

Abstract
The kinetics of oxidation of a series of para-substituted N, N'-dibenzyl-1,4-diaminobutanes by the flavoprotein polyamine oxidase from mouse have been determined to gain insight into the mechanism of amine oxidation by this member of the monoamine oxidase structural family. The kcat/Km values are maximal at pH 9, consistent with the singly charged substrate being the active form. The rate constant for flavin reduction, kred, by N,N'-dibenzyl-1,4-diaminobutane decreases about 5-fold below a pKa of ~8; this is attributed to the need for a neutral nitrogen at the site of oxidation. The kred and kcat values are comparable for each of the N, N'-dibenzyl-1,4-diaminobutanes, consistent with rate-limiting reduction. The deuterium kinetic isotope effects on kred and kcat are identical for each of the N, N'-dibenzyl-1,4-diaminobutanes, consistent with rate-limiting cleavage of the substrate CH bond. The kred values for seven different para-substituted N, N'-dibenzyl-1,4-diaminobutanes correlate with a combination of the van der Waals volume and σ value of the substrates, with ρ values of −0.59 at pH 8.6 and −0.09 at pH 6.6. These results are consistent with direct transfer of a hydride from the neutral CN bond of the substrate to the flavin as the mechanism of polyamine oxidase.
The polyamines spermine and spermidine are critical for growth of eukaryotic cells due to the ability of these polycations to bind a variety of macromolecules (1). As a result polyamine metabolism has been a frequent target for the development of anticancer agents (2). Catabolism of spermine and spermidine can occur by two routes (3). In the first pathway to be discovered, spermidine/spermine N1-acetyltransferase converts spermine or spermidine to the N-acetyl form (4). The N1-acetylspermine or N1-acetylspermidine can then be transported out of the cell. Alternatively, the flavoprotein polyamine oxidase (PAO)1 can catalyze the oxidation of N1-acetylspermine and N1-acetylspermidine to spermidine and putrescine, respectively, producing H2O2 (5, 6). The more recently discovered flavoprotein spermine oxidase catalyzes the oxidation of spermine directly to spermidine (7, 8), bypassing the necessity for acetylation. While PAO is a constitutive enzyme, spermine oxidase is induced by polyamine analogues with antitumor activity (7). The relative importance of the two enzymes in polyamine metabolism in normal and cancer cells remains to be established.
PAO belongs to the flavin amine oxidase structural family that includes monoamine oxidase A and B (MAO A/B) (9), lysine-specific demethylase (LSD1) (10), and L-amino acid oxidase (11). At present there is no structure available of a mammalian PAO, and the sequence identity of mouse or human PAO to these enzymes is low (<20%). Still, structures are available of maize PAO and yeast Fms1 that establish that these two enzymes and mammalian PAOs belong to the MAO structural family (12). However, PAO from maize and other plants catalyzes CH bond cleavage on the endo-side of the N4-nitrogen of spermine and N1-acetylspermine (13), whereas mammalian PAOs catalyze CH bond cleavage on the exo-side of the N4-nitrogen, and Fms1 is more accurately a spermine oxidase than a PAO, in that its preferred substrate is spermine rather than N1-acetylspermine (14). The structural basis for the differences in reactivity remains unknown.
The reaction of PAO, like other flavoprotein amine oxidases, can be divided into two half-reactions. In the reductive half-reaction a hydride equivalent is transferred from the amine substrate to the flavin to form the reduced flavin and the oxidized amine; in the oxidative half-reaction the reduced flavin reacts with molecular oxygen to produce hydrogen peroxide and the oxidized flavin. Mouse PAO displays the ping-pong kinetic pattern (5) observed with most flavoprotein oxidases (15), since oxygen only reacts with the reduced enzyme formed upon oxidation of the amine. The mechanism by which electrons are transferred from the amine substrate to the flavin by the flavin amine oxidases has been controversial (16). The simplest mechanism is direct hydride transfer; this is supported by the lack of detectable intermediates in the reductive half-reactions of flavin amine oxidases and by kinetic isotope effects (17–21). The ability of compounds containing cyclopropyl rings to act as mechanism-based inhibitors of MAO has been taken as evidence for radical intermediates in the reactions of that enzyme and other flavin amine oxidases (22). Finally, a mechanism involving nucleophilic addition of the amine substrate to the flavin has been proposed (23). In the present study, para-substituted N, N'-dibenzyl-1,4-diaminobutanes were characterized as substrates for mouse PAO to gain insight into the substrate specificity and mechanism of amine oxidation by that enzyme.
EXPERIMENTAL PROCEDURES
Materials Reagents for syntheses were routinely purchased from Aldrich Chemical Co. N1-Acetylspermine was from Fluka. All remaining reagents were of the highest purity commercially available.
Syntheses
Deuterated benzaldehydes
Commercially available 4-substituted benzoic acids were converted to the corresponding benzyl-deuterated benzaldehydes by modification of the method of Paik et al. (24). Lithium aluminum deuteride (1.2 g, 28.5 mmol) was added in small portions to a stirred solution of the p-substituted benzoic acid (7.4 mmol) in anhydrous THF (20 ml) at −20 °C over 4 h. The solution was then warmed to room temperature for 1 h. The reaction was quenched with saturated aqueous NH4Cl (30 ml). Twenty ml of 10% NaOH was added; after 1 h 15 ml of water was added. The resulting precipitate was filtered off and washed with ether. The combined organic layers were evaporated under vacuum. The remaining oil was dried under vacuum and dissolved in anhydrous CH2Cl2 (40 ml) at 4 °C. Pyridinium chlorochromate (5.0 g, 23 mmol) was added, and the reaction mixture was stirred for 40 min in an ice bath and then quenched with 75 ml of saturated aqueous NaHCO3. The combined organic layers from three extractions with ethyl acetate (50 ml each) were washed with water and brine and then dried over anhydrous Na2SO4. Column chromatography on silica gel with 5% ethyl acetate in hexane yielded the deuterated benzaldehyde as a colorless oil.
N,N'-dibenzyl-1,4-butanediamines
The synthesis was adapted from Burns et al. (25). The appropriate benzaldehyde (12.5 mmol) and triethylamine (1.74 ml, 12.5 mmol) were added to a solution of 1,4-diaminobutane (0.5 g, 5.7 mmol) in 20 ml of dichloromethane, followed by 0.5 g of MgSO4. The reaction mixture was stirred overnight at room temperature and then filtered. The solvent was removed under reduced pressure. The residue was dissolved in 20 ml of methanol, and NaBH4 (0.56 g, 13.4 mmol) was added. After stirring overnight at room temperature, 25 ml of water was added, followed by sufficient 2 M NaOH to yield pH 10–11. The solution was extracted three times with 25 ml dichloromethane. The combined organic phases were dried over MgSO4 and filtered. The volume was decreased to ~ 5 ml under reduced pressure. Concentrated HCl was added dropwise until a white precipitate appeared. The precipitate was collected by filtration, washed with dry diethyl ether, and recrystallized from water/isopropanol. For the deuterated compounds, the respective deuterated benzaldehyde and NaBD4 were used.
N,N'-dibenzyl-1,4-butanediamine dihydrochloride Yield 1.5 g (75%); MP 327; 1H-NMR (D2O) δ 7.38 (10H), 4.66 (4H, s), 2.98 (4H, m), 1.64 (4H, m); HRMS (m+H): theor: 269.19, found: 269.21.
