

Abstract
The following review was constructed as a concept paper based on a recent workshop on neurodegenerative disease sponsored by the National Institute on Aging (NIA), the American Geriatric Society (AGS), and the John A. Hartford Foundation. The meeting was entitled “Thinking, moving and feeling: Common underlying mechanisms? 4th Annual Bedside-to-Bench Conference” and had the purpose to connect current basic and clinical findings on common brain-related alterations occurring with aging such as depression, movement disorders, and cognitive decline. Many prominent researchers expressed their opinion on aging and it was revealed that age-related brain dysfunction of any kind seems to share several risk factors and/or pathways. But can something be done to actively achieve “successful aging”? In this review, based largely on the workshop and current literature, we have summarized some of the current theories for depression, movement and cognitive impairment with aging, as well as potential preventive measures. We have also summarized the emerging need for relevant animal models and how these could be developed and utilized.
Keywords: Neurodegeneration, memory, cognitive impairment, movement disorders, age-related depression, oxidative stress, neuroinflammation
“Thinking, Moving, Feeling”: What Do They Have in Common?
Aging has been associated with an increased risk of developing depression (Late Life Depression, LLD) and neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease (PD) [1]. Both AD (classically defined as a cognitive disorder) and PD (typically described as a movement disorder) are associated with increased risk for depression and other commonalities such as similar risk factors and age distribution. Furthermore, as AD and PD progress, it becomes evident that their symptoms are not limited to one neurotransmitter system. Because of these findings, investigators have started to investigate the possibility of a biological and/or clinical connection between the triad of symptoms (emotion, cognition and movement). This analysis was the core of a recent workshop on neurodegenerative disease sponsored by the National Institute on Aging (NIA), the American Geriatric Society (AGS), and the John A. Hartford Foundation. The meeting was entitled “Thinking, moving and feeling: Common underlying mechanisms? 4th Annual Bedside-to-Bench Conference” and had the purpose to connect current basic and clinical findings on common brain-related alterations occurring with aging such as depression, movement disorders, and cognitive decline.
Although it is understood that more people develop depression with normal aging, the biochemical or morphological basis for this is not known. It could potentially be as simple as reduced neurotransmitter function with age; both serotonin (5-HT) and norepinephrine (NE) are intimately involved in mood and are known to decline with aging, as well as in AD and PD patients. In the workshop, David Bennett (Rush University Medical Center) presented data from the Religious Order Study demonstrating a significant co-morbidity of AD and PD, as well as a relationship between gait and pathological findings in AD brains such as tangles in the substantia nigra (SN), regardless of PD diagnosis/pathology [2,3]. It is also interesting to note that brain-stem NE neurons have been said to degenerate prior to the cholinergic (ACh) neurons in AD and prior to SN dopamine (DA) neurons in PD [4]. Thus, clinical and basic science studies both unequivocally suggest that AD and PD are complex disorders with common pathology and symptomatology. But it is not clear how these processes converge, or why certain individuals develop one or the other, or why both AD and PD seem to be connected to mood alterations in general.
One possible explanation may be that mood in general, and on a deeper level personality, drives the other functions (cognition and movement) negatively or positively. Studies of centenarians have clearly shown that unifying factors of these “successful agers” are optimism and strong social interactions [5,6]; thus, a strong social network and a positive outlook on life might be the key to help avoid age-related impairment in cognition and/or movement. It is difficult to perform “mood” studies in animal models, but it is possible to link clinical studies to findings regarding environmental enrichment and its powerful effects on animals. Our group and others have shown that environmental enrichment can reduce age-related memory loss, increase cholinergic markers, hippocampal neurogenesis, and growth factors, and also reduce microglial activation in the aged brain [7-9]. This may be as close to “happiness” and social interaction as we can get in animal models and also clearly shows the importance of quality of life even among animals. If possible, factors such as motivation should be examined in animal models to determine its role on movement and cognitive performance.
Clinical studies have shown that there is also a connection between cognitive and movement testing in aged individuals. In a presentation by Jeffrey Hausdorff (Harvard University, MA), it was demonstrated that when older adults were presented with a dual task (walking and cognition) and were offered help with one of the tasks, they tended to first choose a walking aid rather than a memory aid. Further, patients with AD were more likely to have stride alterations when asked to perform dual tasks and patients with early PD demonstrated freezing in pedaling or walking exercises when presented with a simple cognitive task [10]. Pamela Duncan from Duke University presented data demonstrating that stroke victims undergo a significant reorganization of cortical function on parallel cognitive tasks leading to effects on movement. Based on these findings it was suggested that the “brain reserve”, when depleted in one functional modality such as movement, can “leak” and have effects on other functional modalities. Other studies have shown that plaque formation can be altered by exercise in mouse models of AD, suggesting that activity can certainly reorganize the brain even in terms of pathological alterations [11]. Thus, the aged brain is trying, albeit sometimes unsuccessfully, to repair itself and neuronal networks still have some plasticity even after a major ischemic event or degenerative process.
