

Abstract
Duchenne and Becker muscular dystrophies are caused by a large number of different mutations in the dystrophin gene. Outside of the deletion/duplication “hot spots,” small mutations occur at unpredictable positions. These account for about 15 to 20% of cases, with the major group being premature stop codons. When the affected male is deceased, carrier testing for family members and prenatal diagnosis become difficult and expensive. We tailored a cost-effective and reliable strategy to discover point mutations from stored DNA samples in the absence of a muscle biopsy. Samples were amplified in combinatorial pools and tested by denaturing high-performance liquid chromatography analysis. An anomalous elution profile belonging to two different pools univocally addressed the allelic variation to an unambiguous sample. Mutations were then detected by sequencing. We identified 121 mutations of 99 different types. Fifty-six patients show stop codons that represent the 46.3% of all cases. Three non-obvious single amino acid mutations were considered as causative. Our data support combinatorial denaturing high-performance liquid chromatography analysis as a clear-cut strategy for time and cost-effective identification of small mutations when only DNA is available.
Duchenne (DMD [MIM 310200]) and Becker muscular dystrophies (BMD [MIM 300376]) are allelic inherited disorders of muscle. They affect males in >99% of cases, being transmitted as X-linked recessive traits.1 The DMD gene spans 2.2 million bp of genomic DNA on the X chromosome, and the 14-kb transcript encodes a full-length protein (dystrophin) of 427 kd (Dp427m). Both DMD and BMD arise due to mutations at the dystrophin gene locus, which comprises 79 exons and eight tissue-specific promoters. The most common mutations are large intragenic deletions or duplications, encompassing one or more exons, but point mutations are about 15 to 20% of cases, with the major group being premature stop codons.2,3,4,5,6,7,8,9
Patients and their families confer great value to mutation detection for genetic counseling, but also for therapeutic options, since there are claims of novel mutation-targeted treatments.10,11,12 Unfortunately, very often muscle biopsies are not possible because the affected family member is deceased. We have tailored a cost-effective and reliable strategy to discover point mutations from DNA samples. Based on the sensitivity of denaturing high-performance liquid chromatography (DHPLC) to detect mutations, especially in A/T-rich sequences, such as the dystrophin gene,6,7 we developed a combinatorial DHPLC approach to screen pooled samples.
Materials and Methods
Patients
We used archive DNA samples from six different centers: Laboratory of Molecular Biology, Scientific Institute E. Medea, Lecco; Department of Neurological and Psychiatric Sciences, University of Padua; Institute of Neurology, Catholic University, Policlinico Gemelli, Rome; Muscular and Neurodegenerative Disease Unit, Giannina Gaslini Institute, University of Genova; Department of Experimental Medicine, Cardiomyology and Medical Genetics, Second University, Naples; and Centro de Estudos do Genoma Humano, Instituto de Biociências Universidade de São Paulo, Brasil. Diagnosis was determined by clinical features consistent with DMD or BMD, along with an X-linked family history. Informed consent was obtained from patients, when possible, according to the guidelines of Eurobiobank or Telethon.
Archive Samples
One hundred fifty-three DNA archive samples were stored in Tris-EDTA at 4°C. Fifteen were extracted by phenol-chloroform before 1994, whereas 31 were extracted from 1994 to1999, and 46 from 2000 to 2004 (Figure 1). More recent samples (from 2005 to 2007) were extracted using a FlexiGene DNA kit (Qiagen, Hamburg, Germany). Old samples were often recovered as dry pellets. In this case, we rehydrated the pellet. We evaluated the DNA integrity by 0.6% agarose gel electrophoresis. We did not re-precipitate any of the samples. When required, we performed a preamplification step using the GenomiPhi HY DNA amplification kit (GE Healthcare, Chalfont St. Giles, UK), according to the manufacturer's instruction. This kit provides microgram quantities of DNA from nanogram amounts of starting material in only a few hours. The limit of polymerase chain reaction (PCR) product size using this archived DNA was about 1000 bp.
Figure 1.
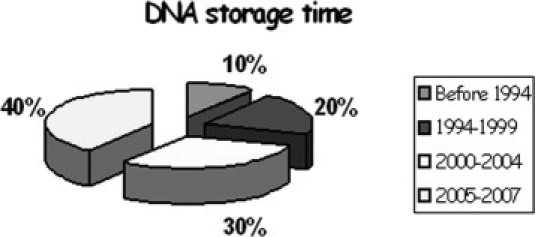
Extraction dates of DNA samples.
Sample Optimization
Each DNA sample was diluted to a final concentration of 30 ng/μl, and 1 μl was used in each pool. To control for the possibility of unequal PCR product yield, short tandem repeat (STR) polymorphic markers DXS8015-HEX and DXS1204-FAM (Table 1) were amplified from single and pooled DNA templates, in a final reaction volume of 20 μl, by using 0.5 μmol/L each marker primer, buffer LB 10× [200 mmol/L Tris; 100 mmol/L Hepes; 25 mmol/L MgSO4 × 7 H2O; 100 nm KCl; 100 mmol/L (NH4)2 SO4], 0.25 μmol/L each dNTP, 0.5 U AmpliTaq Gold (Applied Biosystems, Foster City, CA).
Table 1.
STR Markers
| DXS1204-FAM | DXS8015-HEX |
|---|---|
| F Primer: 5′-ATGAACCCTTAACTCATTTAGCAGG-3′ | F Primer: 5′-AGTCTTCTCAGGCCAGAGC-3′ |
| R Primer: 5′-AGCNTGCACCAACATGCC-3′ | R Primer: 5′-AGGACCAACTTTCACATGC-3′ |
| Length: 237–251 bp | Length: 174–190 bp |
F, forward; R, reverse.
Primer Design
Genomic sequence for Dp427m, the main dystrophin isoform found in muscle, was obtained from GenBank (NM 004006.1). Its exon 1 encodes a unique N-terminal MLWWEEVEDCY amino acid sequence and is expressed in the skeletal muscle and heart.
For each dystrophin exon and muscular promoter a primers pair was designed using the Primer 3 software package with the following criteria: product size between 200 and 400 bp, primer size between 24 and 28 nucleotides, and melting temperature between 58°C and 62°C (Table 2).
Table 2.
