

Abstract
Polo-like kinase 1 (Plk1) is becoming an increasingly attractive target for cancer management. Plk1 has been shown to be over-expressed in a variety of cancers; however its role in skin cancers is not well-understood. We recently demonstrated that Plk1 is over-expressed in human melanoma and gene-knockdown as well as chemical-inhibition of Plk1 resulted in a significant decrease in melanoma cell viability and growth without affecting the growth of the normal human epidermal melanocytes (NHEMs). Further, the observed anti-proliferative response of Plk1 was found to be accompanied with a significant G2/M cell cycle arrest, mitotic catastrophe and induction of apoptosis in melanoma cells. In this study, we determined the expression profile of Plk1 in non-melanoma skin cancers viz. basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). Our data demonstrated that like melanoma, Plk1 is significantly over-expressed in BCC and SCC samples. Further, we also found that compared to normal human epidermal keratinocytes (NHEKs), Plk1 was over-expressed at both the protein and mRNA levels in squamous A253 and A431 cells. In addition, a similar protein expression pattern was found for the downstream targets of Plk1, viz. Cdk1, Cyclin B1, and Cdc25C. We believe that the expression pattern of Plk1 in the various skin cancers, the insusceptibility of normal keratinocytes, to Plk1 inhibition and the easy accessibility for topical applications lends the skin as an attractive tissue for Plk1 based cancer chemoprevention and chemotherapeutic applications.
Keywords: Polo-like kinase, Skin Cancer, BCC, SCC, Mitosis
Introduction
Skin cancer is by far the most diagnosed cancer in the United States. With over one million new cases diagnosed annually, which translates to over one third of all new cancer cases, it is evident that the current educational and preventative measures have not been effective in managing this neoplasm 1. The basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) account for >90% of the skin cancers diagnosed 1. Even with the low incidence of associated mortality, BCC and SCC are associated with a significant health care burden and morbidity that warrants for both novel mechanism-based approaches for their prevention, treatment, and post-operative control for disease reoccurrence.
Cumulative ultraviolet (UV) radiation has been demonstrated to be the primary contributor to the majority of skin cancers. This is reinforced by the fact that most non-melanoma skin cancers develop in sun exposed areas, occur at a higher frequency in fair skinned individuals, incidence increases with a decrease in latitude, and about 50% of all skin cancers show mutations in p53, particularly C → T and CC → TT transitions, which are hallmarks of UV induced mutagenesis 2,3. However, the molecular pathways contributing to BCC and SCC development beyond an initiating mutation in p53 or other genes are not completely understood.
Polo-like kinases (Plks), a family of conserved serine/threonine kinases, are important regulators of cell cycle progression and are gaining increasing attention as a possible target for cancer management 4. Mammalian Plks are a group of four kinases with varying roles throughout the cell cycle. They each contain a conserved N-terminal catalytic domain and one (Plk4) or two (Plks 1-3) C-terminal polo-box domains (PBD) that have been implicated in protein localization to various cellular structures and binding to their targets 4,5. The Plk family encompasses both pro- and anti-proliferative members. Plk1 regulates reentry into the cell cycle after the DNA damage checkpoint, promotes the entrance of the cell through the G2/M transition, and then plays a critical role in bipolar spindle formation, centrosome maturation, chromosome segregation and finally cytokinesis 4,5. Throughout these stages, continued Plk1 expression and activity promote advancement through the cell cycle. Conversely, Plk2 and Plk3 play anti-proliferative roles during centriole duplication and the DNA damage checkpoint 6,7,8. Finally, Plk4 is the most divergent of the Plks with only one PBD and is involved in centriole duplication where loss of Plk4 results in improper centrosome formation and disorganized spindle formation, and over-expression results in multiple centrosome formation which can lead to anueploidy 9.
In a recent study, we demonstrated that Plk1 is significantly over-expressed in i) human malignant melanoma tissues versus normal skin, and ii) human melanoma cells versus normal human epidermal melanocytes (NHEMs) 10. Further, we found that short hairpin RNA (shRNA)-mediated knockdown of Plk1 as well as Plk1 small molecule inhibitor-mediated activity inhibition resulted in a significant decrease in cell viability and proliferation in human melanoma cell lines, without any observable effect on NHEMs 10. This was in line with the findings of Liu and Erikson that show that unless Plk1 expression is completely null or p53 is concomitantly knocked down with Plk1, normal cells are able to continue cell division without any observable mitotic defects or growth disadvantages 11. In addition, we found that inhibition of Plk1 resulted in i) a G2/M cell cycle arrest, multiple mitotic abnormalities (with predominant “polo” phenotype, typically seen in Plk1 mutants or knockouts), and iii) induction of apoptosis, in human melanoma cells 10.
