

Abstract
New arylthioindoles along with the corresponding ketone and methylene compounds were potent tubulin assembly inhibitors. As growth inhibitors of MCF-7 cells, sulfur derivatives were superior or sometimes equivalent to the ketones, while methylene derivatives were substantially less effective. Esters 24, 27–29, 36, 39,and 41 showed ~50% of inhibition on human HeLa and HCT116/chr3 cells at 0.5 μM, and these compounds inhibited the growth of HEK, M14, and U937 cells with IC50's in the 78–220 nM range. While murine macrophage J744.1 cell growth was significantly less affected (20% at higher concentrations), four other nontransformed cell lines remained sensitive to these esters. The effect of drug treatment on cell morphology was examined by time-lapse microscopy. In a protocol set up to evaluate toxicity on the Saccharomyces cerevisiae BY4741 wild type strain, compounds 24 and 54 strongly reduced cell growth, and 29, 36, and 39 also showed significant inhibition.
Introduction
Microtubules are a key target for anticancer agents.1 Tubulin interacting agents interfere with the dynamic equilibrium of microtubules by either inhibition of tubulin polymerization or blocking microtubule disassembly, and both effects lead cells to the arrest of cell division. Colchicine (1), combretastatin A-4 (CSA4,a 2), and the Catharanthus alkaloids vincristine and vinblastine are tubulin assembly inhibitors that interact with α,β-tubulin dimers at distinct interfaces between the two subunits2,3 (Chart 1). These interactions result in microtubule destabilization and subsequent cellular apoptosis. Taxoids and epothilones target a lumenal site on the β-subunit of dimers in microtubules4,5 and may enter the lumen through a binding site6 located at a pore on the microtubule surface formed by two different tubulin dimers. Paclitaxel stimulates microtubule polymerization and stabilization at high concentrations, whereas at lower concentrations the drug inhibits microtubule dynamics with little effect on the proportion of tubulin in polymer.7,8 Independent of precise mechanism of action, clinical use of antitubulin drugs is associated with problems of drug resistance, toxicity, and bioavailability.9
Chart 1.

Structures of Derivatives 3–56 and Reference Compounds 1 and 2
Arylthioindoles (ATIs) are potent tubulin assembly inhibitors that bind to the colchicine site on β-tubulin close to its interface with α-tubulin within the α,β-dimer.10 ATIs efficiently inhibit tubulin polymerization and cancer cell growth, with activities comparable with those of colchicine and CSA4. To rationalize the structure–activity relationship (SAR) studies, we dissected the ATI structure into four regions: (A) the substituent at position 2 of the indole, (B) the sulfur atom bridge, (C) the 3-arylthio group, and (D) the substituent at position 5 of the indole (Chart 1). In our previous papers, we reported SAR and molecular modeling studies on ATI derivatives differing in substituent groups and/or position at the A, C, and D regions.11,12 However, chemical modification of the sulfur bridge (B region) was not exhaustively explored, although five ATIs were transformed into the corresponding sulfur dioxides.11 In contrast to the weak potency displayed by these dioxides,11 several CSA4 analogues, characterized by a carbonyl functionality as a bridging group between the indole nucleus and the trimethoxyphenyl ring, show potent inhibition of tubulin polymerization.13 However, in comparing ATIs with the 3-aroylindoles reported by Hsieh and co-workers,14 we should note that these two groups of compounds are significantly different because there are major differences in our SAR findings and those of Hsieh's group. Moreover, our molecular modeling studies indicate that in the colchicine site of tubulin, ATI derivatives preferentially assume a colchicine-like rather than a CSA4-like binding conformation.10–12
These observations prompted the SAR investigations at the B bridge reported here. To this end, we synthesized new indole derivatives by bioisosteric replacement of the sulfur bridge of the ATI's with either carbonyl or methylene moieties, and other related groups (compounds 3–45, Table 1). In addition, we examined the replacement of the indole ring with differently substituted pyrrole and imidazole analogues (compounds 46–56, Table 2).
Table 1.
Inhibition of Tubulin Polymerization, Growth of MCF-7 Human Breast Carcinoma Cells and Colchicine Binding by Compounds 3–45 and Reference Compound 2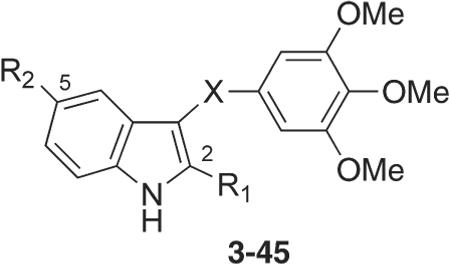
| compd | R1 | R2 | X | tubulina IC50 ± SD (μM) | MCF-7b IC50 ± SD (nM) | inhibition colchicine bindingc (% ± SD) |
|---|---|---|---|---|---|---|
| 3 | H | H | S | 2.6±0.2 | 34±9 | 68±0.8 |
| 4 | H | H | C=O | 3.5±0.07 | 150±50 | 26±0.3 |
| 5 | H | H | CH2 | 28±2 | nd | nd |
| 6 | COOMe | H | S | 2.9±0.1 | 25±1 | 74±2 |
| 7 | COOMe | H | C=O | 2.7±0.3 | 33±10 | 49±10 |
| 8 | COOMe | H | CH2 | 4.2±0.02 | 190±70 | 33±7 |
| 9 | COOEt | H | S | 2.9±0.2 | 40±2 | 19±7 |
| 10 | COOEt | H | C=O | 2.6±0.3 | 80±20 | 51±6 |
| 11 | COOEt | H | CH2 | 5.4±1 | nd | nd |
| 12 | H | 5-Cl | S | 2.6±0.2 | 77±7 | 51±4 |
| 13 | H | 5-C1 | C=O | 2.5±0.5 | 53±10 | 44±9 |
| 14 | H | 5-Cl | CH2 | 24±2 | nd | nd |
| 15 | COOMe | 5-Cl | S | 2.5±0.3 | 42±10 | 73±0.2 |
| 16 | COOMe | 5-Cl | C=O | 1.6±0.1 | 30±6 | 61±4 |
| 17 | COOMe | 5-Cl | CH2 | 1.7±0.5 | 87±30 | 49±4 |
| 18 | COOEt | 5-Cl | S | 2.2±0.2 | 110±2 | 53±3 |
| 19 | COOEt | 5-Cl | C=O | 1.4±0.2 | 110±10 | 49±3 |
| 20 | COOEt | 5-Cl | CH2 | 2.5±0.5 | 230±60 | 39±4 |
| 21 | COOEt | 5-Cl | COCH2 | >40 | nd | nd |
| 22 | COOEt | 5-Cl | COCO | >40 | nd | nd |
| 23 | COOEt | 5-Cl | CH2CH2 | >40 | nd | nd |
| 24 | H | 5-Br | S | 1.6±0.3 | 43±7 | 65±3 |
| 25 | H | 5-Br | C=O | 1.9±0.3 | 60d | 45±5 |
| 26 | H | 5-Br | CH2 | 13±0.8 | nd | nd |
| 27 | COOMe | 5-Br | S | 0.99±0.1 | 33±10 | 75±3 |
| 28 | COOMe | 5-Br | C=O | 1.3±0.08 | 18±4 | 67±4 |
| 29 | COOMe | 5-Br | CH2 | 1.3±0.08 | 30±9 | 59±7 |
| 30 | COOEt | 5-Br | S | 1.6±0.2 | 83±20 | 62±7 |
| 31 | COOEt | 5-Br | C=O | 1.6±0.05 | 67±10 | 58±2 |
| 32 | COOEt | 5-Br | CH2 | 1.7±0.2 | 100d | 53±4 |
| 33 | H | 5-OMe | S | 4.1±0.6 | 22±2 | 61±4 |
| 34 | H | 5-OMe | C=O | 3.4±0.4 | 200d | 32±7 |
| 35 | H | 5-OMe | CH2 | 14±0.5 | nd | nd |
| 36 | COOMe | 5-OMe | S | 2.0±0.2 | 13±3 | 93±0.8 |
| 37 | COOMe | 5-OMe | S=O | >40 | nd | nd |
| 38 | COOMe | 5-OMe | S(=O)2 | >40 | nd | nd |
| 39 | COOMe | 5-OMe | C=O | 0.67±0.02 | 17±6 | 78±6 |
| 40 | COOMe | 5-OMe | CHOH | >40 | nd | nd |
| 41 | COOMe | 5-OMe | CH2 | 1.4±0.2 | 33±10 | 59±1 |
| 42 | COOEt | 5-OMe | S | 2.4±0.2 | 46±3 | 71±2 |
| 43 | COOEt | 5-OMe | C=O | 2.6±0.04 | 90±10 | 49±4 |
| 44 | COOEt | 5-OMe | CHOH | >40 | nd | nd |
| 45 | COOEt | 5-OMe | CH2 | 2.8±0.3 | 93±10 | 42±4 |
| 2 | 1.1±0.1 | 2.5±0.6 | 99±0.7 |
Inhibition of tubulin polymerization. Tubulin was 10 μM during polymerization.
Inhibition of growth of MCF-7 human breast carcinoma cells.
Inhibition of [3H]colchicine binding. Tubulin was at 1 μM, both [3H]colchicine and inhibitor were at 5 μM.
Same value obtained in all experiments.
Table 2.
Inhibition of Tubulin Polymerization, Growth of MCF-7 Human Breast Carcinoma Cells by Compounds 46–56, and Reference Compound 2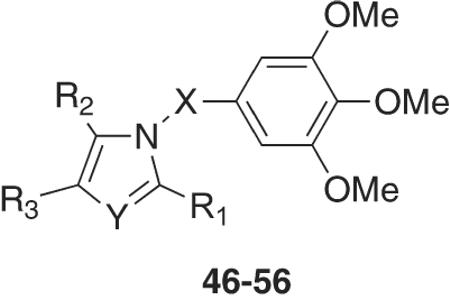
| compd | R1 | R2 | R3 | X | Y | tubulind IC50 ± SD (μM) | MCF-7a IC50 ± SD (nM) |
|---|---|---|---|---|---|---|---|
| 46 | COOEt | H | H | CH2 | CH | >40 | nd |
| 47 | COOEt | H | Me | CH2 | C-Me | >40 | nd |
| 48 | COOEt | Me | Me | CH2 | C-Me | >40 | nd |
| 49 | COOEt | Et | Me | CH2 | C-Et | >40 | nd |
| 50 | H | COOEt | H | CH2 | N | >40 | nd |
| 51 | COOEt | H | H | C=O | CH | 32±0.6 | >2500 |
| 52 | H | COOEt | H | C=O | N | >40 | >2500 |
| 53 | H | H | COOEt | C=O | N | >40 | >2500 |
| 54 | COOH | H | H | CH2 | CH | >40 | >2500 |
| 55 | COO(CH2)2O(CH2)2OH | Me | Me | CH2 | C-Me | >40 | nd |
| 56 | H | COOH | H | CH2 | N·HCl | >40 | nd |
| 2 | 1.1 ± 0.1 | 2.5 ± 0.6 |
Inhibition of tubulin polymerization. Tubulin was 10 μM during polymerization.
Inhibition of growth of MCF-7 human breast carcinoma cells.
Chemistry
Compounds 4, 7, 10, 13, 16, 19, 25, 28, 31, 34, 39, and 43 were synthesized in a microwave (MW) reactor by treating the appropriate indole with 3,4,5-trimethoxybenzoyl chloride in the presence of aluminum chloride in 1,2-dichloroethane at 110 °C (150 W) for 2 min (Scheme 1). Similarly, ethyl 5-chloro-3-[2-(3,4,5-trimethoxyphenyl)acetyl]-1H-indole-2-carboxylate 21 was prepared by MW synthesis using ethyl 5-chloro-1H-indole-2-carboxylate and 2-(3,4,5-trimethoxyphenyl)acetyl chloride. The latter reagent was obtained by treating the appropriate carboxylic acid with oxalyl chloride in the presence of a catalytic amount of DMF in anhydrous THF at 0 °C for 10 min (Scheme 2). Sodium borohydride (10 equiv) reduction of 4, 13, 25, and 34 in boiling ethanol for 3 h furnished the corresponding benzyl derivatives 5, 14, 26, and 35, respectively (Scheme 1). Alternatively, benzyl (8, 11, 17, 20, 29, 32, 41, 45) and phenylethyl (23) derivatives were obtained in good yield by treating benzoyl (7, 10, 16, 19, 28, 31, 39, and 43) (Scheme 1) or oxalyl (22) (Scheme 2) compounds with triethylsilane in trifluoroacetic acid at 25 °C for 12 h. Reduction of ketones 39 and 43 with sodium borohydride (1 equiv) in refluxing aqueous THF for 2 h gave the corresponding alcohols 40 and 44. MW oxidation of ethyl 3-[2-(3,4,5-trimethoxyphenyl)acetyl]-1H-indole-2-carboxylate (21) with selenium(IV) oxide in dimethylsulfoxide (DMSO) at 150 °C (150 W) for 2 min furnished the corresponding 2-oxo derivative 22.