Primer Design and DHPLC Conditions
| Primers |
DHPLC |
||||
|---|---|---|---|---|---|
| Forward | Reverse | bp | %A | Melt(°C) | |
| Pm | 5′-GGTAGACAGTGGATACATAACAAATGCATG-3′ | 5′-TTCTCCGAAGGTAATTGCCTCCCAGATCTGAGTCC-3′ | 531 | 49% | 58.8 |
| 2 | 5′-TTTAATTTGGATGCCCCAAACCAG-3′ | 5′-AATGACACTATGAGAGAAATAAAACGG-3′ | 347 | 45% | 53 |
| 3 | 5′-GATAATCGTGAAAATGTATCATTGGA-3′ | 5′-CAGTTTCTGGTCTGAAATTCTACTAAGTTT-3′ | 222 | 53% | 57.6 |
| 4 | 5′-TTGTCGGTCTCCTGCTGGTCAGTG-3′ | 5′-CAAAGCCCTCACTCAAACATGAAGC-3′ | 194 | 50% | 59.9 |
| 5 | 5′-TTGCAACTAGGCATTTGGTCTCTTACC-3′ | 5′-AGATTAATGTTACCCAAAAGGAAACC-3′ | 193 | 50% | 55.3 |
| 6 | 5′-TCTATTTATCACTGAAGATCAAGGAC-3′ | 5′-TGGGGAAAAATATGTCATCAGAGTC-3′ | 344 | 50% | 57.9 |
| 7 | 5′-ACACTCAAGACTTAAGGACTATGGGC-3′ | 5′-TACCATACTAAAAGCAGTGGTAGTCCAG-3′ | 287 | 53% | 59.5 |
| 8 | 5′-ATATAGAAACCAAAAATTGATGTGTAG-3′ | 5′-ATGCATATAAAACAGAAAACATCTTG-3′ | 280 | 49% | 57.3 |
| 9 | 5′-TTCTACCATGTTGGAAAGTAGTCCT-3′ | 5′-AAGCAGTGTTAGATTATCTTGGAAGC-3′ | 283 | 50% | 59.9 |
| 10 | 5′-TTGTGCAGCATTGGAAGCTCCTGA-3′ | 5′-TAGTTTACCTCATGAGTATGAAACTGGTC-3′ | 205 | 52% | 58.8 |
| 11 | 5′-ACCACACCGATTTACCTAGAG-3′ | 5′-CACAAGCTTCCAAAACTTGTT-3′ | 292 | 49% | 57.4 |
| 12 | 5′-GATAGTGGGCTTTACTTACATCCTTC-3′ | 5′-GAAAGCACGCAACATAAGATACACCT-3′ | 332 | 48% | 56.9 |
| 13 | 5′-GCAGAAATAAATTTCACCATTTGAGAGC-3′ | 5′-ACTTCAGCTGATTATGAGTGTGTG-3′ | 362 | 48% | 54.2 |
| 14 | 5′-GATACTTTGGCAAATTATTCATGCC-3′ | 5′-CGTGTCTTTTACAGCTAGTTTCTCAC-3′ | 227 | 51% | 58.4 |
| 15 | 5′-GTGAGAAACTAGCTGTAAAAGACACG-3′ | 5′-TGGGTTTTTATAAGACCATTGAAAGC-3′ | 244 | 50% | 56.1 |
| 16 | 5′-CTATAGTGGTGTATGGAATGCAACC-3′ | 5′-TGAGATAGTCTGTAGCATGATAATTGG-3′ | 276 | 48% | 57.3 |
| 17 | 5′-GTCTGACCTCTGTTTCAATACTTCTCAC-3′ | 5′-AAGCTTGAGATGCTCTCACCTTTTCC-3′ | 225 | 50% | 58.3 |
| 18 | 5′-GTGTCAGGCAGGAGTCTCAGATTGAGA-3′ | 5′-GCACGGAGTTTACAAGCAGCACAAAATGAG-3′ | 301 | 50% | 56.4 |
| 19 | 5′-TGAATTACTCATCTTTGCTCTCATGCTG-3′ | 5′-CCCTAAGAAGATTATCTAAATCAACTCGTG-3′ | 156 | 56% | 60.1 |
| 20 | 5′-GCTTTCAGATCATTTCTTTCAGTCTG-3′ | 5′-CCAAGAAATACCTATTGATTATGCTC-3′ | 360 | 50% | 59.5 |
| 21 | 5′-CTTGCCTTACTGCTTTTTAATACCTTC-3′ | 5′-TTATTGTTTCATGTTAGTACCTTCTGG-3′ | 360 | 47% | 57.3 |
| 22 | 5′-GAGTTTGCTGACAATTTAGGAAAACATGGC-3′ | 5′-GATAAGCGTGCTTTATTGTTTTGAC-3′ | 270 | 52% | 60.1 |
| 23 | 5′-GTTTGAATCATATAGATTTCAAGTACAG-3′ | 5′-AACAAGTAAATAAAAATGAGGGTAG-3′ | 357 | 50% | 57.6 |
| 24 | 5′-ACCAGTAATGCCTTATAACGGGTCTCG-3′ | 5′-ATCCACCCCAGCTGTAAAACACTGATC-3′ | 233 | 52% | 57.4 |
| 25 | 5′-ATCCAATATGCAATGCCATCAGTTCCC-3′ | 5′-CTTAGTTAAGTACGTTGAGGCAAGC-3′ | 315 | 50% | 58.1 |
| 26a | 5′-GTCTATGCCAGAAAGGAGGCCTTGA-3′ | 5′-ACCAGGAAAGAGCAGACTGTATACGAC-3′ | 274 | 51% | 56.2 |
| 26b | 5′-TCTAAGCTTTCTGTTATTTACATACTGATG-3′ | 5′-TTCAACTGCTTTCTGTAATTCATCTGGAG-3′ | 258 | 51% | 56.4 |
| 27 | 5′-CTCATTCTAACTGGATGTTGTGAGAAAG-3′ | 5′-CACTATGCCTCACATATGACCATG-3′ | 355 | 50% | 58.6 |
| 28 | 5′-CTGTCTGCTGCATTTTGAATTACCTGC-3′ | 5′-TTCTATTTGGTACTTGACCTCTTTTA-3′ | 356 | 50% | 55.8 |
| 29 | 5′-TCAGAAGATACTGAGCATTTGCTGATAATCC-3′ | 5′-CTGAGAGCTGTATCTGCTATACATTAATGC-3′ | 300 | 51% | 58.4 |
| 30 | 5′-CAGGATTACAGAAAAGCTATCAAGAGT-3′ | 5′-AAGAATGGAAGCTGATTCCCAGATGTAC-3′ | 259 | 51% | 59.6 |
| 31 | 5′-GTTGTTCTTTGTAGAGCATGCTGACT-3′ | 5′-TGCCCAACGAAAACACGTTCCTTAG-3′ | 203 | 50% | 56.3 |
| 32 | 5′-GACCAGTTATTGTTTGAAAGGCAAA-3′ | 5′-GTACCTGCGTATTTGCCACCAGAAAT-3′ | 265 | 49% | 58.2 |
| 33 | 5′-CAAACATGGAATAGCAATTAAGGGGATCTC-3′ | 5′-GAAGTGTTTGTGGTCTCAGCATGC-3′ | 293 | 50% | 57 |
| 34 | 5′-ACAGAAATATAAAAGTTCCAAATAAGT-3′ | 5′-ACGTATGTTCAAAATAACCTTCAGTG-3′ | 299 | 49% | 55 |
| 35 | 5′-ACAAGACATTACTTGAAGGTCAATGC-3′ | 5′-AAGCTTCTAGCCTTTTCTCTTACC-3′ | 243 | 50% | 58.3 |
| 36 | 5′-CCAATAATGCCATGGTATGTCTCTG-3′ | 5′-GGACAAAGATGATTGAAGTAACTGGTG-3′ | 229 | 52% | 57.7 |
| 37 | 5′-CTTCAAGTCCTATCTCTTGCTCATGG-3′ | 5′-CACAAGTTTCCACCTTGGAGTAGATC-3′ | 237 | 52% | 60.6 |