This study was designed to evaluate the expression patterns of Plk1 in non-melanoma skin cancers viz. BCC and SCC. Our data demonstrated that compared to normal skin, Plk1 is significantly over-expressed in BCC as well as SCC clinical samples as well as in squamous carcinoma cell lines viz. A253 and A431.
Results and Discussion
In order to ascertain the expression profile of Plk1 in non-melanoma skin cancers, employing commercial tissue micro-arrays (TMA) and immunohistochemical analysis, we evaluated the expression pattern of Plk1 in BCC and SCC tumor samples compared to normal skin. As shown by immunostaining, we found that Plk1 was significantly over-expressed in both BCC (Fig. 1) and SCC (Fig. 2) samples when compared to normal skin. Though the increased over-expression in both cancer types was statistically significant, it is interesting to note that the number of cores with increased Plk1 expression compared to control does noticeably differ between the BCC and SCC TMAs and between the number of positive cores we previously found in melanoma 10. Only about 37% of BCC samples were scored as either 2+ or 3+, whereas about 59% of SCC (in this study) and melanoma samples (previously) were scored similarly.
Figure 1. Plk1 is over-expressed in basal cell carcinoma versus normal skin tissue.
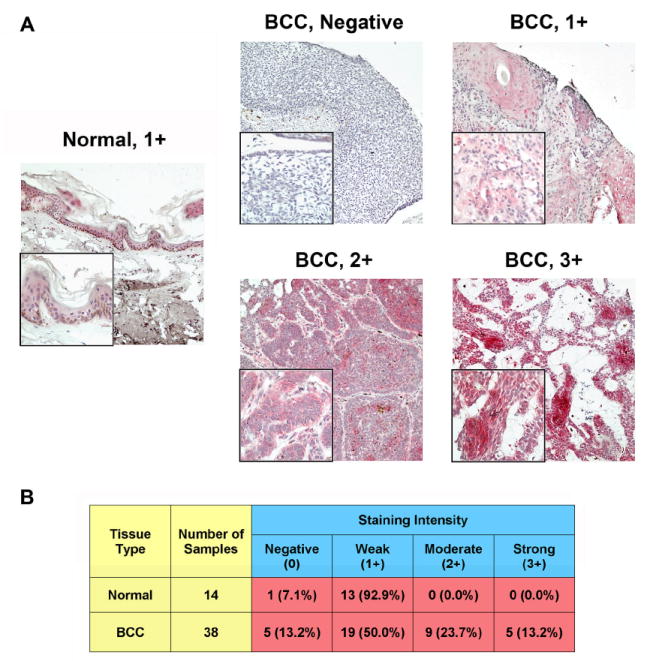
A) Immunohistochemical staining for Plk1. Paraffin embedded human BCC and normal skin tissue arrays were obtained from Biomax USA (Rockville, MD). The tissue arrays were deparaffinized with xylenes and ethanol series and antigen retrieval performed by heating in 1 mM EDTA (pH 8.0) at 85°C. Slides were blocked in 10% normal goat serum (Caltag, CA) in PBS for 1 hour at room temperature followed by incubation with Plk1 antibody (Upstate, MA) or IgG2b control anti-sera (data not shown) (Upstate, MA) diluted 1:100 in 10% normal goat serum in PBS overnight at 4°C in a humidified chamber. The following day, slides were incubated with biotin conjugated secondary antibody (Invitrogen, CA) (1:100 in blocking buffer) and then fresh ABC-Alkyline Phosphatase reagent (Vector Labs, CA) for 1 hour each at room temperature in a humidified chamber. Tissues were then washed with PBS and exposed to fresh Vector Red reagent (Vector Labs, CA) for 20 minutes giving the pink to deep red color for positive staining. Tissues were then counterstained with hematoxylin, dehydrated with ethanol and xylenes and mounted. Cores were analyzed and imaged on a Olympus BX41 bright field microscope and images were obtained with a digital camera (model 14.2 color Mosaic, Diagnostic Instruments, Inc., MI) and Spot software (Windows: Version 4, Diagnostic Instruments, Inc., MI). Images shown are 10X magnification, insets are 40X; B) Quantitation of Plk1 scoring intensity. Cores were scored blindly for staining intensity as negative (0), weak (1+), moderate (2+), or strong (3+). Normal and BCC cores were divided into two groups, those with a score of negative or 1+ versus those scored as 2+ or 3+. Data for was compared using a two-tailed Fisher’s exact test (P-value=0.0107). A P-value < 0.05 was considered statistically significant.