Scheme 1.

Synthesis of Compounds 4, 5, 7, 8, 10, 11, 13, 14, 16, 17, 19, 20, 25, 26, 28, 29, 31, 32, 34, 35, 39–41 and 43–45a
a Reagents and reaction conditions: (a) 3,4,5-trimethoxybenzoyl chloride, AlCl3, 1,2-dichloroethane, closed vessel, 110 °C, 150 W, Pmax = 250 PSI, 2 min, 40–68%; (b) (5, 14, 26, and 35) NaBH4 (10 equiv), EtOH, reflux, 3 h, 42–68%; (c) (8, 11, 17, 20, 29, 32, 41, and 45)Et3SiH, CF3COOH, 25 °C, overnight, 46–90%; (d) (40 and 44) NaBH4 (1 equiv), THF/H2O, reflux, 2 h, 29–53%.
Scheme 2.

Synthesis of Compounds 21, 22, and 23a
a Reagents and reaction conditions: (a) oxalyl chloride, anhydrous DMF cat., anhydrous THF, 0 °C, 10 min; (b) ethyl 5-chloro-1H-indole-2-carboxylate, AlCl3, 1,2-dichloroethane, closed vessel, 110 °C, 150 W, Pmax = 250 PSI, 2 min, 32%; (c) selenium(IV) oxide, DMSO, closed vessel, 150 °C, 150 W, Pmax = 250 PSI, 2 min, 56%; (d) Et3SiH, CF3COOH, 25 °C, overnight, 40%.
Methyl (27) and ethyl (30) 5-bromo-3-(3,4,5-trimethoxyphenylthio)-1H-indole-2-carboxylate were obtained by MW reaction of 5-bromo-1H-indole-2-carboxylic acid (59) with bis-(3,4,5-trimethoxyphenyl)disulfide10 in the presence of sodium hydride in anhydrous DMF at 110 °C (150 W) for 2 min (Scheme 3). The crude carboxylic acid was transformed into the corresponding methyl ester 27 with trimethylsilyldiazomethane (TMSDM) in dichloromethane/methanol at 25 °C for 30 min or into the ethyl ester 30 with thionyl chloride in absolute ethanol at 65 °C for 2 h.
Scheme 3.

Synthesis of Compounds 27 and 30a
a Reagents and reaction conditions: (a) NaH, bis(3,4,5-trimethoxyphenyl)disulfide, anhydrous DMF, closed vessel, 110 °C, 150 W, Pmax = 250 PSI, PowerMAX, 2 min; (b) (27) TMSDM, CH2Cl2/MeOH, 25 °C, 30 min, 79%; (c) (30) SOCl2, absolute EtOH, 65 °C, 2 h, Ar stream, 70%.
Oxidation of methyl 5-methoxy-3-(3,4,5-trimethoxyphenylthio)-1H-indole-2-carboxylate (36)10 with 1 or 2 equiv of 3-chloroperoxybenzoic acid (MCPBA) in chloroform at 25 °C for 1.5 h gave the corresponding sulfoxide (37)or sulfone (38) derivatives, respectively (Scheme 4). Ethyl 5-bromo-1H-indole-2-carboxylate (59) was prepared by intra-molecular cyclization of the corresponding ethyl pyruvate 4-bromophenylhydrazone (57) in polyphosphoric acid (PPA) as a catalyst, according to Fischer's indole synthesis. The needed hydrazone 57 was obtained by MW reaction of 4-bromophenylhydrazine hydrochloride with ethyl pyruvate and sodium acetate in ethanol at 100 °C (250 W) for 5 min. MW hydrolysis of ester 58 with 3 N sodium hydroxide at 110 °C (150 W) for 1 min furnished 5-bromo-1H-indole-2-carboxylic acid (59), which was transformed into the corresponding methyl ester 50 by treatment with thionyl chloride in anhydrous methanol at 65 °C for 2 h (Scheme 5).
Scheme 4.
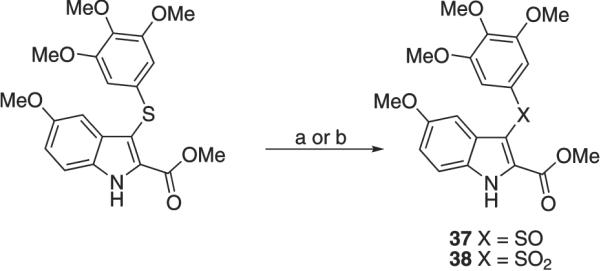
Synthesis of compounds 37 and 38a
a Reagents and reaction conditions: (a) (37) MCPBA (1 equiv), CHCl3,25 °C, 1.5 h, 99%; (b) (38) MCPBA (2 equiv), CHCl3,25 °C, 1.5 h, 96%.
Scheme 5.

Synthesis of compounds 57–60a
a Reagents and reaction conditions: (a) ethyl pyruvate, CH3COONa, EtOH, open vessel, 100 °C, 250 W, Pmax, 5 min, 90%; (b) PPA, 110 °C, 30 min, 60%; (c) 3 N NaOH, closed vessel, 110 °C, 150 W, 1 min, 95%; (d) SOCl2, absolute MeOH, 65 °C, 2 h, Ar stream, 96%.
Compounds 46–49 were synthesized by phase-transfer reaction from an appropriate ethyl 1H-pyrrole-2-carboxylate and 3,4,5-trimethoxybenzyl chloride15 in the presence of tetrabutylammoniun hydrogen sulfate (TBAHS) in 50% sodium hydroxide/dichloromethane. Reaction of 3,4,5-trimethoxybenzyl alcohol with ethyl 1H-imidazole-4-carboxylate in the presence of diisopropyl azodicarboxylate (DIPAD) according to the Mitsunobu reaction16 afforded ethyl 1-(3,4,5-trimethoxybenzyl)-1H-imidazole-4-carboxylate (50). Reaction of 3,4,5-trimethoxybenzoyl chloride with ethyl 1H-pyrrole-2-carboxylate in the presence of potassium tert-butoxide and 18-crown-6 or with ethyl 1H-imidazole-4-carboxylate gave ethyl 1-(3,4,5-trimethoxybenzoyl)-1H-pyrrole-2-carboxylate (51) or ethyl 1-(3,4,5-trimethoxybenzoyl)-1H-imidazole-4-carboxylate (52) and ethyl 1-(3,4,5-trimethoxybenzoyl)-1H-imidazole-5-carboxylate (53), respectively. The esters 46 and 50 were hydrolyzed into the corresponding carboxylic acids (54 and 56) by reacting the appropriate ester with potassium hydroxide in aqueous methanol or 37% hydrochloric acid, respectively. Ester 48 was treated with potassium hydroxide in diethylene glycol at 140 °C for 3 h to obtain the corresponding carboxylic acid, which underwent a transesterification reaction to furnish 2-(2-hydro-xyethoxy)ethyl 3,4,5-trimethyl-1-(3,4,5-trimethoxybenzyl)-1H-pyrrole-2-carboxylate (55) (Scheme 6). Attempts to obtain 3-(3,4,5-trimethoxybenzoyl)-1H-pyrrole by Friedel–Crafts acylation of 1-tosyl-1H-pyrrole with 3,4,5-trimethoxybenzoyl chloride in the presence of anhydrous aluminum chloride (2 equiv) invariably furnished (4-hydroxy-3,5-dimethoxyphenyl)-(1-tosyl-1H-pyrrol-3-yl)methanone (scheme not shown).
Scheme 6.

Synthesis of Compounds 46–56a
a Reagents and reaction conditions: (a) (46–49,X = H2,Y = Cl) appropriate ethyl 1H-pyrrole-2-carboxylate, 3,4,5-trimethoxybenzyl chloride, TBAHS, 50% KOH/DCM, 25 °C, overnight, 45–57%; (b) (50,X = H2,Y = OH) ethyl 1H-imidazole-4-carboxylate, 3,4,5-trimethoxybenzyl alcohol, P(Ph3)3, DIPAD, anhydrous THF, 25 °C, overnight, Ar stream, 10%; (c) (51, X = O,Y = Cl) ethyl 1H-pyrrole-2-carboxylate, 3,4,5-trimethoxybenzoyl chloride, 18-crown-6, t-BuOK, anhydrous THF, 25 °C, 4 h, 8%; (d) (52 and 53, X = O,Y = Cl) ethyl 1H-imidazole-4-carboxylate, 3,4,5-trimethoxybenzoyl chloride, anhydrous DCM, reflux temperature, 2.5 h, 13–15%; (e) (54) KOH, MeOH/H2O, reflux temperature, 3 h, 74%; (f) (55) diethylene glycol, KOH, 140 °C, 3 h, 34%; (g) (56) 37% HCl, reflux temperature, 1.5 h, 68%.
Results and Discussion
Biological data for inhibition of tubulin polymerization, binding of [3H]colchicine to tubulin (more active compounds only), and growth of MCF-7 human breast carcinoma cells by arylthioindoles 3–45 and reference compound 2 are summarized in Table 1. MCF-7 cell growth was quantitated by measuring cell protein with the dye sulforhodamine B after 48 h in the presence of various compound concentrations.
With few exceptions, tested derivatives showed potent tubulin polymerization inhibitory activity. Replacement of the sulfur atom with a carbonyl functionality led to compounds endowed with comparable ability to inhibit tubulin assembly (compare 3 with 4, 6 with 7, 9 with 10, 12 with 13, 15 with 16, 18 with 19, 24 with 25, 27 with 28, 30 with 31, 33 with 34, 36 with 39, and 42 with 43). Replacement of the carbonyl with a methylene group led to inhibitors of tubulin assembly whose potency was dependent on the substituent at position 2 of the indole. In particular, methylene compounds bearing a methoxy- or ethoxycarbonyl group at this position differed little in activities from the corresponding sulfur/carbonyl compounds (compare 8 with 6 and 7, 11 with 9 and 10, 17 with 15 and 16, 20 with 18 and 19, 29 with 27 and 28, 32 with 30 and 31, 41 with 39 and 36, and 45 with 43 and 42). In contrast, when there was a hydrogen atom at position 2, there was a 3–11-fold reduction in inhibitory activity (compare 5 with 3 and 4, 14 with 12 and 13, 26 with 24 and 25, and 35 with 33 and 34). Seven compounds (21–23, 37, 38, 40, and 44) were completely inactive. These results clearly indicate forbidden chemical modifications at the B region, namely (i) elongation of the bridging group from 1 to 2 atomic units (compounds 21–23), (ii) oxidation of the sulfur atom to oxide (37) or dioxide (37 and 38), as we also observed in our previous paper,11 and (iii) reduction of the carbonyl bridge to an alcohol (40 and 44).
As growth inhibitors of MCF-7 human breast carcinoma cells, the sulfur derivatives were superior (compare 3 with 4, 9 with 10, 24 with 25, 33 with 34, and 42 with 43) or equivalent (compare 6 with 7, 12 with 13, 15 with 16, 18 with 19, 27 with 28, 30 with 31, and 36 with 39) to the corresponding ketones, while methylene derivatives were usually less effective (compare 8 with 6 and 7, 17 with 15 and 16, 20 with 18 and 19, 29 with 27 and 28, 32 with 30 and 31, 41 with 39 and 36, and 45 with 42 and 53). (Generally, only inhibitors of tubulin polymerization with IC50<5 μM were evaluated for inhibition of MCF-7 cell growth). The greatest inhibitory effects on MCF-7 cells were observed with either sulfur or carbonyl derivatives bearing a 2-methoxycarbonyl group at position 2 of the indole.
