

Abstract
A recent genomewide association study reported association between schizophrenia and the ZNF804A gene on chromosome 2q32.1. We attempted to replicate these findings in our Irish Case-Control Study of Schizophrenia (ICCSS) sample (N=1021 cases, 626 controls). Following consultation with the original investigators we genotyped 3 of the most promising SNPs from the Cardiff study. We replicate association with rs1344706 (trend test one tailed p=0.0113 with the previously associated A allele) in ZNF804A. We detect no evidence of association with rs6490121 in NOS1 (one tailed p=0.21), and only a trend with rs9922369 in RGRIP1L (one tailed p=0.0515). Based on these results, we completed genotyping of 11 additional LD-tagging SNPs in ZNF804A. Of 12 SNPs genotyped, 11 pass QC criteria and 4 are nominally associated, with our most significant evidence of association at rs7597593 (p=0.0013) followed by rs1344706. We observe no evidence of differential association in ZNF804A based on family history or sex of case. The associated SNP rs1344706 lies in ~30 bp of conserved mammalian sequence and the associated A allele is predicted to maintain binding sites for the brain-expressed transcription factors MYT1L and POU3F1/OCT-6. In controls, expression is significantly increased from the A allele of rs1344706 compared to the C allele. Expression is increased in schizophrenic cases compared to controls, but this difference does not achieve statistical significance. This study replicates the original reported association of ZNF804A with schizophrenia and suggests that there is a consistent link between the the A allele of rs1344706, increased expression of ZNF804A and risk for schizophrenia.
INTRODUCTION
Schizophrenia (MIM 181500) is a severe psychiatric illness with complex etiology and lifetime prevalence of ~1% worldwide. Family, twin and adoption studies have consistently demonstrated an important genetic component to schizophrenia, together with developmental and environmental influences1,2.
The genomewide association study (GWAS), with up to 1 million single nucleotide polymorphisms (SNPs) genotyped, is currently the most powerful, systematic and unbiased genetic approach to the study of the common disease/common variant (CDCV) hypothesis of complex disorders like schizophrenia. Since the initial suggestion by Risch and Merikangas of the power of association methods for human diseases3, GWAS have successfully identified novel associations and/or replicated prior susceptibility genes for a number of complex traits, including age-related macular degeneration4, Type 15 and Type 26–11 diabetes, episodic memory12, Crohn’s disease13–16, prostate cancer17–19 and obesity20.
So far, 3 independent schizophrenia GWAS using individual genotyping have been published21–23. Sample sizes (and power) are modest given the range of effect sizes and allele frequencies expected. These studies have not provided either convergence between new loci identified or strong support for loci previously identified (for example, through positional cloning). Direct replication studies of current GWAS findings have provided mixed results.
The first of these used a sample of 178 cases and 144 controls, assessed 500K SNPs and reported one experiment-wide significant association at rs4129148 (P= 3.7×10−7) in the X/Y pseudoautosomal region near the CSF2RA and IL3R genes21. Cytokines have been suggested as possible candidate genes previously, and one replication attempt supported association in IL3R24. The second used the CATIE25 sample of 738 cases and 733 controls and reported no genomewide significant results in stage 1 analysis22. The third used a multi-stage design of discovery in a newly genotyped sample of 479 cases compared with existing data from the 2937 UK controls used in the Wellcome Trust Case/Control Consortium studies26 and targeted replication in 6666 cases and 9897 controls. This study reported consistent evidence of association with 1 SNP in the zinc-finger protein ZNF804A gene23. Although the discovery sample in this study was modest, it is drawn from an Anglo-Celtic population (from Wales and the UK) broadly similar to the population from which we have sampled cases and controls (from Ireland and Northern Ireland) and the total sample size was substantial due to the use of existing control data. We therefore set out to replicate the findings from the Cardiff study in our Irish case/control sample.
MATERIALS, METHODS AND SAMPLES
The Irish Case/Control Study of Schizophrenia (ICCSS) Sample
Cases were ascertained from in-patient and out-patient psychiatric facilities in Ireland and Northern Ireland. Cases completed the same diagnostic instrument used in our prior family study, the Structured Clinical Interview for DSM III-R, Patient version with an expanded psychosis section27; DSM-III-R criteria were used for consistency with our family collection28. Detailed personal interviews and hospital record rating forms were completed for each proband. Subjects with a field diagnosis of schizophrenia or poor-outcome schizoaffective disorder were eligible if all four grandparents were born in Ireland or the UK. The ICCSS sample includes 1021 cases.
Controls (N=626) were recruited from donors at the Northern Ireland Blood Transfusion Service (N=554) and from the Irish national police (N=38) and army reserve (N=34). Controls were briefly screened and were eligible if they reported no history of schizophrenia and all four grandparents born in Ireland or the UK. All participants gave appropriate informed consent. Recorded sex was verified by X/Y genotypes; cases were 68% male and 32% female, controls were 55% male, 45% female. This sample has ≥78% power to detect effects with minor allele frequency (MAF) ≥20% and allelic odds ratio (OR) ≥1.3.
