47–4.39 (1 H, m, 4-H), 1.99 (2 H, dd, J 6.0, 13.8, 3-H′, 5-H′), 1.77–1.68 (2 H, m, 3-H″, 5-H″) and 1.04 [9 H, s, C(CH3)3]; δC (75 MHz, CDCl3) 135.6, 134.0, 129.7, 127.6, 94.0, 78.8, 72.7, 41.0, 26.9 and 19.1; m/z (EI) 368 (M, 100). After further elution of the column 8 (0.5 g, 5%) was obtained: IR νmax (NaCl; cm−1) 2827, 2726, 1427, 1113 and 703; δH (300 MHz, CDCl3) 7.66–7.62 (4 H, m, ArH), 7.46–7.35 (6 H, m, ArH), 5.17 (2 H, d, J 7.8, 2-H′, 4-H′), 4.70 (2 H, d, J 7.8, 2-H″, 4-H″), 4.52–4.43 (3 H, m, 5a-H, 7-H, 8a-H), 2.15–2.08 (2 H, m, 6-H′, 8-H′), 1.93–1.85 (2 H, m, 6-H″, 8- H″) and 1.06 [9 H, s, C(CH3)3]; δC (75 MHz, CDCl3) 135.6, 133.8, 129.7, 127.7, 96.1, 82.5, 71.7, 41.1, 26.9 and 19.1; m/z (CI) 397 (M – H, 100); HRMS (M – H)+ calcd for C23H29O4Si, 397.1832; found, 397.1832.
(4α,1β,2β)-4-Hydroxy-1,2-(methylenedioxy)cyclopentane (9)
A mixture of 7 (0.47 g, 1.3 mmol) and n-Bu4N+F− (1.0 M solution in THF, 1.4 mL, 1.4 mmol) in dry THF (10 mL) was stirred at 23 °C for 16 h. To the reaction mixture was added a saturated solution of NaHCO3, the solvent was removed in vacuo and the aqueous phase extracted with Et2O. The organic extracts were dried (Na2SO4) and evaporated and the residue was purified by flash-chromatography (1 : 1 EtOAc–Hex) to yield 9 (0.16 g, 96%) as a colorless oil: IR νmax (NaCl; cm−1) 3044, 2792, 2602, 1065, 821 and 602; δH (300 MHz, CDCl3) 4.89 (1 H, s, OCH HO), 4.59 (1 H, s, OCHHO), 4.50 (2 H, d, J 6.0, 1-H, 2-H), 4.41–4.32 (1 H, m, 4-H), 3.13 (1 H, br. s, OH), 2.09 (2 H, dd, J 5.6, 14.0, 3-H′, 5-H′) and 1.61–1.51 (2 H, m, 3-H″, 5-H″); δC (75 MHz, CDCl3) 94.1, 78.9, 70.8 and 40.6; m/z (CI) 129 (M – H, 100); HRMS (M – H)+ calcd for C6H9O3, 129.0552; found, 129.0556.
(5aα,7β,8aα)-7-Hydroxytetrahydrocyclopenta[f ]-1,3,5-trioxepane (10)
The title compound was obtained as described for 9 in 83% yield. Flash-chromatography was performed using EtOAc: IR νmax (NaCl; cm−1) 3036, 2649, 1424, 1118 and 930; δH (300 MHz, CDCl3) 5.15 (2 H, d, J 7.2, 2-H′, 4-H′), 4.67 (2 H, d, J 7.2, 2-H″, 4-H″), 4.47–4.40 (3 H, m, 5a-H, 7-H, 8a-H), 2.07–2.02 (4 H, m, 6-H2, 8-H2) and 1.86 (1 H, br. s, OH); δC (75 MHz, CDCl3) 96.1, 82.3, 70.0 and 40.8; m/z (EI) 160 (M, 100); HRMS (M)+ calcd for C7H12O4,160.0736; found, 160.0738.
(1β,2β,4β)-4-Hydroxy-1,2-(methylenedioxy)cyclopentane (11)
To a mixture of 9 (100 mg, 0.77 mmol), p-nitrobenzoic acid (250 mg, 1.5 mmol), and triphenylphosphine (450 mg, 1.5 mmol), was added diisopropylazodicarboxylate (300 μL, 1.5 mmol) dropwise and the resulting mixture was stirred at 23 °C. After 16 h, the solvent was removed under reduced pressure and the residue purified by flash-chromatography (1 : 2 EtOAc–Hex). The resulting ester was dissolved in a 3 : 2 : 1 mixture of THF, methanol, and water (10 mL) and LiOH·H2O (162 mg, 3.8 mmol) was added. The yellow mixture was stirred at 23 °C for 5 h and then the solvent was removed in vacuo. The residue was diluted with water and the aqueous phase extracted with Et2O. The organic extracts were dried (Na2SO4) and the solvent was evaporated. Purification of the residue by flash-chromatography (1 : 1 EtOAc–Hex) afforded 11 (57 mg, 57%) as a colorless oil: IR νmax (NaCl; cm−1) 3052, 2804, 2577, 1164, 1096, 1011 and 924; δH (300 MHz, CDCl3) 5.17 (1 H, s, OCHHO), 4.68 (1 H, s, OCHHO), 4.61 (2 H, d, J 4.8, 1-H, 2-H), 4.27 (1 H, t, J 4.7, 4-H), 2.33 (1 H, br. s, OH), 2.21 (2 H, d, J 15.3, 3-H′, 5-H′) and 1.85–1.77 (2 H, m, 3-H″, 5-H″); δC (75 MHz, CDCl3) 94.7, 81.5, 74.0 and 41.0; m/z (EI) 129 (M – H, 100); HRMS (M – H)+ calcd for C6H9O3, 129.0611; found, 129.1012.