Compounds bearing either a bromine atom or a methoxy group at position 5 and a 2-methoxycarbonyl group at position 2 of the indole were found to be potent inhibitors in both the tubulin polymerization and MCF-7 cell growth assays, with potencies comparable to those of reference compound 2. Worthy of note, among these esters, the methylene derivatives 29 and 41 were also highly active in both assays. In previous studies, ATIs showed comparable11,12 to greater10 inhibition of tubulin polymerizarion than 1 and adopted a binding orientation similar to that of DAMA-colchicine.10
Compounds that inhibited tubulin assembly with IC50's < 5 μM were also evaluated for inhibition of the binding of [3H]colchicine to tubulin. Although none was as potent as CSA4, significant inhibition was observed with all active agents. As usually occurs with a series of antitubulin compounds, there was not a linear correlation between colchicine binding inhibition and inhibition of tubulin assembly. The most active inhibitor of colchicine binding was compound 36, which was almost as active as CSA4, although the difference was more noticeable when the inhibitor concentration was 1 μM instead of 5 μM (data not presented). Compound 36 was about half as active as CSA4 as an inhibitor of assembly (IC50's of 2.0 and 1.1 μM, respectively). Compounds 27 and 39, both more active than CSA4 as inhibitors of assembly (IC50's of 0.99 and 0.67 μM, respectively), were less active than 36 as inhibitors of colchicine binding.
We designed derivatives 46–56 for correlation with the structure of the ATI's by replacing the indole ring with differently substituted pyrrole and imidazole analogues (Table 2). Predictive docking studies suggested the synthesis of 46–56 because we observed a close overlap between their trimethoxyphenyl group and the corresponding ring A of the cocrystallized DAMA-colchicine in the colchicine site on tubulin. However, only 51 showed any inhibition of tubulin polymerization and, in keeping with its weak activity, 51 did not inhibit MCF-7 cell growth. An explanation of these findings is suggested in the modeling section (see below). Surprisingly, compound 54, which was inactive against bovine tubulin and MCF-7 cells, effectively inhibited growth of the Saccharomyces cerevisiae BY4741 strain. Perhaps 54 might interact with yeast tubulin, and this hypothesis is under investigation in our laboratories.
The potent esters 24, 27–29, 36, 39, and 41 were evaluated at 0.5 and 1.0 μM for cell growth inhibition on human HeLa and HCT116/chr3 cells, derived from a cervix uterine carcinoma and a colon carcinoma, respectively. All compounds caused about 50% growth inhibition at the lower concentration tested (Table 3).
Table 3.
Effects of 24,27–29,36,39, and 41 on HeLa and HCT116/chr3 Cell Proliferation
| % of cell proliferationa,b |
||||
|---|---|---|---|---|
| HeLac |
HCT116/chr3d |
|||
| compd | 0.5 μM | 1 μM | 0.5 μM | 1 μM |
| 24 | 47±0.5 | 56±1.2 | 60±0.8 | 60±3.9 |
| 27 | 53±0.1 | 60±0.1 | 57±0.9 | 62±1.9 |
| 28 | 41±1.1 | 47±0.4 | 59±0.2 | 66±3.5 |
| 29 | 52±0.4 | 45±1.6 | 51±4.6 | 51±1.6 |
| 36 | 57±1.4 | 43±0.3 | 63±1.0 | 54±1.0 |
| 39 | 55±0.7 | 54±0.3 | 57±3.4 | 55±1.2 |
| 41 | 52±0.7 | 51±0.2 | 69±6.7 | 69±0.3 |
Data are expressed as % mean values ± SD; control cells are considered as 100% (MTT method).
Treatments were performed for 24 h at the indicated concentrations.
HeLa cervical cancer cells.
HCT116/chr3 human colon cancer cells.
Compounds 24, 27–29, 36, 39, and 41 also reduced the viability of HEK, M14, and U937 cells in a dose- and time-dependent manner with IC50 values ranging from 78 nM (28, M14 cells) to 220 nM (24, HEK cells) (Table 4). In contrast, at 300 nM, these compounds caused only 20% reduction in murine macrophage J744.1 cell growth compared with untreated controls (Figure 1S, Supporting Information). Thus these compounds were active in six cancer cell lines, but they showed a reduced cytototoxic effect on the nonmalignant murine macrophage J744.1 cells.
Table 4.
Inhibition of Growth of HEK, M14 and U937 Cells by Compounds 24, 27–29, 36, 39, and 41a
| IC50 ± SD (nM) |
|||
|---|---|---|---|
| compd | HEKb | M14c | U937d |
| 24 | 220±3 | 194±3 | 100±3 |
| 27 | 189±5 | 155±9 | 177±10 |
| 28 | 131±3 | 78±3 | 191±7 |
| 29 | 160±5 | 120±10 | 159±10 |
| 36 | 128±7 | 111±6 | 122±6 |
| 39 | 140±1 | 100±5 | 155±10 |
| 41 | 181±3 | 137±5 | 189±5 |
Growth inhibition of the indicated cell lines (MTT method); incubation time was 48 h.
HEK, human embryonic kidney 293 cells;
M14, human melanoma cells;
U937, human leukemic monocyte lymphoma cells.
This observation with the J744.1 cells raised the possibility that there might be a differential effect of esters 24, 27–29, 36, 39, and 41 in malignant and nontransformed cells, which in turn would suggest the possibility of a potential improvement in the therapeutic index for the ATI's versus other antitubulin agents. Accordingly, we expanded our study of nontrans-formed cells to an epithelial line (PtK2, Potorus tridactylis kidney epithelial cells), two aortic smooth muscle lines (human and rat), and an endothelial line (human umbilical vein endothelial cells). We also determined the effects of CSA4 and paclitaxel on the growth of these four lines (Table 5).
Table 5.
Inhibition of Growth and MCF-7 Selectivity Index of HAOSMC, PtK2, A10, and HUVEC Cells by Compounds 24, 27–29, 36, 39, and 41 and Reference Compounds 2 and Paclitaxela
| IC50 ± SD (nM) |
|||||
|---|---|---|---|---|---|
| compd | MCF-7 | HAOSMCb (MCF-7 SI)f | PtK2c (MCF-7 SI) | A10d (MCF-7 SI) | HUVECe (MCF-7 SI) |
| 24 | 43 ± 7 | 300 ± 200 (7.0) | 230 ± 100 (5.3) | 210 ± 100 (4.9) | 400 ± 100 (9.3) |
| 27 | 33 ± 10 | 100 ± 30 (3.0) | 280 ± 30 (8.5) | 230 ± 100 (7.0) | 140 ± 20 (4.2) |
| 28 | 18 ± 4 | 55 ± 40 (3.1) | 110 ± 60 (6.1) | 280 ± 40 (16) | 150 ± 0g (8.3) |
| 29 | 30 ± 9 | 35 ± 7 (1.2) | 270 ± 100 (9.0) | 230 ± 100 (7.7) | 140 ± 20 (4.7) |
| 36 | 13 ± 3 | 15 ± 7 4 (1.2) | 78 ± 50 (6.0) | 70 ± 70 (5.4) | 38 ± 30 (2.9) |
| 39 | 17 ± 6 | 35 ± 7 (2.1) | 190 ± 20 (11) | 95 ± 80 (5.6) | 110 ± 60 (6.5) |
| 41 | 33 ± 10 | 70 ± 10 (2.1) | 190 ± 200 (5.8) | 130 ± 100 (3.9) | 170 ± 50 (5.2) |
| 2 | 2.5 ± 0.6 | 18 ± 4 (7.2) | 1.0 ± 1 (0.4) | 23 ± 10 (9.2) | 5.0 ± 5 (2.0) |
| Ptxh | 3.0 ± 0.5 | 6.0 ± 6 (2.0) | 21 ± 8 (7.0) | 38 ± 10 (13) | 7.0 ± 4 (2.3) |
Growth inhibition of the indicated cell lines.
HAOSMC: human aortic smooth muscle cells (ATCC CLR-1999).
PtK2: potorous tridactylis kidney epithelial cells (ATCC CLL-56).
A10, rat embryonic aortic smooth muscle cells (ATCC CRL-1476).
HUVEC: human umbilical vein endothelial cells (ATCC CRL-2873).
MCF-7 SI: selectivity index (SI) for each agent in each cell line versus the MCF-7 cells (IC50 in specific cell line divided by IC50 in MCF-7 cells; the higher the SI, the more resistant the nontransformed cell line relative to the MCF-7 cells).
Same value obtained in both experiments.
Ptx: paclitaxel.
In all cases, except for compound 24 with the umbilical vein endothelial cells, the ATI's were more inhibitory than they had been with the J744.1 cells (that is, all IC50's were less than 300 nM). The cytotoxicity pattern with each agent was different, although compound 36 was the most cytotoxic and compound 24 was overall the least cytotoxic with these four nontransformed cell lines. We devised the MCF-7 SI as a test for the idea that the ATI's would have selective toxicity for a tumor cell line as compared with nonmalignant cells and as compared with previously described antitubulin agents. This hope, raised with the J744.1 macrophage cells, was not confirmed with the additional cell lines. The average SI was 4.8 with CSA4 and 6.1 with paclitaxel, although even with these two agents specific SI's differed from each other (the kidney epithelial cells were highly sensitive to CSA4, while both muscle lines were relatively resistant; with paclitaxel, the human muscle cells were most sensitive, the rat muscle cells most resistant). The average SI with the esters ranged from 4.3 with compound 41 to 7.3 with compound 28. We conclude that there is no significant difference between compounds 24, 27–29, 36, 39, and 41 as compared with CSA4 and paclitaxel.
The effect of drug treatment on cell morphology was examined by time-lapse microscopy (TLM) using a Leica CTR6500 microscope. Cells were kept at 37 °C under a 5% carbon dioxide atmosphere for 48 h. The cytotoxic effects of compounds 24, 27–29, 36, 39, and 41 on HEK and M14 cell growth increased in a concentration- and time-dependent manner. Figure 1 shows the effects of 24, 39, and 41 after a 48 h treatment. Control HEK and M14 cell cultures displayed elongated bipolar or polygonal morphology, while a significant effect on cell morphology and 12–30% growth inhibition was already observed as early as 12 h after compound addition (data not shown). The treatment for 48 h with 24, 39, or 41 induced significant morphologic changes in both cell lines, which became rounded and developed large vacuoles. Moreover, cells of both lines were less elongated than the untreated controls, and some cells that became rounded also had a tendency to detach from the substrate. Similar changes were observed after treatment with 27–29 and 36 (data not shown).
Figure 1.

Effects of compounds 24, 39, and 41 on HEK (top) and M14 (bottom) cell morphology and growth as seen by TLM (magnification 10×).
Saccharomyces cerevisiae budding yeast has been used to enhance understanding of fundamental cellular and molecular processes occurring in mammalian cells, including DNA replication, DNA recombination, cell division, protein turnover, vesicular trafficking, and mechanisms involved in longevity of cell life and cell death. Approximately 31% of yeast genes have a mammalian homologue, and an additional 30% of yeast genes show domain similarity.17 Potentially, yeast can be a powerful model for the development of cell death-directed drugs. For example, paclitaxel, arsenic, bleomycin, and valproate induce apoptotic phenotypes in yeast.18 Yeast has increased our understanding of the pathogenic role of human proteins in neurodegenerative diseases.19 Finally, Cassidy-Stone and co-workers20 identified MDIVI-1 (mitochondrial division inhibitor-1) by yeast screens of chemical libraries.
Here we set up a protocol to evaluate the toxicity of compounds 24, 27, 28, and 54 on BY4741, a standard laboratory wild type strain of S. cerevisiae. As shown in Figure 2, 24 and 54 strongly reduced the viability of the BY4741 strain cells, which were cell growth limited at the first dilution (the second showed limited cell growth). Compound 29 also showed significant BY4741 inhibition (cell growth was limited to the third dilution).
Figure 2.