N,N'-bis(p-N,N-dimethylaminobenzyl)-1,4-butanediamine tetrahydrochloride Yield 1.1 g (78 %); MP 244; 1H-NMR (D2O) δ 7.72 (4H, d), 7.68 (4H, d), 4.33 (4H, s), 3.31 (12H, s), 3.16 (4H, t), 1.81 (4H, m); HRMS (m+H): theor: 355.28, found: 355.21
N,N'-bis(p-methoxybenzyl)-1,4-butanediamine dihydrochloride Yield 1.03 g (61 %); MP 301; 1H-NMR (D2O) δ 7.32 (4H, d), 6.93 (4H, d), 3.84 (6H, s), 3.81 (4H, s), 2.65 (4H, t), 1.57 (4H, m, CH2); HRMS (m+H): theor: 329.22, found: 329.25
N,N'-bis(p-methylbenzyl)-1,4-butanediamine dihydrochloride Yield 1.4 g (67 %); MP 331; 1H-NMR (D2O) δ 7.24 (4H, d), 7.18 (4H, d), 4.23 (4H, s), 2.42 (6H, s), 3.06 (4H, t), 1.92 (4H, m); HRMS (m+H): theor: 297.23, found: 297.21
N,N'-bis(p-chlorobenzyl)-1,4-butanediamine dihydrochloride Yield 1.31 g (62 %); MP 334; 1H-NMR (D2O) δ 7.42 (4H, d), 7.33 (4H, d), 4.11 (4H, s), 2.99 (4H, t), 1.67 (4H, m); HRMS (m+H): theor: 337.12, found: 337.1
N,N'-bis(p-bromobenzyl)-1,4-butanediamine dihydrochloride Yield 1.31; g (62 %); MP 333 1H-NMR (D2O) δ 7.45 (4H, d), 7.36 (4H, d), 4.15(4H, s), 3.09 (4H, t), 1.85 (4H, m); HRMS (m+H): theor: 425.01, found: 425.04
N,N'-bis(p-trifluoromethylbenzyl)-1,4-butanediamine dihydrochloride Yield 1.35 g (66 %); MP 327; 1H-NMR (D2O) δ 7.57 (4H, d), 7.45 (4H, d), 3.84 (4H, s), 2.64 (4H, t), 1.57 (4H, m); HRMS (m+H): theor: 405.17, found: 405.12
N,N'-bis(p-N,N-dimethylamino-α,α-dideuterobenzyl)-1,4-butanediamine tetrahydrochloride Yield 0.85 g; (89 %); 1H-NMR (D2O) δ 7.74 (4H, d), 7.69 (4H, d), 3.35 (12H, s), 3.19 (4H, t), 1.83 (4H, m); HRMS (m+H): theor: 359.28, found: 359.01
N,N'-bis(p-methoxy-α,α-dideuterobenzyl)-1,4-butanediamine dihydrochloride Yield 0.93 g (87 %); 1H-NMR (D2O) δ 7.33 (4H, d), 6.98 (4H, d), 3.87 (6H, s), 2.68 (4H, t), 1.60 (4H, m); HRMS (m+H): theor: 333.22, found: 333.04
N,N'-bis(p-methyl-α,α-dideuterobenzyl)-1,4-butanediamine dihydrochloride Yield 0.85 g (87 %); 1H-NMR (D2O) δ 7.22 (4H, d), 7.18 (4H, d), 2.45 (6H, s), 3.12 (4H, t), 1.95 (4H, m); HRMS (m+H): theor: 300.23, found: 301.05
N,N'-bis(α,α-dideuterobenzyl)-1,4-butanediamine dihydrochloride Yield 0.63 g (85 %); 1H-NMR (D2O) δ 7.41 (10H, m), 3.05 (4H, t), 1.65 (4H, m); HRMS (m+H): theor: 273.22, found: 273.11
N,N'-bis(p-chloro-α,α-dideuterobenzyl)-1,4-butanediamine dihydrochloride Yield 0.74 g (84 %); 1H-NMR (D2O) δ 7.44 (4H, d), 7.34 (4H, d), 3.01 (4H, t), 1.68 (4H, m); HRMS (m+H): theor: 341.12, found: 341.00
N,N'-bis(p-bromo-α,α-dideuterobenzyl)-1,4-butanediamine dihydrochloride Yield 0.85 g (87 %); 1H-NMR (D2O) δ 7.46 (4H, d), 7.37 (4H, d), 3.11 (4H, t), 1.86 (4H, m); HRMS (m+H): theor: 429.01, found: 429.91
N,N'-bis(p-trifluoromethyl-α,α-dideuterobenzyl)-1,4-butanediamine dihydrochloride Yield 0.86 g (84 %); 1H-NMR (D2O) δ 7.58 (4H, d), 7.47 (4H, d), 2.66 (4H, t), 1.58 (4H, m); HRMS (m+H): theor: 409.17, found: 409.04
N,N'-dibenzyl-1,2-diaminoethane was synthesized in a similar fashion from diaminoethane (0.5 g) and benzaldehyde (1.9 g). Yield 1.3 g (65%); 1H-NMR (D2O) δ 7.39 (10H, m), 4.20 (4H, s), 3.37 (4H, t); HRMS (m+H): theor: 241.16, found: 241.27
N,N'-dibenzyl-1,3-diaminopropane was synthesized in a similar fashion from diaminopropane (1.0 g) and benzaldehyde (3.2 g). Yield 2.5 g (66%); 1H-NMR (D2O) δ 7.39 (10H, m), 4.15(4H, s) 3.05 (4H, t), 2.05 (2H, p); HRMS (m+H): theor: 255.18, found: 255.19
Expression and Purification of PAO
His-tagged mouse PAO was purified from BL21(DE3) E. coli cells as described previously (26). The concentration of active enzyme was determined using an ε458 value of 10,400 M−1 cm−1 (20).
Steady-State Assays
Steady-state kinetic parameters for PAO were determined using a computer-interfaced Clark oxygen electrode (Hansatech Instruments) to measure the initial rate of oxygen consumption. All assays were performed at 30 °C and were initiated by addition of enzyme to a final concentration of 0.1–0.5 μM. The buffers were 50 mM Tris-HCl for pH 7.1–8.6, 50 mM CHES for pH 9.1–9.6, and 50 mM CAPS at pH 10.1. All buffers contained 10 % glycerol. The concentrations of substrates were determined enzymatically, measuring the amount of oxygen required to completely oxidize a limiting amount of the substrate; the concentration of each substrate based on this assay was consistently twice that based on molecular weight, indicating an ability of PAO to recognize and oxidize both sides of N,N'-dibenzyl-1,4-diaminobutanes.
Rapid Reaction Kinetics
Rapid reaction kinetic measurements were performed on an Applied Photophysics SX-18MV stopped-flow spectrophotometer using both single wavelength and photodiode array detection. To establish anaerobic conditions, the instrument was loaded with anaerobic buffer containing 5 mM glucose and 36 nM of glucose oxidase and left overnight. Enzyme solutions (~ 40 μM) containing 5 mM glucose were made anaerobic by applying cycles of argon and vacuum; substrate solutions containing 5 mM glucose were made anaerobic by bubbling with argon. Glucose oxidase was added to all anaerobic solutions at a final concentration of 36 nM. For each substrate, the rate constant for reduction was determined as a function of amine concentration, and the results were fit to the Michaelis-Menten equation to obtain the kred value.