What are some of the physiological processes leading to these age-related brain dysfunctions? It is well known from animal and human studies that movement disorders and memory loss associated with aging have some of the same pathological hallmarks, but starting in different brain regions and involving different neuronal circuits. Among the best described are: protein aggregation, oxidative stress, microglial activation and specific neurotransmitter loss (see below and Fig. 1). There are also primary factors that can affect the aging brain and have severe implications for normal everyday function, such as sleep disturbances, sedentary lifestyle, high blood pressure, metabolic syndrome, obesity, and diabetes. Studies have shown that all of the above can be considered risk factors for both AD and PD [12-15]. All of these are unfortunate side effects of modern society, undoubtedly leading to a surge in these diseases in future aged generations.
Fig. (1).
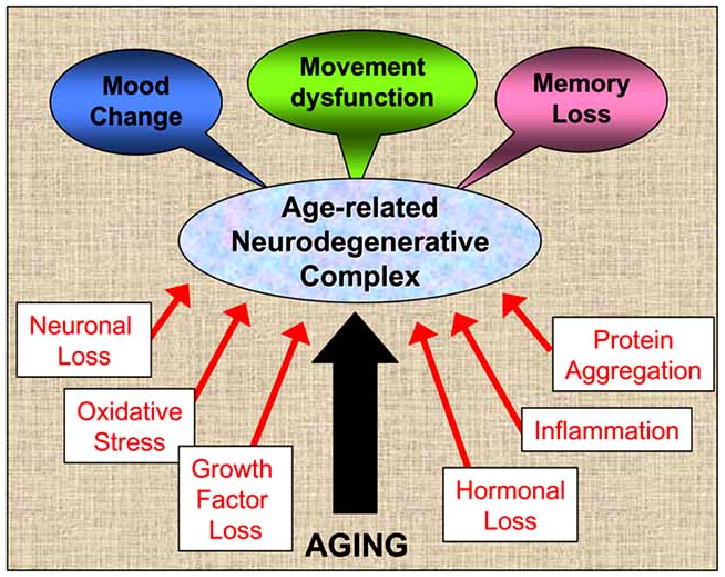
Age-related neurodegenerative complex can be caused by a number of different factors, isolated or in combination. While aging itself is the most common denominator, oxidative stress, inflammation and protein aggregation, for example, are all part of the common pathology seen in PD and AD patients. Studies on movement disorders or memory loss with aging should include at least minimal studies on the other modalities described as well (and listed above in this figure), to reach further understanding of this common co-morbidity in the aged population. For example, most animal models for PD are not tested for cognitive loss or loss of motivation or attention, studies that would definitely add to the validity for future clinical treatment and for more successful drug development.
To summarize, it is clear that mood, cognition, and movement functions are all affected by the normal aging process, and are at least partially the result of convergent pathways. Future studies need to take into consideration these co-morbidities, and relevant animal models have to be developed. There is an imminent need for more standardized animal models, both in regards to movement and cognitive testing. For example, when investigating an animal model for PD, it is important to examine not only movement behavior but also the effects of depletions and/or treatment paradigms on cognitive and, if possible, mood parameters such as motivation. Most often, studies on animal models of PD do not include analysis of memory performance or attention/motivation even though dopamine (DA) has strong effects on working memory, and many patients with PD undergo significant decline in cognitive function.
