

Abstract
Management of patients with acute myeloid leukemia relies on genetic tests that inform diagnosis and prognosis, predict response to therapy, and measure minimal residual disease. The value of genetics is reinforced in the revised 2008 World Health Organization acute myeloid leukemia classification scheme. The various analytic procedures—karyotype, fluorescence in situ hybridization, reverse transcription polymerase chain reaction, DNA sequencing, and microarray technology—each have advantages in certain clinical settings, and understanding their relative merits assists in specimen allocation and in effective utilization of health care resources. Karyotype and array technology represent genome-wide screens, whereas the other methods target specific prognostic features such as t(15;17) PML-RARA, t(8;21) RUNX1-RUNX1T1, inv(16) CBFB-MYH11, 11q23 MLL rearrangement, FLT3 internal tandem duplication, or NPM1 mutation. New biomarkers and pharmacogenetic tests are emerging. The pathologist's expertise is critical in 1) consulting with clinicians about test selection as well as specimen collection and handling; 2) allocating tissue for immediate testing and preserving the remaining specimen for any downstream testing that is indicated once morphology and other pertinent test results are known; 3) performing tests that maximize outcome based on the strengths and limitations of each assay in each available specimen type; and 4) interpreting and conveying results to the rest of the health care team in a format that facilitates clinical management. Acute myeloid leukemia leads the way for modern molecular medicine.
More is known about the molecular basis of leukemia than any other form of cancer, primarily due to the availability of abundant malignant cells for study and because translocations and other gross chromosomal changes are often visible by karyotype. Limited prognostic and predictive ability of traditional morphological, immunophenotypic, and cytogenetic tests has driven research to define more subtle nucleotide-level alterations that not only shed light on pathogenesis but also serve as tumor markers and, in some cases, impart valuable prognostic information. Better understanding of disease biology and pathogenesis is essential to cancer prevention and to design novel interventions that are personalized to the host and tumor genotype.
The World Health Organization classification scheme for acute myeloid leukemia (AML) provides a framework for clinical management. It was revised in 2008 to add three distinct forms of AML with recurrent cytogenetic abnormalities [t(6;9), inv(3) and t(1;22)] and two provisional categories with nucleotide level changes (involving NPM1 and CEBPA genes). These revisions emphasize the importance of genetic test results to define clinically relevant disease entities in conjunction with morphology, immunophenotype, and other clinicopathologic features1 (Table 1). Moreover, management guidelines of the National Comprehensive Cancer Network highlight the added value of genetic tests in combination with more traditional microscopic examination and immunophenotyping (http://www.nccn.org/professionals/physician_gls/f_guidelines.asp, accessed July 14, 2009).
Table 1.
2008 World Health Organization Classification of Acute Myeloid Leukemia and Related Tumors
| AML with recurrent genetic abnormalities |
| AML with t(8;21)(q22;q22) RUNX1-RUNX1T1 (CBFA-ETO) |
| AML with inv(16)(p13q22) or t(16;16)(p13;q22) CBFB-MYH11 |
| APL with t(15;17)(q22;q11–12) PML-RARA |
| AML with t(9;11)(p22;q23) MLLT3-MLL and other balanced translocations of 11q23 (MLL) |
| AML with t(6;9)(p23;q34) DEK-NUP214 |
| AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2) RPN1-EVI1 |
| AML (megakaryoblastic) with t(1;22)(p13;q13) RBM15-MKL1 |
| AML with mutated NPM1* |
| AML with mutated CEBPA* |
| Acute myeloid leukemia with myelodysplasia-related changes |
| Therapy-related myeloid neoplasms |
| AML, not otherwise specified |
| AML with minimal differentiation |
| AML without maturation |
| AML with maturation |
| Acute myelomonocytic leukemia |
| Acute monoblastic/monocytic leukemia |
| Acute erythroid leukemias |
| Acute megakaryoblastic leukemia |
| Acute basophilic leukemia |
| Acute panmyelosis with myelofibrosis |
| Myeloid sarcoma |
| Myeloid proliferations related to Down syndrome (+21) |
| Transient abnormal myelopoiesis |
| Myeloid leukemia associated with Down syndrome |
| Blastic plasmacytoid dendritic cell neoplasms |
Adapted from Swerdlow et al.1
Provisional categories.
The relevant genetic technologies include karyotype, fluorescence in situ hybridization (FISH), polymerase chain reaction (PCR), sequencing, and microarrays. These DNA or RNA assays are widely considered to be the most powerful tools for predicting behavior of AML in response to therapy. Automation and kits are becoming available to facilitate implementing standardized assays in clinical laboratories. Results not only identify disease-specific genetic alterations that are important for diagnosis and upfront management but also provide a mechanism to monitor tumor burden in response to therapy and to detect minimal residual disease that could herald impending relapse.
Definitions of several terms are pertinent. A prognostic test is one used to assess the likelihood of response to standard therapy, while a predictive test is used to assess response to a particular nonstandard intervention. A pharmacogenetic test is a predictive test for a specific pharmaceutical agent or regimen. Worldwide consensus on best practices for managing AML is evolving, and optimal test strategies and intervention for a given patient depend on factors beyond genetic test results.
Genetic Technologies
A brief description of each genetic technology is provided in Table 2. Gross translocations and numerical changes in chromosomes are readily detected by karyotyping. Less commonly recognized (but certainly not less commonly present) are mutations, subtle deletions, and/or gene amplifications. Regardless of which genetic defects initiate tumor cell growth, these defects are passed down to all cellular progeny within a tumor clone. Certain defects are characteristic of distinct clinicopathologic subtypes of cancer, and these defects serve as markers of the malignancy that can be used to assist in diagnosis, classification, and monitoring residual disease after therapy. Furthermore, knowledge of the affected biochemical pathway could help identify therapy targeting the underlying cause of malignant cell growth.
Table 2.
Genetic Test Methods
| Laboratory Test | Description of Methods |
|---|---|
| Karyotype: | Whole chromosomes from cells in the metaphase stage of cell division are stained and visualized by microscopy. |
| Fluorescence in situ hybridization (FISH): | Whole chromosomes (metaphase from dividing cells or interphase from non-dividing cells) are hybridized to complementary probes and visualized on a fluorescence microscope. |
| Polymerase chain reaction (PCR): | DNA is isolated and a specific segment of it is copied a billion-fold for ease of detection and for further analysis. A variant method called reverse transcription PCR (rtPCR) converts RNA into complementary DNA (cDNA) prior to PCR amplification. A variant called quantitative PCR (Q-PCR) can measure the level of target DNA, usually by monitoring product accumulation during each cycle using one or more fluorescent internal probes, and then comparing the time course of product accumulation to a series of standards of known concentration. A fluorescent internal probe combined with “melt curve analysis” detects sequence variants within the amplicon. |
| DNA sequencing: | The nucleotide sequence is determined by replicating one of the DNA strands and monitoring the order in which labeled nucleotides are added. |
| Comparative genomic hybridization array (CGH array): | Patient DNA is hybridized to hundreds or thousands of probes arrayed on a solid surface, and gene dosage is determined for each locus on the array, thus identifying deletions, duplications, and gene amplifications. Single nucleotide polymorphism (SNP) arrays can additionally detect copy-neutral loss of heterozygosity (uniparental disomy). |
| Gene expression array: | Patient RNA is typically amplified and labeled, then mixed with control RNA labeled with a different fluorochrome and hybridized to hundreds or even hundreds of thousands of probes (eg, 60-mers) arrayed on a solid surface. Scans of each spot followed by data analysis permit evaluation of the gene expression profile in the tissue, which can be matched to the pattern of normal or diseased tissues for purposes of diagnosis, or to the pattern of clinical outcome variants to predict response to therapy. |
Karyotype
The karyotype represents a genome wide screen for translocations and other numeric or structural defects that are present in about half of AMLs. Giemsa staining patterns (G-bands) are interpreted for each chromosome in at least 20 fresh dividing cells. Findings are further interpreted in the context of the patient's clinical and histopathological features to help diagnose and classify AML. Even nonspecific karyotypic changes can impact prognosis: a complex karyotype, usually defined as three or more concomitant defects, portends a poor prognosis in children and adults, including patients over 60 years old.2,3,4,5 Recent data suggest that a “monosomal karyotype,” defined by two or more autosomal monosomies or one monosomy plus one or more structural defects, for example, −5 with −20, or −7 with t(3;3)(q21;q26), is indicative of bad outcome in adults less than 60 years old, with an overall survival of only 4% at 4 years.6
Fluorescence in Situ Hybridization
FISH can be applied to either interphase (nondividing) or metaphase (dividing) cells where it is used to: 1) confirm a tumor-related karyotypic defect that can then be monitored over time in blood or marrow specimens, 2) detect cryptic translocation in a tumor suspected to harbor a particular defect based on clinicopathologic findings, 3) detect a deletion or duplication not evident by karyotype, and 4) discover critical cytogenetic information in specimens failing to grow in tissue culture media.
FISH probes are commercially available for relevant targets including t(15;17) PML-RARA, t(8;21) RUNX1-RUNX1T1, inv(16) CBFB-MYH11, and 11q23 MLL.7 Various probe strategies (eg, single fusion, dual fusion, or break-apart) are used depending on the technical and clinical circumstances.8 For example, a dual fusion strategy has better analytic sensitivity for finding low level PML-RARA, while a break-apart probe strategy detects any of the relevant RARA translocations irrespective of the partner gene. Centromere probes are used to enumerate chromosomal gains or losses such as trisomy 8 or monosomy 7. Published guidelines for validating FISH assays include recommendations on how to set a cutoff for interpreting results as normal versus abnormal.8
Sensitivity for detecting minimal residual disease depends on the probe strategy, specimen quality, and the number of cells that are scored. A typical interphase FISH is performed on 200 cells and reliably detects a leukemic clone involving at least 5% of cells in the specimen. This level of sensitivity is comparable with that of a 20-cell karyotype, although karyotype sensitivity varies depending on the relative growth rate of leukemic versus nontumor cells ex vivo. In dividing cells, metaphase FISH or hypermetaphase FISH can be applied to resolve complex karyotypes and identify partner genes fused by translocation.
A variation of FISH known as spectral karyotyping or multiplex FISH applies multiple labeled probes along whole chromosomes, essentially “painting” each of the 24 chromosomes a different color. Results are interpreted in conjunction with G-banded karyotype to sort out complex rearrangements. The gene copy number information is similar to, albeit with less resolution than, comparative genomic hybridization arrays.