Ireland is relatively isolated at the North Western extreme of Europe, which may be advantageous in investigating a complex genetic disorder such as schizophrenia. The current Irish population, though not a genetic isolate, is more homogeneous than the US population29–31. Fewer ancestral haplotypes increase LD and the power to detect association. Studies of Y chromosome and mtDNA point to a common genetic legacy in Ireland that probably extends back to the repopulation of the island, after the last Ice Age approximately 15,000 years ago, from population centres in the Iberian peninsula and south western France32–34. The genetic structure of the population has been minimally influenced by more recent human migrations over the last three millennia34. In the experience of the blood bank staff, non-Irish donors are very rare, in agreement with the history of minimal in-migration to Ireland, and would have been excluded on the basis of questions about their grandparents.
Family History
Family history research diagnostic criteria (FH-RDC)35 were assessed by having affected participants complete the FH-RDC interview36, the best validated instrument available, to report on psychotic illness in relatives. Detailed study of the reliability, sensitivity and specificity of the FH-RDC versus personal interview of first-degree family members in the Roscommon Family Study37 showed that between informants, the kappa±SE was 0.49±0.02; between FH-RDC and best-estimate diagnoses, the kappa±SE was 0.43±0.02. Validated against the best-estimate diagnosis (which included personal interview and hospital records in most cases), the sensitivity was 0.37 and the specificity was 0.996. The FH-RDC criteria38 and instrument36 thus miss true illness in relatives but almost never produce a false positive.
Family history information is available for 739 cases (72.4%). We defined positive family history (FH+) by report of 1 or more first-degree relatives with schizophrenia or unspecified functional psychosis. We restrict the definition to first-degree relatives because we expect greater reliability of reporting for immediate family members. Unspecified functional psychosis required positive report of one or more specific symptoms (delusions, hallucinations, incoherence, or bizarre behavior) not due to a mood disorder. Under this definition, there are 196 FH+ cases. We compare these to the most conservatively defined 478 FH− cases reporting no psychotic illness in either first or second degree relatives. This approach has ≥65% power to detect differences between the FH+ and FH− subsamples assuming the same parameters as in our case/control power calculation (MAF ≥20%, allelic OR ≥1.3 and alpha=0.05).
Marker Selection and Genotyping
Following discussion with the Cardiff group, we genotyped 3 of the most promising SNPs from the report of genomewide association in the Cardiff sample, rs1344706 in ZNF804A, rs6490121 in NOS1 and rs9922369 in RGRIP1L. Based on the results in these markers, we then completed genotyping of 11 additional LD-tagging SNPs (tSNPs) in ZNF804A. We first manually included rs1344706 (associated in the original study) in the test alleles set and then selected additional tSNPs using the TAGGER39 algorithm with default criteria of r2≥0.8 and minor allele frequency (MAF)≥0.2 as implemented in Haploview 4.040 using HapMap data release 22/Phase II, Apr 07. Including rs1344706, we genotyped a total of 12 SNPs in ZNF804A. Markers are shown in Table 1; orientation and alleles are reported on the genomic (+) strand (rs1344706 is reported here as A/C, not T/G as in the original report23).
Table 1. Results of single marker analyses for SNPs in ZNF804A, NOS1 and RGRIP1L.
2-tailed p-values are shown for all tests and 1-tailed p-values are shown for the three previously reported SNPs
| Locus | Mbp | SNP | Allele | fCases | fControls | Allelic OR (95% CI) | CA (Z) | 1t p | 2t p | HWE p |
|---|---|---|---|---|---|---|---|---|---|---|
| Chr 2 ZNF804A | 185.19 | rs13393273 | G | 0.44 | 0.4 | 1.17 (1.01–1.36) | −2.107 | 0.0351 | 0.2684 | |
| 185.19 | rs17508595 | G | 0.31 | 0.27 | 1.21 (1.02 − 1.43) | −2.285 | 0.0230 | 0.0809 | ||
| 185.20 | rs12613195 | C | 0.7 | 0.67 | 1.15 (0.98 − 1.35) | −1.687 | 0.0917 | 0.7266 | ||
| 185.22 | rs12693385 | T | 0.57 | 0.54 | 1.11 (0.96 − 1.29) | −1.421 | 0.1553 | 0.5129 | ||
| 185.24 | rs7597593 | T | 0.44 | 0.38 | 1.28 (1.10 − 1.49) | −3.212 | 0.0013 | 0.8647 | ||
| 185.26 | rs1480481 | C | 0.41 | 0.35 | 1.13E-10 | |||||
| 185.41 | rs7605689 | T | 0.81 | 0.79 | 1.12 (0.93 − 1.35) | −1.222 | 0.2216 | 0.4775 | ||
| 185.47 | rs3931790 | T | 0.83 | 0.81 | 1.10 (0.91 − 1.34) | −0.998 | 0.3181 | 0.8894 | ||
| 185.48 | rs7603001 | A | 0.51 | 0.47 | 1.16 (1.00 − 1.34) | −1.929 | 0.0537 | 0.7286 | ||
| 185.49 | rs1344706 | A (+) | 0.65 | 0.61 | 1.20 (1.03 − 1.40) | −2.279 | 0.0113 | 0.0227 | 0.0153 | |
| 185.51 | rs4667001 | G | 0.44 | 0.41 | 1.14 (0.98 − 1.32) | −1.661 | 0.0966 | 0.6028 | ||
| 185.51 | rs12477430 | A | 0.33 | 0.29 | 1.17 (1.00 − 1.38) | −1.909 | 0.0563 | 0.7632 | ||
| Mbp | SNP | Allele | fCases | fControls | Allelic OR (95% CI) | CA (Z) | 1t p | 2t p | HWE p | |
| Chr 12 NOS1 | 116.19 | rs6490121 | A | 0.66 | 0.65 | 1.07 (0.91 − 1.25) | −0.806 | 0.210 | 0.420 | 0.7591 |
| Mbp | SNP | Allele | fCases | fControls | Allelic OR (95% CI) | CA (Z) | 1t p | 2t p | HWE p | |
| Chr 16 RGRIP1L | 52.21 | rs9922369 | A (+) | 0.03 | 0.02 | 1.57 (0.91 − 2.71) | −1.631 | 0.0515 | 0.1029 | 1 |
Note that rs1344706 is here reported on the genomic (+) strand, where its alleles are A or C. Associated SNPs are shown in bold, (+) indicates the same allele as observed in the original study. Mbp: physical position in megabasepairs; fCases/Controls: Frequency of indicated allele in cases and controls respectively; CA (Z): Cochran-Armitage test statistic; 1t p, 2t p: one- and two-tailed p-values respectively; HWE p: Hardy-Weinberg equilibrium p-value.
