

Abstract
Real-time polymerase chain reaction (PCR) is the current method of choice for detection and quantification of nucleic acids, especially for molecular diagnostics. Complementarity between primers and template is often crucial for PCR applications, as mismatches can severely reduce priming efficiency. However, little quantitative data on the effect of these mismatches is available. We quantitatively investigated the effects of primer-template mismatches within the 3′-end primer region on real-time PCR using the 5′-nuclease assay. Our results show that single mismatches instigate a broad variety of effects, ranging from minor (<1.5 cycle threshold, eg, A–C, C–A, T–G, G–T) to severe impact (>7.0 cycle threshold, eg, A–A, G–A, A–G, C–C) on PCR amplification. A clear relationship between specific mismatch types, position, and impact was found, which remained consistent for DNA versus RNA amplifications and Taq/Moloney murine leukemia virus versus rTth based amplifications. The overall size of the impact among the various master mixes used differed substantially (up to sevenfold), and for certain master mixes a reverse or forward primer-specific impact was observed, emphasizing the importance of the experimental conditions used. Taken together these data suggest that mismatch impact follows a consistent pattern and enabled us to formulate several guidelines for predicting primer-template mismatch behavior when using specific 5-nuclease assay master mixes. Our study provides novel insight into mismatch behavior and should allow for more optimized development of real-time PCR assays involving primer-template mismatches.
During the past decade, real-time polymerase chain reaction (PCR) has established itself as an essential technique for reliable detection and quantification of nucleic acids.1,2,3 The result is a widespread application of real-time PCR assays in both research1,2,3,4 and diagnostic4,5,6 laboratories. Vital to the specificity, sensitivity, and efficiency of real-time PCR are the primers. The most important primer characteristics contributing to a successful amplification are primer-template association and dissociation kinetics, possible secondary structures, and primer-template complementarity (Watson-Crick base-pairing).7,8 Full complementarity between primer and template sequences is generally considered crucial for the specific amplification of a nucleic acid sequence, but can be difficult to achieve, in particular for applications depending on highly heterogenic nucleic acid input for amplification (eg, diagnostic assays for influenza virus and human immunodeficiency virus). Conserved regions are often too small to accommodate a typical real-time PCR assay (50 to 150 bp), exhibit inferior G-C contents or are prone to the formation of secondary structures. Primer-template mismatches can therefore be unavoidable.
Unfortunately, mismatches between primers and template are known to affect both the stability of the primer-template duplex and the efficiency with which the polymerase extends the primer,7,8,9,10,11,12,13 potentially leading to biased results or even PCR failure.14,15 Even apparently small effects on nucleic acid quantification (0.5 to 1.0 log underestimation of initial copy number) can have serious consequences, as illustrated by studies on the relation between viral load and disease prognosis in HIV-1.16
The detrimental effects of primer-template mismatches can however also prove beneficial. They provide a discriminative force that can be used for PCR assays opting to distinguish between different nucleic acids (eg, single nucleotide polymorphism detection, allele-specific PCR), which have become important tools for modern molecular diagnostics.4
Every mismatch, irrespective of its location within the primer sequence, will result in a decreased thermal stability of the primer-template duplex, thus potentially affecting PCR specificity. However, mismatches located in the 3′ end region (defined as the last 5 nucleotides of the 3′ end region) of a primer have significantly larger effects on priming efficiency than more 5′ located mismatches,9,11,13,14,15 since 3′ end mismatches can disrupt the nearby polymerase active site.17,18
Strategies to alter mismatch impact, eg, degenerate/modified bases or extensive adaptation of PCR conditions, can prove helpful in specific situations, but these strategies often require a lot of time-consuming optimization and can result in unwanted side effects (eg, increased primer-dimer formation). Quantitative data on the effects of 3′ end mismatches is necessary to improve knowledge and reliable prediction of mismatch behavior, which is beneficial for the development and optimization of real-time PCR assays involving mismatches.
Several studies on the effects of 3′ end primer-template mismatches have been published.9,10,19,20,21,22 However, only few systematically examined the behavior of 3′ end primer-template mismatches (including the relationship between these effects and the position of the mismatch) using modern quantitative methods. In this study, we comprehensively investigate the effects of 3′ end primer-template mismatches using different commercially available 5′-nuclease assay master mixes. Diagnostic laboratories often employ such optimized pre-mixed reagents, which are generally directly used with few adaptations. Our approach therefore provides a relevant system for quantification of mismatch impact on diagnostic real-time PCR assays. Our experiments resulted in a large quantitative dataset from which different aspects of mismatch effects on PCR amplification were further analyzed, ultimately leading to the formulation of a set of general guidelines for improved prediction of primer-template mismatch impact.
Materials and Methods
Vector Construction
A pGA4 vector containing a 148 bp region from the HIV-1 5′ long terminal repeat (LTR) and a 75 bp region from the nucleoprotein (NP) gene in human metapneumovirus (hMPV) was constructed (the HIV-hMPVpGA4 model vector). Two real-time Taqman PCR assays were defined on the insert: i) a previously described and commonly used HIV-1 LTR assay23 and ii) an in-house developed hMPV NP assay. The primers and probes (synthesized by Eurogentec, Seraing, Belgium) used for mismatch analysis, together with their location within the HIV-HMPV insert, are depicted in Figure 1A1B.