Polymerase Chain Reaction
PCR has tremendous analytic sensitivity and has become a mainstay of molecular pathology. PCR can find “a needle in a haystack” because, after 30 cycles of amplification, each DNA or cDNA target sequence has been copied 230 times, yielding a billion amplicons. These amplicons can be quantified using precise real-time instrumentation, and/or amplicons can be further evaluated using various analytic methods like sequencing, melt curve analysis, or electrophoresis. In typical clinical assays, a dilution of one leukemic cell per 100,000 normal cells is identifiable, facilitating detection of minimal residual disease. Paucicellular specimens and partially degraded nucleic acid can often be accommodated, which is helpful in unusual samples like cerebrospinal fluid or biopsied myeloid sarcoma. Molecular protocols have been published, and commercial primers and probes are available for selected fusion transcripts, mutations, and controls.9,10,11,12,13,14,15,16,17
DNA Sequencing
Determining the order of nucleotide bases is useful for genes like CEBPA having multiple alternative mutations at different nucleotide positions. Traditional assays relying on dideoxynucleotide incorporation can identify a variant comprising at least 20% of alleles in the specimen (equivalent to 40% of cells). Pyrosequencing, which detects pyrophosphate release, is potentially more sensitive and also more quantitative. High throughput sequencing methods are now being validated to expand coverage and in some instances to improve assay sensitivity.
Microarrays
Array technology is being validated for use in clinical laboratories so that many simultaneous analyses may be performed, such as gene expression profiling, gene copy number measurement, methylation, or allele-specific mutation detection. The massive amount of data generated by arrays requires bioinformatic tools to present the data in a manner that facilitates interpretation. Quality assurance and assay validation are especially challenging when so many tests are performed simultaneously.
Prognostic Applications of Genetic Technologies
Karyotype Is Prognostic
Karyotype is recommended in every suspected AML for proper diagnosis and classification. In the 2008 World Health Organization classification scheme, over two-thirds of AML cases are categorized based on genetic tests compared with only one-third in the 2001 World Health Organization scheme.1 Even when blast percentage in blood or marrow does not exceed the 20% usually required for diagnosis of AML, presence of t(15;17), t(8;21), or inv(16) in the face of abnormal hematopoiesis is considered sufficient for a diagnosis of AML.
National Comprehensive Cancer Network guidelines refer to cytogenetics as the “single most important prognostic factor for predicting remission rate, relapse, and overall survival” (Figure 1). The choice of which therapy to deliver rests largely on grouping of AMLs into one of three categories: favorable, intermediate, or unfavorable risk.18,24,25,26 Among 1213 AML patients treated on CALGB protocols, the 5-year survival rate was 55% for favorable, 24% for intermediate, and 5% for unfavorable cytogenetic categories.27 Outcomes have improved since 2002 when these data were published, and now it has become routine to supplement karyotype with assays for molecular-level defects as described below.
Figure 1.
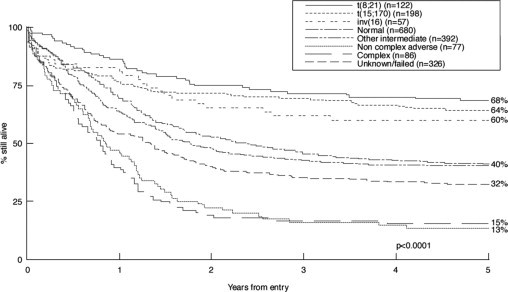
Prognosis in AML strongly correlates with cytogenetic findings.18,19,20,21 Favorable prognosis is associated with t(15;17), t(8;21), or inv(16) whether alone or in combination with other chromosomal abnormalities, with the possible exception of inv(16) or t(8;21) with complex karyotype. An intermediate prognosis is associated with normal karyotype or select abnormalities: +6, +8, −7q, −9q, −12p, +21, +22, −Y. Adverse outcome is associated with abnormal 3q, 11q, 17p, 20q, 21q, or with −5, −5q, −7, −7q, −9q, t(6;9), t(9;22), or with complex karyotype. Abnormalities of the MLL gene on 11q23 impart a dismal prognosis.22,23 In children, poor survival is associated with monosomies or complex karyotype.5 (Adapted from Grimwade18 with permission).
Assays for Cryptic Translocation and MLL Gene Defects
In the ∼45% of adult AML having a normal karyotype, FISH or rtPCR may help detect cryptic translocation whenever the clinical presentation (eg, DIC), morphology and immunophenotype is suggestive of one of the three favorable subtypes, t(8;21), t(15;17), or inv(16).7,28,29 Another defect that is missed by karyotype involves partial tandem duplication of the MLL gene on chromosome 11q23.30,31 Whether MLL is altered by partial tandem duplication or by translocation with any of >70 partners, MLL rearrangement portends a poor outcome in AML, except for t(9;11), which imparts an intermediate risk.32,33,34 Patients with poor prognosis MLL rearrangement or partial tandem duplication are candidates for allogeneic stem cell transplant.
Acute Promyelocytic Leukemia with t(15;17) PML-RARA
Acute promyelocytic leukemia (APL) is an archetypal example of how genetic technologies are used in cancer diagnosis and management. It is important to recognize leukemia harboring t(15;17) PML-RARA because of the unique treatment strategies and monitoring assays that are available to affected patients. Retinoic acid receptor α (RARA) gene structure is altered by translocation to thwart RARA transcription factor function and arrest differentiation at the promyelocyte stage. The molecular defect in most cases can be overcome by treating with high doses of retinoic acid (all-trans-retinoic acid, ATRA), providing a prime example of cancer therapy specifically targeting a gene product involved in tumorigenesis. Introduction of retinoic acid therapy represented a paradigm shift in managing leukemia because this drug operates by overcoming the effect of the translocation rather than by eliminating the malignant clone. To diminish the likelihood that secondary mutation renders the tumor resistant to single agent ATRA, combination therapy with an anthracycline-based regimen or with arsenic trioxide is recommended and is curative in about 80% of cases, representing one of the greatest advances in the history of cancer therapy.
A small subset of patients with morphological and clinical features overlapping those of classic t(15;17) APL have variant translocations: t(11;17)(q23;q12) ZBTB16-RARA (previously called PLZF-RARA), t(5;17)(q35;q12) NPM1-RARA, t(11;17)(q13;q12) NUMA1-RARA, t(4;17)(q12;q21) FIP1L1-RARA, interstitial duplication of chromosome 17 resulting in STAT5B-RARA fusion, or occult rearrangement of chromosome 17 resulting in PRKAR1A-RARA fusion.35,36,37 Unraveling the genetics has therapeutic implications since defects involving ZBTB16 or STAT5B may be resistant to ATRA. Nonetheless, when a morphological diagnosis of APL is made, it is reasonable to begin targeted therapy pending genetic testing, and adjust the therapeutic regimen if a relevant genetic defect is not identified.38
More than 95% of APLs harbor a PML-RARA translocation detectable by karyotype, FISH, or rtPCR.28,39,40,41 The most sensitive of these is rtPCR in which RNA extracted from blood or marrow is converted to cDNA and then primers flanking the PML-RARA breakpoint specifically amplify the chimeric sequence (Figure 2). As with any assay targeting RNA, special precautions are needed to avoid RNA degradation. Negative results are reported only when a control test of a “housekeeping” transcript shows that amplifiable cDNA was achieved, and results are reported in the context of assay sensitivity so that it is clear whether the assay can detect minimal residual disease.
Figure 2.
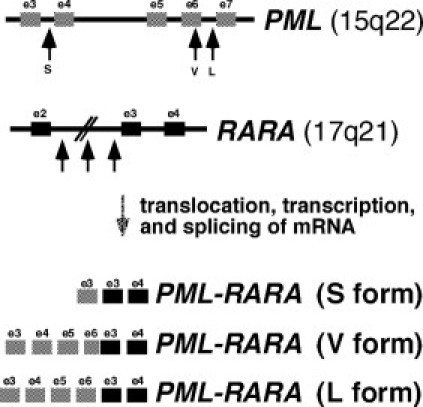
The PML and RARA genes are shown, along with three forms of PML-RARA fusion transcripts. Breaks in PML are clustered into three regions: intron 3, the distal half of exon 6, or intron 6. RARA breakpoints are spread across intron 2. In the PML-RARA fusion transcript, either exon 3 or exon 6 of PML is juxtaposed with exon 3 of RARA to produce short (S), long (L), or variable (V) length coding sequence. The short form, also called the bcr3 isoform, may carry a worse prognosis.
After chemotherapeutic induction, consolidation therapy is used to achieve a durable molecular remission. Patients are likely to relapse if they do not achieve molecular remission in the marrow as assessed by rtPCR after consolidation.42 National Comprehensive Cancer Network guidelines suggest that a positive rtPCR test result should be confirmed, and repeat positivity is treated as if the patient had hematological relapse (AML Clinical Practice Guideline Version 2009.1, accessed at http://www.nccn.org/professionals/physician_gls/f_guidelines.asp on July 14, 2009) In contrast, patients having consistently negative results after consolidation therapy enjoy prolonged survival and may even be cured.26,43
To detect early relapse, the National Comprehensive Cancer Network guidelines recommend that rtPCR be performed on either blood or marrow at 3-month intervals for the next 2 years, and then every 6 months for 2 to 3 years. Confirmed positive results, as defined by persistent positive rtPCR within a month of the first positive result, with at least one of these positive results being in marrow, warrant treatment for relapse. Patients who achieve molecular remission (negative by rtPCR) after being treated for relapse have better outcomes than those who remain positive. Readers are referred to alternative practice guidelines for further opinions on testing strategies in patient management.38,44,45,46,47
Prognosis of AML with Normal Karyotype
Studies have implicated a number of molecular abnormalities as being prognostic in cytogenetically normal AML (Figure 3). The various defects are not necessarily mutually exclusive, implying that each tumor probably harbors multiple genetic abnormalities, and any one lesion may be insufficient for malignant transformation.48,49 The combinations of abnormalities are not random, suggesting that co-acquisition of selected defects synergizes in leukemogenesis. One defect blocking differentiation and a second defect inducing proliferation appears to be a potent recipe for acute leukemia.50,51,52 Sorting out the pathogenic defects from benign “passenger defects” is a daunting task given the propensity of cancers to acquire secondary genetic alterations. Some abnormalities are prognostic only in certain subsets of cancer (Table 3).
Figure 3.
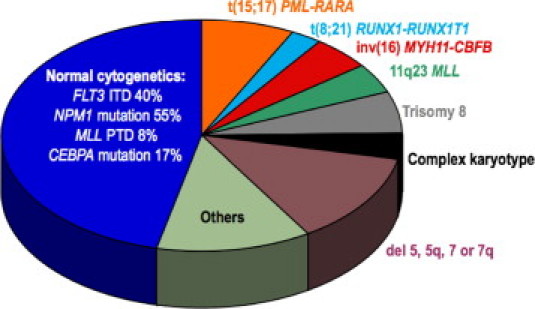
Relative frequencies of common recurrent genetic abnormalities in acute myeloid leukemia.
Table 3.