All markers were genotyped with Taqman Assays-on-Demand (Applied Biosystems, Foster City, CA). To ensure uniformity and accuracy, all reaction steps were performed using the Eppendorf 5075 automated liquid handling platform. Genotypes were called using an automated allele scoring platform41. For quality control, we exclude any individual with >50% missing genotypes. We analyzed duplicate genotype data for 35 duplicate pairs (11 cases and 24 controls) and use discordant genotypes in these duplicates to estimate genotyping error rates.
Statistical Analysis
Primary Analyses
Single marker analyses were performed in SAS42 and followed the original study in assessing the Cochran-Armitage trend test. For GWAS data, the trend test is widely thought to be the most robust and broadly appropriate primary hypothesis test currently available and here it also provides the closest possible test of replication. Previous work demonstrated 1) validity of the trend statistic in the presence of Hardy Weinberg disequilibrium, and 2) asymptotic equivalence of allele and genotype-based trend statistics when Hardy Weinberg equilibrium holds43. We present uncorrected results from one-tailed tests of the three direct replication SNPs (rs1344706 in ZNF804A, rs6490121 in NOS1 and rs9922369 in RGRIP1L). We treat the remaining analyzed SNPs in ZNF804A (10 after one SNP dropped during QC) as independent (based on their selection as tags) and apply Bonferroni correction for 10 tests.
Haplotype analyses were performed assessing Χ2 in Haploview v4.040. The Χ2 is also appropriate given the ethnic homogeneity of our sample and the absence of observed substructure in the Irish population44. We also use HAPLOVIEW to identify significant (p<0.001) departures from Hardy-Weinberg Equilibrium (HWE) and compare linkage disequilibrium (LD) against HapMap data. Haplotypes were analyzed within blocks defined by the default confidence-interval method45. In one additional analysis undertaken on the basis of single marker association results, we analyzed rs7597593 (not included in any confidence-interval defined block) with the confidence-interval defined SNP set that includes rs1344706. We assessed empirical significance of haplotype results with 5000 permutations of case and control status.
Secondary Analyses
To assess potential gender differences in the association signal, we tested for SNP × Sex interaction using logistic regression, coding each SNP in an additive framework, including gender and a SNP × Sex interaction term. To test for differences in evidence for association between sets of cases defined by FH status, we stratified the sample by the presence (FH+) or absence (FH−) of first degree relatives with schizophrenia or unspecified psychosis as described above (1st degree broad definition). Genotypes at rs1344706 were missing for 23 individuals with FH data, and We tested for allelic (1df) association by assessing Χ2 between FH+ and FH− cases. Both analyses were performed using SAS42.
Gene Expression Analysis
Post-mortem Brain Tissue Samples
The Stanley Medical Research Institute (SMRI) provided genomic DNA and postmortem brain tissue samples from dorsolateral prefrontal cortex (Brodmann’s area 46) from 35 individuals with schizophrenia (SCH) and 35 controls (CON). Exclusion criteria for these samples included: (1) significant structural brain pathology, (2) history of pre-existing central nervous system disease, (3) poor RNA quality, (4) documented IQ < 70, (5) age less than 30 and (6) substance abuse within one year of death46. RNA was isolated using the Ambion miRvana kit (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. We obtained poor yields of RNA from 1 CON sample, which was excluded from further study, leaving 35 SCH and 34 CON samples.
Quantitative Real-Time PCR
cDNA was reverse transcribed using the Ready-To-Go Kit from Amersham Bioscience (Piscataway, NJ) following manufacturer’s instructions. ZNF804A expression was analyzed on the StepOne Plus instrument (ABI, Foster City, CA) using a Taqman quantitative real-time PCR expression assay (ABI, Foster City, CA). Each sample was assayed in triplicate and mean values from the triplicates were used for all analyses. Gene expression was normalized against two reference genes47 (HPRT and TBP), and analyzed by the 2−ΔΔCT algorithm48. The 2−ΔΔCT algorithm, widely used to compare differences in gene expression, is valid only if PCR efficiency of studied genes is equal48. PCR efficiencies (measured by standard curve) of all three genes assayed were very similar (ZNF804A: 91%, HPRT: 93%, TBP: 90%).