Figure 1.
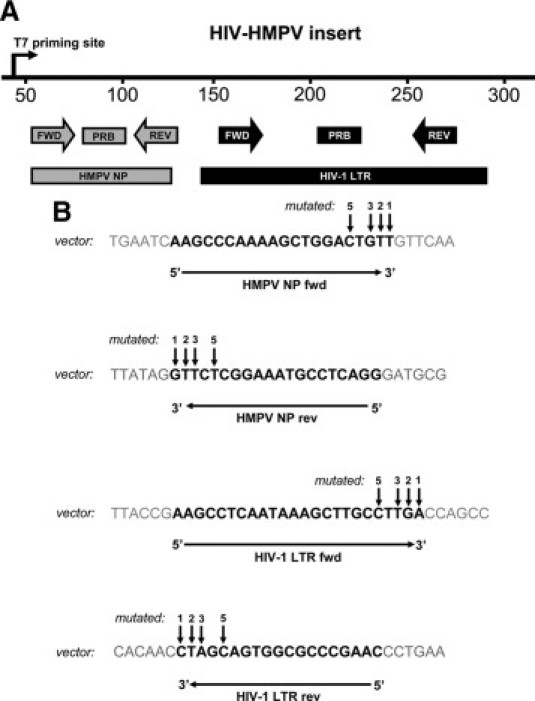
Oligonucleotides used to study the effects of 3′ end primer-template mismatches on nucleic acid detection and quantification with real-time Taqman PCR. Panel (A) represents a linear map of the HIV-HMPV insert, in which the location of both viral DNA sequences and corresponding primers and probes are depicted. Panel (B) shows the nucleotide sequences of the primers (black) as located in the vector. Arrows indicate the positions on the HIV-hMPVpGA4 vector that have been subjected to site-directed mutagenesis. fwd = forward primer, rev = reverse primer, PRB = probe.
Mutagenesis and Generation of PCR Templates
Site-directed mutagenesis (QuikChange XL Site-Directed Mutagenesis Kit from Stratagene, La Jolla, CA) was used to generate 48 single bp mutations in the HIV-HMPV insert at 4 positions: the 3′ terminal base (position 1), the penultimate base (position 2), and the third (position 3), and fifth base (position 5) from the 3′ primer terminus (Figure 1B, indicated in the vector sequence by arrows). Mutations were individually introduced in only one primer, resulting in constructs containing a single mismatch in one of the four primer sequences. This strategy allowed the utilization of the non-mutated PCR as a control reaction for calculating mismatch effects. Additionally, three constructs with multiple mutations in the last 3 bases were generated (all located in the hMPV NP forward primer, including the 3′ terminal base).
Mutations were verified by colony PCR using M13 (−20) primers (forward: 5′-TGTAAAACGACGGCCAGT-3′, reverse: 5′-GGTCATAGCTGTTTCCTG-3′) and subsequent sequencing using the BigDye Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) with the previously mentioned M13 (−20) primers. Sequencing reactions were performed on a 3100 Genetic Analyzer (Applied Biosystems).
For real-time Taqman RT-PCR assays, sense RNA was transcribed from the HIV-hMPVpGA4 vector and its mutated derivatives using the T7 RiboMAX Express Large Scale RNA production System kit (Promega, Madison, WI) according to the manufacturer's instructions. RNA synthesis reactions were subsequently incubated at 37°C for 15 minutes in the presence of 2 units of TURBO DNase (Applied Biosystems) to remove any DNA contaminations. DNA removal was validated by testing log10 serial dilutions of RNA using the 2× Taqman Universal PCR Mastermix, as described below.
Real-Time PCR Amplifications
Sensitivity, efficiency, and Ct-values for both the HIV-1 LTR and hMPV NP assay were determined by testing log10 serial dilutions of DNA or RNA template using three different commercially available real-time Taqman PCR setups: i) the 2× Taqman Universal PCR Mastermix (Applied Biosystems, catalog # 4304437), ii) the Taqman PCR Core Reagents Kit (Applied Biosystems, catalog # N8080228), and iii) the Taqman EZ RT-PCR Kit (Applied Biosystems, catalog # N8080236). Efficiencies were calculated using the equation E = [(10−1/slope)/2] * 100% (using the slope of the serial dilution's standard curve).
Plasmid DNA was amplified in 50 μl reaction volumes containing 15 pmol primers (HIV-1 LTR forward/reverse or hMPV NP forward/reverse primers, Figure 1B), 5 pmol probe (the HIV-1 LTR or the hMPV NP probe, Figure 1B), and 2× Taqman Universal PCR Mastermix, containing Taq polymerase, dUTP, and uracil N-glycosylase. Samples were amplified by running the following PCR program on an ABI 7500 real-time PCR system (Applied Biosystems): 2 minutes at 50°C (uracil N-glycosylase activity), 10 minutes at 95°C, and 40 cycles consisting of 15 seconds at 95°C and 60 seconds at 60°C. This setup was also used to test transcribed RNA for DNA contamination, by testing log10 serial dilutions of RNA stocks ranging from 10−3 to 10−11. RNA stocks were considered DNA-free when these dilutions failed to generate a positive amplification curve.
