

Abstract
The proximal promoter region of many human growth-related genes contains a polypurine/polypyrimidine tract that serves as a multiple binding site for Sp1 or other transcription factors. These tracts often contain a guanine-rich sequence consisting of four runs of three or more contiguous guanines separated by one or more bases, corresponding to a general motif known for the formation of an intramolecular G-quadruplex. Recent results provide strong evidence that specific G-quadruplex structures can be formed naturally by the G-rich sequence within these polypurine/polypyrimidine tracts of many human promoter regions, raising the possibility that the transcriptional control of these genes can be modulated by G-quadruplex-interactive agents. In this chapter, we describe three general biochemical methodologies, electrophoretic mobility shift assay (EMSA), dimethylsulfate (DMS) footprinting, and the DNA polymerase stop assay, which can be useful for initial characterization of G-quadruplex structures formed by G-rich sequences.
Keywords: G-quadruplex, transcriptional regulation, DMS footprinting, EMSA, DNA polymerase stop assay
1. INTRODUCTION
G-rich sequences have been reported to form non-canonical four-stranded secondary structures called G-quadruplexes, which consist of two or more G-tetrads in the presence of monovalent cations such as Na+ and K+, as shown in Figure 1 (1). The G-rich sequences capable of forming G-quadruplexes were initially found in telomeric sequences, the insulin gene, the control region of the retinoblastoma susceptibility gene, fragile X syndrome triplet repeats, and HIV-1 RNA (2–6) and were later also found in the proximal promoter region of many TATA-less mammalian genes, including c-Myc, Hmga2, EGF-R, VEGF, BCL-2, PDGF-A, c-Myb, malic enzyme, I-R, AR, c-Src, c-Ki-Ras, TGFβ, and PDGF A-chain (reviewed in 7). In particular, the G-rich sequences from the promoter region of these genes have been proposed to be very dynamic in their conformation, easily adopting non-B-DNA conformations, such as melted DNA, hairpin structures, slipped helices, or others, under physiological conditions, provided that there is conformational or torsional stress (8–9). Direct evidence for the existence of G-quadruplexes in vivo is beginning to emerge, and the ability of these important sequences to form very stable G-quadruplex structures in vitro suggests that G-quadruplex DNA may play an important role in several biological events, including telomere maintenance, DNA replication, and transcription. For instance, a recent study provided compelling evidence that a specific G-quadruplex structure formed in the c-Myc promoter functions as a transcriptional repressor element (10), establishing the principle that c-Myc transcription could be controlled by ligand-mediated G-quadruplex stabilization (Figure 2). Recent studies of both crystal and solution structures of various G-quadruplexes revealed that their structures are very stable under physiological conditions and very diverse in their folding patterns (11–13). Therefore, there are high expectations that specific interactions can be achieved between different types of G-quadruplexes and small molecular weight ligands.
Figure 1.

G-tetrad and G-quadruplexes. (A) Four guanine residues form a planar structure G-tetrad through Hoogsteen hydrogen bonding to form an intramolecular parallel G-quadruplex. Models are shown for an intramolecular antiparallel basket (B), an intramolecular parallel heptad-tetrad (C), an intramolecular antiparallel chair (D), and a mixed-type intramolecular quadruplex (E). Each parallelogram in (B–F) represents a G-tetrad.
Figure 2.

Model for the transition of a duplex strand into atypical secondary structures and repression of gene transcription by the stabilization of a G-quadruplex structure with a small ligand.
With improved understanding of the structures and potential biological functions of G-quadruplexes, there is increased demand for simple but reproducible and reliable biochemical tools best suited for studying G-quadruplexes. Our previous studies on the structures and functions of G-quadruplex structures suggested that the combined use of EMSA with DMS footprinting and the DNA polymerase stop assay is very useful for initial characterization of G-quadruplex structures from any origin (14–18). Therefore, in this chapter, we will discuss the application of these biochemical techniques in studying the formation of G-quadruplex structures from various G-rich sequences and their potential application in studying the effects of novel classes of small molecular weight compounds, based on their ability to bind to and stabilize G-quadruplexes.
2. MATERIALS
2.1. EMSA and DMS footprinting
2.1.1. Labeling 5′-termini of nucleic acids with [32P]
T4 polynucleotide kinase (Fermentas)
Kinase buffer (10X): 500 mM Tris-HCl (pH 7.6), 100 mM MgCl2, 50 mM DTT, 1 mM spermidine, and 1 mM EDTA.