Statistical Analysis of Expression Data
To achieve a normal distribution, ZNF804A expression values were log transformed, and normality of the transformed data was verified by the Anderson-Darling empirical distribution function test49,50, a powerful statistic to detect departure from normality even in a relatively small (N≤100) sample. Samples (N=7, 3 SCH and 4 CON) with values greater than ±2SD from the group mean were omitted from further analyses, reducing the totals to 32 SCH and 30 CON. Mean expression levels in SCH and CON groups were compared using Welch’s corrected unpaired t-test. The potential confounding effects of diagnosis, age, gender, pH, post-mortem interval, refrigerator interval, smoking and drug abuse in the analysis of ZNF804A expression were assessed by analysis of variance (ANOVA).
Statistical Analysis of Expression by rs1344706 Genotype
We assessed sequence around SNP rs1344706 in the UCSC Genome Browser (http://genome.ucsc.edu). Based on evidence of conservation, we sought to identify transcription-factor bindingsites altered by the alternative rs1344706 alleles. A 53 bp sequencesurrounding rs1344706 (chr2:185,486,646-185,486,698) was analyzed using MatInspector v7.7.3.1 and SNPinspector v2.251 (http://www.genomatix.de/). In both cases the vertebrate transcription factor matrices and optimized matrix threshold were used to reduce the incidence of false-positives.
Based on the results of these predictions, the SMRI CON samples were genotyped for rs1344706 for analysis of expression differences due to variation at this position. Genotypes are collected exactly as described above using the same instrumentation and reagents.
For the allelic test, expression values were binned by rs1344706 allele and the means of the bins were then compared52. Each individual’s expression value is used twice: for homozygotes, twice in the same bin, for heterozygotes, once in each. As a result, the variation in the homozygotes may be less than that in heterozygotes, and we therefore compare the two group means using the unpaired t-test with Welch’s correction to account for possible heteroscedastic variances.
RESULTS
Genotyping: Missingness, Error Rates and LD
A total of 28 case (2.7%) and 56 (8.9%) control samples were excluded from analysis with >50% missing genotypes. All subsequent results are based on a total of 1563 samples (993 cases and 570 controls). Of these, 1499 (95.9%) are missing 2 or fewer genotypes (1270 samples (81.3%) missing zero, 195 (12.5%) missing one and 36 (2.3%) missing two genotypes). Higher missing data rates (3–7 genotypes) were present in 62 samples (4.0%).
The rates of sample exclusion due to missing genotypes are higher than usual due to technical issues during genotypic data collection. Degradation of a filter in a fluorescence reader reduced signal at the detector, thus artificially increasing the minimum signal necessary for detection. This attenuation of signal did not affect robustly performing samples, but did cause a higher fail rate among more weakly amplifying samples. Prior to exclusion of poorly genotyped samples, average genotyping completion by marker was 93.5% (90.5–96.4%); following exclusions, it was 97.3% (95.2–99.2%). Following identification of the problem and replacement of the filter, 4 SNPs (rs13393273, rs7597593, rs1344706 and rs4667001, including the two most strongly associated) were completely regenotyped. This repeated data collection yields a higher genotyping completion rate (mean 98.5%) and a low rate (0.52%) of discordant genotypes for these four markers.
We genotyped 14 SNPs × 35 samples in duplicate (N=490 genotype pairs); both genotypes were available for 465 (94.9%), of which 0 were discordant, estimating our overall genotyping error rate at less than 1 in 465 (<0.21%). All markers except rs1480481 (p=1.13×10−10 and not further analyzed) satisfied our HWE cut-off for inclusion. Linkage disequilibrium patterns in this sample are virtually identical to those in HapMap data (Figure S1).
Single Marker Analysis
ZNF804A
Results of single marker analyses are shown in Table 1. In the data from ZNF804A, rs1344706 (associated in the original report in discovery and combined replication samples) is also associated in the ICCSS (one-tailed p=0.0113). The same allele (A) is increased in frequency in cases compared to controls as in the original study. In total, we observe nominal evidence of association between schizophrenia and 4 of the 11 SNPs passing QC criteria. The T allele of rs7597593 yields substantially more significant evidence of association (two-tailed p=0.0013), but this SNP lacks the evidence of possible functional significance observed for rs1344706 (see below). The single marker results from rs7597593 (p=0.0013) remain significant after Bonferroni correction for 10 tests. Allele and genotype counts are shown in Table S1.
NOS1 and RGRIP1L
No significant difference between cases and controls was observed for rs6490121 in NOS1. Results for rs9922369 in RGRIP1L show only a trend towards significance (one-tailed p=0.0515), with the same allele as originally reported observed to be more common in cases in our study. Allele and genotype counts are shown in Table S1.
Haplotype Analysis
Results of haplotype analyses within confidence interval defined blocks are shown in Table 2. Compared to single marker analyses, we observe an increased association in block 3 (containing rs1344706) on a haplotype (frequency 0.188 in controls) bearing the common alleles of rs7605689 (T) and rs3931790 (T), the rare allele of rs7603001 (A) and the previously associated common (A) allele of rs1344706 (Χ2= 8.626, p<0.003). The increased significance is due to a larger case/control difference (5%) in the frequency of this TTAA haplotype compared to the difference for the A allele of rs1344706 (4%).