Prognostic Genetic Characteristics in Acute Myeloid Leukemia
| Favorable risk factors |
| t(15;17)(q22;q12) PML-RARA |
| t(8;21)(q22;q22) RUNX1-RUNX1T1 |
| inv(16)(p13;q22) or t(16;16)(p13;q22) CBFB-MYH11 |
| NPM1 mutation when FLT3 internal tandem duplication is absent and cytogenetics are normal |
| CEBPA mutation (correlates with erythroid differentiation and higher hemoglobin) |
| Intermediate risk group |
| Normal karyotype* |
| FLT3 internal tandem duplication with NPM1 mutation and normal cytogenetics |
| KIT mutation with t(8;21) or inv(16) |
| +8 only |
| t(9;11) AF9-MLL only |
| Abnormalities not otherwise listed |
| Unfavorable risk factors |
| Complex karyotype (≥3 abnormalities) |
| Monosomal karyotype (≥2 autosomal monosomies, or a single one plus ≥1 structural defect) |
| −5, −7 or other autosomal monosomy |
| del(5q) or del(7q) |
| 11q23 MLL translocation, excluding t(9;11) AF9-MLL |
| MLL partial tandem duplication with normal cytogenetics |
| inv(3)(q21;q26) or t(3;3)(q21;q26) RPN1-EVI1 or MDS1-EVI1 |
| EVI1 overexpression |
| 17p abnormality or TP53 mutation |
| FLT3 internal tandem duplication when NPM1 mutation is absent and cytogenetics are normal |
| t(9;22)(q34;q11) BCR-ABL1 |
| t(6;9)(p23;q34) DEK-CAN |
| ERG overexpression without FLT3 ITD when cytogenetics are normal |
| BAALC overexpression with normal cytogenetics |
| MN1 overexpression with normal cytogenetics |
| WT1 mutation with normal cytogenetics |
| TET2 mutation |
Prognostic categorization may vary by analytic method, patient population, study design, and other variables.
Loss of X or Y chromosome is not considered an abnormality for purposes of prognosis.
Molecular tests are increasingly applied to reveal tumor characteristics that refine prognosis in cytogenetically normal AML. Test results are used to select those patients who may benefit from chemotherapy and those who should be considered for an allogeneic stem cell transplant if a suitable donor is available and if patient age, comorbidities, and other factors are amenable for allografting.48
FLT3 Internal Tandem Duplication Confers a Worse Prognosis
Activating mutation of the FMS-related tyrosine kinase 3 (FLT3) gene is associated with a higher risk of relapse and a worse prognosis.53,54 The relevant mutation is an in-frame internal tandem duplication (ITD) within the coding sequence of the juxtamembrane domain that causes constitutive activation of the encoded FLT3 tyrosine kinase. Signaling through the MAPK, PI3K, and STAT5 pathways contributes to proliferation, resistance to apoptosis, and blocked differentiation. FLT3 ITD testing is recommended in all cytogenetically normal AML patients who are candidates for allogeneic transplantation or investigational therapies.55 FLT3 ITD is also present in a subset of APLs, but the implications for patient management are unclear.56,57,58
To identify the FLT3 ITD, DNA from leukemic cells is first amplified using PCR and then sized by capillary electrophoresis to detect an abnormally large amplicon associated with ITD59 (Figure 4). The extent of amplicon enlargement varies from 3 to about 400 bp, and the insertion is always “in frame” to preserve the translation and function of the remaining FLT3 domains. A high ratio of mutant to wild-type FLT3 has been linked to a worse outcome.60
Figure 4.
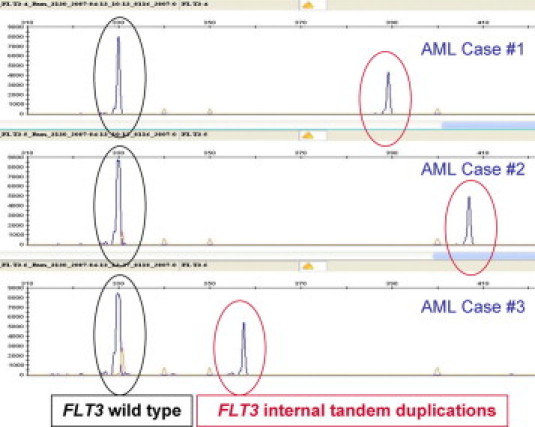
FLT3 internal tandem duplication is identified in genomic DNA that has been amplified across exons 14 and 15 using PCR and then sized by capillary electrophoresis. Amplicons representing the wild-type allele are 325 bp in length (circled in black). In each of three AML cases, a larger amplicon is also seen (circled in red), consistent with FLT3 internal tandem duplication in the leukemic cells.
While the ITD is strongly prognostic in both adults and children, point mutation in the kinase domain of FLT3 does not seem to have a major influence on outcome.61,62 Mis-sense mutations can rarely occur in the juxtamembrane domain and may confer activation similar to that of the ITD.63 Testing for activating point mutations in FLT3 is currently recommended only in clinical trials where efficacy of a putative FLT3 inhibitor is being examined.
NPM1 Mutation Confers a Better Prognosis
Nucleophosmin (NPM1) mutation is associated with a good prognosis when FLT3 ITD is absent and an intermediate prognosis when FLT3 ITD is present.60 The NPM1 mutation results from insertion (or combined insertion and deletion) in one allele of NPM1. The gene encodes a nuclear shuttle protein that, when mutated, aberrantly localizes to the cytoplasm, affecting its regulation of the ARF-p53 pathway.64,65 NPM1 mutation is found in 30% of all adult AML,66 and it is enriched for in those with normal karyotype (55% of cases), which is the recommended target population for prognostic testing. It is much less common in childhood AML (8%) where it is most informative of outcome in cytogenetically normal tumors without FLT3 ITD.67,68
Two predictive aspects of NPM1 mutation have been proposed and must now be independently validated: 1) Patients with NPM1 mutation without FLT3 ITD do not necessarily benefit from allogeneic stem cell transplant following conventional anthracycline and cytarabine-based induction therapy54; and 2) older patients with NPM1 mutation without FLT3 ITD might benefit from adding ATRA to their chemotherapy regimen.69
Laboratory testing for NPM1 mutation typically relies on PCR followed by capillary electrophoresis to detect a small insertion in one allele in exon 1270 (Figure 5). Alternative molecular methods are feasible.12,71,72 At least 40 molecular variants exist, most of which result in a 4-bp enlargement, although rarely an amplicon up to 11 bp larger than normal is seen. Frameshift mutations interfere with the nucleolar localization motif in the C-terminal end of the protein. Mislocalized NPM1 protein can often be visualized using immunohistochemistry except in blasts having scant cytoplasm. Molecular testing for NPM1 mutation is a reasonable alternative to immunohistochemistry when one considers that interpretation of NPM1 results is usually done in conjunction with FLT3 results, and FLT3 is tested using molecular methods.
Figure 5.

NPM1 mutation is identified in genomic DNA from four cases of AML by amplification of a segment of exon 12 followed by sizing of the amplicons using capillary electrophoresis. Amplicons representing the normal, wild-type allele are 267 ± 1 bp in length, while abnormal amplicons are usually 4 bp larger, consistent with insertional mutation in the leukemic cell DNA.
In adult AML, type A NPM1 mutation (a 4-base TCTG duplication) comprises three quarters of mutated cases, while two alternate 4-bp insertions at the same position (type B is CATG, and type D is CCTG) comprise an additional 15% of mutated cases. Type B insertion is more common in children. Each of these common variants has been targeted using allele-specific amplification to detect minimal residual disease and to predict relapse.73,74,75 Unfortunately, lack of the mutation in 10% of relapsed AML patients limits the reliability of these allele-specific assays for monitoring tumor burden over time.73
CCAAT/Enhancer Binding Protein α (CEBPA) Mutation Confers a Better Prognosis
“AML with a mutated CEBPA gene” is a provisional category of the World Health Organization classification comprising 17% of cytogenetically normal AML. CEBPA mutation is a favorable prognostic indicator when it is biallelic and when it occurs in isolation of other prognostic genetic defects.76,77,78 Moreover, CEBPA mutation portends a better prognosis than does wild-type CEBPA in AMLs with FLT3 ITD.78 Despite the absence of FLT3 ITD, the subset of patients with so called “triple negative” results (negative for CEBPA mutation, NPM1 mutation, and FLT3 ITD) do poorly and may be considered for allogeneic transplant.54
CEBPA can be silenced by either mutation or by promoter hypermethylation, 76,79 implying that DNA sequencing in combination with methylation analysis is required to capture all of the relevant prognostic information. CEBPA encodes a transcription factor important in neutrophil differentiation. Mutation down-regulates HOX gene expression leading to decreased expression of myeloid differentiation factors, induction of miR181, and increased expression of erythroid differentiation genes leading to elevated hemoglobin.80,81 There is slow uptake of CEBPA testing, in part because rather extensive sequencing and methylation analysis is required to detect the relevant defects.
ERG, BAALC, WT1, EVI1, MN1, microRNA, and Integrated Panels of Prognostic Factors
In addition to the gene rearrangements and mutations described so far, transcriptional dysregulation of selected genes (eg, ERG, BAALC, WT1, EVI1, MN1, miR181) seems to confer prognostic information.51,82 For example, EVI1 overexpression in AML (or rearrangement of the EVI1 gene on 3q26) is associated with lack of response to current treatments and a dismal prognosis.82
Applying a large panel of microRNAs reveals patterns of expression that are associated with outcome in AML.80 The microRNA signature has been purported to add prognostic value beyond what is achievable with FLT3 and NPM1 testing. A panel of just seven microRNAs could distinguish the major karyotypic categories of AML.83 A panel of 12 microRNAs can divide cytogenetically normal AML into poor and intermediate risk categories independently of FLT3 ITD.84
The list of prognostic factors seems to be growing rapidly, making it difficult to discern which prognostic factors are independent of the others, and which panel of tests to perform in a given patient. Prognostic factors are most useful when they impact on therapeutic response, in other words, when they are predictive of outcome.54,55 It would be helpful to have a tiered algorithm for ordering various genetic tests based on cost effectiveness data in various clinical scenarios that account for the available therapeutic options (eg, stem cell transplant). Alas, long lists of putative prognostic tests and therapeutic regimens, combined with a paucity of clinical trial data for various settings, render it difficult to achieve international consensus on a testing algorithm. A useful integrated predictor has recently been proposed by Dutch/Belgian/Swiss investigators that relies on combined FLT3, NPM1, ERG, CEBPA, and BAALC genotypes to place patients into one of four groups with respect to the risks and benefits of stem cell transplantation.85
Predictive Applications of Genetic Technologies
KIT Mutation and Drug Responsiveness
KIT is a receptor tyrosine kinase that functions in normal hematopoiesis. Gain of function mutations in KIT have been found in 2% of AML overall and in a third of the “core binding factor” leukemias [AML with t(8;21) or inv(16)], as well as in systemic mastocytosis and several non-hematopoietic malignancies.86 KIT mutation (especially D816V encoded by exon 17) is associated with a worse prognosis in AML with t(8;21) RUNX1-RUNX1T1, in contrast to the good prognosis normally associated with t(8;21).20 Some KIT-mutated malignancies respond to tyrosine kinase inhibitors, although response depends on the type of KIT mutation and on the mechanism and site of action of the drug.87 A number of tyrosine kinase inhibitors such as imatinib, dasatinib, and PKC412 are being tested for efficacy against KIT-mutated AML.88
KIT mutation is generally detected by sequencing exons 8 and 17 in leukemic cells. Alternatively, allele-specific PCR can detect exon 17 D816V, the most relevant mutation in AML.89
RAS Mutation and Drug Responsiveness
Among the RAS family of genes, NRAS and KRAS are more frequently mutated in AML than is HRAS.90 Overall, RAS mutation is present in about 15% of AMLs and is enriched for in cases having inv(16) or inv(3).91,92 RAS mutation may enhance response to high dose cytarabine,54,93 while response to a farnesyl transferase inhibitor (tipifarnib, a drug that shuts down activated RAS) was predicted by the RASGRP1 to APTX gene expression ratio.94
Emerging Array Technologies
Microarray-Based Gene Expression Profiles
As the list of prognostic and predictive tests becomes longer, it is reasonable to consider whether massive parallel transcriptional analysis might be more cost effective than panels of disparate ancillary methods (eg, karyotype, FISH, mutational analysis, immunophenotype). Gene expression profiling uses arrays to detect and semiquantify expression of all ∼25,000 human genes at once. Smaller or custom arrays can be created to order. The microarray method requires extracting RNA from a fresh or frozen specimen containing a high proportion of malignant cells to identify patterns of gene expression that are characteristic of prognostic or predictive subsets of disease. Distinct profiles are seen in most AML categories of clinical importance, such as t(8;21), t(15;17), inv(16), and the monocytic subclasses.95,96,97,98 CEBPA mutation or methylation-related silencing has a distinct profile, highlighting the ability of arrays to detect both genetic and epigenetic forms of CEBPA dysfunction. Not surprisingly, the group of AMLs with a complex karyotype lacks a distinct profile, in keeping with the diversity of the genetic defects comprising complex karyotypes. Interestingly, NPM1 and FLT3 defects are not readily identified by array signatures, suggesting that mutational effects are diverse or else the same effects are seen in non-mutated cases.97
Once validated, it is likely that expression profiles will be used for patient care, either in place of traditional assays or to select the next round of ancillary tests that are most appropriate for managing that patient initially and after treatment when residual disease testing is relevant. Novel array-based prognostic algorithms have been proposed. In one study of adult AML, a 133-gene algorithm predicted survival independent of the usual clinical predictors.99 In another study, 86 probes targeting 66 genes yielded a prognostic score that was independent of FLT3 and NPM1 status in cytogenetically normal AML.100 MicroRNA signatures are also informative and may complement mRNA signatures in classifying AML and predicting outcome.95,98,101,102 It is likely that array signatures will be interpreted in combination with more traditional clinical data (eg, age, cell counts) and independent prognostic factors. Expression profiling does not currently have a role in monitoring residual disease; however, this does not exclude a role for expression profiles in monitoring response to treatment.