RNA template was amplified in 50 μl reaction volumes containing 15 pmol primers and 5 pmol probe. Both the Taqman PCR Core Reagents Kit (containing TaqDNA polymerase) and the Taqman EZ RT-PCR Kit (containing rTth polymerase) were used for amplification according to the manufacturer's description: 12.5 U Multiscribe Reverse Transcriptase (recombinant Moloney murine leukemia virus reverse transcriptase, Applied Biosystems) and 10 U RNase inhibitor (Applied Biosystems) were added to the Taqman PCR Core Reagents Kit reactions. Taqman PCR Core Reagents Kit samples were amplified by the following PCR program: 2 minutes at 50°C (uracil N-glycosylase activity), 30 minutes at 48°C (reverse transcription), 10 minutes at 95°C, and 40 cycles consisting of 15 seconds at 95°C and 60 seconds at 60°C. Taqman EZ RT-PCR Kit samples were amplified by the following PCR program: 2 minutes at 50°C, 30 minutes at 60°C, 5 minutes at 95°C, and 40 cycles consisting of 20 seconds at 94°C and 60 seconds at 62°C. For both reverse transcription-PCR assays, the reverse primer was used as the reverse transcribing primer.
PCR Characteristics and Mismatch Analysis
Amplification efficiencies of the two targets in the three experimental setups were always between 94% and 105%, and never more than 2.5% apart for both PCR assays (data not shown). The maximum difference in standard curve cycle threshold (Ct)-values between both assays was never greater than 0.35 Ct, with the exception of reactions run with the rTth DNA polymerase based PCR, which displayed Ct-values differing 0.9 Ct at maximum (data not shown). Sensitivities of both assays were similar for all three methods used, with a lower detection limit of approximately 30 (DNA) to 300 (RNA) copies of target template.
Mismatch impact was measured by quantifying Ct 25 dilutions (±0.5 Ct, corresponding to approximately 30,000 copies of DNA or 300,000 copies of RNA input) of the plasmid/RNA stocks in different real-time Taqman PCR assays (as described above), hereby ensuring amplification of equal amounts of nucleic acid. In the same experiment, both the HIV-1 LTR and the hMPV NP primer sets were used in separate reactions to amplify the mutated nucleic acids, resulting in a set of fully complementary and a set of mismatched amplifications (four replicates per primer set) for each. No alternative primers were used to form the mismatches, to prevent single nucleotide differences in primer sequences from altering original primer dynamics and possibly influencing experimental outcomes. Since standard curve Ct-values of both PCR assays were similar, mismatch effects were calculated by subtracting the average Ct value generated by the mismatched amplification from the average Ct value generated by the complementary amplification. Statistical significance (P < 0.05) was determined by a Student t-test. When directly comparing two effects of different mismatches, a minimum difference of three times the average SD of the control amplifications between two effects was considered significant. The average SD of all control amplifications in the dataset was 0.57 Ct, setting the criterion of significance for such comparisons at 3 * 0.57 Ct = 1.71 Ct (a 0.52 log difference in copy number quantification).
Results
Real-Time PCR Amplification in the Presence of Mismatches
To investigate the effects of primer-template mismatches on real-time Taqman PCR, we first tested the constructs using a commonly used real-time Taqman PCR master mix based on Taq polymerase. The results (Figure 2A2B2C2D2E2F2G2H2I2J2K2L) showed a strong correlation between the severity of the effect and the position of the mismatch: the most detrimental effects were present at the 3′-terminal position (average effect of 5.19 Ct, almost all statistically significant). Mismatch impact rapidly declined at positions further away from the 3′-terminal nucleotide. Single mismatches at position 2 (nt 2) had an average effect of 1.87 Ct (with only A–C and T–G mismatches showing statistically insignificant effects), while effects at position 3 (nt 3) and 5 (nt 5) were only statistically significant for less than half of the 12 mismatches (displaying average effects of 0.53 and 0.71 Ct respectively). However, individual mismatches at non-terminal positions could occasionally induce large effects (up to 5.96 Ct).
Figure 2.

Effects of primer-template mismatches on the quantification of nucleic acids using real-time Taqman PCR. Each panel represents the effects of one single mismatch (depicted as a primer-template mismatch) at different positions on the HIV-hMPVpGA4 construct. The position of the mismatch and the primer in which it is located are shown on the x axis, while the y axis shows the average increase in detected Ct-value as compared with the complementary situation. Amplifications were performed using the 2× Taqman Universal PCR Mastermix. Error bars represent the SD (n = 4). Differences in Ct-values were tested for statistical significance using a Student t-test. Significant values (*P < 0.05) are indicated nt = nucleotide, rev = reverse primer, fwd = forward primer.