Pathological Markers for Age-Related Neurodegenerative Complex
It is well known that basal forebrain cholinergic (ACh) neurons and mesencephalic DA neurons deteriorate with normal aging, and a specific loss of these neurons is seen in AD and PD, respectively. Both of these neuronal populations are spontaneously active electrophysiologically with high-energy demands and rapid metabolism. In addition, monoamines are highly reactive molecules and it is thought that excessive adaptation in DA neurons to age-related or disease-related cell loss can actually exacerbate the DA degeneration seen in Parkinson's disease due to increased intracellular dopamine levels [16]. Studies from animal models as well as humans with degenerative disease have shown a progressive increase in oxidative stress and mitochondrial damage in these two neuronal populations with aging [17-19]. Some investigators, like workshop participant Eric Schon (Columbia University), proposed that mitochondrial dysfunction is the key pathological process giving rise to both AD and PD. Yet, is oxidative stress a cause or a result of the mitochondrial dysfunction? This is also one of the key questions in research of AD and PD today. Adding to that pathological picture, the basal forebrain and mesecephalon have an unusually high density of microglial cells, which are increasingly activated with both normal aging and degenerative diseases [20]. Neuroinflammation is a key factor of aging, and studies suggest that pro-inflammatory cytokines can accumulate progressively throughout life. An interesting example of this life-long event is that a one-time prenatal exposure to an endotoxin such as lipopolysaccharide (LPS) during a sensitive embryonic developmental stage gives rise to a loss of DA neurons coupled with increased “inflammatory state” in the brain throughout life [21]. Based on these studies and others, an intriguing question is whether the infections we contract during our lifetime, both before and after birth, in combination with a slow escalation/accumulation of cytokines or other inflammatory triggers finally results in movement or cognitive impairments with aging (see Fig. 2 below). It is possible that inflammatory processes during fetal development could be the cause of AD and PD many decades later. This could be caused by for example bacterial vaginosis during a specific stage of fetal development, as discussed above. Both AD and PD exhibit elevated microglial activation, increased cytokine levels, and common inflammation-related risk factors. Infarction and other cerebrovascular diseases are high risk factors for AD. It is likely that ischemic events give rise to a break in the blood-brain barrier, in turn leading to increased levels of cytokines in brain parenchyma. Nonhuman primate studies have shown microglial activation in the SN persisting many years after a single exposure to the DAergic neurotoxin 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP; see [22]) that induces a long-term parkinsonian syndrome [23] suggesting that neurotoxins can also trigger a persistent inflammatory state. As described above, there are parallel events in basal forebrain and SN with aging, as well as in specific neurodegenerative diseases. Another interesting parallel between AD and PD is the conspicuous and early loss of locus coeruleus (LC) noradrenergic (NE) neurons [4] compared to the steady, slow decline of NE during normal aging. Animal studies have demonstrated that systemic LC denervation leads to elevated microglial activation in areas innervated by NE neurites, including the basal forebrain and SN, suggesting that NE innervation is a regulator of microglial activity in certain regions of the brain. Apart from a common loss of both serotonergic and LC-NE neurons, both PD and AD also include loss of basal forebrain cholinergic neurons, again showing converging pathways in development of these two neurological diseases [24,25].
Fig. (2).
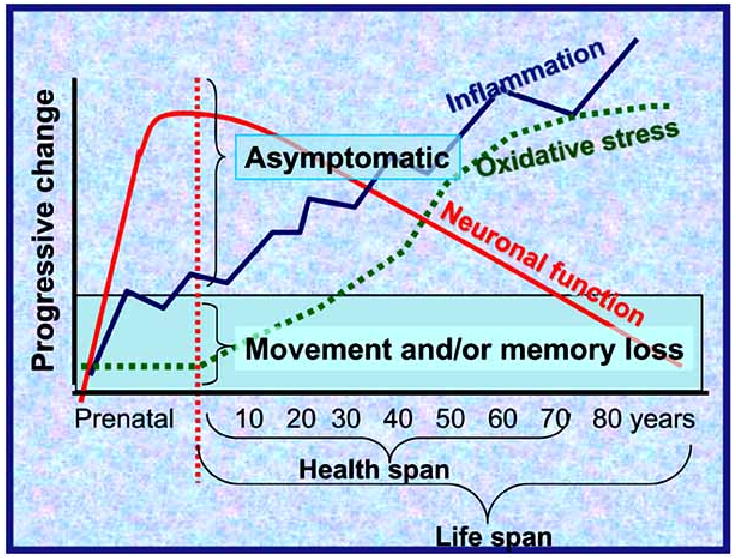
Aging leads to a series of parallel processes, which all affect behavior to different extents. Inflammation, oxidative stress and reduced neuronal function can all have detrimental outcomes on “thinking, moving, feeling”. As the oxidative stress and inflammation increase and neuronal function decreases with age, eventually the individual will develop clinical symptoms. It is important to note, though, that symptoms do not occur (“asymptomatic”) until significant damage has already occurred and the individual crosses the threshold (green bar). It is not known why certain individuals are prone to AD rather than PD, or why certain individuals have PD earlier in life than others. Better animal models are needed to explore these processes.