| 38 | 5′-GCATGTGATTAGTTTAGCAACAGGAGG-3′ | 5′-CAGTTGGAGACTTATCTAAGTTCTTTCC-3′ | 311 | 50% | 55.7 |
| 39 | 5′-TGAAGACTGTACTTGTTGTTTTTGATCAG-3′ | 5′-GTTTCTGATGACTAAGAGTCTGAAGCAG-3′ | 276 | 51% | 56.3 |
| 40 | 5′-ATAACTGCAGCCAGAAGTGCACTATAC-3′ | 5′-GTATAATAAAATCTGGTATTGACATTC-3′ | 261 | 50% | 56.2 |
| 41 | 5′-ATGTGGTTAGCTAACTGCCCTGGGC-3′ | 5′-CATACGTGGGTTTGCCAGTAACAACTC-3′ | 260 | 54% | 63.2 |
| 42 | 5′-GGAGGAGGTTTCACTGTTAGGAAGC-3′ | 5′-ATGATCACCTTGTAAAATACGAATG-3′ | 297 | 47% | 56.4 |
| 43 | 5′-GCAACACCATTTGCTACCTTTGGGA-3′ | 5′-CCTGAAAACAAATCATTTCTGCAAG-3′ | 331 | 48% | 54.8 |
| 44 | 5′-CTTGATCCATATGCTTTTACCTGCA-3′ | 5′-TCCATCACCCTTCAGAACCTGATCT-3′ | 268 | 48% | 56 |
| 45 | 5′-AGTACAACTGCATGTGGTAGCACACTG-3′ | 5′-CATTCCTATTAGATCTGTCGCCCTAC-3′ | 296 | 48% | 58.2 |
| 46 | 5′-ATTGCCATGTTTGTGTCCCAGTTTGC-3′ | 5′-TAACCTAATGGGCAGAAAACCAATG-3′ | 336 | 47% | 55 |
| 47 | 5′-AAAGACAAGGTAGTTGGAATTGTGCTG-3′ | 5′-TTAACACATGTGACGGAAGAGATGG-3′ | 252 | 49% | 57.9 |
| 48 | 5′-GCTTATGCCTTGAGAATTATTTACCT-3′ | 5′-TCCTGAATAAAGTCTTCCTTACCACACT-3′ | 372 | 48% | 55.6 |
| 49 | 5′-TTGCTAACTGTGAAGTTAATCTGCAC-3′ | 5′-TGATTATAAATAGTCCACGTCAATGG-3′ | 243 | 49% | 57.4 |
| 50 | 5′-CACCAAATGGATTAAGATGTTCATGAAT-3′ | 5′-TCTCTCTCACCCAGTCATCACTTCATAG-3′ | 271 | 51% | 59.3 |
| 51 | 5′-GAAATTGGCTCTTTAGCTTGTGTTTC-3′ | 5′-GGAGAGTAAAGTGATTGGTGGAAAATC-3′ | 388 | 49% | 58.8 |
| 52 | 5′-GTAAAAGGAATACACAACGCTGAAG-3′ | 5′-AAATGTGAGGGGGATATATGAACTTAAG-3′ | 265 | 50% | 58.3 |
| 53 | 5′-TTTAAAATGTCTCCTCCAGACTAGC-3′ | 5′-GTCTACTGTTCATTTCAGCTTTAACGTG-3′ | 410 | 47% | 54.3 |
| 54 | 5′-GACCTGAGGATTCAGAAGCTGTTTACGA-3′ | 5′-CACCACCCCATTATTACAGCCAACAG-3′ | 312 | 49% | 57.2 |
| 55 | 5′-TGAGTTCACTAGGTGCACCATTCTGA-3′ | 5′-CACAAGAGTGCTAAAGCGGAAATGCC-3′ | 288 | 48% | 59.3 |
| 56 | 5′-GCACATATTCTTCTTCCTGCTGTCCTG-3′ | 5′-GTGGCCTTTTTGCTCCACATCTTTTCC-3′ | 233 | 49% | 58.2 |
| 57 | 5′-ACTTCTAGATATTCTGACATGGATCGC-3′ | 5′-TGTGCTTAACATGTGCAAGGCACGAG-3′ | 243 | 49% | 60 |
| 58 | 5′-GAATGCCACAAGCCTTTCTTAGCACTTC-3′ | 5′-TGCTCCGTCACCACTGATCCTTCTATC-3′ | 225 | 50% | 57.4 |
| 59 | 5′-ATGTGGCCTAAAACCTTGTCATATTGCC-3′ | 5′-TTGTGGGAAGATAACACTGCACTCAAG-3′ | 392 | 47% | 60.9 |
| 60 | 5′-CCTAAAGAGAATAAGCCCAGGTATC-3′ | 5′-TCCTATCCTCACAAATATTACCATGA-3′ | 353 | 49% | 57.4 |
| 61 | 5′-GAGAACATAATTTCTCTCCTTTTCCTCCC-3′ | 5′-CAAGATGCAATAAAGTTAAGTGATAAAAGC-3′ | 154 | 55% | 58 |
| 62 | 5′-TGGAGATTAATGTTGTCTTTCCTGTTTGCGA-3′ | 5′-TACTCACTTGTGAATATACAGGTTAGTCAC-3′ | 207 | 52% | 57.1 |
| 63 | 5′-TCCTGTTTTCTTGACTACTCATGGTAAATGC-3′ | 5′-TAACTTGGAGGAAACATGGCCATGTCC-3′ | 154 | 53% | 56.6 |
| 64 | 5′-TATTTCTGATGGAATAACAAATGCTC-3′ | 5′-TAGTATCAAGATCTTCAAATACTGGCCAATAC-3′ | 157 | 53% | 56.9 |
| 65 | 5′-GAGTCCTAGCTAGGATTCTCAGAGG-3′ | 5′-CTAAGCCTCCTGTGACAGAGCCC-3′ | 341 | 49% | 59.7 |
| 66 | 5′-AGAAGTGTTTACCCTCTAGGAAAGGGTC-3′ | 5′-TCCCATCTAGAACTAGGGTAATTAGCCAAC-3′ | 216 | 51% | 56.4 |
| 67 | 5′-CCACTACTGTGGAAATACTGGCTACTC-3′ | 5′-CCTACTGCCTACTGAAGAGCTAATATGAG-3′ | 391 | 48% | 59.6 |
| 68 | 5′-GATATACACCTCCTTTGCCATCTTGCC-3′ | 5′-AACTAACAGCAACTGGCACAGGAGA-3′ | 342 | 53% | 62.5 |
| 69 | 5′-TGGTAGAAGGTTTATTAAAGAGTGTTCTTTGGG-3′ | 5′-TGAACTAACTCTCACGTCAGGCTGGCGTC-3′ | 230 | 51% | 58 |
| 70 | 5′-CATCCTGTCCTAAATCTGATCTCACC-3′ | 5′-TGGGAGTGAAAGGAGGGTGTTCAGCT-3′ | 262 | 50% | 59.8 |
| 71 | 5′-TGCGTGTGTCTCCTTCACCACCTCA-3′ | 5′-GCGAGCGAATGTGTTGGTGGTAGCAGCACCC-3′ | 131 | 54% | 58.2 |
| 72 | 5′-CATAACTGTGTGGTGGGTTTTTTCTCCA-3′ | 5′-TATTTGCCTGGCATACAACTAGCCTCA-3′ | 168 | 54% | 59.6 |
| 73 | 5′-TTTCAGGAATGTTCGATTAGGTCTTGAA-3′ | 5′-TCCTGTGCTATCCTACCTCTAAATCCCTC-3′ | 226 | 50% | 56.4 |
| 74 | 5′-CTGAGTCCCTAACCCCCAAAGCA-3′ | 5′-GTGCAAGTGTATGCACTCTGCATACC-3′ | 280 | 50% | 59 |
| 75 | 5′-CCATGGTATATAAAATTTGGTGATGA-3′ | 5′-GCACCTATAAAAAGTGCTCTCTGAGG-3′ | 429 | 50% | 60.8 |
| 76 | 5′-TAATTCTGTTTTCTTTTGGATGACTTAGCC-3′ | 5′-GGCCAAATATTCATGTCCCTGTAATACG-3′ | 230 | 54% | 61.9 |