Adenosine 5′-gamma 32P triphosphate (γ-32P ATP), triethylammonium salt (10 mCi/mL, GE, Healthcare).
Micro Bio-Spin™ 30 Columns (Bio-Rad)
2.1.2. Native polyacrylamide gel electrophoresis (PAGE)
TBE electrophoresis buffer (10X): 0.89 M Tris-HCl (pH 8.0), 0.89 M boric acid, 20 mM EDTA. Store at room temperature.
Sixteen percent acrylamide/bisacrylamide (29:1 with 3.3% C) and N,N,N′,N′-tetramethylethylenediamine (TEMED) (Bio-Rad).
Ammonium persulfate: prepare 10% solution in water. Store at 4 °C up to 1 month.
A gel loading buffer (10X): 50% glycerol by volume, 0.005% bromophenol blue (w/v). Store at 7minus;20 °C.
A gel elution buffer: 0.4 M ammonium acetate, 1 mM MgCl2, 0.2% SDS. Store at room temperature.
100% and 75% ethanol
2.1.3. Chemical DNA sequencing and DMS footprinting
Formic acid (Sigma-Aldrich); hydrazine (Sigma-Aldrich).
DNA sequencing stop solution: 0.5 M sodium acetate (pH 6.0) and 50 μg/mL calf thymus DNA. Store at 4 °C.
1 M piperidine solution in water (freshly prepared).
10% dimethylsulfate solution in 50% ethanol.
2.1.4. Denaturing PAGE
TBE electrophoresis buffer (10X): 0.89 M Tris-HCl (pH 8.0), 0.89 M boric acid, 20 mM EDTA. Store at room temperature.
Sixteen percent acrylamide/bisacrylamide (29:1 with 3.3% C) with 8 M urea and N,N,N′,N′-TEMED, Bio-Rad, Hercules, CA.
Ammonium persulfate: prepare 10% solution in water store at 4 °C up to 1 month.
Alkaline gel loading dye (1X): 80% formamide by volume, 10 mM NaOH, 0.005% bromophenol blue (w/v). Store at −20 °C.
2.2. DNA Polymerase Stop Assay
2.2.1. Labeling 5′-termini of primer with [32P] (see section 2.1.1)
T4 polynucleotide kinase (Fermentas).
Kinase buffer (10X): 500 mM Tris-HCl (pH 7.6), 100 mM MgCl2, 50 mM DTT, 1 mM spermidine, 1 mM EDTA.
Adenosine 5′-gamma 32P triphosphate, triethylammonium salt (10 mCi/mL, GE, Healthcare).
Micro Bio-Spin™ 30 Columns (Bio-Rad).
2.2.2. Native PAGE (see items 1–4 of section 2.1.2)
TBE electrophoresis buffer (10X): 0.89 M Tris-HCl (pH 8.0), 0.89 M boric acid, 20 mM EDTA. Store at room temperature.
Eight percent acrylamide/bisacrylamide (29:1 with 3.3% C) and N,N,N′,N′-TEMED (Bio-Rad).
Ammonium persulfate: prepare 10% solution in water store at 4 °C up to 1 month.
A gel loading buffer (10X): 50% glycerol by volume, 0.005% bromophenol blue (w/v). Store at −20 °C.
2.2.3. DNA polymerase reaction
Taq DNA Polymerase (Fermentas)
DNA polymerase buffer (10X): 500 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 50 mM DTT.
dNTP solution: 2 mM of dATP, dGTP, dTTP, and dCTP.
2.2.4. Dideoxy sequencing reaction
A termination mix: 2 μM dATP, 250 μM ddATP, 800 μM dGTP, 800 μM dTTP, and 800 μM dCTP.
G termination mix: 2 μM dGTP, 250 μM ddGTP, 800 μM dATP, 800 μM dTTP, and 800 μM dCTP.
T termination mix: 2 μM dTTP, 250 μM ddTTP, 800 μM dATP, 800 μM dGTP, and 800 μM dCTP.
C termination mix: 2 μM dCTP, 250 μM ddCTP, 800 μM dATP, 800 μM dGTP, and 800 μM dTTP.