Figure 2. Plk1 is over-expressed in squamous cell carcinoma versus normal skin tissue.
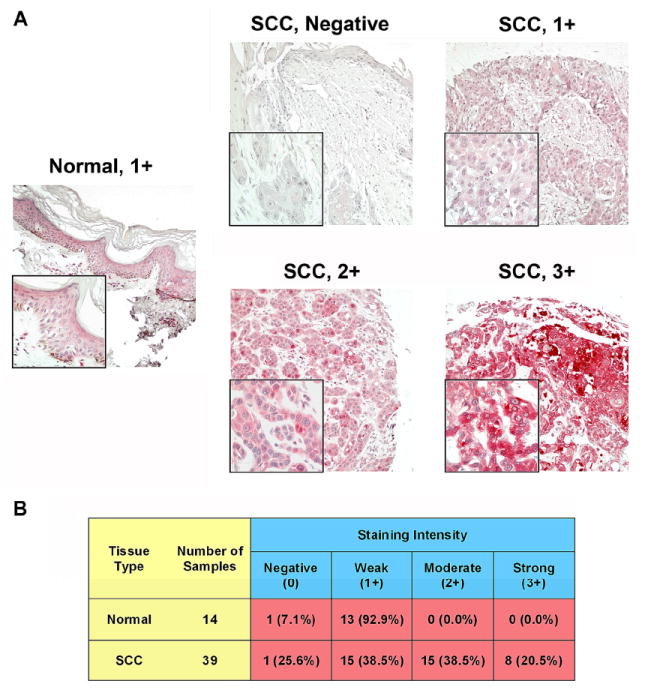
A) Immunohistochemical staining for Plk1. Paraffin embedded human SCC and normal skin tissue arrays were obtained from Biomax USA (Rockville, MD). The tissue arrays were stained for Plk1 similarly to the BCC tissue microarray described above in Fig. 4. Cores were analyzed and imaged on a Olympus BX41 bright field microscope and images were obtained with a digital camera (model 14.2 color Mosaic, Diagnostic Instruments, Inc., MI) and Spot software (Windows: Version 4, Diagnostic Instruments, Inc., MI). Images shown are 10X magnification, insets are 40X; B) Quantitation of Plk1 scoring intensity. Cores were scored blindly for staining intensity as negative (0), weak (1+), moderate (2+), or strong (3+). Normal and SCC cores were divided into two groups, those with a score of negative or 1+ versus those scored as 2+ or 3+. Data for was compared using a two-tailed Fisher’s exact test (P-value=7.5×10-5). A P-value < 0.05 was considered statistically significant.
In vitro we found that Plk1 is over-expressed at both the protein and mRNA levels in the squamous cell carcinoma lines (A253 and A431) when compared to normal keratinocytes (NHEK) (Fig. 3A & 3B). Further, a similar protein expression pattern was found for the downstream targets of Plk1, viz Cdk1, Cyclin B1, and Cdc25C (Fig. 3C). These data are not surprising given the enhanced cell proliferation kinetics found in cancer cells versus NHEKs, but do signify that modulating Plk1, or other mitotic specific proteins, may serve as a possible target to manage these skin cancers.
Figure 3. Plk1 and its downstream targets are over-expressed in squamous carcinoma A253 and A431 cells.