These many commonalities in pathological cascades between PD and AD may simply be different manifestations of the same disease or accelerated processes occurring with normal aging. But it is uncertain why certain individuals get one or the other, or a combination of both [2, 3, 18]. A possible explanation could be, for example, polymorphisms in growth factor expression. Studies have shown that individuals with AD have altered brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) levels and may also express BDNF polymorphisms [26], studies of PD patients have primarily reported alterations in BDNF and glial cell line-derived neurotrophic factor (GDNF) levels [27], showing an interesting overlap but partial selectivity in growth factor alterations. BDNF is a growth factor for serotonergic, NE, DA, and ACh basal forebrain neurons [28-31] and is therefore a strong candidate for degeneration occurring in both of these diseases. We have recently demonstrated that mice with a partial deletion of the GDNF gene exhibit progressive motor dysfunction and DA cell loss with aging [32], and possibly also a parallel loss in spatial memory, presenting an interesting model to pursue for examination of convergent brain dysfunction with age. Recent work has suggested that BDNF and NGF may have cleavage alterations from their larger pro-neurotrophins with aging or neurodegenerative disease [33, 34]. In vitro studies have suggested that pro-NGF and pro-BDNF can in fact be damaging rather than protective for neurons, via signaling through the sortilin/p75 receptor complex [35], suggesting that age-related degenerative disease may be aggravated by a large-scale problem with protein cleavage, trafficking, aggregation, and/or degradation. The many similarities in growth factor regulation, as well as inflammatory and oxidative stress indicators that are found in animal models suggest that we should look for commonalities in behavioral assessments, treatments and biological mechanisms of disease. It would be interesting to focus future studies on why this happens in certain individuals, what underlies an inclination towards cholinergic vs. DAergic degeneration in certain individuals, and if there are early markers that can be utilized to design treatment or prevention paradigms that would address the entire triad of symptoms described above.
Genetic Factors for Aging and Longevity
Genetics have been said to account for at least 30% of longevity in animal models and in humans. It is thought that a general stability of gene expression, as well as the expression of more specific longevity genes, is associated with longevity both in animal models and in humans [36, 37]. Interestingly, caloric restriction leads to more stable gene expression than normally fed aged animals, and involves elevated expression of longevity-related genes as well [38-40]. Caloric restriction increases the life span, as well as health span (ie. length of life without major pathological processes, see Fig. 2 below) of many organisms, from yeast to mammals. In yeast, one life span gene affected by caloric restriction is Sir2 (silent information regulator 2), and in mammals, the ortholog of Sir2 is Sirt1. Caloric restriction activates Sirt1 to inhibit peroxysome proliferator-activator receptor gamma (PPARgamma) receptor, which is a nuclear receptor promoting adipogenesis, leading to lipolysis and loss of fat. Thus, lowering of adiposity appears to be one mechanism whereby caloric restriction affects life span [40]. It should be noted that PPARgamma agonists have anti-inflammatory properties, suggesting that decreased PPARgamma activity may also affect caloric restriction positive effects [41]. It is possible that neuroprotective and anti-inflammatory effects of the LC-NE neurons noted above may be mediated via the PPAR receptors as well, according to recent data from Feinstein and collaborators [42], thus providing a possible substrate for NE effects on neuro-inflammation.
Obesity and sedentary life-styles are known risk factors for most age-related diseases, as discussed above. With our current “epidemic” of obesity in the United States, we are sure to see a surge of cardiovascular and metabolic diseases, such as diabetes and, accordingly, a shortening of life span in the next generations. Based on the risk factors discussed above, we most certainly will see a surge in neurodegenerative diseases as well. Specific genetic factors have been discovered in a subset of AD and PD patients; at least 5-10% of patients have mutations proven to play a role in incidence as well as age of onset. Martin et al. [43] found that the offspring of centenarians were less likely to have age-related diseases. By comparing the genetic profile of the offspring with age-matched controls, three candidate genes for longevity were identified; all of which are involved in regulation of large membrane lipoproteins.
In summary, there are genetic factors for age-related diseases and for longevity itself, but there are also risk factors that can alter the outcome considerably. Clinical and animal studies have shown that factors which affect oxidative stress, inflammation, adiposity, and cholesterol levels, trafficking, and metabolism can have significant effects on outcome and progression of neurodegenerative diseases. It appears overall that prevention rather than treatment is more effective when it comes to reducing the risk and progression of age-related degenerative complexes (see Fig. 2 below).
Prevention of Age-Related Motor Dysfunction and Cognitive Impairment: Is it Possible?