| 77 | 5′-GCTTGAGGGTTTTCTTTGTTATTTATGAGCAAG-3′ | 5′-TGATCCCAGCAAATCTGAGTCCCAC-3′ | 269 | 50% | 55 |
| 78 | 5′-TCCCTTTCTGATATCTCTGCCTCTTCC-3′ | 5′-AGCAGGATGAGACAGACAGAAGCCAT-3′ | 127 | 55% | 56.5 |
| 79 | 5′-AACAGAGTGATGCTATCTATCTGCACC-3′ | 5′-TCTGCTCCTTCTTCATCTGTCATGACTG-3′ | 159 | 54% | 58 |
%A indicates the starting concentration of buffer A (without acetonitrile) used to load samples. Melt(°C) indicates the preferred temperature of analysis.
Primer pairs were chosen to include flanking-intron sequence. Primer sequences were checked by BLASTn to avoid matching with repeated human sequences or covering single nucleotide polymorphisms in the vicinity of exon sequences. Only in the case of exon 26, we designed two primer pairs that split it into two overlapping fragments. Following these requirements, we created a series of amplicons, all with the same melting characteristics. All were amplified using the same PCR conditions. Primers were synthesized by MWG Biotech AG, Ebersberg, Germany. All PCR share the same conditions (95°C 30 seconds, 60°C 90 seconds, 68°C 90 seconds for 33 cycles).
Amplification of Genomic DNA
PCR reactions were set up semiautomatically using an automatic liquid handling Eppendorf epMotion and 384/96-well plates. DNA was amplified in a final reaction volume of 18 μl by using 30 ng of genomic DNA for each pool, buffer LB [20 mmol/L Tris; 10 mmol/L Hepes; 2.5 mmol/L MgSO4 × 7 H2O; 10 nm KCl; 10 mmol/L (NH4)2 SO4], 1.5 mmol/L MgCl2, 0.25 μmol/L each dNTP, 0.5 μmol/L each primers, 0.5 U AmpliTaq Gold (Applied Biosystems).
WAVE System DHPLC Analysis
The dystrophin exons and flanking intronic sequences and the muscular promoter were analyzed using high-throughput denaturing high-performance liquid chromatography (HT-DHPLC). PCR products were directly analyzed. Using pooled samples a preliminary annealing step is not required. The system is based on DHPLC. The WAVE DHPLC system is an ion-pair, reverse-phase HPLC method optimized to separate heteroduplex from homoduplex DNA fragments (Transgenomic Inc., Omaha, NE).
Sequence Analysis
PCR amplicons were purified using the EXOSAP purification kit (GE Healthcare, Chalfont St. Giles, UK): 2 μl of ExoSAP-IT was directly added to 5 μl of PCR product and incubated at 37°C for 15 minutes. ExoSAP-IT was inactivated by heating at 80°C for 15 minutes. The sequence reactions were purified by Applied Biosystems BigDye XTerminator purification kit to remove unincorporated dye and other contaminants. Samples were analyzed using an ABI3130xL and sequencing analysis software (Applied Biosystems).
Results
We screened 153 DNA samples from unrelated DMD or BMD patients. These samples were extracted and studied many years ago without obtaining a genetic diagnosis (Figure 1). We preliminarily excluded deletions or duplications by MLPA and Log-PCR.3,4
Combinatorial Pools
To speed up the analysis and improve sensitivity, we pooled DNA samples in 17 units, each comprising samples from nine male patients. For each unit, we assembled six pools, each one containing DNA from three different patients, so that each DNA sample was present in two different pools and thus analyzed in duplicate (Table 3). This enabled the parallel amplification of three DNA samples in one run and allowed us to detect point mutations without the annealing with control DNA. To avoid pooling samples with significantly different PCR yield, we preliminarily genotyped STR markers in each of the DNA samples with the ABI-Prism 3130 xl using Gene Mapper software. We used two different X markers (DXS8015 and DXS1204) for the amplification of separate samples to determine tandem repeat lengths. On the basis of STR analyses, we created the pools by mixing three DNA samples with a different number of repeats (Figure 2).
Table 3.