Data Analysis
Data were analyzed using the programs KaleidaGraph (Adelbeck Software, Reading, PA) and Igor (WaveMetrics, Lake Oswego, OR). Initial rate data were fit to the Michaelis-Menten equation to determine steady-state kinetic parameters. kcat/Km-pH profiles were fit to eq 1, where y is the kcat/Km value at a given pH, c is its pH-independent value, K1 is the ionization constant for a moiety that must be unprotonated for activity and K2 is the ionization constant for a moiety that must be protonated for activity (27). Kinetic isotope effects on steady-state parameters were determined from fits to eq 2 or 3 (28, 29), where E is the isotope effect on kcat/Km and kcat, s is the concentration of substrate, and Fi is the fraction of heavy isotope in the substrate. Some of the substrates exhibited substrate inhibition and were consequently fit with eq 3, where Kai is the substrate inhibition constant.
| (1) |
| (2) |
| (3) |
Rapid reaction kinetic traces were fit as single or double exponential decays (eqs 4 and 5); here, k1 and k2 are the first order rate constants for the two phases, A1 and A2 are the absorbance changes associated with the two phases, At is the total absorbance change, and A∞ is the final absorbance. The kred-pH profile was fit to eq 6, where y is the value of kred at a given pH, yL is its limiting value at low pH, Δy is the difference in kred at high and low pH, and K1 is the ionization constant for the transition (27).
| (4) |
| (5) |
| (6) |
Results
N, N'-Dibenzyl-diamines As Substrates
Several N, N'-dibenzyl-diamines were previously shown to be substrates for partially purified polyamine oxidase from pig liver, producing benzaldehyde as product; the highest activity was seen with the 1,3-diaminopropane and 1,4-diaminobutane derivatives (30). With the purified recombinant mouse enzyme in hand, we determined the kinetics of oxidation of N, N'-dibenzyl-1,2-diaminoethane, N,N'-dibenzyl-1,3- diaminopropane, and N, N'-dibenzyl-1,4-diaminobutane in air-saturated buffer at pH 8.6. No activity was detected with the diaminoethane derivative. The apparent kcat, Km, and kcat/Km values for N, N'-dibenzyl-1,3-diaminopropane were 6.1 ± 0.2 s−1, 40 ± 6 μM, and 0.15 ± 0.02 μM−1s−1, respectively. For N, N'-dibenzyl-1,4-diaminobutane, the values of these kinetic parameters were 0.80 ± 0.02 s−1, 15 ± 2 μM, and 0.054 ± 0.004 μM−1s−1. The latter Km value is within a factor of two of the value reported for the pig enzyme at 37 °C; the kinetic parameters for N, N'-dibenzyl-1,3-diaminopropane were not reported previously. The lower activity of N, N'-dibenzyl-1,4-diaminobutane suggested that chemistry is more likely to be rate-limiting with this compound. Consequently, analogs of this compound were selected for further study.
Steady-State Kinetics
The steady-state kinetic parameters kcat, kcat/Km, and Km were determined for PAO at pH 8.6 and 30 °C for seven para-substituted N,N'-dibenzyl-1,4-diaminobutanes as substrates (Table 1). Assays were conducted in air-saturated buffers; therefore, the kcat and Km values in the table are apparent values, while the kcat/Km values are independent of the oxygen concentration used.
Table 1.
Kinetic parameters of PAO with para-substituted N,N'-dibenzyl-1,4-diaminobutanes as substrates at pH 8.6, 30 °C.
| Kinetic Parameter | |||
|---|---|---|---|
| para-Substituent | kcat (s−1) | kcat/Km (μM−1 s−1) | Km (μM) |
| (CH3)2N | 10.1 ± 0.5 | 0.12 ± 0.02 | 87 ± 15 |
| CH3O | 4.5 ± 0.1 | 0.24 ± 0.01 | 19 ± 1 |
| CH3 | 3.8 ± 0.3 | 0.35 ± 0.06 | 11 ± 3 |
| H | 0.80 ± 0.02 | 0.054 ± 0.004 | 15 ± 2 |
| Cl | 1.4 ± 0.1 | 0.97 ± 0.30 | 1.4 ± 0.5 |
| Br | 1.8 ± 0.1 | 1.0 ± 0.2 | 1.8 ± 0.4 |
| CF3 | 2.3 ± 0.1 | 0.53 ± 0.10 | 4 ± 2 |
kcat/Km-pH Profiles
The effect of pH on the kcat/Km value was determined for N, N'-dibenzyl-1,4-diaminobutane and the CH3O- and CF3-substituted compounds. These were selected because the CH3O group is among the most electron-donating of the substituents examined here, while the CF3 group is the most electron-withdrawing. The kcat/Km-pH profiles for all three substrates are bell-shaped curves, and the data fit well to eq 1 (Fig. 1A). The pKa values for all three (Table 2) are within error of each other, with an average value of 8.0 for the pKa value for a group that must be deprotonated and of 9.9 for the pKa of a group that must be protonated for activity.
Figure 1.

(A) kcat/Km-pH profiles for PAO with N,N'-dibenzyl-1,4-diaminobutane (●), N,N'-bis(4-CH3O-benzyl)-1,4-diaminobutane (□), and N,N'-bis(4-CF3-benzyl)-1,4-diaminobutane (▲) as substrates. The average error in the data is 15%. The lines are from fits of the data to eq 1. (B) pH distribution of N,N'-benzyl-1,4-diaminobutane with zero, one, or two protonated nitrogens.
Table 2.
pKa values from kcat/Km-pH profiles for para-substituted N,N'-dibenzyl-1,4-diaminobutanes as substrates for PAO
| X | pK1 | pK2 |
|---|---|---|
| H | 8.1 ± 0.1 | 9.8 ± 0.2 |
| CH3O | 8.2 ± 0.2 | 9.9 ± 0.2 |
| CF3 | 7.8 ± 0.1 | 9.9 ± 0.1 |
Steady-State Kinetic Isotope Effects
Deuterium kinetic isotope effects were determined over the pH range 7.1–10.1 for N, N'-dibenzyl-1,4-diaminobutane and the CH3O- and CF3-substituted compounds. The data for all three were fit using eq 2 or 3, which apply for equal isotope effects on Dkcat/KDBDB and Dkcat; alternate equations with different isotope effects on these kinetic parameters did not give improved fits, but did give comparable values for the isotope effects. For all three substrates, the isotope effects are less than two and are pH-independent (Table 3).
Table 3.
Effect of pH on the D(kcat/Km) and Dkcat values for para-substituted N,N'-dibenzyl-1,4-diaminobutanes as substrates for PAO
| pH | Kinetic Isotope Effects | ||
|---|---|---|---|
| X = H | X = CH3O | X = CF3 | |
| 7.1 | 1.3 ± 0.1 | 1.7 ± 0.1 | n.d. |
| 7.6 | 1.7 ± 0.1 | 1.8 ± 0.1 | 1.4 ± 0.1 |
| 8.1 | 1.8 ± 0.1 | 1.7 ± 0.1 | 1.5 ± 0.1 |
| 8.6 | 2.0 ± 0.1 | 1.7 ± 0.1 | 1.6 ± 0.1 |
| 9.1 | 2.1 ± 0.2 | 1.7 ± 0.1 | 1.5 ± 0.1 |
| 9.6 | 2.0 ± 0.2 | 1.6 ± 0.1 | 1.6 ± 0.1 |
| 10.1 | 1.9 ± 0.1 | 1.6 ± 0.1 | 1.6 ± 0.1 |
| Average | 1.8 ± 0.3 | 1.7 ± 0.1 | 1.5 ± 0.1 |
n.d. not determined
Flavin Reduction Kinetics
Stopped-flow experiments were conducted to determine directly the effects that para-substituents have on kred, the rate constant for flavin reduction at saturating concentrations of substrate. These results are summarized in Table 4. The reactions were routinely monitored at 458 nm as a function of substrate concentration; with the majority of the substrates, the spectral changes upon mixing enzyme and substrate could be fit to a single exponential decay (Fig. 2A). The exception was the (CH3)2N-substituted compound; in this case the traces were fit better to a double exponential decay (results not shown), where the second rate constant is slower than turnover and therefore not catalytically relevant. These experiments were conducted at both pH 8.6, near the pH optimum, and at pH 6.6. In addition, for each substrate a photodiode array detector was used to monitor the spectral changes during the course of the reaction (Fig. 2B). Global analyses of the data showed that flavin reduction occurs with no observable intermediates between fully oxidized and fully reduced flavin (Fig. 2C). Similar analyses were carried out with the deuterated substrates; the resulting isotope effects on the kred values range from 1.3 to 2.9 and are comparable at pH 8.6 and 6.6 (Table 4). The isotope effects on kred are similar to the isotope effects on kcat/Km and kcat for those compounds where these were all measured.