The baby-boomers, who have started to turn 60, grew up during the post-WWII era. Growing up, the majority of these individuals were not exposed to the super-sized meals and sedentary life-style which has become characteristic of the current western societies in the last two decades. As a result of this, studies show that they age relatively successfully. This is shown by epidemiological data; rate of older adults residing in nursing homes as well as those with disabilities have decreased. This may not be the case of future elderly: We do not yet know the long-term population effects of trans-fat diets, excessive television viewing, or video games. Today, thirty percent of school children eat fast food at least once every day in the United States, and it is not known whether this will have effects on incidence of depression, AD, and PD, as well as other age-related disorders, as they age. We live in a reactive society, not a preventive society - when we see the damage we will try to repair damages rather than prevent them, but it may be too late. There is a wealth of information suggesting that preventive strategies for neurodegenerative disorders may be more successful than treatments that exist today, primarily because of the fact that behavioral problems do not manifest until a majority of neurons are already dysfunctional or have undergone cell death, and general disease processes in the brain are already rampant (see Fig. 2 below). If we are focusing on health span instead of life span, it is clear that a number of preventive therapies will enhance quality of life for several, if not many, decades in the patient and these are discussed below.
Antioxidant foods, such as spirulina, blueberries, spinach, and strawberries, have been shown to have remarkable effects on cardiovascular disease, cancer, stroke, AD, and PD in both humans and animal models [44-46, 17]. Nutritional derivatives such as the active ingredients in green tea, resveratrol in grapes and red wine, and polyphenols in blueberries have also been shown to modulate oxidative stress factors and sometimes also microglial activation in vitro and in vivo [47, 48]. We have recently performed studies of Vitamin E effects on pathology and behavior in a mouse model of Down's syndrome, the Ts65Dn mouse [49]. This model can give some answers regarding AD as well, since individuals with Down's syndrome develop pathological hallmarks of AD with age. We found that Vitamin E treatment from 4-8 months of age gave rise to effective prevention of cholinergic cell loss, oxidative stress, and cognitive impairment occurring with age in the Ts65Dn mouse. We have previously reported similar results in the same mouse, using the tetracycline derivative minocycline [50]. Thus, it is possible to prevent significant cognitive impairment at least in animal models, with antioxidant or anti-inflammatory treatment at the right time in the disease process. These data clearly show the powerful effects of preventive treatment and the “spillover” effect of antioxidants or anti-inflammatory supplements on the process not directly targeted. It is possible that finding earlier biomarkers for AD or PD could therefore be utilized to design a prevention strategy for human conditions as well. Further, it would be interesting to utilize animal models to examine effects of these preventive factors on depression or motivation possibly preceding motor dysfunction and/or cognitive impairment.
Another well-studied prevention factor is exercise, and studies by Cotman and collaborators [51] have shown that moderate exercise gives rise to elevated brain BDNF levels, which in turn can overcome age-related cognitive impairment. Michael Zigmond and collaborators have performed seminal studies showing that exercise can affect both memory and motor function in animal models [52]. Several groups have recently demonstrated significant benefits of aerobic exercise for slowing the progression of AD, at least in terms of activities of daily living (ADL) and some cognitive measures [53, 54], suggesting that exercise may also have significant preventive measures in humans with neurodegenerative diseases. Since physicians have been prescribing exercise programs as a documented intervention for cardiovascular disease for some time now, perhaps utilizing this paradigm should also be strongly considered for the prevention of neurodegenerative disorders. It would be possible, for example, to assign in-house “health coaches” to work with afflicted individuals to improve compliance.
Summary
Recent findings suggest that both clinical and animal studies should more carefully consider the triad of symptoms often occurring in the age-related neurodegenerative complex (depression, movement, and cognitive impairment). More appropriate and inclusive animal models need to be developed, taking into consideration the complex processes leading to depression, memory loss, and motor dysfunction in the elderly. Powerful studies have shown effective preventive measures for this triad of symptoms: should efforts be focused on these preventive life-style changes, and be prescribed to patients with AD and PD, such as is the case for cardiovascular disease? These are challenging issues that could represent a truly translational research effort.
Acknowledgments
This review was made possible by funding from the National Institutes on Aging (A-CG: AG023630, AG12122, and AG10755), WNPRC NIH base grant 5P51RR000167 and the Kinetics Foundation. The authors wish to thank Jason Lockrow, Larry Middaugh, and Alfred Moore for valuable discussions.