Combinatorial Pools
| Pool 1 | Pool 2 | Pool 3 |
|---|---|---|
| 1 | 4 | 7 |
| 2 | 5 | 8 |
| 3 | 6 | 9 |
| Pool 4 | Pool 5 | Pool 6 |
| 1 | 2 | 3 |
| 4 | 5 | 6 |
| 7 | 8 | 9 |
Samples were divided into groups of nine. For each group we created six overlapping pools, each one containing three DNA samples from three different patients, so that each sample was present in a unique combination of two different pools.
Figure 2.
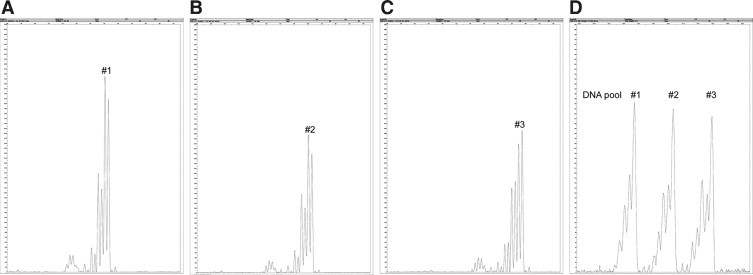
Quality control of PCR yield. A–C: Analysis of each individual sample using the STR DXS8015. D: Analysis of a pool containing three samples.
We analyzed the DMD exons, flanking intronic sequences and the muscle-promoter using HT-DHPLC. Each pool was amplified for all of the 79 dystrophin gene exons and promoter. PCR products were directly analyzed by WAVE system using predetermined temperature and elution buffers concentrations (Table 2). The WAVE system provides rapid, automated scanning for single nucleotide polymorphisms, even when the nature and location of the mutations are unknown.
DHPLC analysis of the pools allowed the unambiguous identification of the mutant sample, avoiding the subsequent screening of three single DNA samples. The presence of a variation within a fragment appears as altered chromatogram shapes of the two different pools sharing the same DNA. This type of scanning unequivocally points out the patient and the fragment for sequence analysis (Figure 3). This approach reduced the turnaround time and was more cost-effective.
Figure 3.
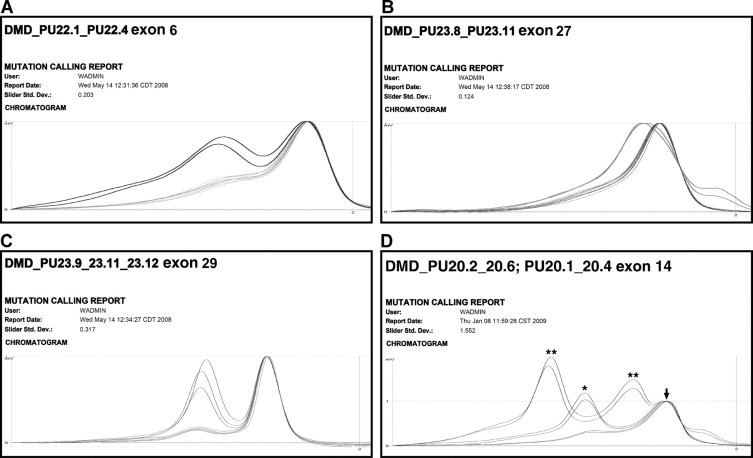
Examples of aberrant DHPLC profiles. The figure shows different DHPLC profiles with growing complexity from A to D. A: Exon 6 showed a heteroduplex in both pools 1 and 4 sharing the DNA sample TU19, in which a frameshift mutation (c.401 404 delCCAA) was detected. B: Exon 27 heteroduplexes in both pools 2 and 6 sharing the DNA sample TU124, in which a splicing mutation (c.3433-1 A>G) was detected. C: Exon 29 heteroduplexes in pools 1, 4, and 5. Pools 1 and 4 shared the DNA sample TU181, pools 1 and 5 shared the DNA sample TU188. The same nonsense mutation (c.3940 C>T) was detected in both samples. D: Three different exon 14 heteroduplexes in pools 2 and 6 and 1 and 4, corresponding to combination of a mutation (**) and a known polymorphism (*). Arrow indicates homoduplexes.
Sequence Analysis
From 153 samples tested, we identified 121 causative mutations of 99 different types. We detected stop codons in the relative majority of patients (56/121 = 46.3%, Table 4), while we found frameshift mutations in 42 cases (34.7%, Table 5), splice mutations in 20 (16.5%, Table 6), and missense mutation in three patients (2.5%, Table 7). In addition, we detected 36 variations classified as polymorphisms or private variants (Table 8).
Table 4.
Nonsense Mutations
| Sample | Exon | DNA change | Stop | Protein | New | Disease |
|---|---|---|---|---|---|---|
| 3761 | 6 | c.409 G>T | TAA | E137X | Yes | DMD |
| TU182-TU294-TU183-TU378 | 6 | c.433 C>T | TGA | R145X | No | DMD |
| TU139-3443 | 7 | c.583 C>T | TGA | R195X | No | DMD |
| TU184 | 10 | c.1062 G>A | TGA | W354X | No | DMD |
| TU86 | 10 | c.1093 C>T | TAA | Q365X | No | DMD |
| TU180 | 11 | c.1292 G>A | TGA | W431X | No | DMD |
| TU318 | 14 | c.1652 G>A | TGA | W551X | Yes | DMD |
| TU187-TU189 | 17 | c.2125 C>T | TAA | Q709X | No | DMD |
| TU05 | 19 | c.2302 C>T | TGA | R768X | No | DMD |
| G11 | 19 | c.2380 G>T | TAG | E794X | Yes | DMD |
| TU70 | 20 | c.2414 C>G | TGA | S805X | Yes | DMD |
| F1 | 20 | c.2521 C>T | TAA | Q841X | No | DMD |
| TU01 | 23 | c.2956 C>T | TAA | Q986X | No | DMD |
| TU107 | 23 | c.3151 C>T | TGA | R1051X | No | DMD |
| TU51-TU185 | 24 | c.3259 C>T | TAG | Q1087X | No | DMD |
| TU32 | 25 | c.3409 C>T | TAG | Q1137X | No | DMD |
| TU12 | 26a | c.3580 C>T | TAG | Q1194X | No | DMD |
| 475 | 27 | c.3625 C>T | TAA | Q1209X | Yes | DMD |
| TU342 | 28 | c.3843 G>A | TGA | W1281X | Yes | BMD |
| TU181-TU188-TU102 | 29 | c.3940 C>T | TGA | R1314X | No | BMD |
| TU218 | 30 | c.4117 C>T | TAG | Q1373X | No | DMD |
| R46 | 33 | c.4600 C>T | TAG | Q1534X | No | DMD |
| TU24 | 34 | c.4690 C>T | TAA | Q1564X | Yes | DMD |
| TU271 | 35 | c.4979 G>A | TGA | W1660X | Yes | BMD |
| TU190 | 35 | c.4996 C>T | TGA | R1666X | No | DMD |
| TU63 | 37 | c.5209 C>T | TAA | Q1737X | Yes | DMD |
| TU266 | 39 | c.5476 G>T | TAA | E1826X | No | DMD Carrier |
| TU194 | 39 | c.5530 C>T | TGA | R1844X | No | DMD |
| TU60 | 41 | c.5773 G>T | TAG | E1925X | No | DMD |
| TU112-TU178-TU84 | 41 | c.5899 C>T | TGA | R1967X | No | DMD |
| TU159 | 42 | c.6023 C>A | TGA | S2008X | Yes | DMD |
| G2-G8-R42 | 46 | c.6678 G>A | TGA | W2226X | Yes | DMD |
| TU186 | 48 | c.7006 C>T | TAG | Q2336X | No | DMD |
| R88 | 57 | c.8422 A>T | TAG | K2808X | Yes | DMD |
| TU152 | 59 | c.8713 C>T | TGA | R2905X | No | DMD |
| 3448 | 59 | c.8880 G>A | TGA | W2960X | Yes | DMD |
| TU02-TU157 | 60 | c.8944 C>T | TGA | R2982X | No | DMD |
| TU87 | 65 | c.9461 T>A | TAG | L3154X | No | BMD |
| TU18 | 68 | c.9829 G>T | TAA | E3277X | Yes | BMD/DMD |
| TU208-G13 | 70 | c.10108 C>T | TGA | R3370X | No | DMD |
| F4 | 70 | c.10135 A>T | TAA | K3379X | No | DMD |
| G3 | 70 | c.10171 C>T | TGA | R3391X | No | DMD |
Resulting TGA stop codons are indicated in bold.