(5aα,7α,8aα)-7-Hydroxytetrahydrocyclopenta[f ]-1,3,5-trioxepane (12)
The title compound 12 was obtained as described for 11 in 69% yield. Flash-chromatography was performed using EtOAc: IR νmax (NaCl; cm−1) 3044, 2832, 2633, 1481, 1116 and 928; δH (300 MHz, CDCl3) 5.18 (2 H, d, J 7.2, 2-H′, 4-H′), 4.67 (2 H, d, J 7.2, 2-H″, 4-H″), 4.31–4.25 (2 H, m, 5a-H, 8a-H), 4.18–4.13 (1 H, m, 7-H), 2.40 (1 H, br. s, OH), 2.17–2.08 (2 H, m, 6-H′, 8-H′) and 2.03–1.96 (2 H, m, 6-H″, 8-H″); δC (75 MHz, CDCl3) 95.3, 82.8, 71.0 and 41.1; m/z (CI) 161 (M + H, 100); HRMS (M + H)+ calcd for C7H12O4, 161.0814; found, 161.0814.
(±)-(1β,2β,4α)-2-(2′-Hydroxyethoxy)-4-(tert-butyldiphenyl-silyloxy)cyclopentane-1-ol (13)
A mixture of 6 (1.4 g, 3.9 mmol) and dibutyltin oxide (0.94 g, 3.9 mmol) in dry toluene (130 mL) was heated under reflux with azeotropic removal of water. After 5 h, the reaction mixture was concentrated to half the initial volume and chloroethanol (2.5 mL, 39 mmol) and n-Bu4N+I− (1.4 g, 3.9 mmol) were added. The resulting mixture was heated under reflux for 19 h. Afterwards the solvent was evaporated and the residue was purified by flash-chromatography (10 : 1 EtOAc–MeOH) to afford 13 (1.3 g, 86%) as a colorless oil: IR νmax (NaCl; cm−1) 3102, 2604, 1589, 1471, 1062, 823 and 612; δH (300 MHz, CDCl3) 7.62 (4 H, d, J 8.7, ArH), 7.44–7.33 (6 H, m, ArH), 4.45–4.40 (1 H, m, 4-H), 4.33–4.28 (1 H, m, 2-H), 4.04–3.98 (1 H, m, 1-H), 3.76–3.71 (2 H, m, CH2O), 3.66–3.55 (2H, m, CH2O), 3.01 (2 H, br. s, 2 × OH), 1.97–1.80 (4 H, m, 3-H2, 5-H2) and 1.04 (s, 9H); δC (75 MHz, CDCl3) 135.6, 134.1, 129.6, 127.6, 80.7, 71.1, 71.0, 70.8, 61.7, 42.3, 39.0, 26.9 and 14.2; m/z (ESI) 423 (M + Na, 100).
(1β,2β,4α)-4-(tert-butyldiphenylsilyloxy)-1,2-(ethylenedioxy)-cyclopentane (14)
A mixture of 13 (1.2 g, 3.0 mmol), p-toluenesulfonyl chloride (1.3 mg, 6.6 mmol), pyridine (1.2 mL, 15 mmol) and a catalytic amount of N,N-dimethylaminopyridine in CH2Cl2 (40 mL) was stirred at 23 °C for 24 h. The reaction mixture was treated with 1 N HCl and the aqueous phase was extracted with CH2Cl2. The organic extracts were dried (Na2SO4) and the solvent removed. Purification of the residue by flash-chromatography (1 : 1 EtOAc–Hex) afforded the tosylated alcohol (990 mg, 60%) as a colorless oil: IR νmax (NaCl; cm−1) 3104, 2992, 2691, 1598, 1359, 1177, 923 and 705; δH (300 MHz, CDCl3) 7.76 (2 H, d, J 8.4, ArH), 7.61 (4 H, d, J 7.8, ArH), 7.42–7.26 (8 H, m, ArH), 4.53–4.25 (1 H, m, CHO), 4.15–4.09 (3 H, m, CHO, CH2O), 3.96–3.91 (1 H, m, CHO), 3.68–3.62 (2 H, m, CH2O), 2.41 (3 H, s, CH3), 1.89–1.75 (4 H, m, 3-H2, 5-H2) and 1.03 [9 H, s, C(CH3)3]; δC (100 MHz, CDCl3) 135.6, 134.0, 132.8, 129.8, 129.6, 127.9, 127.8, 127.6, 80.7, 71.3, 71.0, 69.0, 68.6, 67.0, 42.1, 38.7, 26.8, 21.6 and 18.9. To a solution of the above product (150 mg, 0.27 mmol) in dry THF (12 mL), NaH (60% in mineral oil, 22 mg, 0.54 mmol) was added and the resulting suspension was heated under reflux for 30 min. After cooling to 23 °C, the reaction mixture was quenched with a saturated solution of NH4Cl, the solvent was removed and the aqueous phase was extracted with EtOAc. The organic extracts were dried (Na2SO4) and the solvent was removed in vacuo. The residue was purified by flash-chromatography (1 : 3 EtOAc–Hex) to afford 14 (82 mg, 80%) as a colorless oil: IR νmax (NaCl; cm−1) 2803, 1427, 1136, 957 and 703; δH (300 MHz, CDCl3) 7.65 (4 H, d, J 7.8, ArH), 7.46–7.36 (6 H, m, ArH), 4.55–4.48 (1 H, m, 4-H), 4.18 (2 H, t, J 5.1, 1-H, 2-H), 3.70–3.62 (2 H, m, CH2O), 3.53–3.46 (2 H, m, CH2O), 2.16–2.07 (2 H, m, 3-H′, 5-H′), 1.82–1.74 (2 H, m, 3-H″, 5-H″) and 1.06 [9 H, s, C(CH3)3]; δC (75 MHz, CDCl3) 135.6, 134.1, 129.5, 127.6, 75.2, 71.1, 62.2, 37.5, 27.0 and 19.1; m/z (CI): 383.25 (M + H, 100).