Table 4.
Rate constants and deuterium kinetic isotope effects for reduction of PAO by para-substituted N,N'-dibenzyl-1,4-diaminobutanes.
| pH 6.6 | pH 8.6 | |||
|---|---|---|---|---|
| Substituent | kred (s−1) | Dkred | kred (s−1) | Dkred |
| (CH3)2N | 2.8 ± 0.3 | 1.4 ± 0.2 | 23.9 ± 0.6 | 1.7 ± 0.1 |
| CH3O | 1.19 ± 0.01 | 1.8 ± 0.1 | 4.1 ± 0.6 | 1.6 ± 0.2 |
| CH3 | 0.66 ± 0.01 | 1.7 ± 0.1 | 1.84 ± 0.01 | 1.8 ± 0.1 |
| H | 0.21 ± 0.01 | 1.8 ± 0.1 | 0.42 ± 0.02 | 1.5 ± 0.1 |
| Cl | 0.48 ± 0.01 | 2.2 ± 0.1 | 0.80 ± 0.02 | 2.9 ± 0.1 |
| Br | 0.56 ± 0.02 | 1.7 ± 0.1 | 1.04 ± 0.06 | 1.5 ± 0.1 |
| CF3 | 1.05 ± 0.02 | 1.8 ± 0.1 | 1.58 ± 0.06 | 1.3 ± 0.1 |
Figure 2.

Spectral changes during reduction of PAO by N,N'-bis(4-CH3-benzyl)-1,4-diaminobutane at pH 6.6, 30 °C: (A) changes at 458 nm (only 1/30th of the points are shown for clarity); (B) changes in the entire visible absorbance spectrum; (C) initial (—) and final (…) spectra from a global analysis of the data in B using a single-step irreversible reaction as the model. The line in A is from a fit of the data to eq 4.
The effect of pH on the kred value was examined in greater detail with N, N'-dibenzyl-1,4-diaminobutane as substrate. The kred value has a constant value at low pH and a higher constant value at high pH (Fig. 3). Fitting the data to eq 6 yields values for the rate constants at low and high pH of 0.17 ± 0.03 s−1 and 1.0 ± 0.20 s−1, respectively, with a pKa value of 8.2 ± 0.3 for the transition.
Figure 3.
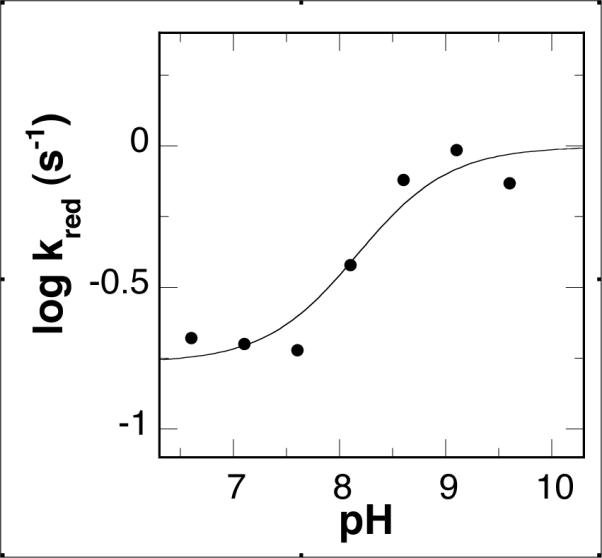
pH dependence of kred for PAO oxidation of N,N'-dibenzyl-1,4-diaminobutane at 30 °C. The line is from a fit to eq 6.
Linear Correlation Analysis
The effect of the substituent at the para-position of the aromatic rings on the kinetic parameters was analyzed using single- and multiple-parameter linear correlations. The effect of the substituent on the electronics of the reaction was examined using the parameters σ, σ+, σ−, and σI (31). The effect of the hydrophobicity of the substituent was examined using the parameter π (32). The effect of the size of the substituent was examined using the van der Waals volume, VW (33), and the Taft steric parameter ES (34). (No ES value is available for the (CH3)2N moiety.)
The results of the analyses of the kred value are summarized in Table 5 and Figure 4. The effect of a single parameter was analyzed initially, fitting the data to eq 7. Here, x is the substituent-specific parameter of interest and C is a constant. The best single-parameter correlation is with the van der Waals volume at both pH 8.6 and 6.6. However, this correlation is clearly much better at pH 6.6 than at pH 8.6. At both pH values the σ+ and the ES values have the next lowest χ2 values. Consequently, the data were fit to eq 8, which describes a two-parameter linear correlation using the electronic parameter σ+ and VW. (A similar analysis was not done with both ES and VW values because Es values reflect the same phenomenon as VW values (35).) At both pH 8.6 and 6.6 the two-parameter analysis with VW and σ+ shows a lower χ2 value than the single-parameter correlation with just VW. A similar analysis with σ and VW yields a slightly better correlation, although the effect is not dramatic. With both σ and σ+, the ρ value is more negative at pH 8.6 than at pH 6.6, whereas the coefficient for VW is essentially unaffected by pH.
| (7) |
| (8) |
Table 5.
Correlation analyses of log kred for para-substituted N,N'-dibenzyl-1,4-diaminobutanes with hydrophobic, steric and electronic parameters.
| pH 6.6 | pH 8.6 | |||
|---|---|---|---|---|
| Parameter | Coefficient | χ2 | Coefficient | χ2 |
| σ | −0.45 ± 0.30 | 0.53 | −1.02 ± 0.36 | 0.77 |
| σ+ | −0.31 ± 0.16 | 0.43 | −0.64 ± 0.17 | 0.53 |
| σ− | −0.14 ± 0.50 | 0.75 | −0.69 ± 0.76 | 1.69 |
| σI | 0.23 ± 0.86 | 0.75 | −0.37 ± 1.38 | 1.94 |
| π | −0.05 ± 0.40 | 0.76 | −0.42 ± 0.62 | 1.80 |
| VW | 0.39 ± 0.03 | 0.02 | 0.58 ± 0.12 | 0.32 |
| ES | −0.20 ± 0.13 | 0.23 | −0.13 ± 0.20 | 0.52 |
| VW | 0.36 ± 0.02 | 0.0088 | 0.40 ± 0.04 | 0.0156 |
| σ+ | −0.06 ± 0.03 | −0.37 ± 0.04 | ||
| VW | 0.37 ± 0.02 | 0.0087 | 0.45 ± 0.02 | 0.00702 |
| σ | −0.09 ± 0.05 | −0.59 ± 0.04 | ||
Figure 4.