Table 5.
Frameshift Mutations
| Sample | Exon | DNA change | Protein | New | Disease |
|---|---|---|---|---|---|
| TU326 | 5 | c.321 delT | G109V fs X1 | Yes | carrier |
| TU19 | 6 | c.401_404 delCCAA | N135V fs X5 | Yes | DMD |
| 3451-3453 | 7 | c.593_594 insA | H198Q fs X19 | Yes | DMD |
| TU65 | 8 | c.713_714 delTT | L239A fs X7 | Yes | DMD |
| TU55 | 11 | c.1188 insT | G397W fs X1 | Yes | DMD |
| TU150 | 11 | c.1181del G | G394A fs X12 | Yes | DMD |
| TU16 | 11 | c.1300_1310 delCTCAGGGTAGC | L434X | Yes | DMD |
| TU07 | 12 | c.1482 delG | K494K fs 7 | Yes | DMD |
| TU03 | 14 | c.delGTA 1603insCT | V535L fs X47 | Yes | DMD |
| TU386 | 16 | c.1859 delT | L620R fs X12 | Yes | DMD |
| TU23 | 22 | c.2880 2884 delCAAAC | K961L fs X5 | Yes | DMD |
| TU267 | 22 | c.2887 del T | S963P fs X40 | Yes | DMD |
| TU137 | 25 | c.3285 3288 delCAGT | S1096_D1097I fs X9 | Yes | DMD |
| TU115 | 25 | c.3420 del C | H1140Q fs X13 | Yes | DMD |
| TU27 | 26 | c.3447 delGGlnsTT | K1149N-E1150X | No | DMD |
| TU177 | 26 | c.3464 3471 del GTTGGAG | G1155E fs X20 | No | DMD |
| TU44 | 30 | c.4100 delA | Q1367R fs X15 | Yes | DMD |
| TU103 | 30 | c.4119 delG | E1374R fs X8 | Yes | DMD |
| G7 | 30 | c.4186 insA | Y1396X fs | Yes | DMD |
| TU29-G10 | 33 | c.4565delT (Stop TAA) | V1522G fs X2 | No | DMD |
| TU08-R49 | 35 | c.4871_4872 delAG | K1625G fs X27 | Yes | DMD |
| TU13 | 36 | c.5091 delG | A1698L fs X22 | Yes | DMD |
| R44 | 37 | c.5272_5280 del TCAGAGCTC ins CCAA | S1758P fs X13 | Yes | DMD |
| TU304 | 40 | c.5606 del G | R1869K fs X4 | Yes | DMD |
| TU06 | 40 | c.5697 dup A | L1900I fs X5 | No | DMD |
| 3488 | 42 | c.5973_5974 ins A | E1992R fs X11 | Yes | DMD |
| TU211 | 44 | c.6353 delA | Q2118R fs X3 | Yes | DMD |
| TU04 | 48 | c.6980del A | K2329S fs X8 | No | DMD |
| TU57 | 55 | c.8081 del G | F2694S fs X31 | Yes | DMD |
| R37 | 56 | c.8284 ins A | I2762N fs X 10 | Yes | DMD |
| TU33 | 58 | c.8597 8598deiTT | L2866R fs X28 | Yes | DMD |
| TU62 | 59 | c.8732 insA | N2912Q fs X2 | No | DMD |
| TU151 | 62 | c.9204_9207 del CAAA | N3068K fs X20 | No | DMD |
| TU179-G1 | 65 | c.9429_9430 del GC | Q3143H fs X9 | Yes | DMD |
| TU192-TU193 | 68 | c.9926_9929 ins AAGC | H3309Q fs X7 | Yes | BMD/DMD |
| G12 | 70 | c.10105 del G | V3369F fs X8 | Yes | DMD |
| TU214 | 73 | c.10386 del T | N3462K fs X3 | Yes | BMD/DMD |
Table 6.
Putative Splicing Defects
| Sample | Position | DNA change | Splice site | New | Disease |
|---|---|---|---|---|---|
| TU22-TU77 | Intron 2 | c.94−1 G>A | Acceptor | No | BMD |
| TU34 | Intron 5 | c.358−2 A>G | Acceptor | No | DMD |
| TU296 | Intron 5 | c.358−2 A>T | Acceptor | No | DMD |
| TU219 | Intron 6 | c.530+1 G>A | Donor | Yes | DMD/BMD |
| TU309 | Intron 11 | c.1331+2 T>C | Donor | Yes | DMD/BMD |
| 2082 | Intron 11 | c.1332−9 A>G | Acceptor | No | DMD |
| TU124 | Intron 26 | c.3433−1 G>A | Acceptor | No | DMD |
| TU164 | Exon 26 | c.3603 G>A | Donor | Yes | DMD |
| TU105 | Intron 35 | c.5026−6 A>G | Acceptor | No | DMD |
| TU332 | Intron 48 | c.7098+1 G>A | Donor | No | DMD |
| TU379 | Exon 58 | c.8668 G>A | Donor | No | DMD |
| TU114 | Intron 58 | c.8668+1 G>A | Donor | Yes | DMD |
| TU209 | Intron 58 | c.8668+3 A>T | Donor | Yes | DMD/BMD |
| TU133 | Exon 65 | c.9560 A>G | Donor | No | DMD |
| TU54-1707 | Intron 65 | c.9563+1 G>A | Donor | No | DMD |
| TU36-TU97 | Intron 70 | c.10223+1 G>A | Donor | No | DMD |
| TU30 | Intron 70 | c.10223+5 G>T | Donor | Yes | DMD |
Table 7.