(1β,2β,4α)-4-Hydroxy-1,2-(ethylendioxy)cyclopentane (15)
The above compound was deprotected as described for 9 to afford 15 in 90% yield as a colorless oil: IR νmax (NaCl; cm−1) 3013, 2797, 2550, 1129 and 664; δH (300 MHz, CDCl3) 4.58–4.51 (1 H, m, 4-H), 4.17 (2 H, t, J 4.8, 1-H, 2-H), 3.78–3.71 (2 H, m, CH2O), 3.58–3.51 (2 H, m, CH2O), 2.34–2.25 (2 H, m, 3-H′, 5-H′) and 1.72–1.66 (3 H, m, 3-H″, 5-H″, OH); δC (75 MHz, CDCl3) 75.1, 69.6, 62.3 and 37.2; m/z (EI) 144 (M, 100).
(1β,2β,4β)-4-Hydroxy-1,2-(ethylendioxy)cyclopentane (16)
Starting from 15 the title compound 16 was obtained as described for 11 in 83% yield as a colorless oil. Flash-chromatography was performed using EtOAc: IR νmax (NaCl; cm−1) 3014, 2571, 1135, 1081 and 875; δH (300 MHz, CDCl3) 4.22–4.16 (1 H, m, 4-H), 4.01 (2 H, t, J 4.2, 1-H, 2-H), 3.88–3.80 (2 H, m, CH2O), 3.63–3.55 (2 H, m, CH2O), 2.57 (1 H, br. s, OH) and 2.10–1.93 (4 H, m, 3-H2, 5-H2); δC (75 MHz, CDCl3) 76.0, 71.4, 62.3 and 37.5; m/z (EI) 144 (M, 100).
(1β,2β,4β)-1,2-(Methylenedioxy)cyclopent-4-yl succinimidylcarbonate (17)
To a solution of 9 (67 mg, 0.52 mmol) in dry acetonitrile (2 mL), N,N′-disuccinimidyl carbonate (198 mg, 0.77 mmol) and triethylamine (145 μL, 1.0 mmol) were added and the resulting mixture was stirred at 23 °C. After 8 h the solvent was removed, the residue was taken up in a saturated solution of NaHCO3 and the aqueous phase was extracted with EtOAc. The organic extracts were dried (Na2SO4) and the solvent was removed in vacuo. Purification of the residue by flash-chromatography (10 :) 1 CHCl3–MeOH) yielded 17 (58 mg, 55%): IR νmax (NaCl; cm−1) 2759, 1787, 1740, 1210, 1090; δH (300 MHz, CDCl3) 5.27 (1 H, t, J 7.2, 4-H), 4.97 (1 H, s, OCHHO), 4.69 (1 H, s, OCHHO), 4.61– 4.59 (2 H, m, 1-H, 2-H), 2.82 (4 H, s, CH2CH2), 2.38 (2 H, dd, J 6.2, 14.2, 3-H′, 5-H′) and 1.99–1.89 (2 H, m, 3-H″, 5-H″); δC (100 MHz, CDCl3) 168.6, 150.8, 94.5, 81.2, 78.1, 37.3 and 25.4; m/z (CI) 270 (M – H, 100); HRMS (M – H)+ calcd for C11H12NO7, 270.0614; found, 270.0607.
(5aα,7β,8aα)-7-(4-nitrophenoxycarbonyloxy)-tetrahydrocyclopenta[f ]-1,3,5-trioxepane (18)
To a solution of 10 (15 mg, 0.094 mmol) and N-methylmorpholine (31 μL, 0.28 mmol) in dry THF (3 mL), p-nitrophenylchloroformate (57 mg, 0.28 mmol) was added and the resulting mixture was stirred at 23 °C. After 1 h, water was added, the solvent was removed under reduced pressure and the aqueous phase was extracted with CHCl3. The organic extracts were dried (Na2SO4) and the solvent evaporated. The residue was purified by flash-chromatography (1 : 4 EtOAc–CHCl3) to afford 18 (31 mg, 95%) as a pale yellow viscous oil: IR νmax (NaCl; cm−1) 2831, 2598, 1766, 1529, 1350, 1116 and 859; δH (300 MHz, CDCl3) 8.27 (2 H, d, J 8.7, ArH), 7.38 (2 H, d, J 8.7, ArH), 5.34–5.31 (1 H, m, 7-H), 5.19 (2 H, d, J 6.9, 2-H′, 4-H′), 4.77 (2 H, d, J 6.9, 2-H″, 4-H″), 4.51–4.47 (2 H, m, 5a-H, 8a-H) and 2.38–2.26 (4 H, m, 6-H2, 8- H2); δC (75 MHz, CDCl3) 155.3, 126.2, 125.3, 121.7, 115.6, 95.5, 81.2, 78.5 and 37.6; m/z (EI) 325 (M, 100).