Correlation of the kred values for para-substituted N,N'-dibenzyl-1,4-diaminobutanes as substrates for PAO with structural and electronic properties of the substituent: (A) correlation of the kred value at pH 6.6 with the van der Waals volume (VW), (B) correlation of the kred value at pH 8.6 with VW, (C) correlation of the kred value at pH 8.6 with σ, and (D) correlation of the kred value at pH 8.6 with σ after correction of the kred value for the contribution of Vw.
The results of similar correlation analyses at pH 8.6 of the effects of the substituents of N,N'-dibenzyl-1,4-diaminobutanes on the steady-state kinetic parameters kcat/Km and kcat are summarized in Table 6. The results of the analyses of the kcat value are very similar to those of the kred value. The best single correlation is with the VW value. Two-parameter analyses with VW and σ or VW and σ+ are significantly better than the single-parameter correlations. The resulting coefficients are similar to those with kred, although in all cases the absolute value of the coefficient is slightly smaller for kcat. The correlations for the kcat/Km value are not as good, in that the low Km of several of the substrates (Table 1) limited the accuracy with which the kcat/Km values could be determined. The best linear correlation for kcat/Km is with the single-parameter π, giving a coefficient of 1.03 ± 0.27. A two-parameter correlation using both π and σI resulted in an improved fit.
Table 6.
Correlation analyses of log kcat/Km and log kcat with para-substituted N,N'-dibenzyl-1,4-diaminobutanes with hydrophobic, steric and electronic parameters at pH 8.6.
| kcat/Km | kcat | |||
|---|---|---|---|---|
| Parameter | Coefficient | χ2 | Coefficient | χ2 |
| σ | 0.63 ± 0.39 | 0.87 | −0.61 ± 0.24 | 0.36 |
| σ+ | 0.31 ± 0.23 | 1 | −0.39 ± 0.13 | 0.28 |
| σ− | 0.76 ± 0.58 | 0.99 | −0.49 ± 0.47 | 0.65 |
| σI | 2.00 ± 0.71 | 0.52 | −0.32 ± 0.87 | 0.78 |
| π | 1.03 ± 0.27 | 0.35 | −0.20 ± 0.40 | 0.76 |
| VW | 0.06 ± 0.23 | 1.3 | 0.36 ± 0.08 | 0.17 |
| ES | −0.35 ± 0.18 | 0.71 | −0.11 ± 0.16 | 0.35 |
| VW | n.d. | 0.26 ± 0.08 | 0.072 | |
| σ+ | n.d. | 0.21 ± 0.09 | ||
| VW | 0.23 ± 0.20 | 0.63 | 0.28 ± 0.06 | 0.060 |
| σ | 0.86 ± 0.41 | −0.34 ± 0.13 | ||
| π | 1.06 ± 0.63 | 0.20 | n.d. | |
| σI | 0.73 ± 0.29 | n.d. | ||
| ES | −0.13 ± 0.21 | 0.34 | n.d. | |
| σI | −0.22 ± 0.85 | n.d. | ||
| π | −0.47 ± 0.54 | 0.28 | n.d. | |
| ES | −0.30 ± 0.28 | n.d. | ||
Discussion
While the physiological substrates for PAO, N1-acetylspermine and N1-acetylspermidine, contain four and three nitrogen atoms, respectively, the present results clearly show that N, N'-dibenzyl-1,4-diaminobutanes, with only two nitrogens, are also substrates. This is consistent with earlier reports for partially purified rat PAO that showed that N, N'-disubstituted diamines can be substrates (36). Our previous studies of mouse PAO established that the oxidation of the physiological substrates requires that the nitrogen at the site of CH bond cleavage be neutral and one other nitrogen in the substrate be positively charged (26). The kcat/Km-pH profiles for N, N'-dibenzyl-1,4-diaminobutanes described here are consistent with such a model. The profiles in Figure 1 match well the pH dependence of the fraction of the N, N'-dibenzyl-1,4-diaminobutane that has only one charged nitrogen (Fig. 1B), without a contribution from a protein residue. Thus, as is the case with other flavin amine oxidases (16), oxidation of the substrate carbon-nitrogen bond requires that the nitrogen be uncharged. The kred-pH profile for N,N'-dibenzyl-1,4-diaminobutane provides further evidence for such a conclusion. With N1-acetylspermine as substrate, the kred-pH profile displays a decrease in activity at acidic pH with a pKa of 7.3 ± 0.1, consistent with the ability of substrate in the incorrectly protonated form to bind to the enzyme, but not react (26). The kred value for N,N'-dibenzyl-1,4-diaminobutane similarly decreases at lower pH, but reaches a limiting value, in contrast to the results with the much faster physiological substrate. This result raises the possibility that both the protonated and deprotonated forms of N,N'-dibenzyl-1,4-diaminobutane can react, although the unprotonated form reacts more rapidly. A more likely rationale for the activity at low pH is that the rate constant of ~0.2 s−1 is due to loss of the proton from the reacting nitrogen to an amino acid side chain or a water molecule in the active site tunnel. Given the rate constant for this step, this pathway is unlikely to be important for the oxidation of the physiological substrate.
For each of the N, N'-dibenzyl-1,4-diaminobutanes, the kcat and kred values are comparable, consistent with the reductive half-reaction being substantially rate-limiting for these substrates.2 This is in contrast to the kinetics for the physiological substrates, where product release is much slower than amine oxidation (26). This change in the rate-determining step can be attributed to the much slower oxidation of N, N'-dibenzyl-1,4-diaminobutanes by PAO, in that the kred values in Table 4 are two orders of magnitude smaller than the value for N1-acetylspermine (26). The kinetic isotope effects provide further evidence for rate-limiting amine oxidation. While extensive steady-state kinetic analyses were only carried out with three of the compounds, the results with these three are quite similar in that the isotope effects on kcat/Km, kcat, and kred are essentially identical. The identity of the isotope effects on the kred and kcat values is most consistent with product release being significantly faster than amine oxidation. The pH independence of the isotope effects on the kcat/Km values establishes that binding steps do not limit amine oxidation (37). This leaves amine oxidation as the slow step in turnover.
All of the isotope effects reported here are significantly less than the semi-classical limit of ~7. This raises the possibility that a first-order step prior to carbon-hydrogen bond cleavage partially limits amine oxidation. The agreement of the isotope effects on the kcat and the kcat/Km values is consistent with the observed isotope effects being the intrinsic ones, but the effects on both kinetic parameters could be suppressed equally by such a step. However, there is no evidence for an intermediate in the PAO-catalyzed oxidation of any amine, including those studied here (20, 26). It should be noted that similar small isotope effects have been seen with the flavoprotein amine oxidases LSD1 (38) and tryptophan monooxygenase (21), both of which are in the MAO structural family, and with D-amino acid oxidase (39), which has a different structure. Thus, the combination of steady-state and rapid-reaction kinetics is most consistent with CH bond cleavage being the rate-limiting step in the oxidation of N, N'-dibenzyl-1,4-diaminobutanes by PAO.
Analysis of the effects of the para substituents on the kinetics of N,N'-dibenzyl-1,4-diaminobutanes provides insights into the mechanism of amine oxidation by PAO. The kred value is of most interest, since this is the rate constant for oxidation of the amine. Qualitatively, similar correlations are seen at both pH 8.6 and 6.6, in that two-parameter linear correlations with VW and σ show the best fits. The σ value is a quantitative measure of the electron donating or withdrawing ability of the substituent in the aromatic ring, becoming greater as the substituent becomes less electron donating (31). The quantitative change in a rate constant as a function of the σ value provides an indication of the extent of charge development in the transition state for the corresponding reaction. In general, positive ρ values are consistent with development of negative charge, reflecting an increase in rate constants as the substituent becomes more electron withdrawing. Negative ρ values are consistent with development of positive change, since the rate constants decrease as the substituent becomes more electron withdrawing. Values close to zero (~−1 to 1) reflect a lack of charge development.