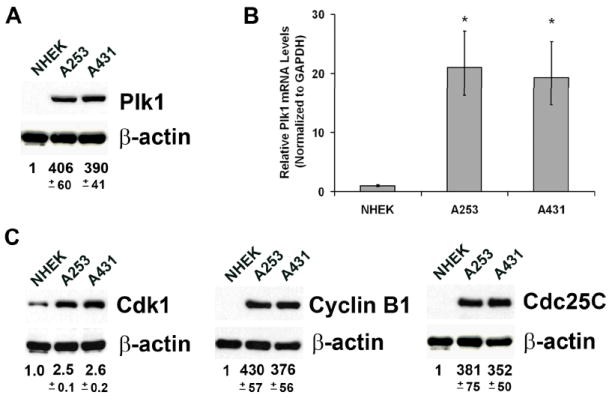
A) Plk1 protein expression. NHEK and human squamous cell carcinoma A253 and A431 cells were trypsinized, washed with ice-cold PBS and lysed for protein with RIPA buffer plus PMSF and protease inhibitor cocktail (Pierce, IL) for immunoblot analysis. Briefly, 30 μg of protein was subjected to SDS-PAGE and transferred onto nitrocellulose membrane. Blots were blocked followed by probing with Plk1 primary antibody (Upstate, MA) and then appropriate HRP-conjugated secondary antibodies followed by enhanced chemiluminescent detection. Blots were subsequently stripped and re-probed with goat anti-β-actin (Santa Cruz, CA) primary antibody followed by appropriate secondary and chemiluminescent detection as a loading control. The quantitation of protein was performed by digital analyses of protein bands (TIFF images) using UN-SCAN-IT software (Silk Scientific, Inc., UT) and the data represented as relative Plk1 protein levels +/- standard deviation of three experiments. All comparisons to NHEK expression were statistically significant with P-values < 0.01 using two-tailed Student’s T-test; B) Plk1 mRNA expression. RNA was isolated using Trizol reagent (Invitrogen, CA), treated with DNAse (Promega, WI), and first strand cDNA created with M-MLV reverse transcriptase (Promega, WI) according to vendor’s protocol. Quantitative real time RT-PCR was performed in triplicate in 20 μl reactions with SYBR® Premix Ex Taq™ Perfect Real Time (Takara, WI) with 50 ng first strand cDNA and 0.2 μg each Plk1 forward (5’-CCCCTCACAGTCCTCAATAA-3’) and reverse (5’-AATAGTCCACCCACTTGCTG-3’) primers or 0.2 μg each GAPDH forward (5’-GGGTGTGAACCATGAGAAGT-3’) and reverse (5’-GTAGAGGCAGGGATGATGTT-3’) primers. Data is expressed as relative Plk1 mRNA using the ΔΔCT method with GAPDH as an endogenous control +/- standard deviation for three separate experiments. *P-value < 0.01; C) Protein expression of Cdk1, Cdc25C or Cyclin B1. NHEKs, A253 and A431 cells were collected, lysed and analyzed using immunoblot analysis as described above. Blots were probed for Plk1 targets viz. Cdk1, Cdc25C or Cyclin B1 using appropriate antibodies (Cell Signaling, MA) and β-actin as loading control. Relative protein expression was quantitated as previously and presented as relative target protein levels +/- standard deviation of three separate experiments. All comparisons to NHEK expression were statistically significant with P-values < 0.01 using two-tailed Student’s T-Test.
The simplest explanation for the observed over-expression of Plk1 in BCC and SCC could be due to the consistent loss of functional p53. Plk1 transcription and enzymatic activity are controlled by p53, both directly and indirectly, and in a cell cycle specific manner and in response to DNA damage. In a normal cell, Plk1 transcriptional levels are negatively controlled by p53 and through the p53 target WAF1/p21, possibly through Plk1’s cell-cycle-dependent (CCD)/cell-cycle-gene homology region (CHR) within the Plk1 promoter region 12. Plk1 transcription is also regulated by the retinoblastoma (Rb) proteins in an E2F dependent manner 12. Further, Plk1 transcription is negatively correlated with DNA damage. The primary regulators of the DNA damage checkpoint are ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) which repress a large number of transcripts to halt cell cycle progression in response to DNA damage 13. And, indeed, they repress Plk1 transcription and activity when the DNA damage checkpoint is activated 14, 15. Further, it appears that Rb family members contribute to the reduced Plk1 transcription in a p53-dependent manner in response to DNA damage as well 16. Interestingly, Plk1 contributes to a negative feedback loop regulating p53, both directly and indirectly. Plk1 and p53 physically interact and Plk1 phosphorylation directly contributes to a reduction in p53 activity 17. Further, Plk1’s activating phosphorylation of Cdc25C further contributes to p53 destabilization. It was found that over-expression of Plk1 decreased the level of Serine 15 phosphorylation of p53 in the presence of UV radiation 18. It was also determined that this dephosphorylation is mediated by Cdc25C as Plk1, p53 and Cdc25C were found to form a complex 18.