2.2.5. Denaturing PAGE (see section 2.1.4)
TBE electrophoresis buffer (10×): 0.89 M Tris-HCl (pH 8.0), 0.89 M boric acid, 20 mM EDTA. Store at room temperature.
Sixteen percent acrylamide/bisacrylamide (29:1 with 3.3% C) with 8 M urea and N,N,N′,N′-TEMED (Bio-Rad).
Ammonium persulfate: prepare 10% solution in water store at 4 °C up to 1 month. 4. Alkaline gel loading dye (1X): 80% formamide by volume, 10 mM NaOH, 0.005% bromophenolblue (w/v). Store at −20 °C.
3. METHODS
The G-rich strand of the promoter of many mammalian oncogenes is characterized by the presence of more than four runs of at least three adjacent guanines (7). To determine which guanine repeats are required for folding into intramolecular G-quadruplex structures, we prepared a series of oligonucleotide DNAs spanning various portions of the G-rich sequence. Each 5′-end-radiolabeled oligonucleotide was subjected to annealing by heating and slowly cooled to room temperature in the presence of KCl, allowing the tandem repeats of guanines to fold into the G-quadruplexes. The resulting structures were treated with DMS to methylate the guanine residues in the oligonucleotides. The methylated oligonucleotides were then subjected to native PAGE to separate intramolecular forms of G-quadruplexes from intermolecular forms or unfolded structures based on differences in the electrophoretic mobility (19). A native PAGE is routinely used for separation of nucleic acids based on the difference in their shape and size, resulting in a difference in the electrophoretic mobility. In general, the mobility of G-quadruplex DNA is determined by the type of G-quadruplex structures as well as the number of DNA strands involved in folding into G-quadruplexes. Often, intramolecular forms of G-quadruplexes showed faster mobility on native PAGE, as is evident in G-quadruplex structures formed from the G-rich sequence of the BCL-2 and PDGF-A genes (16–17). In some cases, intramolecular G-quadruplexes are indistinguishable from unfolded forms in their electrophoretic mobility, although the slowly migrating bands are believed to be an intermolecular G-quadruplex (14, 18). To determine the guanine bases involved in the formation of G-quadruplex structures, each DNA band was excised from the gel and treated with piperidine to produce specific DNA strand breakage at methylated guanine residues, and the cleavage products were resolved on a denaturing PAGE gel. The guanine bases involved in the formation of G-quadruplex structures can be deduced by DMS footprinting, since the N7 position of the guanines involved in Hoogsteen bonding to form the G-tetrad are inaccessible to methylation (19).
We also used a DNA polymerase stop assay to confirm that the G-rich sequence consisting of multiple G-tracts could form intramolecular G-quadruplex structures (20–21). The DNA polymerase stop assay provides a simple and rapid way to identify DNA secondary structures in vitro, based on the principle that DNA polymerase is incapable of traversing these structures. DNA polymerase, traversing toward the 5′-end of the template and unable to efficiently resolve quadruplex DNA, pauses or stops 3′ to the first guanine involved in a stable G-quadruplex. For the DNA polymerase stop assay, the template DNAs containing various G-quadruplex-forming regions are annealed with radiolabeled primers, and the primer-annealed template DNAs are used in a primer extension assay by Taq DNA polymerase, as described below. This assay has also proven useful in identifying potential G-quadruplex-interactive compounds.
An overall strategy to characterize G-quadruplexes formed by G-rich sequences using EMSA, DMS footprinting, and the DNA polymerase stop assay is shown schematically in Figure 3.
Figure 3.

Schematic diagram showing an overall strategy to characterize G-quadruplexes formed by G-rich sequences using EMSA, DMS footprinting, and the DNA polymerase stop assay.
3.1. EMSA and DMS Footprinting
3.1.1. Labeling 5′-termini of oligonucleotides with [32P]
Preparing a reaction mixture (25 μL), containing oligonucleotide (4 μM), 3 μL γ-32P ATP (10 mCi/mL), T4 polynucleotide kinase (10 U), 2.5 μL 10× kinase buffer, and water.
Incubate the reaction mixture at 37 °C for 1 h in water bath for labeling 5′-termini of oligonucleotides with [32P].