Microarray-Based Gene Copy Number Variants and Whole-Genome Sequencing
While normal cells have two copies of every gene (one inherited maternally and the other paternally), many AMLs have fewer or more copies of a given gene, of a whole chromosome, or of an intermediate sized region. While karyotype can detect large additions or deletions, array technology can, depending on the design of the probes and their density, detect smaller copy number changes and even point mutations or segmental uniparental disomy. Uniparental disomy can result in duplication of a mutated locus while the normal locus on the other allele is lost, causing copy neutral loss of heterozygosity with the potential for complete alteration of gene function.
Copy number variation studies show that AMLs contain many alterations that were occult by traditional karyotype. Even balanced translocations are often identifiable based on subtle hybridization variation at the breakpoints in DNA (or in fusion transcripts).103,104,105 Identification of microdeletions or duplications, as well as cryptic translocations, may add value when interpreted in combination with results of traditional metaphase cytogenetics.106
Full-genome sequencing is now feasible and may reveal novel factors responsible for tumor initiation and progression. Full-genome sequencing of an AML showed 10 acquired mutations, two in known AML-related genes and another eight that were unexpected.107 Full genomic or exonic sequencing is considered a discovery tool at this time, but more targeted sequencing or SNP arrays approaches may be useful for finding druggable pathways and markers for monitoring tumor burden.
A Practical Approach to Specimen Allocation and Testing
Given the ever-expanding panels of tests now available, it is timely and important to describe a rational approach to ordering genetic tests at initial diagnosis and in follow-up. For any marrow suspected of harboring AML, karyotype must be done up front.26 If the karyotype is normal, but morphology and immunophenotype suggest one of the prognostically favorable karyotypes, FISH or rtPCR should be done to detect cryptic rearrangement of the relevant locus. This genetic workup is considered sufficient for initial therapeutic decision-making.26 Additional testing to refine prognosis is useful for decision-making at the time of first remission or relapse.108,109,110,111
Normal karyotype AML patients may benefit from an additional panel of prognostic tests (eg, FLT3 ITD, NPM1 mutation) to assist with downstream clinical decisions such as whether to prioritize transplant in first remission.54,109,110,111 Activating KIT mutation (exons 8 or 17) negatively influences prognosis in t(8;21) cases, but otherwise such patients tend to do well with standard high-dose ARA-C-containing consolidation regimens and are not considered candidates for allogeneic transplant in first remission.20 In unfavorable prognosis AML, the benefits of allogeneic transplant may outweigh the risks.109,110 The Dutch-Belgian-Swiss clinical trial experience in adults under age 60 was recently summarized in the context of risk categorization using modern panels of cytogenetic and molecular tests.85,108
Tumor burden can be monitored over time if the malignant clone has a distinct feature that can be accurately and sensitively measured in blood or marrow.26,112 Fusion transcripts, antigen receptor gene rearrangement, and point mutation are examples of these biomarkers. Figure 6 shows the relative sensitivity of various detection methods. Amplification assays can often detect very rare tumor cells in a specimen containing upwards of 100,000 normal cells, permitting relapse to be predicted well before the patient becomes symptomatic.9,10,11 Early detection and treatment of relapsing disease restricts the number of cell divisions and thus limits the risk for secondary genetic hits that might render the tumor less responsive to therapy.
Figure 6.
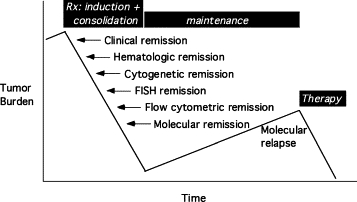
Genetic and phenotypic abnormalities that are unique to the tumor provide a marker by which to measure tumor burden. Up to a billion leukemic cells remain in a patient who is in hematological (morphological) remission. Sensitive assays can detect and measure residual disease to permit early intervention when tumor burden is rising.
A positive molecular test may be the first sign of impending relapse. A negative test result can be interpreted with certainty only when the test is known to detect a valid tumor marker for that patient. Therefore, it is wise to assess tumor markers upfront when the tumor is abundant. A cost-effective alternative is to save residual leukemic specimens for later testing. Assuring proper handling and storage of specimens is critical.
Specimen Collection and Storage
The best available specimen should be used for genetic analysis. Fresh marrow or blood should be collected and handled according to the testing laboratory's recommendations. While heparin is the preferred anticoagulant for cytogenetics, heparin can interfere with DNA amplification, so EDTA is preferred for PCR and rtPCR-based assays. Processing for cytogenetics should be initiated as soon as possible, preferably within 24 hours of collection. Any residual cells remaining after karyotype can be stored for further analysis once the karyotype is known. These leftover cells are typically fixed in Carnoy's solution (methanol and acetic acid) and refrigerated to preserve target analytes for FISH or rtPCR.
Smears and touch preparations are amenable to interphase FISH, and these may be air dried and saved unstained at 4°C for several weeks before analysis, thus allowing for completion of morphological, immunophenotypic, and karyotype studies before ordering FISH. Although interphase FISH can be done on paraffin sections, smear and touch preparations containing single layers of whole nuclei are more readily interpreted compared with paraffin sections where parts of nuclei are often missing.
It should be emphasized that virtually all PCR-based assays are more robust when applied to fresh or frozen cells as compared with fixed cells, since formalin-mediated cross-linking renders nucleic acid less amenable to hybridization. Formalin preservation, although not ideal, is preferred over B5 and other mercury-based fixatives. Decalcification results in acid-mediated degradation of nucleic acid, so clot sections are preferred over decalcified biopsy sections.
RNA is a particularly labile molecule, so handling specifications should be strictly followed and specimens should be delivered promptly to the testing laboratory. Tests for minimal residual disease, whether they target DNA or RNA, require special care to prevent degradation before analysis. As a check on specimen quality, test results are interpreted in conjunction with results of a control assay demonstrating amplifiable housekeeping DNA or cDNA, as appropriate.
A summary of acceptable specimen types for various molecular tests is shown in Table 4. While PCR is typically used to detect point mutations and small duplications or insertions (eg, NPM1 mutation or FLT3 ITD) in extracted DNA, rtPCR is typically used to detect fusion transcripts representing translocations or inversions in extracted RNA. The optimal strategy for designing an assay relies on a thorough understanding of the relevant technologies, specimen types and handling parameters, genetic target(s), and intended use of the assay. Guidelines for assay validation were recently published.113
Table 4.
Tissue Requirements for Genetic Tests
| Laboratory procedure | Recommended sample types |
|---|---|
| Karyotype | Heparinized marrow aspirate (preferred) or blood or fresh biopsy |
| FISH | Fresh cells for metaphase analysis; for interphase analysis, alcohol-fixed cells, smears, touch preparations, or formalin-fixed, non-decalcified tissue sections |
| Southern blot analysis, gene expression array, rtPCR | Fresh blood or marrow (EDTA), frozen nucleated cell pellet, frozen tissue |
| PCR, DNA sequencing, CGH array, microRNA | Fresh blood, marrow aspirate, or body fluid; frozen or paraffin-embedded tissue |
Refer to collection and handling requirements for each assay in each testing laboratory.
Reporting and Quality Assurance of Genetic Tests
College of American Pathologists' recommendations for reporting molecular test results include specifying the technology used and the gene targets.114 To ensure that everyone uses the same term for a given gene, the Gene Nomenclature Committee of the Human Gene Organization has developed a database of the name and symbol for each gene, searchable at http://www.genenames.org (accessed July 14, 2009). Cytogenetic nomenclature rules are used to designate karyotype and FISH findings, while nucleotide-level alterations are described in comparison with a reference sequence.114 So that reports may be deciphered by a general physician, a written explanation of the results and their clinical significance is essential. A molecular genetics pathologist is well suited to interpret and convey results by virtue of expertise in the relevant technologies as applied to diagnosis, prognosis, and prediction.
Molecular results should be interpreted in conjunction with morphological and clinical information to maximize the value for clinical decision-making.114 The 2008 World Health Organization book states, “because of the multidisciplinary approach required to diagnose and classify myeloid neoplasms it is recommended that the various diagnostic studies be correlated with the clinical findings and reported in a single, integrated report.”1 This synthesis is typically done by the consulting hematopathologist at initial diagnosis. In follow-up specimens, the ordering physician (whether the clinician or consulting pathologist) assures that testing is medically necessary and that results are synthesized with relevant clinicopathologic findings. Molecular pathologists and other physicians overseeing laboratory testing are responsible for assisting with test selection, interpreting results, and assuring quality and relevance of laboratory work.