To study the impact of a specific mismatch type on PCR priming efficiency, the effects of mismatches at the 3′ terminal nucleotide were compared. The 12 types of mismatches could be divided into three subgroups, depending on the size of their effect: 1) mismatches that resulted in a >100 fold underestimation of initial copy number, consisting of A–A, A–G, G–A, G–G and C–C mismatches (8.29 to 9.09 Ct, a 320- to 580-fold underestimation); 2) mismatches that resulted in a 10 to 100 fold underestimation of initial copy number, consisting of T–C, C–T and T–T mismatches (3.77 to 4.75 Ct, a 14- to 28-fold underestimation); and 3) mismatches that resulted in a <10-fold underestimation of initial copy number, the A–C, C–A, T–G and G–T mismatches (0.99 to 1.91 Ct, a 2- to 4-fold underestimation).
Mismatch effects were consistent in such a way that the ratios between the effects of the three mismatches at one particular position remained similar throughout the entire dataset. This consistency was present in different ways: i) between the HIV-1 and hMPV assays, ii) within one assay, and iii) between forward and reverse primers. The effects of identical mismatches at the exact same position in different assays were largely consistent as well, as illustrated by the A–A/A–C/A–G mismatches at position 2 (Figure 2A2B2C) and the C–C/C–A/C–T mismatches at position 5 (Figure 2G2H2I). The A–A, A–C, A–G, C–T, and C–A mismatches showed no significant differences (all differences <1.71 Ct) for both assays. Only the two C–C mismatches showed significantly different (3.12 Ct) effects.
Another interesting and unknown aspect of mismatch behavior is the occurrence of symmetry within the effects of 3′ end primer-template mismatches (eg, do C–A and A–C mismatches have similar effects on PCR amplification?). The 3′ terminal mismatches tested using the real-time Taqman PCR setup (Figure 2) presented a strikingly high degree of symmetry within their effects (for both inter- and intra-assay comparisons): the largest difference between two symmetrical mismatches was just 0.86 Ct (the C–A and A–C mismatches). Comparing the effects of symmetrical mismatches at other positions revealed less symmetrical comparisons. The G–A to A–G comparisons at positions 2 and 3 (Figure, 2Cand 2K) showed large significant differences: 3.0 Ct (G–A in the HIV-1 LTR fwd primer − A–G in the hMPV NP rev primer, nt 2) and 4.51 Ct (G–A in the HIV-1 LTR fwd primer − A–G in the HIV-1 LTR rev primer, nt 2). However, no significant differences were found between symmetrical mismatches for the other nine possible comparisons at positions 2, 3, and 5 (<0.8 Ct difference).
None of the plasmids with multiple 3′ end mismatches (all including the 3′ terminal base) generated a positive amplification curve (data not shown). Apparently, mismatches with a modest or low individual impact (eg, T–T, G–T, as seen in Figure 2, D and L) located at both the 3′ terminal and penultimate position had a combined impact large enough to completely abolish PCR amplification.
Real-Time RT-PCR Amplification in the Presence of Mismatches
To investigate whether primer-template mismatches had different effects on a real-time Taqman RT-PCR system, RNA was synthesized from the mutated constructs and used as a template for real-time Taqman RT-PCR. A commercial one-step real-time Taqman PCR setup (a standard TaqDNA polymerase/MMLV reverse transcriptase based approach) was used for measuring the effects of the mismatches (Figure 3). Mismatches between reverse primers and template (Figure 3C and 3D) all resulted in small effects on PCR amplification (<0.7 Ct, with only 1 out of 24 statistically significant), contrary to the large effects the same mismatches had on the standard real-time Taqman PCR reaction (Figure 2).
Figure 3.

Effects of primer-template mismatches on the quantification of nucleic acids using a Taq/MMLV-based real-time Taqman RT-PCR. Each panel represents the effects of the 12 individual mismatches (depicted as primer-template mismatches) per primer. The type and position of the individual mismatches are shown on the x axis, while the y axis shows the average increase in detected Ct-value as compared with the complementary situation. Amplifications were performed using the Taqman PCR Core Reagents Kit, combined with Multiscribe Reverse Transcriptase and RNase inhibitors. Error bars represent the SD (n = 4). Differences in Ct-values were tested for statistical significance using a Student t-test. Significant values (*P < 0.05) are indicated nt = nucleotide, REV = reverse primer, FWD = forward primer.
Forward primer-template mismatches (Figure 3Aand 3B) however displayed a twofold increase in effect (Ct-value shift) as compared with their effects on standard real-time Taqman PCR. Only the C–C, C–T, and C–A mismatches at position 5 of the hMPV NP forward primer exhibited lower effects (0.26-fold decrease in average Ct value shift). The A–A and A–G mismatches at position 1 of the HIV-1 LTR forward primer even resulted in failure of amplification for respectively 25% and 50% of the reactions.