After the completion of the reaction, use Micro Bio-Spin™ 30 Columns (Bio-Rad) to remove unincorporated radioactive γ-32P ATP (10 mCi/mL) from labeled DNA. The instructions for use of Bio-Spin™ 30 Columns are based on recommendations from the manufacturer. In brief, the reaction mixture (25 μL) is loaded at the top of the column after centrifuging the column at 1,000 × g for 4 min in a swinging bucket and removing the packing buffer. The column is then centrifuged for 4 min at 1,000 × g to collect the purified 5′-end-labeled oligonucleotide in water. (Note: columns containing radioactive material should be properly disposed.)
3.1.2. Purification of a desired full-length oligonucleotide using a denaturing 16% polyacrylamide gel
The 5′-labeled oligonucleotides should be purified prior to use in footprinting experiments since oligonucleotides made by automated DNA synthesizers in the laboratory or obtained commercially are often contaminated with products of incomplete synthesis or other unknown impurities. Routinely, a denaturing polyacrylamide-urea gel electrophoresis is used to separate a desired full-length oligonucleotide from other contaminants.
Set up a denaturing 16% polyacrylamide gel of 20 cm × 16 cm × 0.8 mm and 60 mL of gel solution by mixing 6 mL TBE buffer (10×), 24 mL of 40% acrylamide/bisacrylamide (29:1), 30 g urea, and adding water to 60 mL.
After adding 100 μL ammonium persulfate solution and 20 μL TEMED, pour the gel and insert the comb. Allow the gel to polymerize for approximately 30 min.
Once the gel is polymerized, carefully remove the comb and wash the well with TBE buffer (1×) using a pasture pipette. (Note: Be sure to wear safety glasses while pouring the gel since unpolymerized acrylamide is known to be neurotoxic.)
Attach the gel plates to the electrophoresis apparatus and fill both reservoirs of the electrophoresis tank with 1× TBE. Use a DC power supply to pre-run and warm the gel for at least 30 min at 500 V (constant voltage).
Add 20 μL of alkaline gel loading dye to DNA samples, heat the sample at 95 °C for 3 min, and chill the sample on ice before loading.
Run the gel at about 500 V until the desired resolution has been obtained as determined empirically. (Note: avoid excessive heating during electrophoresis to prevent the breakage of the glass plates.)
After the completion of electrophoresis, turn off the power supply, detach the gel plates from electrophoresis apparatus, and carefully separate both plates while the gel is still attached to one plate.
Wrap the gel and plate with plastic wrap. Autoradiography is often used to visualize the location of DNA bands within the gel.
If the amount of DNA is 1 μg or greater, visualize the DNA bands by UV shadowing after wrapping the gel and plate with plastic wrap, inverting, and placing the gel onto a TLC plate containing fluorophores. The DNA fragment of interest can be located with a portable shortwave UV illuminator. (Note: avoid unnecessarily long UV exposure with a shortwave UV light, which will damage the nucleic acids.)
Cut out the desired DNA band with a razor blade, place the gel fragment inside of a 1.5-mL eppendorf tube, and crush the gel fragment into fine pieces by gently touching it with a metal spatula with a narrow blade. Recover the DNA from the gel by adding 400 μL of water and incubating the tube with rotation or in a shaking air incubator at room temperature.
3.1.3. Annealing of the 32P-labeled oligomer DNAs into G-quadruplex structures, DMS methylation, and EMSA
Anneal the 32P-labeled oligomer DNAs by heating at 90 °C for 5 min and then cooling slowly to room temperature in 20 μL of 20 mM Tris-HCl (pH 7.4) buffer with or without 100 mM KCl.
While annealing reaction is in progress, set up a native 16% polyacrylamide gel of 20 cm × 16 cm × 0.8 mm and 60 mL of gel solution by mixing 6 mL TBE buffer (10×), 24 mL of 40% acrylamide/bisacrylamide (29:1), and adding water to 60 mL. Add 100 μL ammonium persulfate solution and 20 μL TEMED, pour the gel, and insert the comb.
Once the gel is polymerized after approximately 30 min, carefully remove the comb, and wash the well with TBE buffer (1×) using a pasture pipette. Attach the gel plates to the electrophoresis apparatus and fill both reservoirs of the electrophoresis tank with 1× TBE. Use a DC power supply to pre-run and warm the gel for a least 30 min at 150 V (constant voltage).
After the annealing reaction is completed, treat each annealed DNA with DMS (0.5%) for 2 min to methylate the DNA.