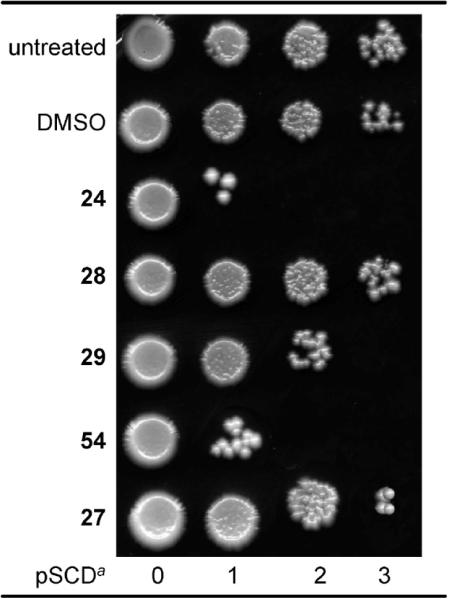
Effect of compounds 24, 27–29, and 54 on BY4741 wild type strain. Growth was recorded after 3 days at 28 °C; final compound concentration was 20 μM. a = serial cell dilution (SCD).
Despite the small test set used in this experiment, the ester functionality of 27–29 clearly caused a drop in activity as compared with the most potent compound 24 lacking the ester group. Even more striking is the contrast between the high activity of 54 with the yeast cells and its inactivity in the mammalian assays. Despite its inactivity with mammalian tubulin, it is unclear from docking studies why 54 does not bind in the colchicine site (see below). The biological behavior of 54 in yeast may indicate an interaction with fungal tubulin, or the activity of the compound in yeast may indicate an alternate target (manuscript in preparation).
Binding of Compounds to β-Tubulin in the Colchicine Site
On the basis of our previous modeling results,10–12 we performed a series of docking simulations on a virtual library of structures with different bridges (B) in order to identify the most promising compounds before their actual synthesis. We also investigated in silico the potential of replacing the indole ring with smaller heterocycles such as pyrrole or imidazole.
The results obtained for the indole series showed that replacement of the sulfur atom in the bridge (B) with a methylene or carbonyl moiety led to virtually identical docking poses as reported previously for ATIs. In particular, the trimethoxyphenyl moiety was situated in close proximity to Cys241, and the indole NH formed a hydrogen bond with Thr179 (Figure 3). These results predicted that these bridge modifications would lead to strong biological activity, and this was confirmed by the experimental results. Conversely, results for compounds with a two-carbon bridge were different. Such structures could not be correctly placed in the colchicine site. The trimethoxyphenyl ring was more distant from Cys241, predicting poor binding and low biological activity. Consequently, we synthesized only three 2-carbon bridge analogues (21–23) to test the model, and the experimental data confirmed the predictions of the docking results (Table 1 data).
Figure 3.

Docking poses for compounds 36, 39, and 41 (DAMA-colchicine in green).
Replacement of the indole ring with differently substituted pyrrole and imidazole analogues led to less clear results. Most of the docked structures showed the trimethoxyphenyl group in a position similar to that of the corresponding ring A of the cocrystallized DAMA-colchicine. However, the heterocycle moieties were placed in different positions compared with the indole ring of the ATIs, and none of the poses obtained showed any interaction with Thr179 (Figure 4). From these results, it was not easy to predict the potential activity of such compounds, and therefore a small series of pyrrole and imidazole analogues was prepared and tested experimentally. The biological data clearly showed that, in our model, the correct position of the trimethoxyphenyl group is necessary, but not sufficient, to predict the activity of novel compounds and that other structural features are required to have an accurate model. It should also be noted that our findings are in accord with the pharmacophore model previously developed for colchicine site compounds.21
Figure 4.

Docking poses for compounds 54 and 56 (compound 36 in green).
Conclusions
Replacement of the sulfur atom of arylthioindoles with a carbonyl functionality led to compounds endowed with comparable inhibition of tubulin assembly. Replacement of the carbonyl with a methylene moiety led to inhibitors of tubulin assembly whose potency was dependent on the substituent at position 2 of the indole ring. As growth inhibitors of MCF-7 human breast carcinoma cells, sulfur derivatives were superior or equivalent to the ketones, while methylene derivatives were generally less effective. Compounds bearing either a bromine atom or a methoxy group at position 5 and the 2-methoxycarbonyl group at position 2 of the indole ring were potent inhibitors of both tubulin polymerization and MCF-7 cell growth, with potencies comparable to those of reference compound 2. Two methylene derivatives, 29 and 41, were also highly active in both assays. Esters 24, 27–29, 36, 39,and 41 showed ~50% inhibition of human HeLa and HCT116/chr3 cell growth at 0.5 μM, and these compounds also reduced cell viability of HEK, M14, and U937 cells, with IC50's in the 78–220 nM range. In contrast, murine macrophage J744.1 cell growth was significantly less affected (20% at the highest concentrations). Four additional nontransformed cell lines, however, did not greatly differ in their sensitivity to compounds 24, 27–29, 36, 39, and 41. The effect of drug treatment on cell morphology was examined by time-lapse microscopy. After 48 h, HEK and M14 cells treated with 24, 27–29, 36, 39, or 41 showed significant morphologic changes (cells became rounded with large vacuoles and with a tendency to detach from the substrate). In a protocol set up to evaluate compound toxicity on S. cerevisiae BY4741 wild type strain, compounds 24 and 54 strongly reduced cell growth, and 29, 36,and 39 also showed significant inhibitory effects. The strong activity of 54, which was inactive against bovine tubulin and MCF-7 cells against BY4741 cells, requires further investigation.
Experimental Section
Chemistry
MW-assisted reactions were performed on Discover LabMate (CEM), setting temperature, irradiation power, maximum pressure (Pmax), PowerMAX (in situ cooling during the MW irradiation), ramp and hold times, and open and closed vessel modes as indicated. Melting points (mp) were determined on a Buchi 510 apparatus and are uncorrected. Infrared spectra (IR) were run on a SpectrumOne FT-ATR spectrophotometer. Band position and absorption ranges are given in cm−1. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Bruker 200 and 400 MHz FT spectrometers in the indicated solvent. Chemical shifts are expressed in δ units (ppm) from tetramethylsilane. Column chromatography was performed on columns packed with alumina from Merck (70–230 mesh) or silica gel from Macherey-Nagel (70–230 mesh). Aluminum oxide TLC cards from Fluka (aluminum oxide precoated aluminum cards with fluorescent indicator visualizable at 254 nm) and silica gel TLC cards from Macherey-Nagel (silica gel pre-coated aluminum cards with fluorescent indicator visualizable at 254 nm) were used for thin layer chromatography (TLC). Developed plates were visualized by a Spectroline ENF 260C/F UV apparatus. Organic solutions were dried over anhydrous sodium sulfate. Evaporation of the solvents was carried out on a Buchi Rotavapor R-210 equipped with a Buchi V-850 vacuum controller and Buchi V-700 and V-710 vacuum pumps. Elemental analyses of the biologically tested compounds were found within ±0.4% of the theoretical values. Purity of tested compounds was >95%. Compounds 3,12 6,10 9,10 12,12 15,10 18,11 24,12 33,12 36,10 and 4211 were prepared as previously reported.
General Procedure for the Synthesis of Compounds 4, 7, 10, 13, 16, 19, 21, 25, 28, 31, 34, 39, and 43. Example: (1H-Indol-3-yl)-(3,4,5-trimethoxyphenyl)methanone (4)
A mixture of AlCl3 (0.57 g; 0.0043 mol), the appropriate 1H-indole (0.5 g, 0.0043 mol) and 3,4,5-trimethoxybenzoyl chloride (0.99 g, 0.0043 mol) in 1,2-dichloroethane (2 mL) was placed into the MW cavity (closed vessel mode, Pmax = 250 PSI). MW irradiation of 150 W was used, the temperature being ramped from 25 to 110 °C while stirring. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min, then cooled, diluted cautiously with water, and extracted with chloroform; the organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate:n-hexane = 1:1 as eluent) to furnish 4 as a white solid (0.71 g, 53%), mp 210–213 °C (from ethanol), differing from lit.22 174–176 °C and lit.23 132–133 °C. 1H NMR (DMSO-d6): δ 3.73 (s, 3H), 3.83 (s, 6H), 7.06 (s, 2H), 7.20–7.24 (m, 2H), 7.48 (d, J = 9.1 Hz, 1H), 8.06 (d, J = 3.0 Hz, 1H), 8.21 (d, J = 8.5 Hz, 1H), 11.99 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1571, 3179 cm−1.
Methyl 3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate (7)
Synthesized as 4, starting from methyl 1H-indole-2-carboxylate, yield 55%, yellow solid, mp 163–168 °C (from ethanol/water). 1H NMR (DMSO-d6): δ 3.52 (s, 3H), 3.73 (s, 6H), 3.74 (s, 3H), 7.03 (s, 2H), 7.17 (t, J = 7.2 Hz, 1H), 7.34 (t, J = 7.2 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 7.60 (d, J =7.3 Hz, 1H), 12.56 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1634, 1710, 3279 cm−1. Anal. (C20H19NO6 (369.37)) C, H, N.
Ethyl 3-(3,4,5-Trimethoxybenzoyl)-1H-indole-2-carboxylate (10)
Synthesized as 4, starting from ethyl 5-methoxy-1H-indole-2-carboxylate, yield 39%, white solid, mp 105–110 °C (ethanol/n-hexane). 1H NMR (CDCl3): δ 1.01 (t, J = 7.1 Hz, 3H), 3.82 (s, 6H), 3.93 (s, 3H), 4.14 (q, J = 7.1 Hz, 2H), 7.18 (s, 2H), 7.24 (t, J = 7.1 Hz, 1H), 7.41 (t, J = 7.1 Hz, 1H), 7.5 (d, J = 8.3 Hz, 1H), 7.73 (d, J = 8.1 Hz, 1H), 9.26 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1648, 1686, 3303 cm−1. Anal. (C21H21NO6 (383.39)) C, H, N.
(5-Chloro-1H-indol-3-yl)-(3,4,5-trimethoxyphenyl)methanone (13)
Synthesized as 4, starting from 5-chloro-1H-indole, yield 68%, white solid, mp 240–245 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.86 (s, 6H), 7.09 (s, 2H), 7.28 (dd, J = 8.7 Hz, 1H), 7.54 (t, J = 8.1 Hz, 1H), 8.19 (s, 1H), 8.23 (d, J = 2.2 Hz, 1H), 12.21 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1600, 3261 cm−1. Anal. (C18H16ClNO4 (345.78)) C, H, Cl, N.
Methyl 5-Chloro-3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate (16)
Synthesized as 4, starting from methyl 5-chloro-1H-indole-2-carboxylate, yield 68%, yellow solid, mp 188–193 °C (from ethanol/water). 1H NMR (CDCl3): δ 3.69 (s, 3H), 3.84 (s, 6H), 3.96 (s, 3H), 7.16 (s, 2H), 7.36 (dd, J = 8.8 Hz, 1H), 7.44 (t, J = 8.8 Hz, 1H), 7.66 (d, J = 1.9 Hz, 1H), 9.50 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1645, 1711, 3259 cm−1. Anal. (C20H18ClNO6 (403.81)) C, H, Cl, N.
Ethyl 5-Chloro-3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate (19)
Synthesized as 4, starting from ethyl 5-chloro-1H-indole-2-carboxylate, yield 38%, yellow solid, mp 160−163 °C (from ethanol). 1H NMR (DMSO-d6): δ 0.86 (t, J = 7.1 Hz, 3H), 3.73 (s, 3H), 3.75 (s, 6H), 3.96 (q, J = 7.1 Hz, 2H), 7.04 (s, 2H), 7.38 (dd, J = 10.1 Hz, 1H), 7.58 (d, J = 7.4 Hz 1H), 7.69 (d, J = 2.1 Hz, 1H), 12.78 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1636, 1684, 3294 cm−1. Anal. (C21H20ClNO6 (417.84)) C, H, Cl, N.