The relative impact of the individual 3′ terminal forward primer-template mismatches was however compatible with the earlier defined subgroups (A–A and A–G > T–T and T–C > A–C and T–G) and mismatch impact rapidly declined when they were positioned further from the 3′ terminal nucleotide. Symmetry and consistency (within the effects of forward primer-template mismatches) diminished somewhat when mismatches were located further away from the 3′ terminal nucleotide, comparable with the real-time Taqman PCR results. RNA transcribed from the constructs containing multiple mismatches (which were all located in the hMPV NP forward primer) could not be amplified (data not shown).
rTth DNA Polymerase-Based Real-Time RT-PCR Amplification in the Presence of Mismatches
To further investigate the influence of different real-time Taqman RT-PCR systems on the effects of 3′ end primer-template mismatches, we also tested the different mismatches using a recombinant Thermus thermophilus (rTth) DNA polymerase-based one-step real-time Taqman RT-PCR setup. The rTth DNA polymerase functions as both a DNA and RNA dependent polymerase, efficiently reverse transcribing RNA at elevated temperatures (<60°C) in the presence of Mn2+ ions.24
Results from these experiments (Figure 4) were surprising: all mismatches between forward primers and template (Figure 4A and 4B) resulted in effects smaller than 2.6 Ct (average effect of 0.85 Ct), with 16 out of 26 mismatches showing no statistically significant effects. Multiple forward primer-template mismatches (eg, a T–T mismatch at both nt 1 and 2) did however have a large detrimental effect on PCR amplification (6.65 to 7.64 Ct, a 105- to 209-fold underestimation of initial copy number) but were still amplified (data not shown), as opposed to their complete abolishment of amplification when tested using the previous two real-time Taqman PCR setups. Reverse primer-template mismatches on the other hand showed severe effects (Figure 4C and 4D). For example, all three mismatches at nt 1 of the hMPV reverse primer (C–C, C–T, and C–A) resulted in PCR failure, while C–T and C–A mismatches at this position yielded only moderate to small effects in both other setups. The detrimental effects of reverse primer-template mismatches were also much more persistent at positions further away from the 3′ terminal base, resulting in a much slower regression of mismatch impact: the average effects of reverse primer-template mismatches on non-terminal positions were more than fivefold higher in the rTth based experiments (5.05 Ct), as compared with standard real-time Taqman PCR data (0.98 Ct).
Figure 4.

Effects of primer-template mismatches on the quantification of nucleic acids using rTth DNA polymerase-based real-time Taqman RT-PCR. Each panel represents the effects of the 12 individual mismatches (depicted as primer-template mismatches) per primer. The type and position of the individual mismatches are shown on the x axis, while the y axis shows the average increase in detected Ct-value as compared with the complementary situation. Amplifications were performed using the Taqman EZ RT-PCR Kit. Error bars represent the SD (n = 4). Differences in Ct-values were tested for statistical significance using a Student t-test. Significant values (P < 0.05) are indicated by an asterisk. nt = nucleotide, REV = reverse primer, FWD = forward primer.
Despite this totally different distribution of impact, mismatch effects on the rTth based RT-PCR amplifications also diminished when mismatches were positioned further away from the 3′ end, and they largely followed the earlier defined classification on severity of impact. Only A–C mismatches deviated from this classification, showing relatively high effects on the rTth DNA polymerase-based PCR reactions. Although not as clear as for the Taq based Taqman PCR setup, symmetry and consistency among the effects (as defined above) of the reverse primer-template mismatches were comparable with the other two setups.
Discussion
Mismatches located in the 3′ end region of the primer are known to be exceptionally detrimental to PCR priming.9,11,13,14,15 As a consequence, these mismatches pose a serious problem for nucleic acid amplification using PCR assays,14,15 but also provide opportunities to discriminate between nucleic acid targets.4,25 Quantitative data on mismatch behavior is scarce, making it difficult to reliably predict the effects of these mismatches. Improved prediction of mismatch behavior could be of great benefit, especially for molecular diagnostic assays, which often have to deal with primer-template mismatches.4,9,14,15 In the present study, we investigated the effects of 3′ end primer-template mismatches on the 5′ nuclease assay using different standard 5′-nuclease assay master mixes. We show that single mismatches instigate a broad but consistent variety of effects, strongly dependent on mismatch type, mismatch position and the master mix used. This consistent pattern enabled us to formulate several guidelines to predict primer-template mismatch behavior on real-time PCR amplification, which should aid real-time PCR design and optimization.
One of the most striking features of mismatch impact observed in this study is its position-dependency. We confirmed that 3′ terminal mismatches are on average by far the most detrimental to PCR amplification, resulting in effects ranging from a twofold underestimation of initial copy number to a complete abolishment of amplification. But despite the fact that the average impact of mismatches at the penultimate, third and fifth base was increasingly diminished, substantial impact (up to 658-fold underestimation of initial copy number) at these positions was still occasionally observed, especially when using an rTth based RT-PCR setup. As little is known about to what extend non-3′ terminal primer-template mismatches can still influence PCR outcome, our observations emphasize that the impact of non-3′ terminal mismatches must not be underestimated, especially in a diagnostic environment.