Stop the DMS modification reaction by adding a tenth volume of a gel loading buffer containing 1 μg calf thymus DNA and immediately load the reactions on a 16% native polyacrylamide gel.
Run the gel until the desired resolution is obtained, detach the gel plates from electrophoresis apparatus, and separate both plates while the gel is still attached to one plate.
Visualize the location of DNA bands within the gel via autoradiography. Figure 4A is an example of the results from EMSA analysis of G-quadruplex structures formed by the G-rich sequence (HIFX) from the polypurine/polypyrimidine tract of the promoter region of the HIF-1α gene (14).
Cut out the desired DNA band from the gel with a razor blade and insert in a 1.5-mL eppendorf tube containing 250 μL of a gel elution buffer. DNA can be eluted from the gel without crushing it by incubating the tube overnight at 37 °C in a water bath.
Recover the supernatant carefully without touching the gel fragment and transfer to a new tube containing 750 μL of 100% ethanol.
Mix the tube well using the vortex and store the samples at −20 °C overnight (or 3 h at −80 °C).
Centrifuge the tubes for 30 min at 12,000 × g at 4 °C to collect the DNA pellet, and wash the recovered DNA pellet once with 250 μL of ice-cold 75% ethanol.
Air-dry DNA pellets, resuspend in 100 μL 1M piperidine solution, and heat at 95 °C for 30 min.
Dry the samples in a speed vac, resuspend dried DNA pellets in 100 μL water, and dry the samples again in a speed vac.
Resuspend dried DNA pellets in 20 μL alkaline sequencing dye, and resolve cleaved DNA products on a 16% denaturing polyacrylamide gel.
Figure 4.
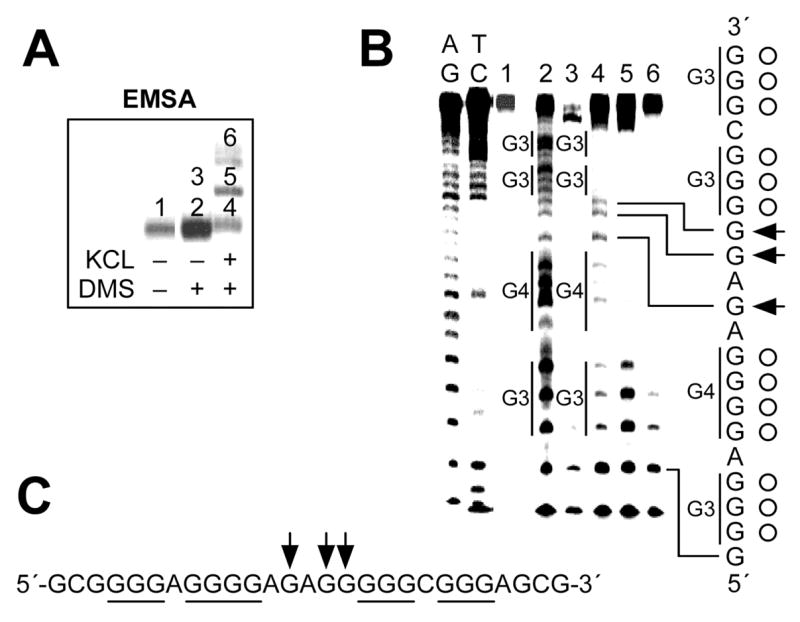
EMSA and DMS footprinting of oligonucleotide HIFX derived from the G-rich sequence of the HIF-1α promoter region. (A) EMSA of HIFX preincubated under the conditions specified in the figure. 5′-End-radiolabeled oligonucleotide HIFX was subjected to annealing by heating and was slowly cooled to room temperature in the presence or absence of KCl, allowing the guanine repeats to fold into G-quadruplexes. The resulting structures were treated with 0.5% dimethylsulfate for 2 min to methylate the guanine residues in the oligonucleotides. The methylated oligonucleotides were then subjected to a 16% native PAGE to separate intramolecular forms of G-quadruplexes from intermolecular forms or unfolded structures by differences in the electrophoretic mobility. The numbers indicate the bands that were excised from the gel and treated with piperidine to induce strand breaks at methylated guanine residues. (B) Pattern of N7 guanine methylation produced by each band (lanes 1–6) isolated from EMSA described in Figure 4A. AG and TC represent chemical cleavage reaction specific to purine and pyrimidine bases, respectively. The vertical bars to the left of lane 4 correspond to DMS-protected guanine repeats. The protected guanines from DMS are indicated by open circles, and arrows indicate the guanine residues hypermethylated by DMS. (C) Summary of DMS footprinting of HIFX in the presence 100 mM KCl. The protected guanines from DMS are underlined, and arrows indicate the guanine residues hypermethylated by DMS.