Ethyl 5-Chloro-3-(2-(3,4,5-trimethoxyphenyl)acetyl)-1H-indole-2-carboxylate (21)
Synthesized as 4, starting from ethyl 5-chloro-1H-indole-2-carboxylate and 2-(3,4,5-trimethoxyphenyl)acetyl chloride, yield 32%, yellow solid, mp 129–134 °C (from ethanol). 1H NMR (CDCl3): δ 1.46 (t, J = 7.1 Hz, 3H), 3.80 (s, 6H), 3.83 (s, 3H), 4.40 (s, 2H), 4.50 (q, J = 7.1 Hz, 2H), 6.46 (s, 2H), 7.32 (dd, J = 8.8 Hz, 1H), 7.33 (dd, J = 9.5 Hz, 1H), 7.96 (d, J = 1.8 Hz, 1H), 9.22 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1641, 1721, 3169 cm−1. Anal. (C22H22ClNO6 (431.87)) C, H, Cl, N.
(5-Bromo-1H-indol-3-yl)(3,4,5-trimethoxyphenyl)methanone (25)
Synthesized as 4, starting from 5-bromo-1H-indole, yield 53%, yellow solid, mp 235–240 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.76 (s, 3H), 3.86 (s, 6H), 7.09 (s, 2H), 7.40 (dd, J = 6.6 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 8.18 (s, 1H), 8.39 (d, J = 2.5 Hz, 1H), 12.19 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1599, 3279 cm−1. Anal. (C18H16BrNO4 (390.23)) C, H, Br, N.
Methyl 5-Bromo-3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate (28)
Synthesized as 4, starting from 49, yield 40%, white solid, mp 207–210 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.68 (s, 3H), 3.83 (s, 6H), 3.95 (s, 3H), 7.14 (s, 2H), 7.38 (d, J = 8.2 Hz, 1H), 7.48 (dd, J = 6.9 Hz, 1H), 7.81 (d, J = 1.8 Hz, 1H), 9.45 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1642, 1711, 3248 cm−1. Anal. (C20H18BrNO6 (448.26)) C, H, Br, N.
Ethyl 5-Bromo-3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate (31)
Synthesized as 4, starting from 48, yield 46%, yellow solid, mp 135–140 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 0.85 (t, J = 7.1 Hz, 3H), 3.73 (s, 3H), 3.75 (s, 6H), 3.98 (q, J = 7.1 Hz, 2H), 7.03 (s, 2H), 7.33 (d, J = 1.2 Hz, 1H), 7.48 (dd, J = 6.9 Hz, 1H), 7.53 (dd, J = 8.8 Hz, 1H), 12.74 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1638, 1702, 3292 cm−1. Anal. (C21H20BrNO6 (462.29)) C, H, Br, N.
(5-Methoxy-1H-indol-3-yl)-(3,4,5-trimethoxyphenyl)methanone (34)
Synthesized as 4, starting from 5-methoxy-1H-indole, yield 42%, yellow solid, mp 202–207 °C (from ethanol), lit.14 194–195 °C.
Methyl 5-Methoxy-3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate (39)
Synthesized as 4, starting from methyl 5-methoxy-1H-indole-2-carboxylate, yield 20%, yellow solid, mp 168–173 °C (from ethanol/water). 1H NMR (DMSO-d6): δ 3.47 (s, 3H), 3.71 (s, 3H), 3.73 (s, 3H), 3.74 (s, 6H), 6.99–7.02 (m, 3H), 7.07 (d, J = 2.3 Hz, 1H), 7.44 (d, J = 8.95 Hz, 1H), 12.49 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1635, 1710, 3274 cm−1. Anal. (C21H21NO7 (399.39)) C, H, N.
Ethyl 5-Methoxy-3-(3,4,5-trimethoxybenzoyl)-1H-indole-2-carboxylate (43)
Synthesized as 4, starting from ethyl 5-methoxy-1H-indole-2-carboxylate, yield 60%, yellow solid, mp 150–155 °C (from ethanol). 1H NMR (CDCl3): δ 0.97 (t, J = 7.1 Hz, 3H), 3.82 (s, 3H), 3.84 (s, 6H), 3.94 (s, 3H), 4.09 (q, J = 7.1 Hz, 2H), 7.07 (dd, J = 6.5 Hz, 1H), 7.18 (s, 2H), 7.26 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 9.0 Hz, 1H), 9.21 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1647, 1694, 3353 cm−1. Anal. (C22H23NO7 (413.42)) C, H, N.
General Procedure for the Synthesis of Compounds 5, 14, 26, and 35. Example: 3-(3,4,5-trimethoxybenzyl)-1H-indole (5)
Sodium borohydride (0.80 g, 0.021 mol) was added to a solution of 4 (0.66 g, 0.0021 mol) in ethanol (85 mL). The reaction mixture was refluxed for 3 h, then cooled, cautiously diluted with water, and extracted with ethyl acetate; the organic layer was washed with brine, dried, and filtered. Evaporation of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate:n-hexane = 1:1 as eluent) to furnish 5 as a white solid (0.26 g, 42%), mp 133–136 °C (from ethanol), lit.22 125–126 °C and lit.24 128–130 °C.
5-Chloro-3-(3,4,5-trimethoxybenzyl)-1H-indole (14)
Synthesized as 5 starting from 13, yield 47%, orange solid, mp 145–150 °C (from ethanol). 1H NMR (CDCl3): δ 3.80 (s, 6H), 3.83 (s, 3H), 4.00 (s, 2H), 6.49 (s, 2H), 6.95 (d, J = 2.3 Hz, 1H), 7.14 (dd, J = 8.6 Hz, 1H), 7.28 (d, J = 8.1 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 8.40 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3372 cm−1. Anal. (C18H18ClNO3 (331.79)) C, H, Cl, N.
5-Bromo-3-(3,4,5-trimethoxybenzyl)-1H-indole (26)
Synthesized as 5, starting from 25, yield 58%, white solid, mp 145–150 °C (from ethanol). 1H NMR (CDCl3): 3.81 (s, 6H), 3.84 (s, 3H), 4.01 (s, 2H), 6.50 (s, 2H), 6.94 (d, J = 2.3 Hz, 1H), 7.24–7.28 (m, 2H), 7.69 (d, J = 1.7 Hz, 1H), 8.05 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3353 cm−1. Anal. (C18H18BrNO3)) C, H, Br, N.
5-Methoxy-3-(3,4,5-trimethoxybenzyl)-1H-indole (35)
Synthesized as 5, starting from 34, yield 68%, white solid, mp 110–113 °C (from ethanol). 1H NMR (CDCl3): δ 3.78 (s, 6H), 3.80 (s, 3H), 3.81 (s, 3H), 4.01 (s, 2H), 6.51 (s, 2H), 6.84 (d, J = 8.8 Hz, 1H), 6.88 (t, J = 2.4 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 7.24 (t, J = 4.7 Hz, 1H), 7.90 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 3365 cm−1. Anal. (C19H21NO4 (327.37)) C, H, N.
General Procedure for the Synthesis of Compounds 8, 11, 17, 20, 23, 29, 32, 41, and 45. Example: Methyl 3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate (8)
To a cold solution of 7 (0.31 g, 0.00084 mol) in trifluoroacetic acid (0.96 g, 0.65 mL, 0.0084 mol) was added dropwise triethylsilane (0.22 g, 0.30 mL, 0.0019 mol). The reaction mixture was stirred at 25 °C for 24 h, then neutralized with a saturated solution of sodium hydrogen carbonate and extracted with ethyl acetate; the organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate:n-hexane = 3:2 as eluent) to furnish 8 as a yellow solid (0.15 g, 50%), mp 156–161 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.69 (s, 6H), 3.72 (s, 3H), 3.89 (s, 3H), 4.38 (s, 2H), 6.46 (s, 2H), 7.05 (t, J = 8.1 Hz, 1H), 7.25 (t, J = 8.3 Hz, 1H), 7.33 (d, J = 7.4 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H), 8.75 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1690, 3318 cm−1. Anal. (C20H21NO5 (355.38)) C, H, N.
Ethyl 3-(3,4,5-Trimethoxybenzyl)-1H-indole-2-carboxylate (11)
Synthesized as 8, starting from 10, yield 73%, white solid, mp 115–118 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 1.42 (t, J = 7.1 Hz, 3H), 3.77 (s, 6H), 3.80 (s, 3H), 4.44 (q, J = 7.1 Hz, 2H), 4.47 (s, 2H), 6.55 (s, 2H), 7.13 (t, J = 7.0 Hz, 1H), 7.33 (t, J = 6.9 Hz, 1H), 7.39 (d, J = 7.5 Hz, 1H), 7.64 (d, J = 7.4 Hz, 1H), 8.82 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1672, 3337 cm−1. Anal. (C21H23NO5) (369.41)) C, H, N.
Methyl 5-Chloro-3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate (17)
Synthesized as 8, starting from 16, yield 69%, white solid, mp 172–175 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.70 (s, 6H), 3.73 (s, 3H), 3.89 (s, 3H), 4.33 (s, 2H), 6.42 (s, 2H), 7.21 (d, J = 1.9 Hz, 1H), 7.24 (t, J = 8.7 Hz, 1H), 7.51 (d, J = 1.9 Hz, 1H), 8.74 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1682, 3322 cm−1. Anal. (C20H20ClNO5 (389.83)) C, H, Cl, N.
Ethyl 5-Chloro-3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate (20)
Synthesized as 8, starting from 19, yield 95%, yellow solid, mp 147–150 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 1.41 (t, J = 7.1 Hz, 3H), 3.79 (s, 6H), 3.81 (s, 3H), 4.41 (s, 2H), 4.43 (q, J = 6.4 Hz, 2H), 6.51 (s, 2H), 7.25–7.28 (m, 1H), 7.33 (dd, J = 8.1 Hz, 1H), 7.59–7.60 (m, 1H), 8.91 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1666, 3315 cm−1. Anal. (C21H22ClNO5)) C, H, Cl, N.
Ethyl 5-Chloro-3-(3,4,5-trimethoxyphenethyl)-1H-indole-2-carboxylate (23)
Synthesized as 8, startingfrom 22, yield 40%, white solid, mp 163–165 °C (from ethanol). 1H NMR (CDCl3): δ 1.44 (t, J = 7.5 Hz, 3H), 2.88 (t, J = 7.4 Hz, 2H), 3.35 (t, J = 7.3 Hz, 2H), 3.79 (s, 6H), 3.83 (s, 3H), 4.42 (t, J = 7.1 Hz, 2H), 6.38 (s, 2H), 7.24 (dd, J = 9.2 Hz, 1H), 7.31 (d, J = 8.8 Hz, 1H), 7.49 (d, J = 1.9 Hz, 1H), 9.01 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1669, 3318 cm−1. Anal. (C22H24ClNO5 (417.88)) C, H, Cl, N.
Methyl 5-Bromo-3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate (29)
Synthesized as 8, starting from 28, yield 97%, yellow solid, mp 177-182 °C (from ethanol/n-hexane). 1H NMR (DMSO-d6): δ 3.57 (s, 3H), 3.67 (s, 6H), 3.92 (s, 3H), 4.34 (s, 2H), 6.62 (s, 2H), 7.36–7.36 (m, 2H), 7.90 (d, J = 2.3 Hz, 1H), 11.88 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1675, 3322 cm−1. Anal. (C20H20BrNO5 (434.28)) C, H, Br, N.
Ethyl 5-Bromo-3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate (32)
Synthesized as 8, starting from 31, yield 46%, white solid, mp 150–155 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 1.41 (t, J = 7.1 Hz, 3H), 3.79 (s, 6H), 3.82 (s, 3H), 4.43 (q, J = 7.1 Hz, 2H), 6.51 (s, 2H), 7.28 (d, J = 9.4 Hz, 1H), 7.29 (s, 2H), 7.40 (dd, J = 6.9 Hz, 1H), 7.77 (d, J = 1.8 Hz, 1H), 8.90 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1665, 3317 cm−1. Anal. (C21H22BrNO6 (448.31)) C, H, Br, N.