Our results furthermore provide novel evidence of symmetric mismatches displaying similar effects on PCR amplification. Symmetry was most striking at the 3′ terminal nucleotide, although this somewhat diminished for mismatches at the other positions, possibly due to influence of the different and larger adjacent sequence context on more 5′-located mismatches (eg, increased/decreased duplex stability and/or steric hindrance). This high degree of symmetry cannot be directly explained from a structural point of view, since it has been shown that symmetrical mismatches interact with the polymerase in a unique manner.17 On the other hand, the inherent flexibility of DNA allows for many conformational states, which makes direct comparisons of mismatch impact between structural data and PCR or extension studies not very straightforward.17 Our results do however indicate that the nature of the different active site distortions that two symmetrical mismatches instigate appears similar in terms of their effect on the 5′ nuclease assay.
A persistent relationship between the type of mismatch and the severity of its impact was present, largely independent of the experimental conditions used: purine-purine (A–A, A–G, G–A, and G–G) and C–C mismatches had the most detrimental effect, followed by the remaining pyrimidine-pyrimidine mismatches (T–T, C–T, and T–C) and the relatively minor effects of purine-pyrimidine (A–C, C–A, G–T, and T–G) mismatches. The only exceptions were the A–C mismatches when tested with the rTth based RT-PCR setup, which occasionally showed effects as large as purine-purine mismatches. Previous studies9,10,19,20,21,22 have also reported a general purine-purine > pyrimidine-pyrimidine > purine-pyrimidine hierarchy of mismatch impact, indicating that the classification of mismatch severity is a general phenomenon also observed in a real-time PCR environment. Differences between the effects of certain mismatch types have however been reported. For example, Huang et al10 reported C–T mismatches to be most readily extended, while these mismatches had significant effects in other studies,20,21,22 including our study. The small discrepancies between these studies are not entirely unexpected, since many use different enzymes, stringencies, experimental approaches, and sequence contexts, which can all influence experimental outcome.19 However, these factors do not seem to play a major role in determining this classification, considering that many studies report similar hierarchies of mismatch impact. In our experiments, ratios between the impacts of the three mismatches at one particular position remained similar, further strengthening the general relevance of the defined hierarchy.
Combinations of multiple mismatches (either two or three) led to abolishment of amplification (data not shown) in all experimental setups (with the exception of the rTth-based RT-PCR), despite the modest or even minor impact of the involved mismatches when tested individually. These findings contradict an earlier report using conventional PCR,9 in which multiple mismatches involving a thymidine base in the last three nucleotides of the primer proved efficiently extended. This discrepancy might have been caused by the low stringency conditions and long primers (>30 nt) used in this study. However, our results point toward an extremely detrimental effect of multiple 3′ end mismatches on real-time PCR using standard size primers (18 to 24 nt).
Mismatches displayed a surprisingly different overall size of impact for the various master mixes, often accompanied by a reverse or forward primer-specific impact. Remarkable were the minor effects of reverse primer-template mismatches when amplified using the Taq/MMLV based real-time RT-PCR mastermix. This phenomenon was most likely caused by the fact that the reverse primer-template mismatches only affect the reverse transcription step. Reverse transcription using the reverse primer will generate cDNA identical to the reverse primer, removing any effect of the mismatch on subsequent PCR cycles. When plasmid DNA was used as input, effects of reverse primer-template mismatches were similar to those seen in the standard real-time Taqman PCR experiments (data not shown), indicating that reverse transcription was responsible for the large difference in effect. The reverse transcription step was run at 48°C, which is 12° lower than the PCR itself. This lower reaction temperature permits less specific DNA hybridization, which could have contributed to the smaller effects.13 Another possible explanation is the higher efficiency with which reverse transcriptases extend mismatches as compared with Taq polymerase (100–1000 fold).20,26 The same reverse primer-template mismatches showed completely contradicting effects when amplified using the rTth based RT-PCR; reverse primer-template mismatches did have a large negative impact on rTth-based PCR amplification. Their effects were also much larger than corresponding effects in the real-time Taqman PCR experiments, with even substantial impact of mismatches located at positions 3 and 5. These differences may have been caused by the large difference (48°C versus 60°C, see above) in reaction temperature between both RT-reactions and/or a difference in fidelity between both enzymes.
Also notable are the differences between the effects of forward primer-template mismatches. Forward primer-template mismatches amplified in the Taq/MMLV-based RT-PCR experiments displayed a higher average effect (twofold) than the same mismatches tested with the standard real-time Taqman PCR setup. Perhaps Mg2+ and dNTP concentrations, which are known to influence the effect of mismatches on PCR amplification,27 play a role in causing this phenomenon. Other differences in reaction conditions (eg, concentration of monovalent salts, glycerol concentration) could also have influenced the effects of mismatches.7,8,28 Contradictory, when the forward primer-template mismatches were analyzed using the rTth DNA polymerase based kit, only minimal effects were detected. The two Taqman RT-PCR kits thus showed entirely opposing phenomena. When plasmid DNA was used as a template, similar effects were seen (data not shown), ruling out the possibility of input type and/or the RT-reaction causing the dramatic differences in effect. A probe-based cause was also ruled out, since different reverse complementary probes yielded similar results (data not shown). Even multiple forward primer-template mismatches (which prevented amplification in the other two setups) were elongated, although they did show substantial impact. It appears as if the impact of forward primer-template mismatches was somehow partly ‘quenched’ in the rTth based RT-PCR. This ‘quenching’ was possibly caused by specific characteristics of rTth DNA polymerase DNA dependent DNA polymerase activity, which could have been enhanced by the fidelity-lowering Mn2+ ions and the higher ionic strength in the rTth DNA polymerase buffer. Since the exact structural mechanisms by which rTth DNA polymerase accomplishes its double function are unknown, further analysis of its structural and enzymatic properties is necessary to properly address these questions.