Therefore, it is not unreasonable to hypothesize that a majority of the Plk1 over-expressing skin cancers may be a direct result of their loss of functional p53 (Fig. 4A). In response to UV exposure, a normal cell with functional p53 will halt cell cycle progression until DNA is repaired or the cell undergoes apoptosis. However, with the loss of functional p53, a sufficient DNA damage response and the suppression of Plk1 transcription may not occur. Without proper control of Plk1 transcription the cell may progress into and through mitosis without proper DNA repair, which can lead to aneuploidy and cancer. Further, with Plk1’s negative regulation of p53 itself, this may be further amplified in future cell divisions contributing to increased proliferation and aneuploidy.
Figure 4. Proposed mechanisms for observed Plk1 over-expression in BCC and SCC.
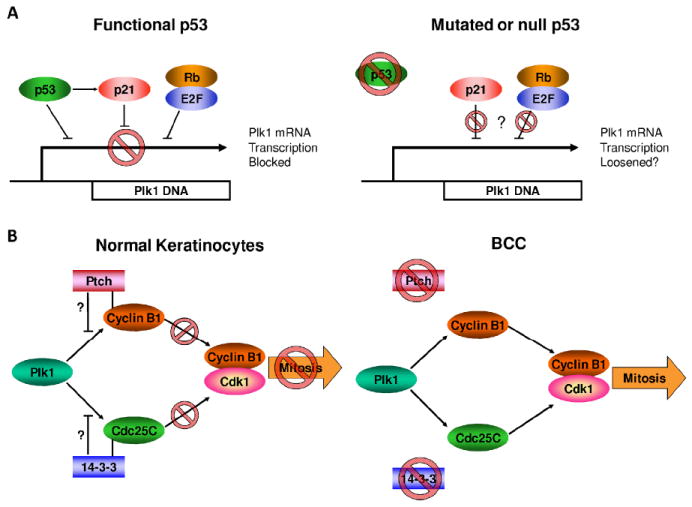
A) p53 regulates Plk1 transcription. Loss of functional p53 is one of the most common traits observed in both BCC and SCC samples. This may contribute to the observed increase in Plk1 because p53 regulates Plk1 transcription in both cell cycle and DNA damage dependent manners. In a cell with functional p53, Plk1 transcription is controlled by p53, WAF1/p21 and Rb-E2F proteins prior to S-phase where Plk1 regularly undergoes an increase in transcription. Further, p53 responds to DNA damage by inhibiting p53 both directly and indirectly through activation of WAF1/p21. With a loss of functional p53, the direct inhibition of Plk1 transcription is definitively lost and the activation and efficiency of WAF1/p21 may be lost. This may lead to loosened restrictions on Plk1 transcription resulting in premature mitotic entry.; B) Loss of function of Ptch and 14-3-3 in BCC may contribute to increased Plk1 signaling. Dysregulated Hedgehog signaling is commonly seen in BCC, and this is frequently due to loss of functional Ptch protein. Another common aberration in BCC is a loss of 14-3-3 protein. Both of these losses may contribute to an increase in Plk1 signaling through a loss of proper inhibition of direct Plk1 targets Cyclin B1 and Cdc25C, respectively.
However, in addition to p53, Hedgehog signaling is thought to be an important contributor to BCC development. This was first demonstrated when Patched (Ptch) was found to be mutated in the familial cancer nevoid basal cell carcinoma syndrome (NBCCS), as well as sporadic BCC 19,20. Further studies demonstrated that mutations causing loss-of-function in Ptch or gain-of-function in Smoothened (Smo) were present in > 70% of sporadic BCC cases 21, 22. In normal development, Sonic Hedgehog signaling (Shh) has been implicated in hair follicle growth and morphogenesis. In the absence of Shh ligand, the tumor suppressor Ptch inhibits Smo, which possesses characteristics of a G-protein coupled receptor 23. In the presence of secreted Shh, inhibition of Smo by Ptch is removed and Smo promotes Gli1 and Gli2 dissociation from an inhibitory complex, leading to the up-regulation of multiple Gli transcriptional targets 24,25. These Gli transcriptional targets include: Cyclin D1, Cyclin D2, Bcl2, IGF2, Ptch, and Gli1, among others 26. Unregulated Hedgehog signaling can be caused either by increased Shh, inactivation of Ptch or release of Smo from Ptch inhibition. Regardless of the mechanism of lost Hedgehog signaling, all scenarios ultimately result in increased cell proliferation.