Heritable Syndromes Predisposing to AML
Inherited predisposition to AML should be considered in patients having a strong family history of cancer (Table 5). The heritable defect often involves a DNA repair protein, in which case affected patients should minimize exposure to radiation and chemotherapeutic drugs that induce DNA damage. Therapy-related leukemia is associated with heritable polymorphisms in drug metabolizing enzymes such as glutathione-S-transferase M1 or T1, N-acetyl transferase 2, quinone oxoreductase, or cytochrome p450 (CYP1A1).115
Table 5.
Genetic Predisposition to Acute Myeloid Leukemia
| Heritable syndrome | Gene symbol | Gene name | Locus |
|---|---|---|---|
| Fanconi anemia A | FANCA | Fanconi anemia, complementation group A | 16q24.3 |
| Fanconi anemia C | FANCC | Fanconi anemia, complementation group C | 9q22.3 |
| Fanconi anemia D2 | FANCD2 | Fanconi anemia, complementation group D2 | 3p26 |
| Fanconi anemia E | FANCE | Fanconi anemia, complementation group E | 6p21-p22 |
| Fanconi anemia F | FANCF | Fanconi anemia, complementation group F | 11p15 |
| Fanconi anemia G | FANCG | Fanconi anemia, complementation group G | 9p13 |
| Fanconi anemia J | BRIP1 | BRCA1 interacting protein C-terminal helicase 1 | 17q22 |
| Fanconi anemia N | PALB2 | Partner and localizer of BRCA2 | 16p12.1 |
| Familial AML | CEBPA | CCAAT/enhancer binding protein (C/EBP), alpha | 19q13.1 |
| Familial platelet disorder with propensity to AML | RUNX1 | Runt-related transcription factor 1 | 21q22.3 |
| Schwachman-Diamond | SBDS | Schwachman-Bodian-Diamond syndrome protein | 7q11 |
| Bloom | BLM | Bloom syndrome | 15q26.1 |
| Li-Fraumeni | TP53 | Tumor protein p53 | 17p13 |
| Down | trisomy 21 | ||
| Ataxia telangiectasia | ATM | Ataxia telangiectasia mutated | 11q22.3 |
Interestingly, studies of families inflicted with multiple myeloid malignancies uncovered rare instances of germline defects in CEBPA or RUNX1, the same genes that are somatically altered in some leukemias.116 The 2008 World Health Organization classification system includes a new category designated as “myeloid leukemia associated with Down syndrome,” defining a group of tumors with distinct clinicopathologic correlates (eg, frequent GATA1 mutation) and altered prognosis compared with sporadic leukemia.
Summary and Future Directions
Genetic technologies are powerful ancillary tools for diagnosing, classifying, and managing acute leukemia. Over 150 different recurrent lesions have been described in AML, and dozens of these influence patient management. The Association for Molecular Pathology Test Directory (http://www.amptestdirectory.org/, accessed July 14, 2009) displays information about testing laboratories, and the following websites (accessed July 14, 2009) also contain reliable information linking cancer genotype and phenotype: http://atlasgeneticsoncology.org (Atlas of Genetics & Cytogenetics in Oncology & Hematology); http://www.ncbi.nlm.nih.gov/Literature/index.html (medical literature); http://www.ncbi.nlm.nih.gov/sites/entrez?db=cancerchromosomes (cancer genetics database of the National Center for Biotechnology Information). On the horizon are pharmacogenetic tests estimating likelihood of response to specific therapy based on knowledge of a patient's germline and/or leukemic genotype. Arrays evaluating dozens to thousands of analytes, when applied to RNA, can survey numerous biochemical pathways by assaying panels of transcripts or miRNAs. When applied to DNA, arrays can simultaneously detect mutations, deletions or amplifications, epigenetic changes, and sometimes even balanced translocations. Prediction scores will emerge as cost-effective strategies for managing reams of disparate clinical and laboratory information. Array results may suggest which additional ancillary test to perform for purposes of identifying a tumor marker to track over time. Finally, arrays hold promise for unraveling the complexity of tumor heterogeneity in a way that drives development of novel therapies and companion assays to predict and track therapeutic efficacy. Classification schemes will regroup patients based on improved understanding of pathobiology and shared response to given interventions. Sensitive genetic tests will evaluate the success of therapy and trigger further intervention when relapse is imminent.
Footnotes
Supported by University of North Carolina Clinical Translational Science Award UL1RR025747 from the National Center for Research Resources and the Department of Pathology and Laboratory Medicine.
This article builds on a presentation from for the Association for Molecular Pathology Companion Meeting at United States and Canadian Academy of Pathology on March 8, 2009.
References
- 1.Swerdlow SH, Campo E, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman J. In: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Swerdlow SH, editor. IARC Press; Lyon, France: 2008. [Google Scholar]
- 2.Wheatley K, Brookes CL, Howman AJ, Goldstone AH, Milligan DW, Prentice AG, Moorman AV, Burnett AK. Prognostic factor analysis of the survival of elderly patients with AML in the MRC AML11 and LRF AML14 trials. Br J Haematol. 2009;145:598–605. doi: 10.1111/j.1365-2141.2009.07663.x. [DOI] [PubMed] [Google Scholar]
- 3.Farag SS, Ruppert AS, Mrozek K, Mayer RJ, Stone RM, Carroll AJ, Powell BL, Moore JO, Pettenati MJ, Koduru PR, Stamberg J, Baer MR, Block AW, Vardiman JW, Kolitz JE, Schiffer CA, Larson RA, Bloomfield CD. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: a cancer and leukemia group B study. J Clin Oncol. 2005;23:482–493. doi: 10.1200/JCO.2005.06.090. [DOI] [PubMed] [Google Scholar]
- 4.Farag SS, Archer KJ, Mrozek K, Ruppert AS, Carroll AJ, Vardiman JW, Pettenati MJ, Baer MR, Qumsiyeh MB, Koduru PR, Ning Y, Mayer RJ, Stone RM, Larson RA, Bloomfield CD. Pretreatment cytogenetics add to other prognostic factors predicting complete remission and long-term outcome in patients 60 years of age or older with acute myeloid leukemia: results from Cancer and Leukemia Group B 8461. Blood. 2006;108:63–73. doi: 10.1182/blood-2005-11-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lange BJ, Smith FO, Feusner J, Barnard DR, Dinndorf P, Feig S, Heerema NA, Arndt C, Arceci RJ, Seibel N, Weiman M, Dusenbery K, Shannon K, Luna-Fineman S, Gerbing RB, Alonzo TA. Outcomes in CCG-2961, a children's oncology group phase 3 trial for untreated pediatric acute myeloid leukemia: a report from the children's oncology group. Blood. 2008;111:1044–1053. doi: 10.1182/blood-2007-04-084293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breems DA, Van Putten WL, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KB, Mellink CH, Nieuwint A, Jotterand M, Hagemeijer A, Beverloo HB, Lowenberg B. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26:4791–4797. doi: 10.1200/JCO.2008.16.0259. [DOI] [PubMed] [Google Scholar]
- 7.Vance GH, Kim H, Hicks GA, Cherry AM, Higgins R, Hulshizer RL, Tallman MS, Fernandez HF, Dewald GW. Utility of interphase FISH to stratify patients into cytogenetic risk categories at diagnosis of AML in an Eastern Cooperative Oncology Group (ECOG) clinical trial (E1900) Leukemia Res. 2007;31:605–609. doi: 10.1016/j.leukres.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 8.Wolff DJ, Bagg A, Cooley LD, Dewald GW, Hirsch BA, Jacky PB, Rao KW, Rao PN. Guidance for fluorescence in situ hybridization testing in hematologic disorders. J Mol Diagn. 2007;9:134–143. doi: 10.2353/jmoldx.2007.060128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diez-Campelo M, Perez-Simon JA, Perez J, Alcoceba M, Richtmon J, Vidriales B, San Miguel J. Minimal residual disease monitoring after allogeneic transplantation may help to individualize post-transplant therapeutic strategies in acute myeloid malignancies. Am J Hematol. 2009;84:149–152. doi: 10.1002/ajh.21340. [DOI] [PubMed] [Google Scholar]
- 10.Picard C, Silvy M, Gabert J. Overview of real-time RT-PCR strategies for quantification of gene rearrangements in the myeloid malignancies. Methods Mol Med. 2006;125:27–68. doi: 10.1385/1-59745-017-0:27. [DOI] [PubMed] [Google Scholar]
- 11.Perea G, Lasa A, Aventin A, Domingo A, Villamor N, Queipo de Llano MP, Llorente A, Junca J, Palacios C, Fernandez C, Gallart M, Font L, Tormo M, Florensa L, Bargay J, Marti JM, Vivancos P, Torres P, Berlanga JJ, Badell I, Brunet S, Sierra J, Nomdedeu JF. Prognostic value of minimal residual disease (MRD) in acute myeloid leukemia (AML) with favorable cytogenetics [t(8;21) and inv(16)] Leukemia. 2006;20:87–94. doi: 10.1038/sj.leu.2404015. [DOI] [PubMed] [Google Scholar]
- 12.Laughlin TS, Becker MW, Liesveld JL, Mulford DA, Abboud CN, Brown P, Rothberg PG. Rapid method for detection of mutations in the nucleophosmin gene in acute myeloid leukemia. J Mol Diagn. 2008;10:338–345. doi: 10.2353/jmoldx.2008.070175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wertheim G, Bagg A. Nucleophosmin (NPM1) mutations in acute myeloid leukemia: an ongoing (cytoplasmic) tale of dueling mutations and duality of molecular genetic testing methodologies. J Mol Diagn. 2008;10:198–202. doi: 10.2353/jmoldx.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, Gottardi E, Rambaldi A, Dotti G, Griesinger F, Parreira A, Gameiro P, Diaz MG, Malec M, Langerak AW, San Miguel JF, Biondi A. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. 1999;13:1901–1928. doi: 10.1038/sj.leu.2401592. [DOI] [PubMed] [Google Scholar]
- 15.Pallisgaard N, Hokland P, Riishoj DC, Pedersen B, Jorgensen P. Multiplex reverse transcription-polymerase chain reaction for simultaneous screening of 29 translocations and chromosomal aberrations in acute leukemia. Blood. 1998;92:574–588. [PubMed] [Google Scholar]