(1β,2β,4β)-4-(4-Nitrophenoxycarbonyloxy)-1,2-(methylenedioxy)-cyclopentane (19)
The title compound 19 was obtained from 11 as described for 18 in 81% yield. Flash-chromatography was performed using 1 : 1 EtOAc–Hex: IR νmax (NaCl; cm−1) 2739, 1764, 1527, 1348 and 1204; δH (300 MHz, CDCl3) 8.27 (2 H, d, J 5.1, ArH), 7.38 (2 H, d, J 5.1, ArH), 5.20–5.16 (2 H, m, OCH2O), 4.83–4.81 (1 H, m, 4-H), 4.68 (2 H, d, J 5.7, 1-H, 2-H), 2.38 (2 H, d, J 14.7, 3-H′, 5-H′) and 2.11–2.02 (2 H, m, 3-H″, 5-H″); δC (100 MHz, CDCl3) 155.4, 126.1, 125.2, 121.8, 115.5, 95.0, 80.7, 80.4 and 38.4; m/z (CI) 296 (M + H, 100); HRMS (M + H)+ calcd for C13H14NO7, 296.0770; found, 296.0769.
(5aα,7α,8aα)-7-(4-Nitrophenoxycarbonyloxy)-tetrahydrocyclopenta[f ]-1,3,5-trioxepane (20)
The title compound was obtained from 12 as described for 18 in 94% yield. Flash-chromatography was performed using 1 : 6 EtOAc–CHCl3: IR νmax (NaCl; cm−1) 2587, 1765, 1594, 1528, 1349 and 858; δH (300 MHz, CDCl3) 8.25 (2 H, d, J 8.0, ArH), 7.39 (2 H, d, J 8.0, ArH), 5.20 (2 H, d, J 7.5, 2-H′, 4-H′), 5.10–5.02 (1 H, m, 7-H), 4.75 (2 H, d, J 7.5, 2-H″, 4-H″), 4.29–4.24 (2 H, m, 5a-H, 8a-H), 2.51–2.41 (2 H, m, 6-H′, 8-H′) and 2.25–2.17 (2 H, m, 6-H″, 8-H″); δC (75 MHz, CDCl3) 155.2, 126.2, 125.2, 121.7, 115.6, 94.6, 80.8, 76.6 and 36.9; m/z (CI) 324 (M – H, 100).
(1β,2β,4α)-4-(4-Nitrophenoxycarbonyloxy)-1,2-(ethylenedioxy)-cyclopentane (21)
The title compound was obtained from 15 as described for 18 in 81% yield. Flash-chromatography was performed using 1 : 4 EtOAc–CHCl3: IR νmax (NaCl; cm−1) 2655, 1757, 1592, 1503, 1337, 852 and 754; δH (300 MHz, CDCl3) 8.29 (2 H, d, J 7.3, ArH), 7.36 (2 H, d, J 7.3, ArH), 5.22–5.18 (1 H, m, 4-H), 3.86–384 (2 H, m, 1-H, 2-H), 3.78–3.63 (4 H, m, CH2O), 2.38–2.24 (4 H, m, 3-H2, 5- H2); δC (100 MHz, CDCl3) 161.8, 126.2, 125.3, 121.7, 115.6, 78.1, 74.3, 62.1 and 33.9; m/z (CI) 310 (M + H, 100).
(1β,2β,4β)-4-(4-Nitrophenoxycarbonyloxy)-1,2-(ethylenedioxy)-cyclopentane (22)
The title compound was obtained from 16 as described for 18 in 95% yield. Flash-chromatography was performed using 1 : 4 EtOAc–CHCl3: IR νmax (NaCl; cm−1) 2588, 1725, 1594, 1222, 1109 and 773; δH (400 MHz, CDCl3) 8.27 (2 H, d, J 7.0, ArH), 7.38 (2 H, d, J 7.0, ArH), 5.14–5.10 (1 H, m, 4-H), 3.99 (2 H, t, J 4.6, 1-H, 2-H), 3.91–3.86 (2 H, m, CH2O), 3.64–3.59 (2 H, m, CH2O) and 2.31–2.18 (4 H, m, 3-H2, 5-H2); δC (100 MHz, CDCl3) 162.5, 126.1, 125.2, 121.7, 115.5, 81.4, 74.3, 62.3 and 32.5; m/z (CI) 310 (M + H, 100).
(1′S,2′R)-{1′-Benzyl-2′-hydroxy-3′-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl} carbamic acid (1β,2β,4β)-1,2-(methylenedioxy)cyclopent-4-yl ester (2)
A solution of 24 (25 mg, 0.05 mmol) in 30% trifluoroacetic acid in CH2Cl2 (4 mL) was stirred at 23 °C for 40 min and then the solvent was removed under reduced pressure. The residue was dissolved in THF (3 mL), a solution of 19 (18 mg, 0.059 mmol) in THF (1 mL) and diisopropylethylamine (100 μL, 0.6 mmol) were added. After 24 h the organic phase was diluted with CHCl3, washed with water, dried (Na2SO4), and evaporated. The residue was purified by flash-chromatography eluting with a 1 : 1 mixture of EtOAc and hexanes to afford 2 (20 mg, 74%) as a white solid: (c 1.2 in CH2Cl2), mp 68 °C(from EtOAc–Hex); IR νmax (NaCl; cm−1) 3129, 2801, 2660, 1711, 1597, 1497, 1155 and 761; δH (300 MHz, CDCl3) 7.71 (2 H, d, J 8.8, ArH), 7.32–7.19 (5 H, m, ArH), 6.98 (2 H, d, J 8.8, ArH), 5.01 (1 H, s, OCHHO), 4.92 (1 H, br. s, NH), 4.80 (2 H, m, 4-H, OCHHO), 4.57 (2 H, d, J 5.4, 1-H, 2-H), 3.87 (3 H, s, OCH3), 3.79 (2 H, m, CHN, CHOH), 3.10–2.76 (6 H, m, CH2N, CH2Ph), 2.11–1.80 [5 H, m, 3-H2, 5-H2, CH (CH3)2], 0.90 (3 H, d, J 6.6, CHCH3) and 0.86 (3 H, d, J 6.6, CHCH 3); δC (75 MHz, CDCl3) 162.9, 155.3, 137.5, 129.9, 129.6, 129.3, 128.5, 126.4, 114.3, 94.7, 80.5, 74.2, 72.3, 58.8, 55.6, 54.9, 53.8, 38.5, 35.4, 27.3, 20.2 and 19.9; m/z (ES) 563 (M + H, 100); HRMS (M + H)+ calcd For C28H39N2O8S, 563.2427; found, 563.2406.