Functional Mutations
| Sample | Exon | DNA change | Protein | New | Disease |
|---|---|---|---|---|---|
| TU118 | 3 | c.160_162 del CTC | L54del | Yes | DMD |
| TU42 | 69 | c.10010 G>A | C3337Y | Yes | DMD |
| TU109 | 70 | c.10101_10103 delAGA | E3367del | No | BMD/DMD |
Table 8.
Variants and Polymorphisms
| Variations | Position | New | Number* |
|---|---|---|---|
| c.32-78 G>T | Intron 1 | No | 1 |
| 94-16 ins T | Intron 2 | Yes | 5 |
| c.832-54 A>G | Intron 8 | No | 1 |
| c.837 G>A T279T | Exon 9 | No | 2 |
| c.853 G>A G285R | Exon 9 | Yes | 1 |
| 1225 A>T T409S | Exon 11 | Yes | 1 |
| c.1603-57 T>C | Intron 14 | No | 2 |
| 1635 A>G R545R | Exon 14 | No | 7 |
| 1869 C>T L623L | Exon 16 | No | 1 |
| c.2176 G>T V726F | Exon 18 | Yes | 1 |
| c.2391 T>G N797K | Exon 20 | No | 1 |
| c.3604-95 delG | Intron 26 | Yes | 2 |
| c.3936 G>C L1312F | Exon 29 | Yes | 1 |
| 4234-13A>G | Intron 30 | Yes | 2 |
| c.4510 H1504Y | Exon 32 | Yes | 1 |
| c.4675-53 G>T | Intron 33 | No | 1 |
| c.4878 G>T V1626V; 5016 T>A N1672K | Exon 35 | Yes | 2 |
| c.5326-54 A>C | Intron 37 | No | 1 |
| 5234 G>A R1745H | Exon 37 | No | 7 |
| 5586+93insCT | Intron 39 | Yes | 1 |
| c.5723 A>T D1908V GAT->GTT | Exon 40 | No | 1 |
| c.5795 A>G Q1932R | Exon 41 | Yes | 1 |
| c.6118-76 ins TA | Intron 42 | Yes | 2 |
| 6290+27 T>A | Intron 43 | No | 2 |
| c.6443 T>C L2148P CTC->CCC | Exon 45 | No | 1 |
| 6913-114A>T | Intron 47 | No | 4 |
| 6913-114A>T | Intron 47 | No | 1 |
| c.3561 A>T 6913-114 | Intron 47 | Yes | 1 |
| c.7200+53 C>G | Intron 49 | No | 2 |
| 8027+11 C>T | Intron 54 | No | 6 |
| c.8571 T>C T2857T | Exon 58 | Yes | 1 |
| c.8810 A>G Q2937R | Exon 59 | No | 1 |
| c.9085-23 C>A | Intron 60 | Yes | 1 |
| 9649+15 T>C | Intron 66 | No | 5 |
| c.10789 L3597L; 10554-30_10554-35 del TTTC | Exon 75/intron 74 | Yes | 1 |
| c.3685*49 c>t | Intron 79 | Yes | 1 |
Number of samples with the same variation.
Discussion
Identification of a pathogenic point mutation in a DMD or BMD patient confirms the clinical diagnosis and allows definitive carrier testing and prenatal diagnosis for family members.8,9 Precise knowledge of the mutation is also required for some of the emerging therapies, such as exon skipping,10 or suppression of premature stop codons.11,12
We found mutations in 121 DMD-BMD patients out of 153 DNA samples tested (Figure 4). In 32 DNA samples (20.9%), no mutation was found. Complete sequence analysis of all exons in these samples confirmed the DHPLC negative results. Considering that 153 DNA samples correspond to 20% of all patients that show no deletions or duplications, about 20% of 20% of patients cannot be diagnosed by DNA analysis alone. This indicates that mRNA13 or CGH array13,14 analyses can be necessary to diagnose about 4% of all DMD/BMD patients.
Figure 4.
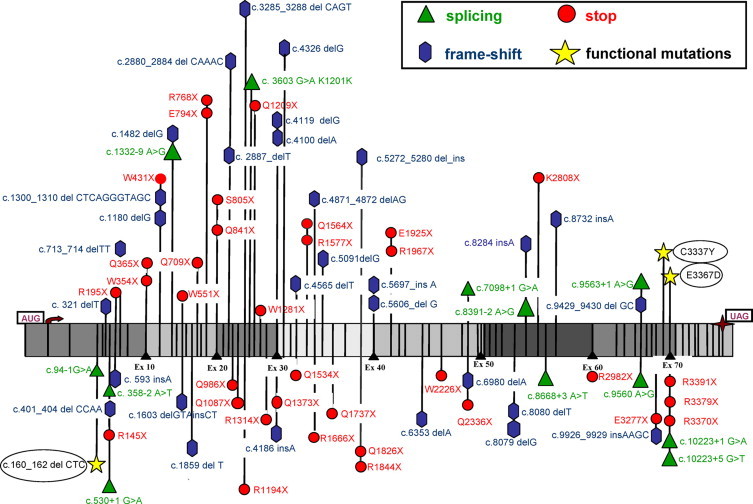
Distribution of all causative point mutations along the dystrophin cDNA. Segments corresponding to groups of 10 exons are indicated in dark and light gray.
Notably, 22 unrelated patients shared the same mutations (Tables 456). Among the 56 nonsense mutations, 34 (60.7%) show the TGA termination codon that is considered optimal for readthrough therapy.11 Notably, among the 37 different frameshift mutations, 31 (83.8%) are absent from the Leiden database.2
We identified three putative functional mutations (Table 7). One is the substitution of a highly conserved cysteine at position 3337 within the second half of the dystroglycan-binding domain. A similar mutation (C3340Y) has been associated with Duchenne muscular dystrophy.15 There is the loss of aspartic acid in position 3368 with the substitution of the glutamic acid in position 3367 with aspartic acid. This produces the loss of glutamic acid in position 3367 that is known to be associated with a particular DMD phenotype.16
The first half of the C terminus and the cysteine-rich (D-domain; amino acid residues 3080–3408) are highly conserved regions of dystrophin. The region is involved in interactions with dystroglycan that mediates attachment of dystrophin to the cytoplasmic surface of the cell membrane. Deletions or chain-terminating nonsense mutations involving the D-domain usually result in DMD.