- 16.Beillard E, Pallisgaard N, Van Der Velden VH, Bi W, Dee R, Van Der Schoot E, Delabesse E, Macintyre E, Gottardi E, Saglio G, Watzinger F, Lion T, Van Dongen JJ, Hokland P, Gabert J. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using “real-time” quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR)—a Europe Against Cancer program. Leukemia. 2003;17:2474–2486. doi: 10.1038/sj.leu.2403136. [DOI] [PubMed] [Google Scholar]
- 17.Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cave H, Pane F, Aerts JL, De Micheli D, Thirion X, Pradel V, Gonzalez M, Viehmann S, Malec M, Saglio G, van Dongen JJ. Standardization and quality control studies of “real-time” quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—a Europe Against Cancer program. Leukemia. 2003;17:2318–2357. doi: 10.1038/sj.leu.2403135. [DOI] [PubMed] [Google Scholar]
- 18.Grimwade D. The clinical significance of cytogenetic abnormalities in acute myeloid leukaemia. Best Pract Res Clin Haematol. 2001;14:497–529. doi: 10.1053/beha.2001.0152. [DOI] [PubMed] [Google Scholar]
- 19.Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, Paietta E, Willman CL, Head DR, Rowe JM, Forman SJ, Appelbaum FR. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–4083. [PubMed] [Google Scholar]
- 20.Mrozek K, Marcucci G, Paschka P, Bloomfield CD. Advances in molecular genetics and treatment of core-binding factor acute myeloid leukemia. Curr Opin Oncol. 2008;20:711–718. doi: 10.1097/CCO.0b013e32831369df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcucci G, Mrozek K, Ruppert AS, Maharry K, Kolitz JE, Moore JO, Mayer RJ, Pettenati MJ, Powell BL, Edwards CG, Sterling LJ, Vardiman JW, Schiffer CA, Carroll AJ, Larson RA, Bloomfield CD. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:5705–5717. doi: 10.1200/JCO.2005.15.610. [DOI] [PubMed] [Google Scholar]
- 22.Haferlach T, Kern W, Schoch C, Schnittger S, Sauerland MC, Heinecke A, Buchner T, Hiddemann W. A new prognostic score for patients with acute myeloid leukemia based on cytogenetics and early blast clearance in trials of the German AML Cooperative Group. Haematologica. 2004;89:408–418. [PubMed] [Google Scholar]
- 23.Schoch C, Schnittger S, Klaus M, Kern W, Hiddemann W, Haferlach T. AML with 11q23/MLL abnormalities as defined by the WHO classification: incidence, partner chromosomes, FAB subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed AML cases. Blood. 2003;102:2395–2402. doi: 10.1182/blood-2003-02-0434. [DOI] [PubMed] [Google Scholar]
- 24.Grimwade D, Walker H, Harrison G, Oliver F, Chatters S, Harrison CJ, Wheatley K, Burnett AK, Goldstone AH. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001;98:1312–1320. doi: 10.1182/blood.v98.5.1312. [DOI] [PubMed] [Google Scholar]
- 25.Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–6295. doi: 10.1200/JCO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Anonymous . NCCN Acute Myelogenous Leukemia Clinical Practice Guidelines in Oncology (Version 2009.1) Edited by National Comprehensive Cancer Network, Inc.; 2009. [Google Scholar]
- 27.Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, Pettenati MJ, Patil SR, Rao KW, Watson MS, Koduru PR, Moore JO, Stone RM, Mayer RJ, Feldman EJ, Davey FR, Schiffer CA, Larson RA, Bloomfield CD. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 28.Iqbal S, Grimwade D, Chase A, Goldstone A, Burnett A, Goldman JM, Swirsky D. Identification of PML/RARalpha rearrangements in suspected acute promyelocytic leukemia using fluorescence in situ hybridization of bone marrow smears: a comparison with cytogenetics and RT-PCR in MRC ATRA trial patients: MRC Adult Leukaemia Working Party. Leukemia. 2000;14:950–953. doi: 10.1038/sj.leu.2401688. [DOI] [PubMed] [Google Scholar]
- 29.Mrozek K, Prior TW, Edwards C, Marcucci G, Carroll AJ, Snyder PJ, Koduru PR, Theil KS, Pettenati MJ, Archer KJ, Caligiuri MA, Vardiman JW, Kolitz JE, Larson RA, Bloomfield CD. Comparison of cytogenetic and molecular genetic detection of t(8;21) and inv(16) in a prospective series of adults with de novo acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol. 2001;19:2482–2492. doi: 10.1200/JCO.2001.19.9.2482. [DOI] [PubMed] [Google Scholar]
- 30.Whitman SP, Liu S, Vukosavljevic T, Rush LJ, Yu L, Liu C, Klisovic MI, Maharry K, Guimond M, Strout MP, Becknell B, Dorrance A, Klisovic RB, Plass C, Bloomfield CD, Marcucci G, Caligiuri MA. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood. 2005;106:345–352. doi: 10.1182/blood-2005-01-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basecke J, Whelan JT, Griesinger F, Bertrand FE. The MLL partial tandem duplication in acute myeloid leukaemia. Br J Haematol. 2006;135:438–449. doi: 10.1111/j.1365-2141.2006.06301.x. [DOI] [PubMed] [Google Scholar]
- 32.De Braekeleer M, Morel F, Le Bris MJ, Herry A, Douet-Guilbert N. The MLL gene and translocations involving chromosomal band 11q23 in acute leukemia. Anticancer Res. 2005;25:1931–1944. [PubMed] [Google Scholar]
- 33.Krauter J, Wagner K, Schafer I, Marschalek R, Meyer C, Heil G, Schaich M, Ehninger G, Niederwieser D, Krahl R, Buchner T, Sauerland C, Schlegelberger B, Dohner K, Dohner H, Schlenk RF, Ganser A. Prognostic factors in adult patients up to 60 years old with acute myeloid leukemia and translocations of chromosome band 11q23: individual patient data-based meta-analysis of the German Acute Myeloid Leukemia Intergroup. J Clin Oncol. 2009;27:3000–3006. doi: 10.1200/JCO.2008.16.7981. [DOI] [PubMed] [Google Scholar]
- 34.Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, Ben Abdelali R, Macintyre E, De Braekeleer E, De Braekeleer M, Delabesse E, de Oliveira MP, Cave H, Clappier E, van Dongen JJ, Balgobind BV, van den Heuvel-Eibrink MM, Beverloo HB, Panzer-Grumayer R, Teigler-Schlegel A, Harbott J, Kjeldsen E, Schnittger S, Koehl U, Gruhn B, Heidenreich O, Chan LC, Yip SF, Krzywinski M, Eckert C, Moricke A, Schrappe M, Alonso CN, Schafer BW, Krauter J, Lee DA, Zur Stadt U, Te Kronnie G, Sutton R, Izraeli S, Trakhtenbrot L, Lo Nigro L, Tsaur G, Fechina L, Szczepanski T, Strehl S, Ilencikova D, Molkentin M, Burmeister T, Dingermann T, Klingebiel T, Marschalek R. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–1499. doi: 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- 35.Kondo T, Mori A, Darmanin S, Hashino S, Tanaka J, Asaka M. The seventh pathogenic fusion gene FIP1L1-RARA was isolated from a t(4;17)-positive acute promyelocytic leukemia. Haematologica. 2008;93:1414–1416. doi: 10.3324/haematol.12854. [DOI] [PubMed] [Google Scholar]
- 36.Catalano A, Dawson MA, Somana K, Opat S, Schwarer A, Campbell LJ, Iland H. The PRKAR1A gene is fused to RARA in a new variant acute promyelocytic leukemia. Blood. 2007;110:4073–4076. doi: 10.1182/blood-2007-06-095554. [DOI] [PubMed] [Google Scholar]
- 37.Zelent A, Guidez F, Melnick A, Waxman S, Licht JD. Translocations of the RARalpha gene in acute promyelocytic leukemia. Oncogene. 2001;20:7186–7203. doi: 10.1038/sj.onc.1204766. [DOI] [PubMed] [Google Scholar]
- 38.Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, Naoe T, Lengfelder E, Buchner T, Dohner H, Burnett AK, Lo-Coco F. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113:1875–1891. doi: 10.1182/blood-2008-04-150250. [DOI] [PubMed] [Google Scholar]
- 39.Gallagher RE, Willman CL, Slack JL, Andersen JW, Li YP, Viswanatha D, Bloomfield CD, Appelbaum FR, Schiffer CA, Tallman MS, Wiernik PH. Association of PML-RAR alpha fusion mRNA type with pretreatment hematologic characteristics but not treatment outcome in acute promyelocytic leukemia: an intergroup molecular study. Blood. 1997;90:1656–1663. [PubMed] [Google Scholar]
- 40.Rennert H, Golde T, Wilson RB, Spitalnik SL, Van Deerlin VM, Leonard DG. A novel, non-nested reverse-transcriptase polymerase chain reaction (RT-PCR) test for the detection of the t(15;17) translocation: a comparative study of RT-PCR cytogenetics, and fluorescence in situ hybridization. Mol Diagn. 1999;4:195–209. doi: 10.1016/s1084-8592(99)80023-x. [DOI] [PubMed] [Google Scholar]
- 41.Grimwade D, Biondi A, Mozziconacci MJ, Hagemeijer A, Berger R, Neat M, Howe K, Dastugue N, Jansen J, Radford-Weiss I, Lo Coco F, Lessard M, Hernandez JM, Delabesse E, Head D, Liso V, Sainty D, Flandrin G, Solomon E, Birg F, Lafage-Pochitaloff M. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party Groupe Francais de Cytogenetique Hematologique, Groupe de Francais d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action “Molecular Cytogenetic Diagnosis in Haematological Malignancies.”. Blood. 2000;96:1297–1308. [PubMed] [Google Scholar]
- 42.Diverio D, Rossi V, Avvisati G, De Santis S, Pistilli A, Pane F, Saglio G, Martinelli G, Petti MC, Santoro A, Pelicci PG, Mandelli F, Biondi A, Lo Coco F. Early detection of relapse by prospective reverse transcriptase-polymerase chain reaction analysis of the PML/RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter “AIDA” trial: GIMEMA-AIEOP Multicenter “AIDA” Trial. Blood. 1998;92:784–789. [PubMed] [Google Scholar]
- 43.Tallman MS, Altman JK. Curative strategies in acute promyelocytic leukemia. Hematology Am Soc Hematol Edu Program. 2008;2008:391–399. doi: 10.1182/asheducation-2008.1.391. [DOI] [PubMed] [Google Scholar]
- 44.Milligan DW, Grimwade D, Cullis JO, Bond L, Swirsky D, Craddock C, Kell J, Homewood J, Campbell K, McGinley S, Wheatley K, Jackson G. Guidelines on the management of acute myeloid leukaemia in adults. Br J Haematol. 2006;135:450–474. doi: 10.1111/j.1365-2141.2006.06314.x. [DOI] [PubMed] [Google Scholar]
- 45.Appelbaum FR, Baer MR, Carabasi MH, Coutre SE, Erba HP, Estey E, Glenn MJ, Kraut EH, Maslak P, Millenson M, Miller CB, Saba HI, Stone R, Tallman MS. NCCN Practice Guidelines for Acute Myelogenous Leukemia. Oncology (Williston Park) 2000;14:53–61. [PubMed] [Google Scholar]
- 46.Gallagher RE. Real-time consensus on relapse risk in acute promyelocytic leukemia. Leukemia Res. 2009;33:1170–1172. doi: 10.1016/j.leukres.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grimwade D, Jovanovic JV, Hills RK, Nugent EA, Patel Y, Flora R, Diverio D, Jones K, Aslett H, Batson E, Rennie K, Angell R, Clark RE, Solomon E, Lo-Coco F, Wheatley K, Burnett AK. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol. 2009 doi: 10.1200/JCO.2008.20.1533. [DOI] [PubMed] [Google Scholar]