(1′S,2′R)-{1′-Benzyl-2′-hydroxy-3′-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl} carbamic acid (1β,2β,4α)-1,2-(methylenedioxy)cyclopent-4-yl ester (26)
A solution of 24 (40 mg, 0.079 mmol) in 30% trifluoroacetic acid in CH2Cl2 (6 mL) was stirred at 23 °C for 40 min and then the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2 (4 mL), a solution of 17 (23 mg, 0.1 mmol) in CH2Cl2 (2 mL) and diisopropylethylamine (140 μL, 0.8 mmol) were added. After 2 h the organic phase was washed with water, dried (Na2SO4) and evaporated. The residue was purified by flash-chromatography (1 : 1 EtOAc–Hex) to afford 26 (34 mg, 76%) as a white foam: (c 1.3 in CH2Cl2); IR νmax (NaCl; cm−1) 3216, 2801, 2670, 1712, 1597, 1497, 1154 and 755; δH (300 MHz, CDCl3) 7.70 (2 H, d, J 8.7, ArH), 7.32–7.21 (5 H, m, ArH), 7.00 (2 H, d, J 8.7, ArH), 5.06 (1 H, t, J 7.0, 4-H), 4.93 (1 H, s, OCHHO), 4.76 (1 H, d, J 8.4, NH), 4.71 (1 H, s, OCHHO), 4.52 (2 H, m, 1-H, 2-H), 3.87 (3 H, s, OCH3), 3.84 (2 H, m, CHN, CH OH), 3.11 (1 H, dd, J 8.0, 14.8, CHHN), 3.04–2.91 (4 H, m, CHHN, CH2N, CH HPh), 2.78 (1 H, dd, J 6.7, 13.1, CHH Ph), 2.17–2.10 (2 H, m, 3-H′, 5-H′), 1.86–1.58 [3 H, m, 3-H″, 5-H″, CH(CH3)2], 0.91 (3 H, d, J 6.6, CHCH 3) and 0.87 (3 H, d, J 6.9, CHCH3); δC (75 MHz, CDCl3) 162.9, 155.8, 137.6, 129.9, 129.7, 129.4, 128.4, 126.5, 114.3, 94.3, 78.5, 74.5, 72.6, 58.8, 55.6, 54.9, 53.7, 37.8, 35.3, 27.3, 20.2 and 19.9; m/z (ES) 585 (M + Na, 100); HRMS (M + Na)+ calcd for C28H38N2NaO8S, 585.2247; found, 585.2228.
(1S,2R)-{1′-Benzyl-2′-hydroxy-3′-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl} carbamic acid (5aα,7β,8aα)-tetrahydrocyclopenta[f ]-1,3,5-trioxaepan-7-yl ester (27)
The title compound was obtained from 24 and 18 as described for 2 in 43% yield. Flash-chromatography was performed with 1 : 4 EtOAc–CHCl3: (c 1.7 in CH2Cl2); IR νmax (NaCl; cm−1) 3118, 2825, 2656, 1712, 1596, 1012 and 771; δH (300 MHz, CDCl3) 7.70 (2 H, d, J 9.0, ArH), 7.32–7.21 (5 H, m, ArH), 6.98 (2 H, d, J 9.0, ArH), 5.15 (2 H, d, J 7.2, 2-H′, 4-H′), 5.05 (1 H, br. s, NH), 4.76 (1 H, d, J 8.4, 7-H), 4.68 (2 H, d, J 7.2, 2-H″, 4-H″), 4.32–4.23 (2 H, m, 5a-H, 8a-H), 3.87 (3 H, s, OCH3), 3.83–3.80 (2 H, m, CHN, CHOH), 3.10 (1 H, dd, J 8.4, 15.3, CHHN), 3.04–2.88 (4 H, m, CHHN, CH2N, CH HPh), 2.78 (1 H, dd, J 6.9, 13.5, CHHPh), 2.09–1.94 (4 H, m, 6-H2, 8-H2), 1.86–1.77 [1 H, m, CH(CH3)2], 0.91 (3 H, d, J 6.9, CHCH 3) and 0.87 (3 H, d, J 6.3, CHCH 3); δC (75 MHz, CDCl3) 163.0, 155.7, 137.6, 129.8, 129.7, 129.4, 128.4, 126.5, 114.3, 95.4, 81.5, 73.6, 72.7, 58.8, 55.7, 54.9, 53.7, 37.8, 35.4, 27.3, 20.2 and 19.9; m/z (ES) 615 (M + Na, 100); HRMS (M + Na)+ calcd for C29H40N2NaO9S, 615.2353; found, 615.2361.