References
- 1.Burgut FT, Benaur M, Hencliffe C. Late-life depression: a neuropsychiatric approach. Expert Rev Neurother. 2006;6(1):65–72. doi: 10.1586/14737175.6.1.65. [DOI] [PubMed] [Google Scholar]
- 2.Allan LM, Ballard CG, Burn DJ, Kenny RA. Prevalence and severity of gait disorders in Alzheimer's and non-Alzheimer's dementias. J Am Geriatr Soc. 2005;53(10):1681–7. doi: 10.1111/j.1532-5415.2005.53552.x. [DOI] [PubMed] [Google Scholar]
- 3.Papapetropoulos S, Lieberman A, Gonzalez J, Mash DC. Can Alzheimer's type pathology influence the clinical phenotype of Parkinson's disease? Acta Neurol Scand. 2005;111(6):353–9. doi: 10.1111/j.1600-0404.2005.00411.x. [DOI] [PubMed] [Google Scholar]
- 4.Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60(3):337–41. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- 5.Tafaro L, Cicconetti P, Piccirillo G, Ettorre E, Marigliano V, Cacciafesta M. Is it possible to predict one-year survival in centenarians? A neural network study. Gerontology. 2005;51(3):199–205. doi: 10.1159/000083994. [DOI] [PubMed] [Google Scholar]
- 6.Clarke DM. Growing old and getting sick: maintaining a positive spirit at the end of life. Aust J Rural Health. 2007;15(3):148–54. doi: 10.1111/j.1440-1584.2007.00867.x. [DOI] [PubMed] [Google Scholar]
- 7.Ickes BR, Pham TM, Sanders LA, Albeck DS, Mohammed AH, Granholm ACh. Long-term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp Neurol. 2000;164:45–52. doi: 10.1006/exnr.2000.7415. [DOI] [PubMed] [Google Scholar]
- 8.Milgram NW, Siwak-Tapp CT, Araujo J, Head E. Neuroprotective effects of cognitive enrichment. Ageing Res Rev. 2006;5(3):354–69. doi: 10.1016/j.arr.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Segovia G, Yague AG, Garcia-Verdugo JM, Mora F. Environmental enrichment promotes neurogenesis and changes the extracellular concentrations of glutamate and GABA in the hippocampus of aged rats. Brain Res Bull. 2006;70(1):8–14. doi: 10.1016/j.brainresbull.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 10.Li KZ, Lindenberger U, Freund AM, Baltes PB. Walking while memorizing: age-related differences in compensatory behavior. Psychol Sci. 2001;12(3):230–7. doi: 10.1111/1467-9280.00341. [DOI] [PubMed] [Google Scholar]
- 11.Nichol KE, Parachikova AL, Cotman CW. Three weeks of running wheel exposure improves cognitive performance in the aged Tg2576 mouse. Behav Brain Res. 2007;184(2):124–32. doi: 10.1016/j.bbr.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayeux R. Epidemiology of neurodegeneration. Ann Rev Neurosci. 2003;26:81–104. doi: 10.1146/annurev.neuro.26.043002.094919. [DOI] [PubMed] [Google Scholar]
- 13.Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med. 2004;82(8):510–29. doi: 10.1007/s00109-004-0552-1. [DOI] [PubMed] [Google Scholar]
- 14.Levy G, Tang MX, Cote LJ, Louis ED, Alfaro B, Mejia H, et al. Do risk factors for Alzheimer's disease predict dementia in Parkinson's disease? An exploratory study. Mov Disord. 2002;17(2):250–7. doi: 10.1002/mds.10086. [DOI] [PubMed] [Google Scholar]
- 15.Brown RC, Lockwood AH, Sonawane BR. Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect. 2005;113(9):1250–6. doi: 10.1289/ehp.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Y, Roth GS. The roles of dopamine oxidative stress and dopamine receptor signaling in aging and age-related neurodegeneration. Antioxid Redox Signal. 2000;2(3):449–460. doi: 10.1089/15230860050192224. [DOI] [PubMed] [Google Scholar]
- 17.Rao AV, Balachandran B. Role of oxidative stress and antioxidants in neurodegenerative diseases. Nutr Neurosci. 2002;5(5):291–309. doi: 10.1080/1028415021000033767. [DOI] [PubMed] [Google Scholar]
- 18.Raule N, Sevini F, Santoro A, Altilia S, Franceschi C. Association studies on human mitochondrial DNA: methodological aspects and results in the most common age-related diseases. Mitochondrion. 2007;7(12):29–38. doi: 10.1016/j.mito.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Mancuso C, Scapagini G, Curro D, Giuffrida Stella AM, De Marco C, Butterfield DA, et al. Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front Biosci. 2007;12:1107–23. doi: 10.2741/2130. [DOI] [PubMed] [Google Scholar]