While the coefficient associated with VW is pH-independent, going from the pH optimum of 8.6 to pH 6.6 results in an increase in the ρ value from −0.59 to −0.09. The ρ value at pH 8.6 is likely to be a better description of the degree of charge development in the transition state for carbon-hydrogen bond cleavage, in that the reaction at this pH involves the correctly protonated form of the substrate and therefore no proton loss is required. In contrast, the ρ value at pH 6.6 reflects both the CH bond cleavage step and the requirement that the reactive nitrogen be unprotonated. In the case of para-substituted benzylamines, the pKa values correlate well with the σ values of the substituents, with the pKa value of the amine decreasing with an increase in the σ value of the substituent, for a ρ value of −1.1 (40, 41); this is due to the pKa value increasing as the electron-donating ability of the substituent increases. A similar effect is likely for the para-substituted N, N'-dibenzyl-1-4-diaminobutanes studied here (42). Since amine oxidation by PAO requires a neutral nitrogen at the site of oxidation, a decrease in the amine pKa will result in a higher concentration of the reactive species at low pH. Consequently, a plot of the fraction of the amine with a neutral nitrogen as a function of the σ value of the substituent would have a positive slope or ρ value. This result would appear to indicate that transition state is electron rich even if the rate constant itself is insensitive to the electron-donating ability of the substituent. This effect of the substituent in the aromatic ring on the amine pKa value provides a reasonable explanation for the pH dependency of the ρ value for PAO reported here. In the present case, it is the pKa of the amine when it is bound to the enzyme that is critical. This pKa is ~8 with N, N'-dibenzyl-1-4-diaminobutane. The 0.5 increase in the ρ value at pH 6.6 can be attributed to the effect of the substituent in the aromatic ring on the pKa value of the nitrogen. This is not a problem at pH 8.6, so that the ρ value of −0.59 at the higher pH provides a more accurate reflection of the transition state for CH bond cleavage by PAO than the value at pH 6.6. These results also suggest that Hammett analyses of enzyme-catalyzed reactions which do not account for the effects of pH can be misleading.
The kcat value at pH 8.6 exhibits a similar correlation to the kred value, with the best correlation being with the volume and the σ value of the para substituent. The absolute values of the coefficients for both parameters are slightly less than those for the kred values. The difference may reflect the lower precision of the steady-state kinetic data or it may reflect attenuation of the effects by a contribution from the product release step. The correlations of the kcat/Km are not as good, possibly due to the low Km values of these compounds limiting the accuracy with which kcat/Km values could be determined. In this case the best correlation is with a combination of π and σI. The correlation with π can be attributed to the importance of hydrophobic interactions in the binding of substrates to PAO. The active forms of the substrates for PAO, whether the N, N'-dibenzyl-1,4-diaminobutanes studied here or the physiological substrate N1-acetylspermine, are fairly hydrophobic in that they contain only one charged atom. The structure of a mammalian PAO is not available, but structures of maize PAO and the yeast spermine oxidase Fms1 have been determined with bound ligands (43, 44). These structures show that the central carbons and nitrogens of substrates, which the present substrates share with spermine, are mostly involved in hydrophobic interactions with the protein.
The mechanism of flavin amine oxidases has been a source of debate for years, with three possible mechanisms regularly discussed. The single electron transfer mechanism proposed by Silverman (22) involves the transient formation of a flavin radical. The polar nucleophilic mechanism proposed by Edmondson (45) involves concerted formation of a 4a-alkylated isoalloxazine ring and proton abstraction of the substrate α-hydrogen. Direct transfer of a hydride from the α-C of the substrate to the N5 position of the flavin is the consensus mechanism for the D-amino acid oxidase structural family of flavin amine oxidases (16, 46); its applicability to the MAO structural family is supported by kinetic isotope effects and the failure to detect intermediates in the reactions of these enzymes (17–20, 38, 45). The ρ value of −0.59 for PAO at pH 8.6 reported here is most consistent with a direct hydride transfer reaction in which there is little charge development in the transition state for carbon-hydrogen bond cleavage. This value would also be consistent with the expectations for abstraction of a hydrogen atom from the carbon of the substrate (47), but such a reaction is unlikely for a flavin given the energetics of such a reaction. Indeed, the various radical mechanisms proposed for flavin-catalyzed amine oxidation involve initial formation of an aminium radical rather than a carbon-based radical (22, 48). The lack of detectable intermediates in the reductive half-reaction of PAO is also consistent with direct hydride transfer from the amine to the flavin.
Hammett analyses have been carried out with other flavin amine oxidases. In the case of yeast D-amino acid oxidase, the kred value for a series of para-substituted phenylglycines correlates well with a combination of the Vw and σ+ values of the substituents, yielding a ρ value of −0.73 (49), in line with the expectations for a hydride transfer mechanism for this structural family of amine oxidase, and very similar to the results reported here. Several analyses of MAO have been described with somewhat contradictory results. With MAO A (45), there is a positive correlation between the kred values for a series of 12 ring-substituted benzylamines and the electronic parameter σ with a ρ value of 2, while the kred values for four additional compounds give a ρ value of 0.5.3 The ρ value of 2 has been interpreted as evidence for removal of the substrate α-hydrogen as a proton (23). In contrast, the kred value for oxidation of substituted phenethylamines by MAO A shows no correlation with any σ parameter; instead the value correlates best with the size of the substituent, measured as either Vw or ES, with larger substrates reacting more slowly (50). As noted at the time, the extra methylene in phenethylamines versus benzylamines is expected to attenuate the electronic effects of substituents. However, this effect typically yields only a 2–3 fold reduction in the ρ value (51). The lack of any correlation of the kred values for oxidation of phenylethylamines by MAO A with σ values was attributed to dominant steric effects masking the electronic effects (50). In the case of MAO B, the kred values for substituted benzylamines correlate best with a combination of π and Vw (52), in good agreement with the results for MAO A and phenethylamines. The present results with PAO are in line with the earlier results for D-amino acid oxidase and MAO B and with the results for MAO A with phenethylamines, but are clearly different from the results with MAO A and benzylamines. This discrepancy can be rationalized in a variety of ways. Oxidation of benzylamines by MAO A could occur by a different mechanism from amine oxidation by MAO B, PAO, and other flavin amine oxidases. This seems unlikely given the structural similarities of these enzymes (12, 23). Alternatively, the electronic effects of the substituents in the oxidation of benzylamines by MAO B could be suppressed due to the restrictive active site of that enzyme preventing the required orbital alignment of the benzyl carbon with the aromatic ring (23). Such an explanation could also be invoked for the results with PAO described here. In that case, the reactions of MAO A and B, and presumably PAO, would involve a cationic transition state, in line with the predictions for a mechanism involving nucleophilic attack of the amine on the flavin. Such an explanation would require different mechanisms of amine oxidation by the D-amino acid oxidase and MAO structural families. However, deuterium and 15N kinetic isotope effects for D-amino acid oxidase are indistinguishable from those for an L-amino acid oxidase (18, 53), suggesting that they have similar mechanisms. L-Amino acid oxidases are in the MAO structural family, and their structures are more consistent with hydride transfer than nucleophilic mechanism (11, 54). Moreover, ab initio calculations show that the 15N isotope effects for both enzymes are inconsistent with nucleophilic mechanisms (19). Thus, the large ρ value for oxidation of benzylamines by MAO A would require that MAO A and B have a mechanism different from structurally similar flavin amine oxidases (23). Another possibility is that the ρ value of 2 with MAO A reflects at least in part the effects of substituents on the pKa values of benzylamines, since the measurements were carried out below the pKa seen in the kred-pH profile for benzylamine (55).