- 48.Mrozek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–448. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dohner H. Implication of the molecular characterization of acute myeloid leukemia. Hematology Am Soc Hematol Edu Program. 2007:412–419. doi: 10.1182/asheducation-2007.1.412. [DOI] [PubMed] [Google Scholar]
- 50.Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–198. doi: 10.1146/annurev.genom.3.032802.115046. [DOI] [PubMed] [Google Scholar]
- 51.Ishikawa Y, Kiyoi H, Tsujimura A, Miyawaki S, Miyazaki Y, Kuriyama K, Tomonaga M, Naoe T. Comprehensive analysis of cooperative gene mutations between class I and class II in de novo acute myeloid leukemia. Eur J Haematol. 2009;83:90–98. doi: 10.1111/j.1600-0609.2009.01261.x. [DOI] [PubMed] [Google Scholar]
- 52.Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P, Preudhomme C. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008;22:915–931. doi: 10.1038/leu.2008.19. [DOI] [PubMed] [Google Scholar]
- 53.Scholl S, Theuer C, Scheble V, Kunert C, Heller A, Mugge LO, Fricke HJ, Hoffken K, Wedding U. Clinical impact of nucleophosmin mutations and Flt3 internal tandem duplications in patients older than 60 yr with acute myeloid leukaemia. Eur J Haematol. 2008;80:208–215. doi: 10.1111/j.1600-0609.2007.01019.x. [DOI] [PubMed] [Google Scholar]
- 54.Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, Habdank M, Spath D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Dohner H. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 55.Bacher U, Haferlach C, Schnittger S, Kern W, Kroeger N, Zander AR, Haferlach T. Interactive diagnostics in the indication to allogeneic SCT in AML. Bone Marrow Transplant. 2009;43:745–756. doi: 10.1038/bmt.2009.54. [DOI] [PubMed] [Google Scholar]
- 56.Gale RE, Hills R, Pizzey AR, Kottaridis PD, Swirsky D, Gilkes AF, Nugent E, Mills KI, Wheatley K, Solomon E, Burnett AK, Linch DC, Grimwade D. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood. 2005;106:3768–3776. doi: 10.1182/blood-2005-04-1746. [DOI] [PubMed] [Google Scholar]
- 57.Yoo SJ, Park CJ, Jang S, Seo EJ, Lee KH, Chi HS. Inferior prognostic outcome in acute promyelocytic leukemia with alterations of FLT3 gene. Leukemia Lymphoma. 2006;47:1788–1793. doi: 10.1080/10428190600687927. [DOI] [PubMed] [Google Scholar]
- 58.Callens C, Chevret S, Cayuela JM, Cassinat B, Raffoux E, de Botton S, Thomas X, Guerci A, Fegueux N, Pigneux A, Stoppa AM, Lamy T, Rigal-Huguet F, Vekhoff A, Meyer-Monard S, Ferrand A, Sanz M, Chomienne C, Fenaux P, Dombret H. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia. 2005;19:1153–1160. doi: 10.1038/sj.leu.2403790. [DOI] [PubMed] [Google Scholar]
- 59.Murphy KM, Levis M, Hafez MJ, Geiger T, Cooper LC, Smith BD, Small D, Berg KD. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn. 2003;5:96–102. doi: 10.1016/S1525-1578(10)60458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, Linch DC. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–2784. doi: 10.1182/blood-2007-08-109090. [DOI] [PubMed] [Google Scholar]
- 61.Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters—an analysis of 3082 patients. Blood. 2008;111:2527–2537. doi: 10.1182/blood-2007-05-091215. [DOI] [PubMed] [Google Scholar]
- 62.Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007;110:1262–1270. doi: 10.1182/blood-2006-04-015826. [DOI] [PubMed] [Google Scholar]
- 63.Reindl C, Bagrintseva K, Vempati S, Schnittger S, Ellwart JW, Wenig K, Hopfner KP, Hiddemann W, Spiekermann K. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood. 2006;107:3700–3707. doi: 10.1182/blood-2005-06-2596. [DOI] [PubMed] [Google Scholar]
- 64.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, La Starza R, Diverio D, Colombo E, Santucci A, Bigerna B, Pacini R, Pucciarini A, Liso A, Vignetti M, Fazi P, Meani N, Pettirossi V, Saglio G, Mandelli F, Lo-Coco F, Pelicci PG, Martelli MF. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 65.Liso A, Bogliolo A, Freschi V, Martelli MP, Pileri SA, Santodirocco M, Bolli N, Martelli MF, Falini B. In human genome, generation of a nuclear export signal through duplication appears unique to nucleophosmin (NPM1) mutations and is restricted to AML. Leukemia. 2008;22:1285–1289. doi: 10.1038/sj.leu.2405045. [DOI] [PubMed] [Google Scholar]
- 66.Haferlach C, Mecucci C, Schnittger S, Kohlmann A, Mancini M, Cuneo A, Testoni N, Rege-Cambrin G, Santucci A, Vignetti M, Fazi P, Martelli MP, Haferlach T, Falini B. AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biological, pathological, immunophenotypic, and prognostic features. Blood. 2009;114:3024–3032. doi: 10.1182/blood-2009-01-197871. [DOI] [PubMed] [Google Scholar]
- 67.Brown P, McIntyre E, Rau R, Meshinchi S, Lacayo N, Dahl G, Alonzo TA, Chang M, Arceci RJ, Small D. The incidence and clinical significance of nucleophosmin mutations in childhood AML. Blood. 2007;110:979–985. doi: 10.1182/blood-2007-02-076604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hollink IH, Zwaan CM, Zimmermann M, Arentsen-Peters TC, Pieters R, Cloos J, Kaspers GJ, de Graaf SS, Harbott J, Creutzig U, Reinhardt D, van den Heuvel-Eibrink MM, Thiede C. Favorable prognostic impact of NPM1 gene mutations in childhood acute myeloid leukemia, with emphasis on cytogenetically normal AML. Leukemia. 2009;23:262–270. doi: 10.1038/leu.2008.313. [DOI] [PubMed] [Google Scholar]
- 69.Schlenk RF, Dohner K, Kneba M, Gotze K, Hartmann F, Del Valle F, Kirchen H, Koller E, Fischer JT, Bullinger L, Habdank M, Spath D, Groner S, Krebs B, Kayser S, Corbacioglu A, Anhalt A, Benner A, Frohling S, Dohner H. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia: results from the AMLSG Trial AML HD98B. Haematologica. 2009;94:54–60. doi: 10.3324/haematol.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen W, Rassidakis GZ, Medeiros LJ. Nucleophosmin gene mutations in acute myeloid leukemia. Arch Pathol Lab Med. 2006;130:1687–1692. doi: 10.5858/2006-130-1687-NGMIAM. [DOI] [PubMed] [Google Scholar]
- 71.Roti G, Rosati R, Bonasso R, Gorello P, Diverio D, Martelli MF, Falini B, Mecucci C. Denaturing high-performance liquid chromatography: a valid approach for identifying NPM1 mutations in acute myeloid leukemia. J Mol Diagn. 2006;8:254–259. doi: 10.2353/jmoldx.2006.050098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Calvo KL, Jorgelina Ojeda M, Ammatuna E, Lavorgna S, Ottone T, Manuel Targovnik H, Lo-Coco F, Ines Noguera N. Detection of the nucleophosmin gene mutations in acute myelogenous leukemia through RT-PCR and polyacrylamide gel electrophoresis. Eur J Haematol. 2009;82:69–72. doi: 10.1111/j.1600-0609.2008.01155.x. [DOI] [PubMed] [Google Scholar]
- 73.Papadaki C, Dufour A, Seibl M, Schneider S, Bohlander SK, Zellmeier E, Mellert G, Hiddemann W, Spiekermann K. Monitoring minimal residual disease in acute myeloid leukaemia with NPM1 mutations by quantitative PCR: clonal evolution is a limiting factor. Br J Haematol. 2009;144:517–523. doi: 10.1111/j.1365-2141.2008.07488.x. [DOI] [PubMed] [Google Scholar]
- 74.Gorello P, Cazzaniga G, Alberti F, Dell'Oro MG, Gottardi E, Specchia G, Roti G, Rosati R, Martelli MF, Diverio D, Lo Coco F, Biondi A, Saglio G, Mecucci C, Falini B. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia. 2006;20:1103–1108. doi: 10.1038/sj.leu.2404149. [DOI] [PubMed] [Google Scholar]
- 75.Ottone T, Ammatuna E, Lavorgna S, Noguera NI, Buccisano F, Venditti A, Gianni L, Postorino M, Federici G, Amadori S, Lo-Coco F. An allele-specific rt-PCR assay to detect type A mutation of the nucleophosmin-1 gene in acute myeloid leukemia. J Mol Diagn. 2008;10:212–216. doi: 10.2353/jmoldx.2008.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ho PA, Alonzo TA, Gerbing RB, Pollard J, Stirewalt DL, Hurwitz C, Heerema NA, Hirsch B, Raimondi SC, Lange B, Franklin JL, Radich JP, Meshinchi S. Prevalence and prognostic implications of CEBPA mutations in pediatric AML: a report from the Children's Oncology Group. Blood. 2009;113:6558–6566. doi: 10.1182/blood-2008-10-184747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Renneville A, Boissel N, Gachard N, Naguib D, Bastard C, de Botton S, Nibourel O, Pautas C, Reman O, Thomas X, Gardin C, Terre C, Castaigne S, Preudhomme C, Dombret H. The favorable impact of CEBPA mutations in patients with acute myeloid leukemia is only observed in the absence of associated cytogenetic abnormalities and FLT3 internal duplication. Blood. 2009;113:5090–5093. doi: 10.1182/blood-2008-12-194704. [DOI] [PubMed] [Google Scholar]
- 79.Hackanson B, Bennett KL, Brena RM, Jiang J, Claus R, Chen SS, Blagitko-Dorfs N, Maharry K, Whitman SP, Schmittgen TD, Lubbert M, Marcucci G, Bloomfield CD, Plass C. Epigenetic modification of CCAAT/enhancer binding protein alpha expression in acute myeloid leukemia. Cancer Res. 2008;68:3142–3151. doi: 10.1158/0008-5472.CAN-08-0483. [DOI] [PubMed] [Google Scholar]
- 80.Marcucci G, Maharry K, Radmacher MD, Mrozek K, Vukosavljevic T, Paschka P, Whitman SP, Langer C, Baldus CD, Liu CG, Ruppert AS, Powell BL, Carroll AJ, Caligiuri MA, Kolitz JE, Larson RA, Bloomfield CD. Prognostic significance of, and gene and microRNA expression signatures associated with. CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. J Clin Oncol. 2008;26:5078–5087. doi: 10.1200/JCO.2008.17.5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27:619–628. doi: 10.1200/JCO.2008.17.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Santamaria CM, Chillon MC, Garcia-Sanz R, Perez C, Caballero MD, Ramos F, Garcia de Coca A, Alonso JM, Giraldo P, Bernal T, Queizan JA, Rodriguez JN, Fernandez-Abellan P, Barez A, Penarrubia MJ, Balanzategui A, Vidriales MB, Sarasquete ME, Alcoceba M, Diaz-Mediavilla J, San Miguel JF, Gonzalez M. Molecular stratification model for prognosis in cytogenetically normal acute myeloid leukemia (CN-AML) Blood. 2009;114:148–152. doi: 10.1182/blood-2008-11-187724. [DOI] [PubMed] [Google Scholar]