3.1.4. Chemical DNA sequencing reactions
Sequence ladders are always required for footprinting experiments, allowing clear assignments of cleaved residues. These ladders can be produced by chemical sequencing of the same DNA fragment used for footprinting experiments. The chemical cleavage of DNA by formic acid or hydrazine at a purine or pyrimidine residue, respectively, is typically used in DMS footprinting experiments to generate a cleavage ladder.
Aliquot DNA (approximately 100,000 cpm) in water into 1.5 mL eppendorf tubes labeled Pu and Py, in which the final volume is adjusted to 20 μL with water.
Add 0.4 μg calf thymus DNA to each tube as a carrier to prevent excessive modification of the bases by chemical reagents.
Add 20 μL of formic acid and hydrazine to purine- and pyrimidine-specific reactions, respectively. Mix the reactions well, and incubate at room temperature for 20 min. (Note: longer incubation is required for the DNA fragments shorter than 20 base pairs.)
Terminate the reactions by adding 60 μL DNA sequencing stop solution. Mix the stopped reaction well with 400 μL of 100% ethanol, and store the samples at −20 °C overnight.
Centrifuge the tubes for 30 min at 12,000 × g at 4 °C to collect the DNA pellet, and wash the recovered DNA pellet once with 75% ethanol.
Air dry DNA, resuspend the pellets in 100 μL 1M piperidine solution, and heat the solutions at 95 °C for 30 min. After piperidine treatment, dry the samples in speed vac, resuspend the dried pellets in 1000 μL water, and dry again in speed vac.
Resuspend cleavage DNA pellets in 20 μL alkaline sequencing dye.
3.1.5. Separation of cleavage products on denaturing PAGE
Set up a denaturing 16% polyacrylamide gel of 30 cm × 30 cm × 0.4 mm and prepare 60 mL of gel solution by mixing 6 mL TBE buffer (10X), 24 mL of 40% acrylamide/bisacrylamide (29:1), and 30 g urea and adding water to 60 mL. After adding 100 μL ammonium persulfate solution and 20 μL TEMED, pour the gel and insert the comb.
Once the gel is polymerized, carefully remove the comb, and wash the well with TBE buffer (1×) using a pasture pipette.
Attach the gel plates to the electrophoresis apparatus, and fill both reservoirs of the electrophoresis tank with 1× TBE. Pre-run and warm the gel for a least 30 minutes at 1600 V (constant voltage) using a DC power supply.
Heat the samples and sequencing ladders at 95 °C for 3 min, and chill the sample on ice before loading. Run the gel at about 1600 V.
After the desired resolution is obtained, detach the gel plates from the electrophoresis apparatus, and carefully separate both plates, leaving the gel attached to one plate.
Place a piece of a thin chromatography paper on top of the gel, and slowly pull back on the paper to transfer gels to the paper.
Place a piece of Whatman paper underneath, and cover the wet gel with plastic wrap on top.
Put the gel sandwich in a dryer between a plastic fiber mat and clear plastic sheet, and dry the gel at 80 °C for at least 1 h with a vacuum.
Place the dried gel in an X-ray film cassette. Obtain an autoradiogram by exposing X-ray film to the dried gel. Alternatively, the image can be obtained by exposing the dried gel to the phosphor screen for an appropriate time and scanning the phosphor screen. Figure 4B is an example of an autoradiogram of a 16% polyacrylamide sequencing gel, showing the results of DMS footprinting experiments carried out with the G-rich sequence (HIFX) from the polypurine/polypyrimidine tract of the promoter region of the HIF-1α gene (14).
3.2. DNA Polymerase Stop Assay
3.2.1. Labeling 5′-termini of primer with [32P]
Label 5′-termini of primer with [32P] by preparing a reaction mixture (25 μL) containing water, kinase buffer (1×), primer (4 μM), 3 μL γ-32P ATP (10 mCi/mL), and T4 polynucleotide kinase (10 U) in a single tube and incubating the reaction mixture at 37 °C for 1 h in a water bath.