(1′S,2′R)-{1′-Benzyl-2′-hydroxy-3′-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl} carbamic acid (5aα,7α,8aα)-tetrahydrocyclopenta[f ]-1,3,5-trioxaepan-7-yl ester (28)
The title compound was obtained from 24 and 20 as described for 2 in 42% yield. Flash-chromatography was performed with 1 : 1 EtOAc–Hex: (c 1.7 in CH2Cl2); IR νmax (NaCl; cm−1) 3117, 2801, 2707, 1711, 1596, 1260 and 1153; δH (300 MHz, CDCl3) 7.70 (2 H, d, J 8.7, ArH), 7.31–7.21 (5 H, m, ArH), 6.97 (2 H, d, J 8.7, ArH), 5.14 (2 H, d, J 6.9, 2-H′, 4-H′), 4.91 (1 H, d, J 7.8, NH), 4.83–4.78 (1 H, m, 7-H), 4.68 (2 H, d, J 6.9, 2-H″, 4-H″), 4.15–4.10 (2 H, m, 5a-H, 8a-H), 3.87 (3 H, s, OCH3), 3.81–3.83 (2 H, m, CHN, CHOH), 3.12–2.85 (5 H, m, 2 × CH2N, CH HPh), 2.77 (1 H, dd, J 6.9, 13.5, CHHPh), 2.34–2.21 (2 H, m, 6-H′, 8-H′), 1.94–1.76 [3 H, m, 6-H″, 8-H″, CH(CH3)2], 0.90 (3 H, d, J 6.6, CHCH 3) and 0.86 (3 H, d, J 6.6, CHCH 3); δC (100 MHz, CDCl3) 162.9, 156.1, 137.5, 129.8, 129.5, 129.4, 128.4, 126.4, 114.3, 94.8, 81.0, 72.3, 71.3, 58.6, 55.6, 55.0, 53.6, 37.1, 35.5, 27.1, 20.1 and 19.8; m/z (ES) 615 (M + Na, 100); HRMS (M + Na)+ calcd for C29H40N2NaO9S, 615.2353; found, 615.2349.
(1′S,2′R)-{1′-Benzyl-2′-hydroxy-3′-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl} carbamic acid (1β,2β,4β)-1,2-(ethylenedioxy)cyclopent-4-yl ester (3)
The title compound was obtained from 24 and 22 as described for 2 in 40% yield. Flash-chromatography was performed with 1 : 1 EtOAc–Hex: (c 0.7 in CH2Cl2); IR νmax (NaCl; cm−1) 3120, 2788, 2656, 2542, 1712, 1596, 1259, 1154 and 755; δH (500 MHz, CDCl3) 7.70 (2 H, d, J 9.0, ArH), 7.31–7.22 (5 H, m, ArH), 6.97 (2 H, d, J 9.0, ArH), 4.90–4.86 (2 H, m, NH, 4-H), 3.87 (3 H, s, OCH3), 3.85–3.79 (7 H, m, 2 × CH2O, 1-H, 2-H, OH), 3.57–3.54 (2 H, m, CHN, CHOH), 3.11 (1 H, dd, J 8.2, 14.7, CHHN), 3.03–2.88 (4 H, m, CHHN, CH2N, CH HPh), 2.78 (1 H, dd, J 6.7, 13.2, CHHPh), 2.17–2.08 (2 H, m, 3-H′, 5-H′), 1.98–1.95 (2 H, m, 3-H″, 5-H″), 1.90 [1 H, dt, J 5.2, 15.0, CH(CH3)2], 0.91 (3 H, d, J 6.5, CHCH 3) and 0.86 (3 H, d, J 6.5, CHCH3); δC (75 MHz, CDCl3) 163.0, 156.2, 137.6, 129.8, 129.6, 129.5, 128.5, 126.5, 114.3, 74.5, 73.2, 72.5, 71.8, 62.5, 62.3, 58.8, 55.6, 55.0, 53.8, 35.5, 33.8, 33.5, 27.3, 20.2 and 19.9; m/z (ES) 599 (M + Na, 100); HRMS (M + Na)+ calcd for C29H40N2NaO8S, 599.2403; found, 599.2394.
(1′S,2′R)-{1′-Benzyl-2′-hydroxy-3′-[isobutyl(4-methoxybenzenesulfonyl)amino]propyl} carbamic acid (1β,2β,4α)-1,2-(ethylenedioxy)cyclopent-4-yl ester (29)
The title compound was obtained from 24 and 21 as described for 2 in 40% yield. Flash-chromatography was performed with 1 : 1 EtOAc–Hex: (c 1.0 in CH2Cl2); IR νmax (NaCl; cm−1) 3121, 2706, 1711, 1596, 1260, 1154 and 757; δH (500 MHz, CDCl3) 7.70 (2 H, d, J 8.7, ArH), 7.31–7.28 (2 H, m, ArH), 7.24–7.22 (3 H, m, ArH), 6.98 (2 H, d, J 8.7, ArH), 5.09 (1 H, br. s, NH), 4.74 (1 H, d, J 8.0, 4-H), 4.06–4.01 (2 H, m, 1-H, 2-H), 3.87 (3 H, s, OCH3), 3.82–3.81 (2 H, m, CH2O), 3.75–3.71 (2 H, m, CH2O), 3.55–3.51 (2 H, m, CHN, CHOH), 3.10 (1 H, dd, J 15.0, 8.5, CHHN), 3.03–2.86 88 (4 H, m, CHHN, CH2N, CH HPh), 2.78 (1 H, dd, J 13.5, 6.5, CHHPh), 2.32–2.23 (2 H, m, 3-H′, 5-H′), 1.81 (1 H, q, J = 6.5, 3-H″), 1.79–1.68 (1 H, m, 5-H″),1.62–1.53 [1 H, m, CH(CH3)2], 0.91 (3 H, d, J 6.6, CHCH 3) and 0.86 (3 H, d, J 6.6, CHCH 3); δC (75 MHz, CDCl3) 163.1, 156.1, 137.6, 129.8, 129.6, 129.5, 128.5, 126.6, 114.4, 74.6, 73.2, 72.7, 62.2, 58.8, 55.7, 54.9, 53.8, 35.4, 34.3, 34.2, 27.3, 20.2 and 19.9; m/z (ES) 599 (M + Na, 100); HRMS (M + H)+ calcd for C29H40N2NaO8S, 599.2403; found, 599.2421.