- 20.Morgan TE, Wong AM, Finch CE. Anti-inflammatory mechanisms of dietary restriction in slowing aging processes. Interdiscip Top Gerontol. 2007;35:83–97. doi: 10.1159/000096557. [DOI] [PubMed] [Google Scholar]
- 21.Carvey PM, Chang Q, Lipton JW, Ling Z. Prenatal exposure to the bacteriotoxin lipopolysaccharide leads to long-term losses of dopamine neurons in offspring: a potential new model of Parkinson's disease. Front Biosci. 2003;8:826–37. doi: 10.2741/1158. [DOI] [PubMed] [Google Scholar]
- 22.Doudet DJ, Holden JE, Jivan S, McGeer E, Wyatt RJ. In vivo PET studies of the dopamine D2 receptors in rhesus monkeys with long-term MPTP-induced parkinsonism. Synapse. 2000;38:105–13. doi: 10.1002/1098-2396(200011)38:2<105::AID-SYN1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 23.Emborg-Knott ME, Domino EF. MPTP-induced hemiparkinsonism in nonhuman primates 6-8 years after a single unilateral intracarotid dose. Exp Neurol. 1998;152:214–20. doi: 10.1006/exnr.1998.6845. [DOI] [PubMed] [Google Scholar]
- 24.Wolters EC, Braak H. Parkinson's disease: premotor clinicopathological correlations. Neural Transm. 2006;70:309–319. doi: 10.1007/978-3-211-45295-0_47. [DOI] [PubMed] [Google Scholar]
- 25.Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110(5):517–36. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- 26.Riemenschneider M, Schwarz S, Wagenpfeil S, Diehl J, Muller U, Forstl H, et al. A polymorphism of the brain-derived neurotrophic factor (BDNF) is associated with Alzheimer's disease in patients lacking the Apolipoprotein E epsilon4 allele. Mol Psychiatry. 2002;7(7):782–5. doi: 10.1038/sj.mp.4001073. [DOI] [PubMed] [Google Scholar]
- 27.Chauhan NB, Siegel GJ, Lee JM. Depletion of glial cell-line derived neurotrophic factor in substantia nigra of Parkinson's disease brain. J Chem Neuroanat. 2001;21:277–88. doi: 10.1016/s0891-0618(01)00115-6. [DOI] [PubMed] [Google Scholar]
- 28.Luellen BA, Bianco LE, Schneider LM, Andrews AM. Reduced brain-derived neurotrophic factor is associated with a loss of serotonergic innervation in the hippocampus of aging mice. Genes Brain Behav. 2007;6(5):482–90. doi: 10.1111/j.1601-183X.2006.00279.x. [DOI] [PubMed] [Google Scholar]
- 29.Traver S, Marien M, Martin E, Hirsch EC, Michel PP. The phenotypic differentiation of locus ceruleus noradrenergic neurons mediated by brain-derived neurotrophic factor is enhanced by corticotropin releasing factor through the activation of a cAMP-dependent signaling pathway. Mol Pharmacol. 2006;70(1):30–40. doi: 10.1124/mol.106.022715. [DOI] [PubMed] [Google Scholar]
- 30.Nagatsu T, Sawada M. Biochemistry of postmortem brains in Parkinson's disease: historical overview and future prospects. J Neural Trans Suppl. 2007;(72):113–20. doi: 10.1007/978-3-211-73574-9_14. [DOI] [PubMed] [Google Scholar]
- 31.Wu H, Friedman WJ, Dreyfus CF. Differential regulation of neurotrophin expression in basal forebrain astrocytes by neuronal signals. J Neurosci Res. 2004;76(1):76–85. doi: 10.1002/jnr.20060. [DOI] [PubMed] [Google Scholar]
- 32.Boger HA, Middaugh LD, Huang P, Zaman V, Smith AC, Hoffer BJ, et al. A partial GDNF depletion leads to earlier age-related deterioration of motor function and tyrosine hydroxylase expression in the substantia nigra. Exp Neurol. 2006;202(2):336–47. doi: 10.1016/j.expneurol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Cuello AC, Bruno MA. The failure in NGF maturation and its increased degradation as the probable cause for the vulnerability of cholinergic neurons in Alzheimer's disease. Neurochem Res. 2007;32(6):1041–5. doi: 10.1007/s11064-006-9270-0. [DOI] [PubMed] [Google Scholar]