Use a Micro Bio-Spin™ 30 Column (Bio-Rad) to remove unincorporated radioactive γ-32P ATP from labeled DNA, as described in section 3.1.1.
3.2.2. Annealing of the 32P-labeled primer DNAs into the template DNA
Mix equimolar amounts of the 32P-labeled primer DNAs and the template DNAs containing various G-quadruplex-forming regions together in a single tube in 25 μL of an annealing buffer.
Anneal the 32P-labeled primer DNAs into the template DNA by heating at 90 °C for 5 min and then cooling slowly to room temperature.
Set up a native 8% polyacrylamide gel of 20 cm × 16 cm × 0.8 mm using 60 mL of gel solution (6 mL TBE buffer (10X), 12 mL of 40% acrylamide/bisacrylamide (29:1), and 42 mL water) as described in section 3.1.3 to separate the primer-annealed template DNAs from excess labeled primers or remaining template DNAs.
Pre-run and warm the gel for at least 30 min at 150 V (constant voltage).
Add a tenth of a gel loading buffer to the annealing reaction mixture, mix well, and load the samples onto a native 8% polyacrylamide gel.
After running the gel to the desired resolution, detach the gel plates from the electrophoresis apparatus, and carefully separate both plates, leaving the gel attached to one plate.
Visualize the location of DNA bands within the gel by autoradiography. Figure 5B is an example of an autoradiogram obtained after exposure of X-ray film to the gel.
Cut out the desired DNA band with a razor blade, and crush the gel fragment using a spatula with a thin blade inside of a 1.5 mL eppendorf tube.
Elute DNA from the gel by incubating the gel fragments overnight in 400 μL annealing buffer at room temperature.
Figure 5.

DNA polymerase stop assay to determine the ability of the VEGF promoter to form G-quadruplex structures in the presence of KCl. (A) Sequence of the primer-annealed template DNA. The template DNA was designed to contain the G-quadruplex-forming region from the G-rich sequence of the VEGF promoter region. (B) Autoradiogram showing the separation of the primer-annealed template DNAs from excess labeled primer or remaining template DNA on an 8% native PAGE. Lanes 1 and 2 represent labeled primer and primer-annealed template DNA, respectively. (C) DNA polymerase stop assay showing the effect of KCl on the formation of G-quadruplex structures in the presence of KCl. DNA polymerase reactions were performed with labeled primer-annealed template DNA at increasing concentrations of K+ (0–150 mM). Arrows indicate the positions of the full-length product of DNA synthesis, the G-quadruplex pause sites, and the free primer. Lanes A, G, T, and C represent dideoxysequencing reactions with the same template as a size marker for the precise arrest sites, and P represents primer without enzyme.
3.2.3. DNA polymerase reaction
Prepare reaction mixtures (20 μL) containing water, DNA polymerase buffer (1×), DNA template (5–10 nM), 200 μM dNTP, and Taq DNA polymerase (1 U), and incubate at 37 °C for 30 min in water bath.
Stop the reactions by adding 20 μL of alkaline dye, and dry the samples down to 20 μL in speed vac.
Dideoxy sequencing reactions with the same DNA template are used for the DNA polymerase stop assay to provide a sequencing ladder for clear assignment of DNA polymerase arrest sites in the DNA polymerase stop assay.
Aliquot 10 μL of A, C, G, and T termination mixes into appropriately labeled tubes, and add 10 μL of remaining reaction mixture, consisting of Taq polymerase (1U) and DNA template (10 nM) in 2X polymerase reaction buffer, to each termination tube.
Mix tubes well, and place in a 37 °C water bath for 30 min.
Terminate the reaction by adding 20 μL of alkaline gel-loading dye to each tube, and heat to 95 °C for 5 min prior to loading onto a denaturing PAGE gel.
Resolve the reaction products and sequencing ladders on a 16% denaturing polyacrylamide gel of 30 cm × 30 cm × 0.4 mm, as described in section 3.1.5. An example result is shown in Figure 5C.
Acknowledgments
This research was supported by grants from the National Institutes of Health (CA109069 and CA94166). We are grateful to David Bishop for preparing, proofreading, and editing the final version of the manuscript and figures.