The studies described here provide further insights into the mechanism of PAO and the entire MAO structural family. The ability to oxidize N, N'-dibenzyl-1,4-diamines establishes that two nitrogens in a substrate are sufficient for oxidation by PAO. The pH dependence of the kcat/Km and kred values confirms the need for a neutral nitrogen at the site of oxidation. The effects of substituents in the aromatic ring on the kinetics of oxidation is most consistent with a direct hydride transfer from the neutral CN bond of the substrate.
Acknowledgments
This work was supported in part by grants from the NIH (R01 GM58698) and The Welch Foundation (AQ-1245)
Footnotes
Abbreviations used: PAO, polyamine oxidase; MAO, monoamine oxidase; LSD1, lysine-specific demethylase.
In several cases the kred values are less than the kcat values. We attribute this to a slight decrease in enzyme activity at the higher enzyme concentrations used in the stopped-flow analysis.
When the kred values for MAO A for all of the substituted benzylamines are combined in a single analysis, the ρ value is 0.93 ± 0.25, closer to the expectations for hydride transfer.
References
- 1.Casero RA, Jr., Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 2.Marton LJ, Pegg AE. Polyamines as targets for therapeutic intervention. Ann.Rev.Pharmacol.Toxicol. 1995;35:55–91. doi: 10.1146/annurev.pa.35.040195.000415. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Casero RA., Jr. Mammalian polyamine catabolism: a therapeutic target, a pathological problem, or both? J. Biochem. (Tokyo) 2006;139:17–25. doi: 10.1093/jb/mvj021. [DOI] [PubMed] [Google Scholar]
- 4.Pegg AE. Spermidine/spermine-N1-acetyltransferase: a key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008;294:E995–1010. doi: 10.1152/ajpendo.90217.2008. [DOI] [PubMed] [Google Scholar]
- 5.Wu T, Yankovskaya V, McIntire WS. Cloning, sequencing, and heterologous expression of the murine peroxisomal flavoprotein, N1-acetylated polyamine oxidase. J. Biol. Chem. 2003;278:20514–20525. doi: 10.1074/jbc.M302149200. [DOI] [PubMed] [Google Scholar]
- 6.Seiler N, Peter M, Yu KFT, Alan AB. Polyamine oxidase, properties and functions. Prog. Brain Res. 1995;106:333–344. doi: 10.1016/s0079-6123(08)61229-7. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Devereux W, Woster PM, Stewart TM, Hacker A, Casero RA., Jr. Cloning and characterization of a human polyamine oxidase that is inducible by polyamine analogue exposure. Cancer Res. 2001;61:5370–5373. [PubMed] [Google Scholar]
- 8.Vujcic S, Diegelman P, Bacchi CJ, Kramer DL, Porter CW. Identification and characterization of a novel flavin-containing spermine oxidase of mammalian cell origin. Biochem. J. 2002;367:665–675. doi: 10.1042/BJ20020720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc. Natl. Acad. Sci. USA. 2006;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pawelek PD, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A. The structure of L-amino acid oxidase reveals the substrate trajectory into an enantiomerically conserved active site. EMBO J. 2000;19:4204–4215. doi: 10.1093/emboj/19.16.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binda C, Mattevi A, Edmondson DE. Structure-function relationships in flavoenzyme dependent amine oxidations. A comparison of polyamine oxidase and monoamine oxidase. J. Biol. Chem. 2002;277:23973–23976. doi: 10.1074/jbc.R200005200. [DOI] [PubMed] [Google Scholar]
- 13.Sebela M, Radová A, Angelini R, Tavladoraki P, Frébort I, Pec P. FAD-containing polyamine oxidases: a timely challenge for researchers in biochemistry and physiology of plants. Plant Sci. 2001;160:197–207. doi: 10.1016/s0168-9452(00)00380-0. [DOI] [PubMed] [Google Scholar]
- 14.Landry J, Sternglanz R. Yeast Fms1 is a FAD-utilizing polyamine oxidase. Biochem. Biophys. Res. Commun. 2003;303:771–776. doi: 10.1016/s0006-291x(03)00416-9. [DOI] [PubMed] [Google Scholar]
- 15.Bright HJ, Porter DJT. Flavoprotein oxidases. In: Boyer P, editor. The Enzymes, 3rd Ed. 3 ed. Vol. XII. Academic Press; New York: 1975. pp. 421–505. [Google Scholar]
- 16.Fitzpatrick PF. Oxidation of amines by flavoproteins. Arch. Biochem. Biophys. 2010 doi: 10.1016/j.abb.2009.07.019. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller JR, Edmondson DE, Grissom CB. Mechanistic probes of monoamine oxidase B catalysis: Rapid-scan stopped flow and magnetic field independence of the reductive half-reaction. J.Am.Chem.Soc. 1995;117:7830–7831. [Google Scholar]
- 18.Ralph EC, Anderson MA, Cleland WW, Fitzpatrick PF. Mechanistic studies of the flavoenzyme tryptophan 2-monooxygenase: Deuterium and 15N kinetic isotope effects on alanine oxidation by an L-amino acid oxidase. Biochemistry. 2006;45:15844–15852. doi: 10.1021/bi061894o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ralph EC, Hirschi JS, Anderson MA, Cleland WW, Singleton DA, Fitzpatrick PF. Insights into the mechanism of flavoprotein-catalyzed amine oxidation from nitrogen isotope effects on the reaction of N-methyltryptophan oxidase. Biochemistry. 2007;46:7655–7664. doi: 10.1021/bi700482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Royo M, Fitzpatrick PF. Mechanistic studies of mouse polyamine oxidase with N1,N12-bisethylspermine as a substrate. Biochemistry. 2005;44:7079–7084. doi: 10.1021/bi050347k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emanuele JJ, Jr., Fitzpatrick PF. Mechanistic studies of the flavoprotein tryptophan 2-monooxygenase. 2. pH and kinetic isotope effects. Biochemistry. 1995;34:3716–3723. doi: 10.1021/bi00011a029. [DOI] [PubMed] [Google Scholar]
- 22.Silverman RB. Radical ideas about monoamine oxidase. Acc. Chem. Res. 1995;28:335–342. [Google Scholar]
- 23.Edmondson DE, Binda C, Mattevi A. Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch. Biochem. Biophys. 2007;464:269–276. doi: 10.1016/j.abb.2007.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paik Y, Y. C, Metaferia B, Tang S, Bane S, Ravindra R, Shanker N, Alcaraz AA, Johnson SA, Schaefer J, O'Connor RD, Cegelski L, Snyder JP, Kingston DGI. Rotational-Echo Double-Resonance NMR Distance Measurements for the Tubulin-Bound Paclitaxel Conformation. J. Am. Chem. Soc. 2007;129:361–370. doi: 10.1021/ja0656604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burns MR, LaTurner S, Ziemer J, McVean M, Devens B, Carlson CL, Graminski GF, Vanderwerf SM, Weeks RS, Carreon J. Induction of apoptosis by aryl-substituted diamines: role of aromatic group substituents and distance between nitrogens. Bioorg. Med. Chem. Lett. 2002;12:1263–1267. doi: 10.1016/s0960-894x(02)00156-7. [DOI] [PubMed] [Google Scholar]