- 34.Tan J, Shepherd RK. Aminoglycoside-induced degeneration of adult spiral ganglion neurons involves differential modulation of tyrosine kinase B and p75 neurotrophin receptor signaling. Am J Pathol. 2006;169(2):528–43. doi: 10.2353/ajpath.2006.060122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen ZY, Leraci A, Teng H, Dall H, Meng CX, Herrera DG, et al. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci. 2005;25(26):6156–66. doi: 10.1523/JNEUROSCI.1017-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arking R. Gene expression and regulation in the extended longevity phenotypes of Drosophila. Ann NY Acad Sci. 2001;928:157–67. doi: 10.1111/j.1749-6632.2001.tb05645.x. [DOI] [PubMed] [Google Scholar]
- 37.Arking R, Novoseltsev V, Novoseltseva J. The human life span is not that limited: the effect of multiple longevity phenotypes. J Gerontol A Biol Sci Med Sci. 2004;59(7):697–704. doi: 10.1093/gerona/59.7.b697. [DOI] [PubMed] [Google Scholar]
- 38.Ingram DK, Anson RM, de Cabo R, Mamczarz J, Zhu M, Mattison J, et al. Development of calorie restriction mimetics as a prolongevity strategy. Ann NY Acad Sci. 2004;1019:412–23. doi: 10.1196/annals.1297.074. [DOI] [PubMed] [Google Scholar]
- 39.Sinclair DA. Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev. 2005;126(9):987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 40.Wolf G. Calorie restriction increases life span: a molecular mechanism. Nutr Rev. 2006;64(2 Pt 1):89–92. doi: 10.1301/nr.2006.feb.89-92. [DOI] [PubMed] [Google Scholar]
- 41.Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–88. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 42.Madrigal JL, Kalinin S, Richardson JC, Feinstein DL. Neuroprotective actions of noradrenaline: effects on glutathione synthesis and activation of peroxisome proliferator activated receptor delta. J Neurochem. 2007;103(5):2092–101. doi: 10.1111/j.1471-4159.2007.04888.x. [DOI] [PubMed] [Google Scholar]
- 43.Martin GM, Bergman A, Barzilai N. Genetic determinants of human health span and life span: progress and new opportunities. PLoS Genet. 2007;3(7):e125. doi: 10.1371/journal.pgen.0030125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau FC, Shukitt-Hale B, Joseph JA. Nutritional intervention in brain aging: reducing the effects of inflammation and oxidative stress. Subcell Biochem. 2007;42:299–318. [PubMed] [Google Scholar]
- 45.Wang Y, Chang CF, Chou J, Chen HL, Deng X, Harvey BK, et al. Dietary supplementation with blueberries, spinach, or spirulina reduces ischemic brain damage. Exp Neurol. 2005;193(1):75–84. doi: 10.1016/j.expneurol.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 46.Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6(3):337–50. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 47.Prior RL, Cao G, Martin A, Sofic E, McEwen J, O'Brien C, et al. Antioxidant capacity as influenced by total phenolic and anthocyanin content, maturity, and variety of Vaccinium Species. J Agric Food Chem. 1998;46:2686–2693. [Google Scholar]
- 48.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25(7):1341–57. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 49.Lockrow J, Huang P, Willis L, Sambamurti K, Granholm ACh. Effects of vitamin E supplementation on cognition in a mouse model for Down syndrome. Neurobiol Dis. submitted 2008. [Google Scholar]
- 50.Hunter CL, Bachman D, Granholm ACh. Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Annal Neurol. 2004;56:675–688. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- 51.Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25(6):295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- 52.Stein DJ, Collins M, Daniels W, Noakes TD, Zigmond M. Mind and muscle: the cognitive-affective neuroscience of exercise. CNS Spectr. 2007;12(1):19–22. doi: 10.1017/s1092852900020484. [DOI] [PubMed] [Google Scholar]
- 53.Yu F, Kolanowski AM, Strumpf NE, Eslinger PJ. Improving cognition and function through exercise intervention in Alzheimer's disease. J Nurs Scholarsh. 2006;38(4):358–65. doi: 10.1111/j.1547-5069.2006.00127.x. [DOI] [PubMed] [Google Scholar]
- 54.Rolland Y, Pillard F, Klapouszczak A, Reynish E, Thomas D, Andrieu S, et al. Exercise program for nursing home residents with Alzheimer's disease: a 1-year randomized, controlled trial. J Am Geriatr Soc. 2007;55(2):158–65. doi: 10.1111/j.1532-5415.2007.01035.x. [DOI] [PubMed] [Google Scholar]