- 83.Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, Chen P, Wang Y, Yan M, Qian Z, Neilly MB, Jin J, Zhang Y, Bohlander SK, Zhang DE, Larson RA, Le Beau MM, Thirman MJ, Golub TR, Rowley JD, Chen J. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci USA. 2008;105:15535–15540. doi: 10.1073/pnas.0808266105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marcucci G, Radmacher MD, Maharry K, Mrozek K, Ruppert AS, Paschka P, Vukosavljevic T, Whitman SP, Baldus CD, Langer C, Liu CG, Carroll AJ, Powell BL, Garzon R, Croce CM, Kolitz JE, Caligiuri MA, Larson RA, Bloomfield CD. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1919–1928. doi: 10.1056/NEJMoa074256. [DOI] [PubMed] [Google Scholar]
- 85.Lowenberg B. Acute myeloid leukemia: the challenge of capturing disease variety. Hematology Am Soc Hematol Edu Program. 2008;2008:1–11. doi: 10.1182/asheducation-2008.1.1. [DOI] [PubMed] [Google Scholar]
- 86.Sritana N, Auewarakul CU. KIT and FLT3 receptor tyrosine kinase mutations in acute myeloid leukemia with favorable cytogenetics: two novel mutations and selective occurrence in leukemia subtypes and age groups. Exp Mol Pathol. 2008;85:227–231. doi: 10.1016/j.yexmp.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 87.Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, Cohen DP, Baum CM, Demetri GD. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–5359. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ustun C, Corless CL, Savage N, Fiskus W, Manaloor E, Heinrich MC, Lewis G, Ramalingam P, Kepten I, Jillella A, Bhalla K. Chemotherapy and dasatinib induce long-term hematologic and molecular remission in systemic mastocytosis with acute myeloid leukemia with KIT D816V. Leukemia Res. 2009;33:735–741. doi: 10.1016/j.leukres.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 89.Schumacher JA, Elenitoba-Johnson KS, Lim MS. Detection of the c-kit D816V mutation in systemic mastocytosis by allele-specific PCR. J Clin Pathol. 2008;61:109–114. doi: 10.1136/jcp.2007.047928. [DOI] [PubMed] [Google Scholar]
- 90.Tyner JW, Erickson H, Deininger MW, Willis SG, Eide CA, Levine RL, Heinrich MC, Gattermann N, Gilliland DG, Druker BJ, Loriaux MM. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113:1749–1755. doi: 10.1182/blood-2008-04-152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Paulsson K, Horvat A, Strombeck B, Nilsson F, Heldrup J, Behrendtz M, Forestier E, Andersson A, Fioretos T, Johansson B. Mutations of FLT3. NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2008;47:26–33. doi: 10.1002/gcc.20502. [DOI] [PubMed] [Google Scholar]
- 92.Shih LY, Liang DC, Huang CF, Chang YT, Lai CL, Lin TH, Yang CP, Hung IJ, Liu HC, Jaing TH, Wang LY, Yeh TC. Cooperating mutations of receptor tyrosine kinases and Ras genes in childhood core-binding factor acute myeloid leukemia and a comparative analysis on paired diagnosis and relapse samples. Leukemia. 2008;22:303–307. doi: 10.1038/sj.leu.2404995. [DOI] [PubMed] [Google Scholar]
- 93.Neubauer A, Maharry K, Mrozek K, Thiede C, Marcucci G, Paschka P, Mayer RJ, Larson RA, Liu ET, Bloomfield CD. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: a Cancer and Leukemia Group B study. J Clin Oncol. 2008;26:4603–4609. doi: 10.1200/JCO.2007.14.0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Raponi M, Lancet JE, Fan H, Dossey L, Lee G, Gojo I, Feldman EJ, Gotlib J, Morris LE, Greenberg PL, Wright JJ, Harousseau JL, Lowenberg B, Stone RM, De Porre P, Wang Y, Karp JE. A 2-gene classifier for predicting response to the farnesyltransferase inhibitor tipifarnib in acute myeloid leukemia. Blood. 2008;111:2589–2596. doi: 10.1182/blood-2007-09-112730. [DOI] [PubMed] [Google Scholar]
- 95.Mrozek K, Radmacher MD, Bloomfield CD, Marcucci G. Molecular signatures in acute myeloid leukemia. Curr Opin Hematol. 2009;16:64–69. doi: 10.1097/MOH.0b013e3283257b42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wouters BJ, Lowenberg B, Delwel R. A decade of genome-wide gene expression profiling in acute myeloid leukemia: flashback and prospects. Blood. 2009;113:291–298. doi: 10.1182/blood-2008-04-153239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Verhaak RG, Wouters BJ, Erpelinck CA, Abbas S, Beverloo HB, Lugthart S, Lowenberg B, Delwel R, Valk PJ. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94:131–134. doi: 10.3324/haematol.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bacher U, Kohlmann A, Haferlach T. Current status of gene expression profiling in the diagnosis and management of acute leukaemia. Br J Haematol. 2009;145:555–568. doi: 10.1111/j.1365-2141.2009.07656.x. [DOI] [PubMed] [Google Scholar]
- 99.Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, Dohner H, Pollack JR. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- 100.Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, Maharry K, Paschka P, Larson RA, Berdel WE, Buchner T, Wormann B, Mansmann U, Hiddemann W, Bohlander SK, Buske C. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–4201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garzon R, Volinia S, Liu CG, Fernandez-Cymering C, Palumbo T, Pichiorri F, Fabbri M, Coombes K, Alder H, Nakamura T, Flomenberg N, Marcucci G, Calin GA, Kornblau SM, Kantarjian H, Bloomfield CD, Andreeff M, Croce CM. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008;111:3183–3189. doi: 10.1182/blood-2007-07-098749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C, Volinia S, Liu CG, Schnittger S, Haferlach T, Liso A, Diverio D, Mancini M, Meloni G, Foa R, Martelli MF, Mecucci C, Croce CM, Falini B. Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA. 2008;105:3945–3950. doi: 10.1073/pnas.0800135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Skotheim RI, Thomassen GO, Eken M, Lind GE, Micci F, Ribeiro FR, Cerveira N, Teixeira MR, Heim S, Rognes T, Lothe RA. A universal assay for detection of oncogenic fusion transcripts by oligo microarray analysis. Mol Cancer. 2009;8:5. doi: 10.1186/1476-4598-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Suela J, Alvarez S, Cigudosa JC. DNA profiling by arrayCGH in acute myeloid leukemia and myelodysplastic syndromes. Cytogenet Genome Res. 2007;118:304–309. doi: 10.1159/000108314. [DOI] [PubMed] [Google Scholar]
- 105.Watson SK, deLeeuw RJ, Horsman DE, Squire JA, Lam WL. Cytogenetically balanced translocations are associated with focal copy number alterations. Hum Genet. 2007;120:795–805. doi: 10.1007/s00439-006-0251-9. [DOI] [PubMed] [Google Scholar]
- 106.Dunbar AJ, Gondek LP, O'Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008;68:10349–10357. doi: 10.1158/0008-5472.CAN-08-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, Cook L, Abbott R, Larson DE, Koboldt DC, Pohl C, Smith S, Hawkins A, Abbott S, Locke D, Hillier LW, Miner T, Fulton L, Magrini V, Wylie T, Glasscock J, Conyers J, Sander N, Shi X, Osborne JR, Minx P, Gordon D, Chinwalla A, Zhao Y, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson M, Baty J, Ivanovich J, Heath S, Shannon WD, Nagarajan R, Walter MJ, Link DC, Graubert TA, DiPersio JF, Wilson RK. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cornelissen JJ, van Putten WL, Verdonck LF, Theobald M, Jacky E, Daenen SM, van Marwijk Kooy M, Wijermans P, Schouten H, Huijgens PC, van der Lelie H, Fey M, Ferrant A, Maertens J, Gratwohl A, Lowenberg B. Results of a HOVON/SAKK donor versus no-donor analysis of myeloablative HLA-identical sibling stem cell transplantation in first remission acute myeloid leukemia in young and middle-aged adults: benefits for whom? Blood. 2007;109:3658–3666. doi: 10.1182/blood-2006-06-025627. [DOI] [PubMed] [Google Scholar]
- 109.Meijer E, Cornelissen JJ. Allogeneic stem cell transplantation in acute myeloid leukemia in first or subsequent remission: weighing prognostic markers predicting relapse and risk factors for non-relapse mortality. Semin Oncol. 2008;35:449–457. doi: 10.1053/j.seminoncol.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 110.Koreth J, Schlenk R, Kopecky KJ, Honda S, Sierra J, Djulbegovic BJ, Wadleigh M, DeAngelo DJ, Stone RM, Sakamaki H, Appelbaum FR, Dohner H, Antin JH, Soiffer RJ, Cutler C. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta-analysis of prospective clinical trials. JAMA. 2009;301:2349–2361. doi: 10.1001/jama.2009.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schlenk RF, Dohner K. Impact of new prognostic markers in treatment decisions in acute myeloid leukemia. Curr Opin Hematol. 2009;16:98–104. doi: 10.1097/MOH.0b013e3283257adb. [DOI] [PubMed] [Google Scholar]
- 112.Freeman SD, Jovanovic JV, Grimwade D. Development of minimal residual disease-directed therapy in acute myeloid leukemia. Semin Oncol. 2008;35:388–400. doi: 10.1053/j.seminoncol.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 113.Jennings L, VanDeerlin VM, Gulley ML. Recommended principles and practices for validating clinical molecular pathology tests. Arch Pathol Lab Med. 2009;133:743–755. doi: 10.5858/133.5.743. [DOI] [PubMed] [Google Scholar]
- 114.Gulley ML, Braziel RM, Halling KC, Hsi ED, Kant JA, Nikiforova MN, Nowak JA, Ogino S, Oliveira A, Polesky HF, Silverman L, Tubbs RR, Van Deerlin VM, Vance GH, Versalovic J. Clinical laboratory reports in molecular pathology. Arch Pathol Lab Med. 2007;131:852–863. doi: 10.5858/2007-131-852-CLRIMP. [DOI] [PubMed] [Google Scholar]
- 115.Seedhouse C, Russell N. Advances in the understanding of susceptibility to treatment-related acute myeloid leukaemia. Br J Haematol. 2007;137:513–529. doi: 10.1111/j.1365-2141.2007.06613.x. [DOI] [PubMed] [Google Scholar]
- 116.Owen C, Barnett M, Fitzgibbon J. Familial myelodysplasia and acute myeloid leukaemia: a review. Br J Haematol. 2008;140:123–132. doi: 10.1111/j.1365-2141.2007.06909.x. [DOI] [PubMed] [Google Scholar]