Table 2. Results of haplotype analyses for SNPs in ZNF804A.
Associated haplotypes are shown in bold.
| Block | HT | Freq. | fCases | fControls | Chi Sq | p | P |
|---|---|---|---|---|---|---|---|
| Block 1 | AC | 0.577 | 0.564 | 0.601 | 3.935 | 0.0473 | 0.558 |
| GG | 0.288 | 0.302 | 0.263 | 5.531 | 0.0187 | 0.284 | |
| GC | 0.135 | 0.134 | 0.137 | 0.062 | 0.8033 | 1 | |
| Block 2 | CT | 0.557 | 0.567 | 0.541 | 1.956 | 0.1619 | 0.952 |
| GC | 0.312 | 0.301 | 0.332 | 3.238 | 0.072 | 0.721 | |
| CC | 0.13 | 0.132 | 0.127 | 0.171 | 0.6796 | 1 | |
| Block 3 | TTAA | 0.304 | 0.322 | 0.272 | 8.626 | 0.0033 | 0.058 |
| CTAA | 0.194 | 0.187 | 0.207 | 1.822 | 0.1771 | 0.964 | |
| TTGC | 0.191 | 0.18 | 0.209 | 3.674 | 0.0553 | 0.611 | |
| TGGC | 0.177 | 0.172 | 0.184 | 0.68 | 0.4094 | 1 | |
| TTGA | 0.133 | 0.137 | 0.126 | 0.753 | 0.3856 | 1 | |
| rs7597593 + Block 3 | TTTAA | 0.221 | 0.24 | 0.188 | 11.221 | 0.0008 | 0.017 |
| CTTGC | 0.186 | 0.175 | 0.204 | 4.018 | 0.045 | 0.582 | |
| CTGGC | 0.176 | 0.171 | 0.183 | 0.625 | 0.4293 | 1 | |
| CCTAA | 0.12 | 0.112 | 0.134 | 3.23 | 0.0723 | 0.749 | |
| TTTGA | 0.118 | 0.123 | 0.109 | 1.171 | 0.2792 | 0.998 | |
| CTTAA | 0.082 | 0.082 | 0.081 | 0.016 | 0.8983 | 1 | |
| TCTAA | 0.075 | 0.075 | 0.076 | 0.011 | 0.915 | 1 | |
| CTTGA | 0.015 | 0.015 | 0.015 | 0.005 | 0.9419 | 1 | |
| Block 4 | AG | 0.57 | 0.558 | 0.59 | 2.958 | 0.0855 | 0.789 |
| GA | 0.315 | 0.327 | 0.295 | 3.329 | 0.0681 | 0.699 | |
| GG | 0.115 | 0.115 | 0.115 | 0 | 0.9901 | 1 | |
fCases/Controls: Frequency of indicated haplotype in cases and controls respectively; Chi-Sq: Chi-square test statistic; p and P: asymptotic and empirical permutation significance levels respectively. The unblocked, associated SNP rs7597593 was included in analysis with block 3 SNPs (including rs 1344706).
In a single targeted secondary haplotype analysis, we included the associated SNP rs7597593 with the 4 markers in the confidence interval defined block 3, containing rs1344706. We observe a further increase in association for the haplotype defined by the associated allele of rs7597593 (T) in conjunction with the TTAA haplotype associated above. This comparison provides the largest observed haplotypic frequency difference between cases and controls (5.2%), and is the only haplotypic comparison to remain significant after permutation testing (P=0.017).
SNP × Sex Interaction Analysis
In logistic regression tests of SNP × Sex interaction (Table S2), 8 markers in ZNF804A yield nominally significant evidence of association including all 4 that were associated in primary analyses (rs17508595, rs13393273, rs7597593 and rs1344706). The SNP × Sex interaction term was not significant for any marker tested, providing no evidence for differential association depending on the sex of the case.
Family History Analysis
Based on the single marker results above, we limited FH analyses to the most significantly associated markers, rs7597593 and rs1344706 (Table S3). Using the categorical definitions described in Methods, there are 196 FH+ and 478 conservatively defined FH− cases (no illness reported in first or second degree family members) in the ICCSS sample. A genotype at rs1344706 was missing for 23 individuals with family history (15 FH− and 8 FH+), leaving a total analyzed sample N=651 (1302 alleles), 463 FH− and 188 FH+. There is no evidence of any difference in association of rs1344706 and rs7597593 between FH+ and FH− cases (Table S3).
Expression analysis
We assessed gene expression differences for ZNF804A in RNA from the prefrontal cortex from the SMRI SCH and CON samples. We excluded 3 SCH and 4 CON outlier samples, as described above, leaving 32 SCH and 30 CON for analysis. Expression was higher in SCH compared to CON samples, but this difference did not achieve statistical significance (Welch-corrected t=1.640, df=52, p=0.107, Figure 1A).
Figure 1.