The third is the c.160_162 CTC deletion in exon 3 in DMD, which resulted in the loss of an evolutionary conserved leucine in position 54 in the actin-binding domain. This is the same amino acid position replaced by an arginine described in a boy with Duchenne muscular dystrophy.17 Interestingly, this missense mutation in position 54 was questioned because it was not completely studied by DNA sequencing. After 16 years, our findings support the causative role of the change.
Two unusual mutations were also identified. Premature stop codons in positions 1281 and 1314 associated with BMD and not DMD. This could be explained by the skipping of exon 28 or 29, respectively.18
Our DNA-based mutation screening strategy is suitable for high-throughput applications in patients for which mRNA is unavailable. Sample pooling, together with identical PCR conditions for all fragments, were set up for the simultaneous detection of any mutations type within exons and exon flanking regions. The preliminary analysis of single and pooled samples by STR markers permitted us to confirm the same PCR efficiency in all combinatorial pools. In the protocol, the pooling was conservative, since we only mixed three samples. With the availability of more sensitive methods of DNA detection (ie, multicolor fluorescence) we can foresee possibilities to pool dozens of samples with further impressive reduction of costs.
Acknowledgements
We thank Marina Fanin, Giuliana Galluzzi, Enzo Ricci, Federico Zara, Claudio Bruno, Sara Scapolan, Maria Teresa Bassi, and Mayana Zatz for DNA samples and Alessandra Ferlini for helpful discussion. We acknowledge the SUN-Naples Human Mutation Gene Bank (Cardiomyology and Medical Genetics), which is partner of the Eurobiobank Network.
Footnotes
Supported by grants from Telethon-UILDMGUP04008 (2005–2007) and TIGEM-11B and TIGEM-C20B, Ministero dell'Istruzione dell'Università e della Ricerca (MIUR: PRIN 2004 and 2006) (to V.N. and C.M.), Ministero della Salute (d.lgs 502/92), Ricerca d'Ateneo (to V.N. and L.P.).
A.T. is a fellow of the Luigi Califano Foundation.
References
- 1.Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 2.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–144. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz M, Duno M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet Test. 2004;8:361–367. doi: 10.1089/gte.2004.8.361. [DOI] [PubMed] [Google Scholar]
- 4.Trimarco A, Torella A, Piluso G, Maria Ventriglia V, Politano L, Nigro V. Log-PCR: a new tool for immediate and cost-effective diagnosis of up to 85% of dystrophin gene mutations. Clin Chem. 2008;54:973–981. doi: 10.1373/clinchem.2007.097881. [DOI] [PubMed] [Google Scholar]
- 5.Deburgrave N, Daoud F, Llense S, Barbot JC, Récan D, Peccate C, Burghes AH, Béroud C, Garcia L, Kaplan JC, Chelly J, Leturcq F. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum Mutat. 2007;28:183–195. doi: 10.1002/humu.20422. [DOI] [PubMed] [Google Scholar]
- 6.Belsito A, Politano L, Piluso G, Comi LI, Nigro V. Dystrophin gene scanning by DHPLC of DMD carriers without deletions or duplications. Acta Myol. 1999;3:221–223. [Google Scholar]
- 7.Bennett RR, den Dunnen J, O'Brien KF, Darras BT, Kunkel LM. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet. 2001;2:17. doi: 10.1186/1471-2156-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nigro V, Politano L, Nigro G, Romano SC, Molinari AM, Puca GA. Detection of a nonsense mutation in the dystrophin gene by multiple SSCP. Hum Mol Genet. 1992;1:517–520. doi: 10.1093/hmg/1.7.517. [DOI] [PubMed] [Google Scholar]
- 9.Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72:931–939. doi: 10.1086/374176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, den Dunnen JT, Koop K, van der Kooi AJ, Goemans NM, de Kimpe SJ, Ekhart PF, Venneker EH, Platenburg GJ, Verschuuren JJ, van Ommen GJ. Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med. 2007;357:2677–2686. doi: 10.1056/NEJMoa073108. [DOI] [PubMed] [Google Scholar]
- 11.Aurino S, Nigro V. Readthrough strategies for stop codons in Duchenne muscular dystrophy. Acta Myol. 2006;25:5–12. [PubMed] [Google Scholar]
- 12.Wilton S. PTC124, nonsense mutations and Duchenne muscular dystrophy. Neuromuscul Disord. 2007;17:719–720. doi: 10.1016/j.nmd.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Bovolenta M, Neri M, Fini S, Fabris M, Trabanelli C, Venturoli A, Martoni E, Bassi E, Spitali P, Brioschi S, Falzarano MS, Rimessi P, Ciccone R, Ashton E, McCauley J, Yau S, Abbs S, Muntoni F, Merlini L, Gualandi F, Ferlini A. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. BMC Genomics. 2008;9:572. doi: 10.1186/1471-2164-9-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.del Gaudio D, Yang Y, Boggs BA, Schmitt ES, Lee JA, Sahoo T, Pham HT, Wiszniewska J, Chinault AC, Beaudet AL, Eng CM. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. Hum Mutat. 2008;9:1100–1107. [Google Scholar]
- 15.Lenk U, Oexle K, Voit T, Ancker U, Hellner KA, Speer A, Hübner C. A cysteine 3340 substitution in the dystroglycan-binding domain of dystrophin associated with Duchenne muscular dystrophy, mental retardation and absence of the ERG b-wave. Hum Mol Gen. 1996;5:973–975. doi: 10.1093/hmg/5.7.973. [DOI] [PubMed] [Google Scholar]
- 16.Becker K, Robb SA, Hatton Z, Yau SC, Abbs S, Roberts RG. Loss of a single amino acid from dystrophin resulting in Duchenne muscular dystrophy with retention of dystrophin protein. Hum Mutat. 2003;21:651. doi: 10.1002/humu.9143. [DOI] [PubMed] [Google Scholar]
- 17.Prior TW, Papp AC, Snyder PJ, Burghes AH, Bartolo C, Sedra MS, Western LM, Mendell JR. A missense mutation in the dystrophin gene in a Duchenne muscular dystrophy patient. Nat Genet. 1993;4:357–360. doi: 10.1038/ng0893-357. [DOI] [PubMed] [Google Scholar]
- 18.Ginjaar IB, Kneppers AL, v d Meulen JD, Anderson LV, Bremmer-Bout M, van Deutekom JC, Weegenaar J, den Dunnen JT, Bakker E. Dystrophin nonsense mutation induces different levels of exon 29 skipping and leads to variable phenotypes within one BMD family. Eur J Hum Genet. 2000;8:793–796. doi: 10.1038/sj.ejhg.5200535. [DOI] [PubMed] [Google Scholar]