Methyl 5-Methoxy-3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate (41)
Synthesized as 8, starting from 39, yield 58%, white solid, mp 125–128 °C (from ethanol/n-hexane). 1H NMR (CDCl3): δ 3.77 (s, 6H), 3.79 (s, 3H), 3.80 (s, 3H), 3.94 (s, 3H), 4.42 (s, 2H), 6.53 (s, 1H), 6.96 (d, J = 2.3 Hz, 1H), 7.00 (dd, J = 6.4 Hz, 1H), 7.28 (d, J = 8.9 Hz, 1H), 8.67 (s, 1H), 11.91 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1687, 3331 cm−1. Anal. (C21H23NO6 (385.41)) C, H, N.
Ethyl 5-Methoxy-3-(3,4,5-trimethoxybenzyl)-1H-indole-2-carboxylate (45)
Synthesized as 8, starting from 43, yield 90%, white solid, mp 132–135 °C (from ethanol). 1H NMR (CDCl3): δ 1.42 (t, J = 7.1 Hz, 3H), 3.78 (s, 9H), 3.81 (s, 3H), 4.44 (q, J = 7.2 Hz, 2H), 4.45 (s, 2H), 6.56 (s, 2H), 6.98–7.03 (m, 2H), 7.28–7.32 (m, 1H), 8.76 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1672, 3330 cm−1. Anal. (C22H25NO6 (399.44)) C, H, N.
General Procedure for the Synthesis of Compounds 40 and 44. Example: Methyl 3-(Hydroxy(3,4,5-trimethoxyphenyl)methyl)-5-methoxy-1H-indole-2-carboxylate (40)
A mixture of 39 (0.34 g, 0.00085 mol) and sodium borohydride (0.03 g, 0.00085 mol) in THF (2.1 mL) and water (0.12 mL) was refluxed for 2 h. After cooling, water and ethyl acetate were added. The organic layer was removed and washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate:n-hexane=1:1 as eluent) to furnish 40 as a brown solid (0.1 g, 29%), mp 100–103 °C (from ethanol). 1H NMR (CDCl3): δ 3.76 (s, 3H), 3.79 (s, 6H), 3.82 (s, 3H), 3.97 (s, 3H), 4.53 (d, J = 1.2 Hz, 1H), 6.50 (d, J = 7.4 Hz, 1H), 6.77 (s, 2H), 6.95 (d, J = 2.4 Hz, 1H), 7.02 (dd, J = 6.5 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 8.78 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1707, 2937, 3322 cm−1. Anal. (C21H23NO7 (401.41)) C, H, N.
Ethyl 3-(Hydroxy(3,4,5-trimethoxyphenyl)methyl)-5-methoxy-1H-indole-2-carboxylate (44)
Synthesized as 40, starting from 43, yield 53%, white solid, mp 115–120 °C(from ethanol). 1H NMR (CDCl3): δ 1.42 (t, J = 7.2 Hz, 3H), 3.76 (s, 3H), 3.79 (s, 3H), 3.81 (s, 3H), 3.83 (s, 3H), 4.43 (q, J = 6.0 Hz, 2H), 4.44 (d, J = 1.2 Hz, 1H), 6.49 (d, J = 7.2 Hz, 1H), 6.77 (s, 2H), 6.95 (d, J = 2.3 Hz, 1H), 7.01 (dd, J = 6.6 Hz, 1H), 7.29 (d, J = 7.3 Hz, 1H), 8.78 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1676, 3209, 3402 cm−1.Anal. (C22H25NO7 (415.44)) C, H, N.
Ethyl 5-Chloro-3-(2-oxo-2-(3,4,5-trimethoxyphenyl)acetyl)-1H-indole-2-carboxylate (22)
A mixture of 21 (0.05 g, 0.00012 mol) and selenium(IV) oxide (0.05 g; 0.00045 mol) in DMSO (2 mL) was placed into the MW cavity (closed vessel mode, Pmax = 250 PSI). MW irradiation of 150 W was used, the temperature being ramped from 25 to 150 °C. Once 150 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min, while stirring, then cooled, diluted with water, and extracted with ethyl acetate; organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate:n-hexane=1:2 as eluent) to furnish 22 as a yellow solid (0.03 g, 56%), mp 177–181 °C (from ethanol). 1H NMR (CDCl3): δ 1.07(t, J = 7.2 Hz, 3H), 3.93 (s, 6H), 3.98 (s, 3H), 4.12 (q, J = 7.1 Hz, 2H), 7.35 (s, 2H), 7.42–7.43 (m, 2H), 8.40 (s, 1H), 9.50 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1650, 1672, 1724, 3275 cm−1. Anal. (C22H20ClNO7 (445.85)).
Methyl 5-Bromo-3-(3,4,5-trimethoxyphenylthio)-1H-indole-2-carboxylate (27)
A mixture of 59 (0.25 g, 0.001 mol), 3,4,5-bis(3,4,5-trimethoxyphenyl)disulfide10 (0.55 g, 0.0014 mol), and sodium hydride (0.071 g, 0.003 mol, 60% in mineral oil) in anhydrous DMF (2 mL) was placed into the MW cavity (closed vessel mode, Pmax = 250 PSI). MW irradiation of 150 W was used, the temperature being ramped from 25 to 110 °C. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min, while stirring, then cooled and quenched on crushed ice and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave crude 5-bromo-3-(3,4,5-trimethoxyphenylthio)-1H-indole-2-carboxylic acid, which was used without further purification. The crude acid was dissolved in methanol (2.5 mL) and dichloromethane (10 mL) and treated with TMSDM (0.78 mL, 0.0015 mol, 2.0 M in hexane) while stirring at 25 °C for 30 min. Removal of the solvent gave a crude product that was purified by silica gel column chromatography (ethyl acetate:n-hexane=1:1) to furnish 27 as a white solid (0.36 g, overall yield 79%), mp 160–162 °C (from ethanol). 1H NMR (CDCl3): δ 3.72 (s, 6H), 3.81 (s, 3H), 3.98 (s, 3H), 6.48 (s, 2H), 7.33 (d, J = 9.3 Hz, 1H), 7.42 (dd, J = 1.9 and 9.3 Hz, 1H), 7.72 (d, J = 1.9 Hz, 1H), 9.20 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1680, 3296 cm−1. Anal. (C19H18BrNO5S(452.32))
Ethyl 5-Bromo-3-(3,4,5-trimethoxyphenylthio)-1H-indole-2-carboxylate (30)
Synthesized as 27, starting from 59 and 3,4,5-bis(3,4,5-trimethoxyphenyl)disulfide.10 The crude acid (0.5 g) was dissolved in absolute ethanol (1.7 mL) and treated with thionyl chloride (0.13 mL) at 0 °C under an Ar stream. The reaction mixture was stirred at 0 °C for 20 min, heated at 65 °C for 2 h, then cooled and diluted with water and ethyl acetate. The organic layer was removed and washed with a saturated solution of sodium hydrogen carbonate and brine, dried, and filtered. Removal of the solvent gave a crude product that was purified by silica gel column chromatography (ethyl acetate:n-hexane = 1:1) to give 30 as a yellow solid (0.33 g, overall yield 70%), mp 150–152 °C (from ethanol). 1H NMR (CDCl3): δ 1.37 (t, J = 7.1 Hz, 3H), 3.72 (s, 6H), 3.80 (s, 3H), 4.43 (q, J = 7.8 Hz, 2H), 6.48 (s, 2H), 7.33 (d, J = 8.7 Hz, 1H), 7.43–7.46 (m, 1H) 7.74–7.75 (m, 1H), 9.23 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1672, 3299 cm−1. Anal. (C20H20BrNO5S (452.32)).
Methyl 5-Methoxy-3-(3,4,5-trimethoxyphenylsulfinyl)-1H-indole-2-carboxylate (37)
To a cold solution of 3610 (0.05 g, 0.00012 mol) in chloroform (5 mL) was added 3-chloroperoxybenzoic acid (0.021 g, 0.00012 mol). The reaction mixture was stirred at 25 °C for 1.5 h, then diluted with water and extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate as eluent) to furnish 37 as a brown solid (0.05 g, yield 99%), mp 161–163 °C (from ethanol). 1H NMR (CDCl3): δ 3.74 (s, 3H), 3.84 (s, 9H), 3.85 (s, 3H), 6.96 (d, J = 9.1 Hz, 1H), 7.10 (s, 2H), 7.31 (dd, J = 1.9 and 9.0 Hz, 1H), 7.42 (d, J = 2.2 Hz, 1H), 9.37 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1708, 2831, 2933 cm−1. Anal. (C20H21NO7S (419.45)) C, H, N, S.
Methyl 5-Methoxy-3-(3,4,5-trimethoxyphenylsulfonyl)-1H-indole-2-carboxylate (38)
Was synthesized as 37, treating 3610 with 3-chloroperoxybenzoic acid (2 equiv), 96%, yellow solid, mp 176–180 °C (from ethanol). 1H NMR (DMSO-d6): δ 3.33 (s, 3H), 3.82 (s, 6H), 3.83 (s, 3H), 3.93 (s, 3H), 7.04 (dd, J = 6.5 Hz, 1H), 7.40 (s, 2H), 7.45 (d, J = 9.0 Hz, 1H), 7.59 (d, J = 2.4 Hz, 1H), 12.99 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1701, 3298 cm−1. Anal. (C20H21NO8S (435.45)) C, H, N, S.
Ethyl 1-(3,4,5-Trimethoxybenzyl)-1H-pyrrole-2-carboxylate (46)
3,4,5-Trimethoxybenzyl chloride15 (1.083 g, 0.005 mol) was added to a well stirred mixture of ethyl 1H-pyrrole-2-carboxylate (0.63 g, 0.0045 mol), tetrabutylammonium hydrogen sulfate (1.53 g, 0.0045 mol), a 50% potassium hydroxide aqueous solution (15 mL) and dichloromethane (22 mL). The reaction mixture was stirred at 25 °C overnight, neutralized with 6 N hydrochloric acid and extracted with chloroform. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (dichloromethane as eluent) to furnish 46 as a yellow oil (0.64 g, 45%). 1H NMR (CDCl3): δ 1.21 (t, J = 7.1 Hz, 3H), 3.68 (s, 6H), 3.71 (s, 3H), 4.15 (q, J = 7.1 Hz, 2H), 5.39 (s, 2H), 6.07 (dd, J = 3.9 and 2.6 Hz, 1H), 6.24 (s, 2H), 6.78 (t, J = 2.1 Hz, 1H), 6.91 ppm (dd, J = 3.9 and 1.8 Hz, 1H). IR: ν 1701 cm−1. Anal. (C17H21NO5 (319.35)) C, H, N.
Ethyl 3,5-Dimethyl-1-(3,4,5-trimethoxybenzyl)-1H-pyrrole-2-carboxylate (47)
Was synthesized as 46, starting from ethyl 3,5-dimethyl-1H-pyrrole-2-carboxylate, yield 56%, pink solid, mp 68–70 °C (from toluene). 1H NMR (CDCl3): δ 1.16 (t, J = 7.1 Hz, 3H), 2.02 (s, 3H), 2.22 (s, 3H), 3.61 (s, 6H), 3.66 (s, 3H), 4.10 (q, J = 7.1 Hz, 2H), 5.37 (s, 2H), 5.71 (s, 1H), 6.03 ppm (s, 2H). IR: ν 1703 cm−1. Anal. (C19H25NO5 (347.41)) C, H, N.
Ethyl 3,4,5-Trimethyl-1-(3,4,5-trimethoxybenzyl)-1H-pyrrole-2-carboxylate (48)
Was synthesized as 46, starting from ethyl 3,4,5-trimethyl-1H-pyrrole-2-carboxylate, yield 45%, yellow solid, mp 82–84 °C (from ethanol). 1H NMR (CDCl3): δ 0.36 (t, J = 7.1 Hz, 3H), 1.03 (s, 3H), 1.17 (s, 3H), 1.36 (s, 3H), 2.81 (s, 6H), 2.86 (s, 3H), 3.30 (q, J = 7.1 Hz, 2H), 4.56 (s, 2H), 5.22 ppm (s, 2H). IR: ν 1710 cm−1. Anal. (C20H27NO5 (361.44)) C, H, N.