References
- 1.Jin RZ, Breslauer KJ, Jones RA, Gaffney BL. Tetraplex formation of a guanine-containing nonameric DNA fragment. Science. 1990;250:543–546. doi: 10.1126/science.2237404. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Patel DJ. Solution structure of the Tetrahymena telomeric repeat d(T2G4)4 G-tetraplex. Structure. 1994;2:1141–1156. doi: 10.1016/s0969-2126(94)00117-0. [DOI] [PubMed] [Google Scholar]
- 3.Hammond-Kosack MC, Kilpatrick MW, Docherty K. The human insulin gene-linked polymorphic region adopts a G-quartet structure in chromatin assembled in vitro. J Mol Endocrinol. 1993;10:121–126. doi: 10.1677/jme.0.0100121. [DOI] [PubMed] [Google Scholar]
- 4.Murchie AI, Lilley DM. Retinoblastoma susceptibility genes contain 5′ sequences with a high propensity to form guanine-tetrad structures. Nucleic Acids Res. 1992;20:49–53. doi: 10.1093/nar/20.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fry M, Loeb LA. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc Natl Acad Sci USA. 1994;91:4950–4944. doi: 10.1073/pnas.91.11.4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majumdar A, Gosser Y, Patel DJ. 1H-1H correlations across N-H···N hydrogen bonds in nucleic acids. J Biomol NMR. 2001;21:289–306. doi: 10.1023/a:1013340227140. [DOI] [PubMed] [Google Scholar]
- 7.Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michelotti GA, Michelotti EF, Pullner A, Duncan RC, Eick D, Levens D. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol Cell Biol. 1996;16:2656–2669. doi: 10.1128/mcb.16.6.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rustighi A, Tessari MA, Vascotto F, Sgarra R, Giancotti V, Manfioletti G. Apolypyrimidine/polypurine tract within the Hmga2 minimal promoter: a common feature of many growth-related genes. Biochemistry. 2002;41:1229–1240. doi: 10.1021/bi011666o. [DOI] [PubMed] [Google Scholar]
- 10.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkinson GN, Lee MP, Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 12.Phan AT, Modi YS, Patel DJ. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J Am Chem Soc. 2004;126:8710–8716. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai J, Chen D, Jones RA, Hurley LH, Yang D. NMR solution structure of the major G-quadruplex structure formed in the human BCL2 promoter region. Nucleic Acids Res. 2006;34:5133–5144. doi: 10.1093/nar/gkl610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Armond R, Wood S, Sun D, Hurley LH, Ebbinghaus SW. Evidence for the presence of a guanine quadruplex forming region within a polypurine tract of the hypoxia inducible factor 1α promoter. Biochemistry. 2005;44:16341–16350. doi: 10.1021/bi051618u. [DOI] [PubMed] [Google Scholar]
- 15.Sun D, Guo K, Rusche JJ, Hurley LH. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005;33:6070–6080. doi: 10.1093/nar/gki917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dexheimer TS, Sun D, Hurley LH. Deconvoluting the structural and drug-recognition complexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter. J Am Chem Soc. 2006;128:5404–5415. doi: 10.1021/ja0563861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin Y, Rezler EM, Gokhale V, Sun D, Hurley LH. Characterization of the G-quadruplexes in the duplex nuclease hypersensitive element of the PDGF-A promoter and modulation of PDGF-A promoter activity by TMPyP4. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm538. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo K, Pourpak A, Beetz-Rogers K, Gokhale V, Sun D, Hurley LH. Formation of pseudo-symmetrical G-quadruplex and i-motif structures in the proximal promoter region of the RET oncogene. J Am Chem Soc. 2007;129:10220–10228. doi: 10.1021/ja072185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akman SA, Lingeman RG, Doroshow JH, Smith SS. Quadruplex DNA formation in a region of the tRNA gene supF associated with hydrogen peroxide mediated mutations. Biochemistry. 1991;30:8648–8653. doi: 10.1021/bi00099a022. [DOI] [PubMed] [Google Scholar]
- 20.Woodford KJ, Howell RM, Usdin K. A novel K(+)-dependent DNA synthesis arrest site in a commonly occurring sequence motif in eukaryotes. J Biol Chem. 1994;269:27029–27035. [PubMed] [Google Scholar]
- 21.Han H, Hurley LH, Salazar M. A DNA polymerase stop assay for G-quadruplex-interactive compounds. Nucleic Acids Res. 1999;27:537–542. doi: 10.1093/nar/27.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]