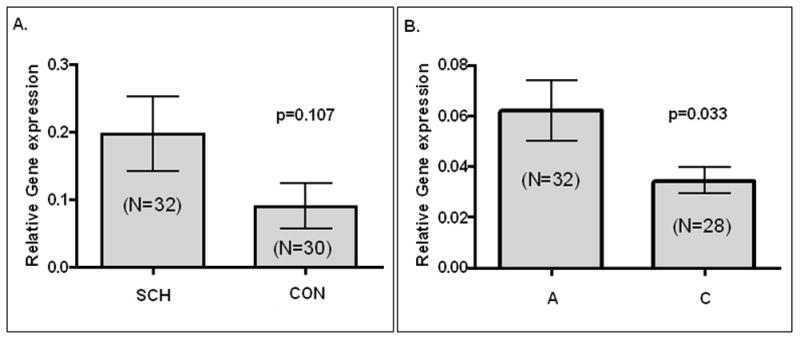
ZNF804A gene expression in the SMRI sample. A. Comparison of ZNF804A expression in schizophrenic (SCH) and control (CON) samples.
B. ZNF804A expression in SMRI control samples stratified by rs1344706 allele.
Bioinformatic Analysis of rs1344706
Inspection of the sequence around SNP rs1344706 shows that it lies in a short region of conserved mammalian sequence in intron 2 of ZNF804A. MatInspector results for the 53 bp of sequence we assessed (chr2:185,486,646-185,486,698) include the A allele of rs1344706 in the predicted binding sites for two brain-expressed transcription factors, Myt1L53,54 and POU3F1/Oct-655, both implicated in multiple CNS developmental processes, particularly in oligodendrocytes54,55. Two additional binding sites are predicted in the presence of the C allele. One of these is for the ubiquitously expressed Homez56 and the other is for Hmx2, which, while expressed in the CNS, is thought to be primarily involved in the development of the inner ear57–59 (Figure S2).
Genotype-specific expression
The SMRI postmortem CON samples were genotyped for rs1344706. Allele frequencies in the N=30 CON samples were A: 32/60, 0.533; C: 28/60, 0.477, and were in good agreement with data from the HapMap (A: 0.567; C: 0.433) and Irish (A: 0.607; C: 0.393) samples. Expression is significantly higher for the associated A allele (t=2.129, df=39, p=0.033, Figure 1B). The observed difference was not due to effects of potential confounder variables assessed by ANOVA (age, post-mortem interval, refrigeration interval, brain pH or smoking, F=0.829; df=12; p=0.62).
DISCUSSION
We studied ZNF804A in a case/control sample appropriate for replication of the original results. First, our sample was drawn from Ireland and should have substantial genetic overlap with the Anglo-Celtic discovery sample. Second, our ascertainment and diagnosis methods were highly homologous with those used to collect the discovery sample. Third, the ICCSS sample has substantial power to detect genetic effects.
Our study provides further support for the association of schizophrenia with common variant alleles and haplotypes in ZNF804A. We replicate the previously reported observation of genetic association between schizophrenia and the A allele of rs1344706. In an extended set of tSNPs, we also observe association with the minor T allele of rs7597593, with a haplotype containing the A allele of rs1344706, and with a haplotype containing the A allele of rs1344706 and the T allele of rs7597593. Only the single marker results from rs7597593 remain significant after Bonferroni correction, and only the results from the longer haplotype including rs7597593 remain significant after permutation testing. LD and haplotypes agree closely with prior observations in other European-descended samples. There is no evidence for differential association with ZNF804A between male and female cases or between cases with and without a positive family history of schizophrenia-like illness.
Although it seems unlikely that SNP rs1344706 directly increases risk for schizophrenia, the association and expression data collectively are consistent with possible functional effects at rs1344706 itself. The A allele shows significantly higher expression than the C allele in control dorsolateral prefrontal cortex. The A allele is associated with schizophrenia, and expression in schizophrenic case dorsolateral prefrontal cortex is increased relative to that in controls, although this last difference does not achieve statistical significance.
Bioinformatic analysis of the source of the mammalian conservation around rs1344706 suggests it may be due to the presence of transcription factor binding sites. The alleles of rs1344706 result in differential prediction of the presence of binding sites for two brain-expressed transcription factors. The Myt1L zinc finger protein is expressed in neural progenitors along with the related family member Myt1 (which is known to modulate proliferation and terminal differentiation of oligodendrocyte progenitors54.) Of note, the MYT1L gene was included in a copy number variant observed in an affected individual in a recent study of structural variation in schizophrenia. The POU3F1/Oct-6 POU-domain transcription factor is involved in oligodendrocyte differentiation55 and the transition of pro-myelinating to myelinating Schwann cells60, and is also normally expressed in adult cortex and hippocampus61. Those transcription factors predicted to bind to the C allele of rs1344706 are widely expressed and not brain-specific (Homez) or involved primarily in the development of the inner ear, and so seem potentially less meaningful in this context.
The pattern of association in published GWAS findings for schizophrenia (and particularly their replication) has been highly variable. Although these studies have shown little support for the results of either other GWAS or prior studies, sample sizes in many cases have not been adequate to deliver reasonable power. In this case, although the discovery sample was somewhat small, the replication sample has reasonable power to detect effects in the ranges modeled. Additional results from a number of large, better powered studies and from meta-analyses of more than 10,000 cases and controls (which have substantial power to detect effects of the kind expected in schizophrenia) are expected in the near future. These results will be critical in determining which of the early results are widely supported. In conclusion, however, our results from the present study of ZNF804A and schizophrenia in a large, ethnically homogeneous sample add further support for the association of the A allele at rs1344706 with schizophrenia.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Mental Health Grant R01-MH41953 to KSK/BR. AF was supported by a Department of Veterans Affairs Merit Review award. We thank X. Chen for genotyping macros and sex determination data and the Northern Ireland Blood Transfusion Services for their invaluable assistance in control sampling.