(1′S,2′R)-{1′-Benzyl-2′-hydroxy-3′-[isobutyl(4- (hydroxymethyl)benzenesulfonyl)amino]propyl} carbamic acid (1β,2β,4β)-1,2-(methylenedioxy)cyclopent-4-yl ester (30)
To a solution of 258 (40 mg, 0.1 mmol) and diisopropylethylamine (150 μL, 0.9 mmol) in THF (3 mL), a solution of 17 (30 mg, 0.11 mmol) was added and the resulting mixture was stirred at 23 °C. After 48 h, the organic phase was diluted with CHCl3, washed with water, dried (Na2SO4) and evaporated. The residue was purified by flash-chromatography (2 : 1 EtOAc–Hex) to afford 30 (35 mg, 63%) as an amorphous solid: (c 1.3 in CHCl3); IR νmax (NaCl; cm−1) 3042, 2996, 2707, 1710, 1530, 1334, 1156 and 755; δH (400 MHz, CDCl3) 7.77 (2 H, d, J 8.1, ArH), 7.52 (2 H, d, J 8.1, ArH), 7.32–7.21 (5 H, m, ArH), 5.00 (1 H, s, NH), 4.92 (1 H, m, 4-H), 4.82–4.80 (4 H, m, OCH2O, CH 2OH), 4.58–4.57 (2 H, m, l-H, 2-H), 3.81–3.79 (2 H, m, CHN, CHOH), 3.11–2.83 (6 H, m, 2 × CH2N, CH2Ph), 6H), 2.10–1.82 [5 H, m, 3-H2, 5-H2, CH(CH3)2], 0.91 (3 H, d, J 6.6, CHCH 3) and 0.83 (3 H, d, J 6.6, CHCH 3); δC (100 MHz, CDCl3) 156.2, 146.2, 137.5, 137.1, 129.5, 128.5, 127.5, 127.1, 126.5, 94.6, 80.5, 75.8, 72.2, 64.0, 58.5, 55.1, 53.5, 38.4, 35.4, 27.1, 20.0 and 19.8; m/z (ES) 585 (M + Na, 100); HRMS (M +Na)+ calcd for C28H38N2NaO8S, 585.2247; found, 585.2246.
X-Ray crystallography
The HIV-1 protease construct with the substitutions Q7K, L33I, L63I, C67A, and C95A to optimize protein stability,20 was expressed and purified as described.21 Crystals were grown by the hanging drop vapor diffusion method using a 1 : 15 molar ratio of protease at 2.0 mg mL−1 and the inhibitor dissolved in dimethylsulfoxide. The reservoir contained 0.1 M sodium acetate buffer (pH = 4.2) and 1.5 M NaCl. Crystals were transferred into a cryoprotectant solution containing the reservoir solution and 20–30% (v/v) glycerol, mounted on a nylon loop and flash-frozen in liquid nitrogen. X-ray diffraction data were collected on the SER-CAT beamline of the Advanced Photon Source, Argonne National Laboratory. Diffraction data were processed using HKL200022 resulting in a Rmerge value of 7.0% (41.8%) for 90 315 unique reflections between 50 and 1.07 Å resolution with a completeness of 88.1% (51.3%), where the values in parentheses are for the final highest resolution shell. Data were reduced in space group P21212 with unit cell dimensions of a = 58.00 Å, b = 86.34 Å and c = 45.83 Å with one dimer in the asymmetric unit. The structure was solved by molecular replacement using the CPP4i suite of programs,23,24 with the structure of the D30N mutant of HIV protease in complex with GRL-98065 (2QCI)19 as the starting model. The structure was refined using SHELX9725 and refitted manually using the molecular graphics programs O26 and COOT.27 Alternate conformations were modeled for the protease residues when obvious in the electron density maps. Anisotropic atomic displacement parameters (B-factors) were refined for all atoms including solvent molecules. Hydrogen atoms were added at the final stages of the refinement. The identity of ions and other solvent molecules from the crystallization conditions was deduced from the shape and peak height of the 2Fo–Fc and Fo–Fc electron density, the hydrogen bond interactions and interatomic distances. The solvent structure was refined with one sodium ion, three chloride ions, and 203 water molecules including partial occupancy sites. The final Rwork was 15.2% and RBfree was 17.7% for all data between 10 and 1.07 Å resolution. The rmsd values from ideal bonds and angle distances were 0.015 Å and 0.034 Å, respectively. The average B-factor was 13.1 and 18.2 Å 2 for protease main chain and side chain atoms, respectively, 12.5 Å2 for inhibitor atoms and 24.0 Å2 for solvent atoms. The X-ray crystal structure of the inhibitor 3 complex with the HIV-1 protease has been deposited in the Protein Databank (PDB)28 with an access code of 3DKJ.