- 26.Henderson Pozzi M, Gawandi V, Fitzpatrick PF. pH Dependence of a Mammalian Polyamine Oxidase: Insights into Substrate Specificity and the Role of Lysine 315. Biochemistry. 2009;48:1508–1516. doi: 10.1021/bi802227m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 28.Schimerlik MI, Grimshaw CE, Cleland WW. Determination of the Rate-Limiting Steps for Malic Enzyme by the Use of Isotope Effects and Other Kinetic Studies. Biochemistry. 1977;16:571–576. doi: 10.1021/bi00623a002. [DOI] [PubMed] [Google Scholar]
- 29.Fitzpatrick PF, Valley MP, Bozinovski DM, Shaw PG, Héroux A, Orville AM. Mechanistic and Structural Analyses of the Roles of Arg409 and Asp402 in the Reaction of the Flavoprotein Nitroalkane Oxidase. Biochemistry. 2007;46:13800–13808. doi: 10.1021/bi701557k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolkenius FN, Seiler N. Acetylderivatives as intermediates in polyamine catabolism. Int. J. Biochem. 1981;13:287–292. doi: 10.1016/0020-711x(81)90080-x. [DOI] [PubMed] [Google Scholar]
- 31.Hansch C, Leo A, Taft RW. A survey of Hammett substituent constants and resonance and field parameters. Chem.Rev. 1991;91:165–195. [Google Scholar]
- 32.Fujita T, Iwasa J, Hansch C. A new substituent constant, π, derived from partition coefficients. J. Am. Chem. Soc. 1964;86:5175–5180. [Google Scholar]
- 33.Bondi A. van der Waals volumes and radii. J. Phys. Chem. 1964;68:441–451. [Google Scholar]
- 34.Hansch C, Leo A, Hoekman D. Exploring QSAR, Volume 2: Hydrophobic, Electronic, and Steric Constants. American Chemical Society; Washington, DC: 1995. [Google Scholar]
- 35.Charton M. Nature of the ortho effect. II. Composition of the Taft steric parameters. J. Am. Chem. Soc. 1969;91:615–618. [Google Scholar]
- 36.Bolkenius FN, Seiler N. New substrates of polyamine oxidase Dealkylation of N-alkyl-α, o-diamines. Biol. Chem. Hoppe. Seyler. 1989;370:525–531. doi: 10.1515/bchm3.1989.370.1.525. [DOI] [PubMed] [Google Scholar]
- 37.Cook PF, Cleland WW. pH variation of isotope effects in enzyme-catalyzed reactions. 1. Isotope- and pH-dependent steps the same. Biochemistry. 1981;20:1797–1805. doi: 10.1021/bi00510a014. [DOI] [PubMed] [Google Scholar]
- 38.Gaweska H, Henderson Pozzi M, Schmidt DMZ, McCafferty DG, Fitzpatrick PF. Use of pH and Kinetic Isotope Effects to Establish Chemistry As Rate-Limiting in Oxidation of a Peptide Substrate by LSD1. Biochemistry. 2009;48:5440–5445. doi: 10.1021/bi900499w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denu JM, Fitzpatrick PF. pH and kinetic isotope effects on the reductive half-reaction of D-amino acid oxidase. Biochemistry. 1992;31:8207–8215. doi: 10.1021/bi00150a013. [DOI] [PubMed] [Google Scholar]
- 40.Blackwell LFF,A, Miller IJ, Topsom RD, Vaughan J. Dissociation of benzylammonium ions. J. Chem. Soc. 1964:3588–3591. [Google Scholar]
- 41.Peters FB, Johnson HW., Jr. Ionization constants of substituted 2-aminoacetanilides and benzylamines. Transmission of electronic effects through amide links. J. Org. Chem. 1975;40:1517–1519. [Google Scholar]
- 42.Clark J, Perrin DD. Prediction of the strengths of organic bases. Q. Rev. Chem. Soc. 1964;18:295–320. [Google Scholar]
- 43.Binda C, Angelini R, Federico R, Ascenzi P, Mattevi A. Structural bases for inhibitor binding and catalysis in polyamine oxidase. Biochemistry. 2001;40:2766–2776. doi: 10.1021/bi002751j. [DOI] [PubMed] [Google Scholar]
- 44.Huang Q, Liu Q, Hao Q. Crystal structures of Fms1 and its complex with spermine reveal substrate specificity. J. Mol. Biol. 2005;348:951–959. doi: 10.1016/j.jmb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 45.Miller JR, Edmondson DE. Structure-activity relationships in the oxidation of para-substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry. 1999;38:13670–13683. doi: 10.1021/bi990920y. [DOI] [PubMed] [Google Scholar]
- 46.Fitzpatrick PF. Carbanion versus hydride transfer mechanisms in flavoproteincatalyzed dehydrogenations. Bioorg. Chem. 2004;32:125–139. doi: 10.1016/j.bioorg.2003.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Fitzpatrick PF, Flory DR, Jr., Villafranca JJ. 3-Phenylpropenes as mechanism-based inhibitors of dopamine ß-hydroxylase: Evidence for a radical mechanism. Biochemistry. 1985;24:2108–2114. doi: 10.1021/bi00330a003. [DOI] [PubMed] [Google Scholar]
- 48.Scrutton NS. Chemical aspects of amine oxidation by flavoprotein enzymes. Nat. Prod. Rep. 2004;21:722–730. doi: 10.1039/b306788m. [DOI] [PubMed] [Google Scholar]
- 49.Pollegioni L, Blodig W, Ghisla S. On the mechanism of D-amino acid oxidase. Structure/linear free energy correlations and deuterium kinetic isotope effects using substituted phenylgylcines. J.Biol.Chem. 1997;272:4924–4934. doi: 10.1074/jbc.272.8.4924. [DOI] [PubMed] [Google Scholar]
- 50.Nandigama RK, Edmondson DE. Structure-activity relations in the oxidation of phenethylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry. 2000;39:15258–15265. doi: 10.1021/bi001957h. [DOI] [PubMed] [Google Scholar]
- 51.Taft RW, Lewis IC. Evaluation of Resonance Effects on Reactivity by Application of the Linear Inductive Energy Relationship. V. Concerning a σR Scale of Resonance Effects. J. Am. Chem. Soc. 1969;81:5343–5352. [Google Scholar]
- 52.Walker MC, Edmondson DE. Structure-activity relationships in the oxidation of benzylamine analogues by bovine liver mitochondrial monoamine oxidase B. Biochemistry. 1994;33:7088–7098. doi: 10.1021/bi00189a011. [DOI] [PubMed] [Google Scholar]
- 53.Kurtz KA, Rishavy MA, Cleland WW, Fitzpatrick PF. Nitrogen isotope effects as probes of the mechanism of D-amino acid oxidase. J. Am. Chem. Soc. 2000;122:12896–12897. [Google Scholar]
- 54.Faust A, Niefind K, Hummel W, Schomburg D. The structure of a bacterial L-amino acid oxidase from Rhodococcus opacus gives new evidence for the hydride mechanism for dehydrogenation. J. Mol. Biol. 2007;367:234–248. doi: 10.1016/j.jmb.2006.11.071. [DOI] [PubMed] [Google Scholar]
- 55.Dunn RV, Marshall KR, Munro AW, Scrutton NS. The pH dependence of kinetic isotope effects in monoamine oxidase A indicates stabilization of the neutral amine in the enzyme-substrate complex. FEBS Journal. 2008;275:3850–3858. doi: 10.1111/j.1742-4658.2008.06532.x. [DOI] [PubMed] [Google Scholar]