Footnotes
CONFLICT OF INTEREST: The authors declare they have no biomedical financial interests or potential conflicts of interest.
Supplementary information is available at the Molecular Psychiatry website.
Reference List
- 1.Kendler KS, Diehl SR. The genetics of schizophrenia: A current, genetic-epidemiologic perspective. Schizophrenia Bulletin. 1993;19:261–285. doi: 10.1093/schbul/19.2.261. [DOI] [PubMed] [Google Scholar]
- 2.Maki P, Veijola J, Jones PB, Murray GK, Koponen H, Tienari P, et al. Predictors of schizophrenia--a review. Br Med Bull. 2005;73–74:1–15. doi: 10.1093/bmb/ldh046. [DOI] [PubMed] [Google Scholar]
- 3.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 4.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 6.Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, et al. Genome-Wide Association Analysis Identifies Loci for Type 2 Diabetes and Triglyceride Levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 7.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. A Genome-Wide Association Study of Type 2 Diabetes in Finns Detects Multiple Susceptibility Variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 9.Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007 doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- 10.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of Genome-Wide Association Signals in U.K. Samples Reveals Risk Loci for Type 2 Diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groves CJ, Zeggini E, Minton J, Frayling TM, Weedon MN, Rayner NW, et al. Association analysis of 6,736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes. 2006;55:2640–2644. doi: 10.2337/db06-0355. [DOI] [PubMed] [Google Scholar]
- 12.Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, et al. Common Kibra alleles are associated with human memory performance. Science. 2006;314:475–478. doi: 10.1126/science.1129837. [DOI] [PubMed] [Google Scholar]
- 13.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 15.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, et al. Novel Crohn Disease Locus Identified by Genome-Wide Association Maps to a Gene Desert on 5p13.1 and Modulates Expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 18.Haiman CA, Patterson N, Freedman ML, Myers SR, Pike MC, Waliszewska A, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007;39:638–644. doi: 10.1038/ng2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 20.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lencz T, Morgan TV, Athanasiou M, Dain B, Reed CR, Kane JM, et al. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12:572–580. doi: 10.1038/sj.mp.4001983. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan PF, Lin D, Tzeng JY, van den OE, Perkins D, Stroup TS, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol Psychiatry. 2008;13:570–584. doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 24.Sun S, Wang F, Wei J, Cao LY, Wu GY, Lu L, et al. Association between interleukin-3 receptor alpha polymorphism and schizophrenia in the Chinese population. Neurosci Lett. 2008;440:35–37. doi: 10.1016/j.neulet.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 25.Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 26.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spitzer RL, Williams JB, Gibbon J. Structured Clinical Interview for DSM-III-R - Patient Version (SCID-P, 4/1/87) New York State Psychiatric Institute; New York: 1987. [Google Scholar]
- 28.Kendler KS, O’Neill FA, Burke J, Murphy B, Duke F, Straub RE, et al. Irish study on high-density schizophrenia families: field methods and power to detect linkage. Am J Med Genet. 1996;67:179–190. doi: 10.1002/(SICI)1096-8628(19960409)67:2<179::AID-AJMG8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 29.Cavalli-Sforza LL, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton University Press; Princeton, NJ: 1994. [Google Scholar]
- 30.Relethford JH. Genetic structure and population history of Ireland: a comparison of blood group and anthropometric analyses. Ann Hum Biol. 1983;10:321–333. doi: 10.1080/03014468300006481. [DOI] [PubMed] [Google Scholar]
- 31.Sunderland E, Tills D, Bouloux C, Doyl J. In: Genetic Variation in Britain. Roberts DF, Sunderland E, editors. XII. Taylor & Francis Ltd; London: 1973. pp. 141–170. [Google Scholar]
- 32.Hill EW, Jobling MA, Bradley DG. Y-chromosome variation and Irish origins. Nature. 2000;404:351–352. doi: 10.1038/35006158. [DOI] [PubMed] [Google Scholar]
- 33.Semino O, Passarino G, Oefner PJ, Lin AA, Arbuzova S, Beckman LE, et al. The genetic legacy of Paleolithic Homo sapiens sapiens in extant Europeans: a Y chromosome perspective. Science. 2000;290:1155–1159. doi: 10.1126/science.290.5494.1155. [DOI] [PubMed] [Google Scholar]
- 34.McEvoy B, Richards M, Forster P, Bradley DG. The Longue Duree of genetic ancestry: multiple genetic marker systems and Celtic origins on the Atlantic facade of Europe. Am J Hum Genet. 2004;75:693–702. doi: 10.1086/424697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andreasen NC, Endicott J, Spitzer RL, Winokur G. The family history method using diagnostic criteria. Reliability and validity. Arch Gen Psychiatry. 1977;34:1229–1235. doi: 10.1001/archpsyc.1977.01770220111013. [DOI] [PubMed] [Google Scholar]