Supplementary Material
Acknowledgments
The research was supported by grants from the National Institutes of Health (GM53386, AKG, and GM62920, IW). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and in part by a Grant-in-aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan (Kosei Rohdosho: H15-AIDS-001), and a Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re-emerging Infectious Diseases (Renkei Jigyo: No. 78, Kumamoto University) of Monbu-Kagakusho.
Footnotes
Electronic supplementary information (ESI) available: HPLC and HRMS data of inhibitors 2–3 and 26–30; crystallographic data collection and refinement statistics. See DOI: 10.1039/b809178a
Notes and references
- 1.(a) Roberts NA, Martin JA, Kinchington D, Broadhurst AV, Craig JC, Duncan IB, Galpin SA, Handa BK, Kay J, Krohn A. Science. 1990;248:358. doi: 10.1126/science.2183354. [DOI] [PubMed] [Google Scholar]; (b) Meek TD, Lambert DM, Dreyer GB, Carr TJ, Tomaszek TA, Moore ML, Strickler JE, Debouck C, Hyland LJ, Matthews TJ, Metcalf BW, Petteway SR. Nature. 1990;343:90. doi: 10.1038/343090a0. [DOI] [PubMed] [Google Scholar]; (c) McQuade TJ, Tomasselli AG, Liu L, Karacostas V, Moss B, Sawyer TK, Heinrikson RL, Tarpley WG. Science. 1990;247:454. doi: 10.1126/science.2405486. [DOI] [PubMed] [Google Scholar]
- 2.(a) Flexner C. N Engl J Med. 1998;338:1281. doi: 10.1056/NEJM199804303381808. [DOI] [PubMed] [Google Scholar]; (b) Cihlar T, Bischofberger N. Annu Rep Med Chem. 2000;35:177. [Google Scholar]
- 3.(a) Wainberg MA, Friedland G. JAMA, J Am Med Assoc. 1998;279:1977. doi: 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]; (b) Grabar S, Pradier C, Le Corfec E, Lancar R, Allavena C, Bentata M, Berlureau P, Dupont C, Fabbro-Peray P, Poizot-Martin I, Costagliola D. AIDS. 2000;14:141. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]; (c) Hertogs K, Bloor S, Kemp SD, Van den Eynde C, Alcorn TM, Pauwels R, Van Houtte M, Staszewski S, Miller V, Larder BA. AIDS. 2000;14:1203. doi: 10.1097/00002030-200006160-00018. [DOI] [PubMed] [Google Scholar]
- 4.On June 23, 2006, the FDA approved a new HIV treatment for patients who do not respond to existing drugs. http://www.fda.gov/bbs/topics/NEWS/2006/NEW01395.html
- 5.(a) Surleraux DLNG, Tahri A, Verschueren WG, Pille GME, de Kock HA, Jonckers THM, Peeters A, De Meyer S, Azijn H, Pauwels R, de Bethune M-P, King NM, Prabu-Jeyabalan M, Schiffer CA, Wigerinck PBTP. J Med Chem. 2005;48:1813. doi: 10.1021/jm049560p. [DOI] [PubMed] [Google Scholar]; (b) Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2003;47:3123. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ghosh AK, Pretzer E, Cho H, Hussain KA, Duzgunes N. Antiviral Res. 2002;54:29. doi: 10.1016/s0166-3542(01)00209-1. [DOI] [PubMed] [Google Scholar]
- 6.(a) Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. Bioorg Med Chem Lett. 1998;8:687. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]; (b) Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. J Virol. 2002;76:1349. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. Bioorg Med Chem Lett. 1998;8:687. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]; (b) Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. J Virol. 2002;76:1349. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J Med Chem. 2006;49:5252. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 9.Ashkenazi P, Kalo J, Rüttimann A, Ginsburgh D. Tetrahedron. 1978:2161. [Google Scholar]
- 10.Godjoian G, Wang VR, Ayala AM, Martínez RV, Martínez-Bernhardt R, Gutiérrez CG. Tetrahedron Lett. 1996;37:433. [Google Scholar]
- 11.Ghosh AK, Duong TT, McKee SP, Thompson WJ. Tetrahedron Lett. 1992;33:2781. doi: 10.1016/S0040-4039(00)78856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Fidanze S. J Org Chem. 1998;63:6146. doi: 10.1021/jo980159i. [DOI] [PubMed] [Google Scholar]
- 13.Toth MV, Marshall GR. Int J Pept Protein Res. 1990;36:544. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 14.Perno CF, Yarchoan R, Cooney DA, Hartman NR, Webb DSA, Hao Z, Mitsuya H, Johns DG, Broder S. J Exp Med. 1989;169:933. doi: 10.1084/jem.169.3.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. FEBS J. 2005;272:5265. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gustchina A, Sansom C, Prevost M, Richelle J, Wodak S, Wlodawer A, Weber I. Protein Eng. 1994;7:309. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- 17.Panigrahi S, Desiraju G. Proteins: Struct, Funct, Bioinf. 2007;67:128. doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 18.Steiner T. Biophys Chem. 2002;95:195. doi: 10.1016/s0301-4622(01)00256-3. [DOI] [PubMed] [Google Scholar]
- 19.Wang YF, Tie Y, Boross PI, Tozser J, Ghosh AK, Harrison RW, Weber IT. J Med Chem. 2007;50:4509. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louis JM, Clore GM, Gronenborn AM. Nat Struct Biol. 1999;6:868. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 21.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Proteins: Struct, Funct, Genet. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Collaborative Computational Project, Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 24.Potterton E, Briggs P, Turkenburg M, Dodson E. Acta Crystallogr. 2003;D59:1131. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 25.Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319. [PubMed] [Google Scholar]
- 26.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr. 1991;A47:110. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Acta Crystallogr. 2004;D60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.