Ethyl 3,4-Diethyl-5-methyl-1-(3,4,5-trimethoxybenzyl)-1H-pyr-role-2-carboxylate (49)
Was synthesized as 46, starting from ethyl 3,4-diethyl-5-methyl-1H-pyrrole-2-carboxylate, yield 57%, yellow oil. 1H NMR (CDCl3): δ 0.99 (t, J = 7.5 Hz, 3H), 1.07 (t, J = 7.3 Hz, 3H), 1.21 (t, J = 7.1 Hz, 3H), 2.02 (s, 3H), 2.35 (q, J = 7.5 Hz, 2H), 2.68 (q, J = 7.4 Hz, 2H), 3.64 (s, 6H), 3.71 (s, 3H), 4.15 (q, J = 7.1 Hz, 2H), 5.44 (s, 2H), 5.96 ppm (s, 2H). IR: ν 1708 cm−1. Anal. (C22H31NO5 (389.49)) C, H, N.
Ethyl 1-(3,4,5-Trimethoxybenzyl)-1H-imidazole-4-carboxylate (50)
A solution of DIPAD (1.29 g, 1.25 mL, 0.0064 mol) in anhydrous THF (18 mL) was added dropwise to a mixture of 3,4,5-trimethoxybenzyl alcohol (1.26 g, 0.0064 mol), ethyl 1-H-imidazole-4-carboxylate26 (1.0 g, 0.0071 mol), and anhydrous triphenylphosphine (1.86 g, 0.0071 mol) in the same solvent (35 mL). The reaction was stirred at 25 °C overnight under an Ar stream. The reaction mixture was concentranted in vacuo, diluted with water, and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate:methanol = 45:5 as eluent) to furnish 50 as a colorless oil (0.2 g, 10%). 1H NMR (CDCl3): δ 1.26 (t, J = 7.4 Hz, 3H), 3.72 (s, 6H), 3.75 (s, 3H), 4.22 (q, J = 7.1 Hz, 2H), 5.36 (s, 2H), 6.33 (s, 2H), 7.54 (s, 1H), 7.69 ppm (s, 1H). IR: ν 1695 cm−1. Anal. (C16H20N2O5 (320.34)) C, H, N.
Ethyl 1-(3,4,5-Trimethoxybenzoyl)-1H-pyrrole-2-carboxylate (51)
To a stirred mixture of potassium tert-butoxide (0.26 g, 0.0023 mol) and 18-crown-6 (0.47 g, 0.0018 mol) in anhydrous THF (25 mL) was added dropwise a solution of ethyl 1H-pyrrole-2-carboxylate (0.25 g, 0.0018 mol) in the same solvent (25 mL). After cooling to 0 °C for 15 min, a solution of 3,4,5-trimethoxybenzoyl chloride (0.41 g, 0.0018 mol) in the same solvent (25 mL) was added dropwise. The reaction mixture was stirred at 25 °C for 4 h and then concentranted to a small volume and extracted with ethyl acetate. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (dichloromethane as eluent) to furnish 51 as an orange oil (0.05 g, 8%). 1H NMR (CDCl3): δ 1.09 (t, J = 7.1 Hz, 3H), 3.78 (s, 6H), 3.82 (s, 3H), 4.02 (q, J = 7.1 Hz, 2H), 6.24 (m, 1H), 6.94 (s, 2H), 6.99 (m, 1H), 7.18 ppm (m, 1H). IR: ν 1670, 1703 cm−1. Anal. (C17H19NO6 (333.34)) C, H, N.
Ethyl 1-(3,4,5-Trimethoxybenzoyl)-1H-imidazole-4-carboxy-late (52) and Ethyl 1-(3,4,5-Trimethoxybenzoyl)-1H-imidazole-5-carboxylate (53)
To a solution of ethyl imidazole 4-carboxylate26 (0.5 g, 0.0035 mol) in anhydrous dichloromethane (10 mL) was added dropwise a solution of 3,4,5-trimethoxybenzoyl chloride (0.41 g, 0.0018 mol) in the same solvent (10 mL). The reaction mixture was refluxed for 2.5 h and, after cooling, quenched on crushed ice. The organic layer was removed and washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate as eluent). The initial eluate furnished 52 as a white solid (0.17 g, 15%), mp 130–133 °C (from ethanol). 1H NMR (CDCl3): δ 1.34 (t, J = 7.1 Hz, 3H), 3.84 (s, 6H), 3.90 (s, 3H), 4.34 (q, J = 7.1 Hz, 2H), 6.96 (s, 2H), 8.06 (s, 1H), 8.09 ppm (s, 1H). IR: ν 1672, 1702 cm−1. Anal. (C16H18N2O6 (334.32)) C, H, N. Further elution with the same solvent afforded 53 as a white solid (0.12 g, 13%), mp 140–143 °C (from ethanol). 1H NMR (CDCl3): δ 1.40 (t, J = 7.1 Hz, 3H), 3.90 (s, 6H), 3.97 (s, 3H), 4.40 (q, J = 7.1 Hz, 2H), 7.02 (s, 2H), 8.16 ppm (s, 2H). IR: ν 1674, 1707 cm−1. Anal. (C16H18N2O6 (334.32)) C, H, N.
1-(3,4,5-Trimethoxybenzyl)-1H-pyrrole-2-carboxylic acid (54)
A mixture of 46 (0.23 g, 0.00072 mol) and potassium hydroxide (0.12 g, 0.0022 mol) in methanol:water (1:1) (10 mL) was refluxed for 3 h. After cooling, the reaction mixture was washed with dichloromethane; the aqueous layer was made acidic with 2 N hydrochloric acid (pH = 2) and extracted with dichloromethane. The organic layer was washed with water, dried, filtered, and evaporated to give 54 as a brown solid (0.15 g, 74%), mp 80–81 °C (from ethanol). 1H NMR (CDCl3): δ 3.68 (s, 6H), 3.71 (s, 3H), 5.36 (s, 2H), 6.12 (dd, J = 3.85 and 2.6 Hz, 1H), 6.27 (s, 2H), 6.85 (t, J = 2.0 Hz, 1H), 7.04 (dd, J = 3.9 and 1.7 Hz, 1H), 10.69 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1703, 2995 cm−1. Anal.(C15H17NO5 (291.30)) C, H, N.
2-(2-Hydroxyethoxy)ethyl 3,4,5-Trimethyl-1-(3,4,5-trimethoxybenzyl)-1H-pyrrole-2-carboxylate (55)
A mixture of 48 (0.05 g, 0.0014 mol) and potassium hydroxide (0.043 g, 0.00077 mol) in diethylene glycol (1 mL) was heated at 140 °C for 3 h. After cooling, water and ethyl acetate were added and the layers separated. The organic layer was washed with brine, dried, and filtered. Removal of the solvent gave a residue that was purified by silica gel column chromatography (ethyl acetate as eluent) to give 55 as a yellow oil (0.02 g, 34%). 1H NMR (CDCl3): δ 1.89 (s, 3H), 2.02 (s, 3H), 2.23 (s, 3H), 3.50 (m, 2H), 3.62 (m, 2H), 3.66 (m, 3H), 3.68 (s, 6H), 3.73 (s, 3H), 4.28 (m, 2H), 5.41 (s, 2H), 6.06 ppm (s, 2H). IR: ν 1705, 2836 cm−1. Anal. (C22H31NO7 (421.28)) C, H, N.
1-(3,4,5-Trimethoxybenzyl)-1H-imidazole-4-carboxylic Acid Hydrochloride (56)
A mixture of 50 (0.08 g, 0.00025 mol) and 37% hydrochloric acid (1.5 mL) was refluxed for 1.5 h. After cooling, the reaction mixture was washed with ethyl acetate. The aqueous layer was evaporated in vacuo. The residue was dissolved in 2-propanol (1.5 mL) and cooled to −78 °C. Upon addition of n-hexane, the suspension was filtered to give 56 as a white solid (0.05 g, 68%), mp 163–166 °C (from methanol). 1H NMR (CD3OD): δ 3.77 (s, 3H), 3.85 (s, 6H), 5.73 (s, 2H), 6.82 (s, 2H), 8.24 (s, 1H), 9.22 (s, 1H), 12.10 (broad s, disappeared on treatment with D2O, 1H). IR: ν 1705, 3100 cm−1. Anal. (C14H16N2O5 · HCl (328.75)) C, H, Cl, N.
Ethyl 2-(2-(4-Bromophenyl)hydrazono)propanoate (57)
A mixture of 4-bromophenylhydrazine hydrochloride (5.7 g, 0.0255 mol), ethyl pyruvate (2 g, 1.91 mL, 0.017 mol), and sodium acetate (2.79 g, 0.034 mol) in ethanol (40 mL) was placed into the MW cavity (open vessel mode). MW irradiation at 250 W was used, the temperature being ramped from 25 to 100 °C. Once 100 °C was reached, taking about 2 min, the reaction mixture was held at this temperature for 5 min while stirring. The reaction mixture was cooled at 0 °C, with stirring, and filtered to give 57, yield 90%, mp 142–144 °C (from ethanol). 1H NMR (CDCl3): δ 1.39 (t, J = 7.2 Hz, 3H), 2.12 (s, 3H), 4.33 (q, J = 7.1 Hz, 2H), 7.10 (d, J = 6.7 Hz, 2H), 7.40 (d, J = 6.9 Hz, 2H), 7.64 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1576, 1675, 3290 cm−1. Anal. (C11H13BrN2O2 (285.14)).
Ethyl 5-Bromo-1H-indole-2-carboxylate (58)
Compound 57 (35.15 g, 0.123 mol) was added in portions to PPA (350 g) preheated at 110 °C. The mixture was stirred at the same temperature for 30 min and then quenched with ice–water. The solid was filtered, washed with water, dried, and recrystallized from ethanol to give 58 as a brown solid (32.95 g, 60%), mp 160–161 °C. 1H NMR (CDCl3): δ 1.43 (t, J = 7.1 Hz, 3H), 4.43 (q, J = 7.1 Hz, 2H), 7.15–7.16 (m, 1H), 7.31 (d, J = 8.1 Hz, 1H), 7.41 (dd, J = 7.0 Hz, 1H), 7.84 (m, 1H), 9.98 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1690, 3311 cm−1. Anal. (C11H10BrNO2 (268.11)).
5-Bromo-1H-indole-2-carboxylic Acid (59)
A mixture of 58 (0.25 g, 0.001 mol) in 3 N sodium hydroxide (2 mL) was placed into the MW cavity (closed vessel mode, Pmax = 250 PSI). MW irradiation at 150 W was used, the temperature being ramped from 25 to 110 °C. Once 110 °C was reached, taking about 1 min, the reaction mixture was held at this temperature for 2 min while stirring. After cooling, the reaction mixture was made acidic with 3 N hydrochloric acid (pH = 2) and extracted with ethyl acetate; the organic layer was washed with brine, dried, filtered, and evaporated to give 59 as a brown solid (0.23 g, 95%), mp 188 °C (from ethanol). 1H NMR (DMSO-d6): δ 7.05 (s, 1H), 7.32 (dd, J = 8.9 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 7.85 (d, J = 2.1 Hz, 1H), 11.94 (broad s, disappeared on treatment with D2O, 1H), 12.61 ppm (broad s, disappeared on treatment with D2O, 1H). IR: ν 1652, 2850, 3422 cm−1. Anal. (C9H6BrNO2 (240.05)).
Methyl 5-Bromo-1H-indole-2-carboxylate (60)
To a cold suspension of 59 (3.4 g, 0.014 mol) in anhydrous methanol (13.4 mL) was added thionyl chloride (1.6 mL) dropwise. The reaction mixture was stirred at the same temperature for 20 min and then heated at 65 °C for 2 h under an Ar stream, then cooled and diluted with water and ethyl acetate. The layers were separated, and the organic phase was washed with a saturated solution of sodium hydrogen carbonate and brine, dried, filtered, and evaporated to give 60 as a white solid (3.43 g, 96%), mp 217–220 °C (from ethanol/cyclohexane), lit.25 211.8–213.6.