In this regard, the findings related to Hedgehog signaling and Plk1 are worth mentioning. Ptch has been found to bind to and inhibit Cyclin B1 activation indicating another possible role Ptch may play in inhibiting cell proliferation and another mechanism by which loss of Ptch may contribute to tumor development 27. This also presents an additional attractive role Plk1 may play in regulation of mitosis where Ptch binding to Cyclin B1 may be a mechanism to prevent or regulate Plk1 phosphorylation of Cyclin B1 at the G2/M transition. Plk1 contributes to both Cyclin B1 and Cdc25C activation and localization at the G2/M transition 4. With a loss of functional Ptch, it may be possible that the regulation of Cyclin B1 is disturbed (Fig. 4B). The loss of Ptch/Cyclin B1 binding alone should not be sufficient for cell cycle progression, as Cyclin B1 requires Plk1 phosphorylation for proper localization. But if the function of Ptch/Cyclin B1 binding is to prevent Plk1 or a priming kinase’s phosphorylation of Cyclin B1, loss of Ptch may contribute to premature mitotic entry. Another alteration found in some BCC cases relevant to Plk1 and mitotic regulation is the loss of the tumor suppressor 14-3-3σ 28. The loss of 14-3-3 protein occasionally seen in BCC samples may also be associated with or contribute to the increased Plk1 expression or activity (Figure 4B). Normally, 14-3-3 binds Cdc25C to regulate the G2/M transition; however, with a loss of functional 14-3-3, the regulation Cdc25C may be loosened in a manner similar to that proposed for Cyclin B1. This would then allow for more efficient phosphorylation and activation by Plk1, thus also contributing to premature mitotic entry.
While BCCs possess an abrogation of the Hedgehog signaling, SCCs appear to proceed in a step-wise fashion involving subsequent mutations and genetic aberrations after UV exposure. However, there is not a canonical mechanism that contributes to SCC development. There is some evidence showing a loss of heterozygosity (LOH) of chromosome 9p in SCCs 29. This has lead some to speculate the loss of p16INK4 may contribute to SCC development, however immunohistochemical data has been contradictory so far 29. Further, studies have found LOH in chromosomes 3p, 13p, 17p and 17q and some gain of function translocations in 3q, 9q, and 11q , but any significant and consistent downstream consequences of this have not yet been elucidated 29-31. Therefore, the biggest difference between SCC and their precancerous lesions, actinic keratoses, may be an increase in genomic instability 29. This suggests that the loss of functional p53 may be the only consistent trait of SCC. The loss of proper DNA damage repair then contributes to further non-lethal genomic mutations or rearrangements, but there may not be a “typical” SCC profile.
Further in-depth studies are needed to decipher the functional relevance mechanism of Plk1 in skin cancers.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009 doi: 10.3322/caac.20006. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 2.Giglia-Mari G, Sarasin A. TP53 mutations in human skin cancers. Hum Mutat. 2003;21:217–228. doi: 10.1002/humu.10179. [DOI] [PubMed] [Google Scholar]
- 3.Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, Remington L, Jacks T, Brash DE. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 4.Schmit TL, Ahmad N. Regulation of mitosis via mitotic kinases: new opportunities for cancer management. Mol Cancer Ther. 2007;6:1920–1931. doi: 10.1158/1535-7163.MCT-06-0781. [DOI] [PubMed] [Google Scholar]
- 5.Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009;10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 6.Bahassi eM, Conn CW, Myer DL, Hennigan RF, McGowan CH, Sanchez Y, Stambrook PJ. Mammalian Polo-like kinase 3 (Plk3) is a multifunctional protein involved in stress response pathways. Oncogene. 2002;21:6633–6640. doi: 10.1038/sj.onc.1205850. [DOI] [PubMed] [Google Scholar]