- 36.Endicott J, Andreasen N, Spitzer RL. Family History Research Diagnostic Criteria. Biometrics Research Department, New York State Psychiatric Institute; New York: 1978. [Google Scholar]
- 37.Roy MA, Walsh D, Kendler KS. Accuracies and inaccuracies of the family history method: a multivariate approach. Acta Psychiatr Scand. 1996;93:224–234. doi: 10.1111/j.1600-0447.1996.tb10639.x. [DOI] [PubMed] [Google Scholar]
- 38.Andreasen NC, Endicott J, Spitzer RL, Winokur G. The family history method using diagnostic criteria. Reliability and validity. Arch Gen Psychiatry. 1977;34:1229–1235. doi: 10.1001/archpsyc.1977.01770220111013. [DOI] [PubMed] [Google Scholar]
- 39.de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 40.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 41.Van den Oord EJCG, Jiang Y, Riley BP, Kendler KS, Chen X. FP-TDI SNP genotype scoring by manual and statistical procedures: A study of error rates and types. Biotechniques. 2003;34:610–624. doi: 10.2144/03343dd04. [DOI] [PubMed] [Google Scholar]
- 42.SAS Institute. SAS. v9. Cary; NC: 2002. Ref Type: Computer Program. [Google Scholar]
- 43.Sasieni PD. From genotypes to genes: doubling the sample size. Biometrics. 1997;53:1253–1261. [PubMed] [Google Scholar]
- 44.O’Dushlaine CT, Dolan C, Weale ME, Stanton A, Croke DT, Kalviainen R, et al. An assessment of the Irish population for large-scale genetic mapping studies involving epilepsy and other complex diseases. Eur J Hum Genet. 2007 doi: 10.1038/sj.ejhg.5201938. [DOI] [PubMed] [Google Scholar]
- 45.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumensteil B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 46.Torrey EF, Webster M, Knable M, Johnston N, Yolken RH. The stanley foundation brain collection and neuropathology consortium. Schizophr Res. 2000;44:151–155. doi: 10.1016/S0920-9964(99)00192-9. [DOI] [PubMed] [Google Scholar]
- 47.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:research0034.1–research0034.11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 49.Anderson TW, Darling DA. Asymptotic theory of certain “goodness-of-fit” criteria based on stochastic processes. Annals of Mathematical Statistics. 1952;23:193–212. [Google Scholar]
- 50.Stephens MA. EDF Statistics for Goodness of Fit and Some Comparisons. Journal of the American Statistical Association. 1974;69:730–737. [Google Scholar]
- 51.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 52.Page GP, Amos CI. Comparison of linkage-disequilibrium methods for localization of genes influencing quantitative traits in humans. Am J Hum Genet. 1999;64:1194–1205. doi: 10.1086/302331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JG, Armstrong RC, Agoston D, Robinsky A, Wiese C, Nagle J, et al. Myelin transcription factor 1 (Myt1) of the oligodendrocyte lineage, along with a closely related CCHC zinc finger, is expressed in developing neurons in the mammalian central nervous system. J Neurosci Res. 1997;50:272–290. doi: 10.1002/(SICI)1097-4547(19971015)50:2<272::AID-JNR16>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 54.Nielsen JA, Berndt JA, Hudson LD, Armstrong RC. Myelin transcription factor 1 (Myt1) modulates the proliferation and differentiation of oligodendrocyte lineage cells. Mol Cell Neurosci. 2004;25:111–123. doi: 10.1016/j.mcn.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 55.Collarini EJ, Kuhn R, Marshall CJ, Monuki ES, Lemke G, Richardson WD. Down-regulation of the POU transcription factor SCIP is an early event in oligodendrocyte differentiation in vitro. Development. 1992;116:193–200. doi: 10.1242/dev.116.1.193. [DOI] [PubMed] [Google Scholar]
- 56.Bayarsaihan D, Enkhmandakh B, Makeyev A, Greally JM, Leckman JF, Ruddle FH. Homez, a homeobox leucine zipper gene specific to the vertebrate lineage. Proc Natl Acad Sci U S A. 2003;100:10358–10363. doi: 10.1073/pnas.1834010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W, Chan EK, Baron S, Van de WT, Lufkin T. Hmx2 homeobox gene control of murine vestibular morphogenesis. Development. 2001;128:5017–5029. doi: 10.1242/dev.128.24.5017. [DOI] [PubMed] [Google Scholar]
- 58.de Geus EJ, Posthuma D, Kupper N, van den BM, Willemsen G, Beem AL, et al. A whole-genome scan for 24-hour respiration rate: a major locus at 10q26 influences respiration during sleep. Am J Hum Genet. 2005;76:100–111. doi: 10.1086/427267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang W, Lufkin T. Hmx homeobox gene function in inner ear and nervous system cell-type specification and development. Exp Cell Res. 2005;306:373–379. doi: 10.1016/j.yexcr.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 60.Jaegle M, Mandemakers W, Broos L, Zwart R, Karis A, Visser P, et al. The POU factor Oct-6 and Schwann cell differentiation. Science. 1996;273:507–510. doi: 10.1126/science.273.5274.507. [DOI] [PubMed] [Google Scholar]
- 61.Ubhi K, Price J. Expression of POU-domain transcription factor, Oct-6, in schizophrenia, bipolar disorder and major depression. BMC Psychiatry. 2005;5:38. doi: 10.1186/1471-244X-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.