Finally, we translated the generated data into a plausible set of guidelines for handling primer-template mismatches during real-time PCR development (Table 1). Although sequence context and reaction conditions (especially stringency) can influence the impact of mismatches, results from this study are coherent enough to suggest that primer-template mismatch impact on real-time PCR is relatively predictable. Using three commonly used 5′-nuclease assay setups and standard size primers makes the presented results even more relevant for (diagnostic) real-time PCR applications. These conclusions are further strengthened by the considerable symmetry and consistency we observed among individual primer-template mismatches. The presented guidelines can, under general real-time PCR conditions, help to minimize the detrimental effects of primer-template mismatches or increase their discriminative capabilities and should therefore allow more flexible design and easier optimization of real-time PCR assays involving primer-template mismatches.
Table 1.
General Guidelines for Handling 3′ End Primer-Template Mismatches during Real-Time PCR Development
| Recommendations per Experimental Setup (for detection/quantification of nucleic acids) |
||||
|---|---|---|---|---|
| Mismatch (single) | Position (primer) | Standard real-time PCR (Taq DNA polymerase-based) | Real-time RT-PCR using specific reverse primer (Taq DNA polymerase) | rTth DNA polymerase-based real-time PCR using specific reverse primer |
| A–A/A–G/G–A/G–G/C–C | 3′ terminal | Avoid | Avoid in FWD, | Acceptable in FWD, |
| (both primers) | Acceptable in REV | Avoid in REV | ||
| 3′ penultimate | Avoid | Avoid in FWD, | Acceptable in FWD, | |
| (both primers) | Acceptable in REV | Avoid in REV | ||
| 3′ positions 3-5 | Acceptable | Avoid in FWD, | Acceptable in FWD, | |
| (both primers) | Acceptable in REV | Avoid in REV | ||
| T–T/T–C/C–T | 3′ terminal | Avoid | Avoid in FWD, | Acceptable in FWD, |
| (both primers) | Acceptable in REV | Avoid in REV | ||
| 3′ penultimate | Acceptable | Avoid in FWD, | Acceptable in FWD, | |
| (both primers) | Acceptable in REV | Avoid in REV | ||
| 3′ positions 3-5 | Acceptable | Acceptable | Acceptable in FWD, | |
| (both primers) | (both primers) | Avoid in REV | ||
| C–A/A–C/G–T/T–G | 3′ terminal | Acceptable | Avoid in FWD, | Acceptable in FWD, |
| (both primers) | Acceptable in REV | Avoid in REV | ||
| 3′ penultimate | Acceptable | Acceptable | Acceptable in FWD, | |
| (both primers) | (both primers) | Avoid in REV | ||
| 3′ positions 3-5 | Acceptable | Acceptable | Acceptable in FWD, | |
| (both primers) | (both primers) | Avoid in REV | ||
| Best discriminating properties | - A–A/A–G/G–A/G–G/C–C at 3′ terminal position of the primer | |||
| - A–A/A–G/G–A/G–G/C–C at 3′ penultimate position of the primer | ||||
| - T–T/T–C/C–T at 3′ terminal position of the primer | ||||
| (primer choice will depend on the type of experimental setup used) | ||||
Mismatches are depicted as primer–template mismatches. Mismatches were designated as acceptable (for general applications) when their effect was generally <2,0 Ct. FWD = forward primer, REV = reverse primer.
Acknowledgements
We thank Dr. Ron Fouchier, Theo Besteboer, and Dr. Thomas Myers for helpful discussions. We are grateful to the reviewers for their constructive comments.