- 7.Cizmecioglu O, Warnke S, Arnold M, Duensing S, Hoffmann I. Plk2 regulated centriole duplication is dependent on its localization to the centrioles and a functional polo-box domain. Cell Cycle. 2008;7:3548–3555. doi: 10.4161/cc.7.22.7071. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Xie S, Chen J, Fukasawa K, Naik U, Traganos F, Darzynkiewicz Z, Jhanwar-Uniyal M, Dai W. Cell cycle arrest and apoptosis induced by human Polo-like kinase 3 is mediated through perturbation of microtubule integrity. Mol Cell Biol. 2002;22:3450–3459. doi: 10.1128/MCB.22.10.3450-3459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–1146. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 10.Schmit TL, Zhong W, Setaluri V, Spiegelman VS, Ahmad N. Targeted Depletion of Polo-Like Kinase (Plk) 1 Through Lentiviral shRNA or a Small-Molecule Inhibitor Causes Mitotic Catastrophe and Induction of Apoptosis in Human Melanoma Cells. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.172. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol Cell Biol. 2006;26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin BT, Strebhardt K. Polo-like kinase 1: target and regulator of transcriptional control. Cell Cycle. 2006;5:2881–2885. doi: 10.4161/cc.5.24.3538. [DOI] [PubMed] [Google Scholar]
- 13.Hurley PJ, Bunz F. ATM and ATR: components of an integrated circuit. Cell Cycle. 2007;6:414–417. doi: 10.4161/cc.6.4.3886. [DOI] [PubMed] [Google Scholar]
- 14.Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000;2:672–676. doi: 10.1038/35023629. [DOI] [PubMed] [Google Scholar]
- 15.van Vugt MA, Smits VA, Klompmaker R, Medema RH. Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J Biol Chem. 2001;276:41656–41660. doi: 10.1074/jbc.M101831200. [DOI] [PubMed] [Google Scholar]
- 16.Jackson MW, Agarwal MK, Yang J, Bruss P, Uchiumi T, Agarwal ML, Stark GR, Taylor WR. p130/p107/p105Rb-dependent transcriptional repression during DNA-damage-induced cell-cycle exit at G2. J Cell Sci. 2005;118:1821–1832. doi: 10.1242/jcs.02307. [DOI] [PubMed] [Google Scholar]
- 17.Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, Fukuzawa M, Nakagawara A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem. 2004;279:25549–25561. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Dai G, Wang YQ, Wang S, Pan FY, Xue B, Zhao DH, Li CJ. Polo-like kinase 1 regulates mitotic arrest after UV irradiation through dephosphorylation of p53 and inducing p53 degradation. FEBS Lett. 2006;580:3624–3630. doi: 10.1016/j.febslet.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 19.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, Leffell DJ, Gerrard B, Goldstein AM, Dean M, Toftgard R, Chenevix-Trench G, Wainwright B, Bale AE. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 20.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH, Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 21.Gailani MR, Stahle-Backdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, Unden AB, Dean M, Brash DE, Bale AE, Toftgard R. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- 22.Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH, Jr, de Sauvage FJ. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 23.Philipp M, Caron MG. Hedgehog signaling: is Smo a G protein-coupled receptor? Curr Biol. 2009;19:R125–R127. doi: 10.1016/j.cub.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 24.Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- 25.Dahmane N, Lee J, Robins P, Heller P, Altaba A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389:876–881. doi: 10.1038/39918. [DOI] [PubMed] [Google Scholar]
- 26.Tsai KY, Tsao H. The genetics of skin cancer. Am J Med Genet C Semin Med Genet. 2004;131C:82–92. doi: 10.1002/ajmg.c.30037. [DOI] [PubMed] [Google Scholar]
- 27.Barnes EA, Kong M, Ollendorff V, Donoghue DJ. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001;20:2214–2223. doi: 10.1093/emboj/20.9.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lodygin D, Yazdi AS, Sander CA, Herzinger T, Hermeking H. Analysis of 14-3-3sigma expression in hyperproliferative skin diseases reveals selective loss associated with CpG-methylation in basal cell carcinoma. Oncogene. 2003;22:5519–5524. doi: 10.1038/sj.onc.1206854. [DOI] [PubMed] [Google Scholar]
- 29.Boukamp P. Non-melanoma skin cancer: what drives tumor development and progression? Carcinogenesis. 2005;26:1657–1667. doi: 10.1093/carcin/bgi123. [DOI] [PubMed] [Google Scholar]
- 30.Jin Y, Jin C, Salemark L, Wennerberg J, Persson B, Jonsson N. Clonal chromosome abnormalities in premalignant lesions of the skin. Cancer Genet Cytogenet. 2002;136:48–52. doi: 10.1016/s0165-4608(01)00517-9. [DOI] [PubMed] [Google Scholar]
- 31.Rehman I, Takata M, Wu YY, Rees JL. Genetic change in actinic keratoses. Oncogene. 1996;12:2483–2490. [PubMed] [Google Scholar]