References
- 1.Valasek MA, Repa JJ. The power of real-time PCR. Adv Physiol Educ. 2005;29:151–159. doi: 10.1152/advan.00019.2005. [DOI] [PubMed] [Google Scholar]
- 2.Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol. 2002;30:503–512. doi: 10.1016/s0301-472x(02)00806-8. [DOI] [PubMed] [Google Scholar]
- 3.Lie YS, Petropoulos CJ. Advances in quantitative PCR technology: 5′ nuclease assays. Curr Opin Biotechnol. 1998;9:43–48. doi: 10.1016/s0958-1669(98)80082-7. [DOI] [PubMed] [Google Scholar]
- 4.Gibson NJ. The use of real-time PCR methods in DNA sequence variation analysis. Clin Chim Acta. 2006;363:32–47. doi: 10.1016/j.cccn.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 5.Mackay IM, Arden KE, Nitsche N. Real-time PCR in virology. Nucleic Acids Res. 2002;30:1292–1305. doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esp MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JDC, Wengenack NL, Rosenblatt JE, Cockerill FR, III, Smith TF. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19:165–256. doi: 10.1128/CMR.19.1.165-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cha RS, Thilly WG. Specificity, efficiency, and fidelity of PCR. PCR Methods Appl. 1992;3:18–29. doi: 10.1101/gr.3.3.s18. [DOI] [PubMed] [Google Scholar]
- 8.Arnheim N, Erlich H. Polymerase chain reaction strategy. Annu Rev Biochem. 1992;61:131–156. doi: 10.1146/annurev.bi.61.070192.001023. [DOI] [PubMed] [Google Scholar]
- 9.Kwok S, Kellogg DE, McKinney N, Spasic D, Godal L, Levenson C, Sninsky JJ. Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res. 1990;18:999–1005. doi: 10.1093/nar/18.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang M, Arnheim N, Goodman MF. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 1992;20:4567–4573. doi: 10.1093/nar/20.17.4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bru D, Martin-Laurent F, Philippot L. Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl Environ Microbiol. 2008;74:1660–1663. doi: 10.1128/AEM.02403-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith S, Vigilant L, Morin PA. The effects of sequence length and oligonucleotide mismatches on 5′ exonuclease assay efficiency. Nucleic Acids Res. 2002;20:43–49. doi: 10.1093/nar/gnf110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christopherson C, Sninsky JJ, Kwok S. The effects of internal primer template mismatches on RT-PCR: hIV-1 model studies. Nucleic Acids Res. 1997;25:654–658. doi: 10.1093/nar/25.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein D, Leutenegger CM, Bahula C, Gold P, Hofmann-Lehmann R, Salmons B, Lutz H, Gunzburg WH. Influence of preassay and sequence variations on viral load determination by a multiplex real-time reverse transcriptase-polymerase chain reaction for feline immunodeficiency virus. J Acquir Immune Defic Syndr. 2001;26:8–20. doi: 10.1097/00126334-200101010-00002. [DOI] [PubMed] [Google Scholar]
- 15.Whiley DM, Sloots TP. Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J Clin Virol. 2005;34:104–107. doi: 10.1016/j.jcv.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Mellors JW, Rinaldo CR, Jr, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 17.Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 18.Beard WA, Shock DD, Wilson SH. Influence of DNA structure on DNA polymerase beta active site function: extension of mutagenic DNA intermediates. J Biol Chem. 2004;279:31921–31929. doi: 10.1074/jbc.M404016200. [DOI] [PubMed] [Google Scholar]
- 19.Joyce CM, Sun XC, Grindley ND. Reactions at the polymerase active site that contribute to the fidelity of Escherichia coli DNA polymerase I (klenow fragment) J Biol Chem. 1992;267:24485–24500. [PubMed] [Google Scholar]
- 20.Creighton S, Huang M, Cai H, Arnheim N, Goodman MF. Base mispair extension kinetics. Binding of avian myeloblastosis reverse transcriptase to matched and mismatched base pair termini. J Biol Chem. 1992;4:2633–2639. [PubMed] [Google Scholar]
- 21.Mendelman LV, Petruska J, Goodman MF. Base mispair extension kinetics. Comparison of DNA polymerase alpha and reverse transcriptase. J Biol Chem. 1990;265:2338–2346. [PubMed] [Google Scholar]
- 22.Day JP, Bergstrom D, Hammer RP, Barany F. Nucleotide analogs facilitate base conversion with 3′ mismatch primers. Nucleic Acids Res. 1999;27:1810–1818. doi: 10.1093/nar/27.8.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drosten C, Panning M, Drexler JF, Hänsel F, Pedroso C, Yeats J, de Souza Luna LK, Samuel M, Liedigk B, Lippert U, Stürmer M, Doerr HW, Brites C, Preiser W. Ultrasensitive monitoring of HIV-1 viral load by a low-cost real-time reverse transcription-PCR assay with internal control for the 5′ long terminal repeat domain. Clin Chem. 2006;52:1258–1266. doi: 10.1373/clinchem.2006.066498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myers TW, Gelfand DH. Reverse transcription and DNA amplification by a Thermus thermophilus DNA polymerase. Biochemistry. 1991;30:7661–7666. doi: 10.1021/bi00245a001. [DOI] [PubMed] [Google Scholar]
- 25.Pas SD, Noppornpanth S, van der Eijk AA, de Man RA, Niesters HG. Quantification of the newly detected lamivudine resistant YSDD variants of Hepatitis B virus using molecular beacons. J Clin Virol. 2005;32:166–172. doi: 10.1016/j.jcv.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Zinnen S, Hsieh J, Modrich P. Misincorporation and mispaired primer extension by human immunodeficiency virus reverse transcriptase. J Biol Chem. 1994;269:24195–24202. [PubMed] [Google Scholar]
- 27.Eckert KA, Kunkel TA. DNA polymerase fidelity and the polymerase chain reaction. PCR Methods Appl. 1991;1:17–24. doi: 10.1101/gr.1.1.17. [DOI] [PubMed] [Google Scholar]
- 28.Cheng S, Fockler C, Barnes WM, Higuchi R. Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci. 1994;91:5695–5699. doi: 10.1073/pnas.91.12.5695. [DOI] [PMC free article] [PubMed] [Google Scholar]