2-(3,4,5-Trimethoxyphenyl)acetyl Chloride
This compound was synthesized by treating a cold solution of 3,4,5-trimethoxyphenylacetic acid (0.25 g, 0.0011 mol) in anhydrous THF (5 mL) with oxalyl chloride (0.28 g, 0.19 mL, 0.0022 mol). Immediately afterward, a catalytic amount (2 drops) of anhydrous DMF was added, and vigorous bubbling ensued. After 10 min, the reaction mixture was carefully concentrated in vacuo to provide a viscous oil. Fresh anhydrous THF was added, and the solution was concentrated in vacuo again to furnish 2-(3,4,5-trimethoxyphenyl)acetyl chloride (0.27 g, yellow oil), which was used without further purification.
Molecular Modeling
All molecular modeling studies were performed on a MacPro dual 2.66 GHz Xeon running Ubuntu 8. The tubulin structure was downloaded from the PDB (http://www.rcsb.org/: PDB code 1SA0).3 Hydrogen atoms were added to the protein, using Molecular Operating Environment (MOE) 2007.09,27 and minimized, keeping all the heavy atoms fixed until a rmsd gradient of 0.05 kcal mol−1 Å−1 was reached. Ligand structures were built with MOE and minimized using the MM-FF94x forcefield until a rmsd gradient of 0.05 kcal mol−1 Å−1 was reached. The docking simulations were performed using FlexX28 with the MOE interface. The rmsd of the trimethoxyphenyl moiety for each of the compounds evaluated was calculated in comparison with ring A of DAMA-colchicine, and the results from the docking were scored using this value.29 The images presented here were created with Zodiac.30
Biology. Tubulin Assembly
The reaction mixtures contained 0.8 M monosodium glutamate (pH 6.6 with hydrochloric acid in 2 M stock solution), 10 μM tubulin, and varying concentrations of drug. Following a 15 min preincubation at 30 °C, samples were chilled on ice, GTP to 0.4 mM was added, and turbidity development was followed at 350 nm in a temperature controlled recording spectrophotometer for 20 min at 30 °C. Extent of reaction was measured. Full experimental details were previously reported.31
[3H]Colchicine Binding Assay
The reaction mixtures contained 1.0 μM tubulin, 5.0 μM [3H]colchicine, and 5.0 μM inhibitor and were incubated 10 min at 37 °C. Complete details were described previously.32
Cell Lines
Methodology for the evaluation of the growth of human MCF-7 breast carcinoma cells was previously described.32 Human HeLa cells (from carcinoma of the uterine cervix) and HCT116/chr3 cells (from colon carcinoma) were grown at 37 °C in a humidified atmosphere containing 5% carbon dioxide in D-MEM (Gibco BRL, UK) supplemented with 10% fetal calf serum, 4 mM glutamine, 2 mM sodium pyruvate, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (all reagents were from Celbio, Italy). For the HCT116/chr3 cells, the medium also included 500 μg/mL of the selection compound G418 (Gibco). Cells were trypsinized when subconfluent and seeded in T75 flasks at a concentration of 105/mL. U937 (human monocytic leukemia cell line) and J774.1 cells (murine macrophage cell line) were cultured in RPMI 1640 medium containing 10% fetal calf serum, 50 μg/mL each of penicillin and streptomycin, and 2 mM glutamine. Human melanoma M14 cells and human embryonic kidney (HEK) 293 cells were grow at 37 °C in Dulbecco's modified Eagle's medium containing 10 mM glucose (DMEM-HG) supplemented with 10% fetal calf serum and 50 μg/mL each of penicillin and streptomycin.
The P. tridactylis PtK2 cells, the A10 rat embryonic aortic smooth muscle cells, the human umbilical vein endothelial cells, and the human aortic smaooth muscle cells were obtained from the American Type Tissue Collection and grown as recommended by the supplier, except that a 5% CO2 atmosphere was used with all cells.
Morphological Analysis
Cells grown in T75 flasks and treated with 100 μM 24 for 24 h were observed with an Olympus IX71 microscope and photographed with a Cool SNAPESES Photo-metric digital camera.
Cell Viability Assay
HeLa and HCT116/chr3 cells. Cells were seeded in 96-well plates at a density of 1 × 103 cells/100 μL well. Twenty-four hours later, cells were treated with the test compound (stock solution: 10 mM in DMSO, kept at room temperature in the dark) used at increasing concentrations (0.1–100 μM) for 24 h. Cells were harvested at the end of the treatment or washed with phosphate-buffered saline and incubated in drug-free medium for an additional 24 h. To rule out a possible effect of the solvent, parallel samples were incubated in complete medium containing 1% DMSO. At the end of the treatments, 20 μL of CellTiter96 Aqueous One Solution Reagent (Promega) was added to each well. The plates were then incubated for 4 h at 37 °C and analyzed with a microplate reader (Gio. De Vita, Roma, Italy) at 492 nm. Experiments were performed in quadruplicate and repeated three times.
M14, HEK, U937, and J774.1 cells. Cell viability was determined using the 3-[4,5-demethylthiazol-2,5-diphenyl-2H-tetrazolium bromide (MTT) colorimetric assay. The test is based on the ability of mitochondrial dehydrogenase to convert, in viable cells, the yellow MTT reagent into a soluble blue formazan dye. Cells were seeded into 96-well plates to a density of 105 cells/100 μL well. After 24 h of growth to allow attachment to the wells, compounds were added at various concentrations (10, 30, 50, 100, 150, and 200 nM). After 24 or 48 h of growth and after removal of the culture medium, 100 μL/well of medium containing 1 mg/mL of MTT was added. Cell cultures were further incubated at 37 °C for 2 h in the dark. The MTT solution was then removed, and 100 μL DMSO was added to dissolve formazan crystals. Absorbance at 550 nm was measured using a plate reader. Experiments were performed in triplicate. As a control, 0.5% DMSO was added to untreated cells.
Time-Lapse Microscopy
Effect of drug treatment on HEK and M14 cell morphology was determined by TLM using a Leica CTR6500 microscope. Images were captured every hour for a 48 h period. Plates (96-well) were incubated under standard culture conditions and kept at 37 °C in a 5% carbon dioxide atmosphere for the observation period (up to 48 h).
Yeast
S. cerevisiae yeast wild type strain BY4741 (Mat a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) was used. Cells were grown at 28 °C in minimal medium (0.67% yeast nitrogen base without amino acids) containing 2% glucose (synthetic dextrose, SD) supplemented with 20 μg/μL of the appropriate nutritional supplements. A fresh exponential culture (5 × 106 cells/mL) of the yeast were incubated for 18 h at 28 °C with the compound of interest at 20 μM. The cells were then centrifuged for 5 min at 13000g at 4 °C, washed twice with distilled water, and resus-pended in SD (1 mL). Cell suspensions (15 μL) were transferred in 60 μL of fresh SD medium, serially diluted 10-fold, incubated overnight at 28 °C, and spotted onto YP plates (1% yeast extract, 2% peptone) supplemented with 2% glucose (YPD). The plates were incubated at 28 °C for three days before recording cell growth.
Supplementary Material
Acknowledgment
This research was funded in part by Istituto Pasteur—Fondazione Cenci Bolognetti, and Progetti di Ricerca di Università, Sapienza Università di Roma. A.C. thanks Istituto Pasteur—Fondazione Cenci Bolognetti for his Borsa di Studio per Ricerche all'Estero. G.L.R. was supported by a fellowship from FIRC. L.M. thanks Progetti di Ricerca di Università, Sapienza Università di Roma, for her Contratto di Collaborazione Esterna per Progetti di Ricerca (Bando 2/2007, Dipartimento di Studi Farmaceutici). This research was also partially supported by a grant of the Fondazione Banca del Monte di Lombardia (Pavia, Italy) to A.I.S. V.G. is a Ph.D. student from the University of Pavia (Dottorato in Scienze biomolecolari e biotecnologie).
Footnotes
Abbreviations: ATI, arylthioindole; CSA4, combretastatin A-4; SAR, structure–activity relationship; MW, microwave; Pmax, maximum pressure; TMSDM, trimethylsilyldiazomethane; MCPBA, 3-chloroperoxybenzoic acid; PPA, polyphosphoric acid; TBAHS, tetrabutylammoniun hydrogen sulfate; DIPAD, diisopropyl azodicarboxylate; DMSO, dimethylsulfoxide; THF, tetrahydrofuran; MCF-7, human breast carcinoma cells; HeLa, cervical cancer cells from Henrietta Lacks; HCT116/chr3, human colon cancer cells; HEK, human embryonic kidney 293 cells; M14, human melanoma cells; U937, human monocytic leukemia cells; J774.1, murine macrophage cells; TLM, time-lapse microscopy, a slow process technique of photographing, which allows watching cellular growth; MTT, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PDB, protein data bank, DAMA-colchicine, N-deacetyl-N-(2-mercaptoacetyl)colchicine.
Supporting Information Available: Effects of compounds 24, 27–29, 36, 39, and 41 on murine macrophage J744.1 cell morphology. Elemental analyses of new derivatives 7, 8, 10, 11, 13, 14, 16, 17, 19–23, 25–32, 35, 37–41, and 43–56. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Teicher BA. Newer cytotoxic agents: attacking cancer broadly. Clin. Cancer Res. 2008;14:1610–1617. doi: 10.1158/1078-0432.CCR-07-2249. [DOI] [PubMed] [Google Scholar]
- (2).Bhattacharyya B, Panda D, Gupta S, Banerjee M. Antimitotic acticity of colchicine and the structural basis for its interaction with tubulin. Med. Res. Rev. 2008;28:155–183. doi: 10.1002/med.20097. [DOI] [PubMed] [Google Scholar]
- (3).Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- (4).Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- (5).Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. The binding mode of epothilone A on alpha,beta-tubulin by electron crystallography. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- (6).Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R, Cerezo G, Vanderwal CD, Day BW, Sorensen EJ, Lopez JA, Andreu JM, Hamel E, Diaz JF. Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat. Chem. Biol. 2007;3:117–125. doi: 10.1038/nchembio853. [DOI] [PubMed] [Google Scholar]
- (7).Lin MC, Ho HH, Pettit GR, Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding to colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- (8).Beckers T, Mahboobi S. Natural, semisynthetic and synthetic microtubule inhibitors for cancer therapy. Drugs Future. 2003;28:767–785. [Google Scholar]
- (9).Sridhare M, Macapinlac MJ, Goel S, Verdier-Pinard D, Fojo T, Rothenberg M, Colevas D. The clinical development of new mitotic inhibitors that stabilize the microtubule. Anticancer Drugs. 2004;15:553–555. doi: 10.1097/01.cad.0000131681.21637.b2. [DOI] [PubMed] [Google Scholar]
- (10).De Martino G, La Regina G, Coluccia A, Edler MC, Barbera MC, Brancale A, Wilcox E, Hamel E, Artico M, Silvestri R. Arylthioindoles, potent inhibitors of tubulin polymerization. J. Med. Chem. 2004;47:6120–6123. doi: 10.1021/jm049360d. [DOI] [PubMed] [Google Scholar]
- (11).De Martino G, Edler MC, La Regina G, Coluccia A, Barbera MC, Barrow D, Nicholson RI, Chiosis G, Brancale A, Hamel E, Artico M, Silvestri R. Arylthioindoles, potent inhibitors of tubulin polymerization. 2. Structure–activity relationships and molecular modeling studies. J. Med. Chem. 2006;49:947–954. doi: 10.1021/jm050809s. [DOI] [PubMed] [Google Scholar]
- (12).La Regina G, Edler MC, Brancale A, Kandil S, Coluccia A, Piscitelli F, Hamel E, De Martino G, Matesanz R, Díaz JF, Scovassi AI, Prosperi E, Lavecchia A, Novellino E, Artico M, Silvestri R. New arythioindoles inhibitors of tubulin polymerization. 3. Biological evaluation, SAR and molecular modeling studies. J. Med. Chem. 2007;50:2865–2874. doi: 10.1021/jm061479u. [DOI] [PubMed] [Google Scholar]
- (13).Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 2006;49:1–12. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- (14).Liou J-P, Chang Y-L, Kuo F-M, Chang C-W, Tseng H-Y, Wang C-C, Yang Y-N, Chang J-Y, Lee S-J, Hsieh H-P. Concise synthesis and structure–activity relationships of combretastatin A-4 analogues, 1-aroylindoles, and 3-aroylindoles as novel classes of potent antitubulin agents. J. Med. Chem. 2004;47:4247–4257. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]
- (15).Mori K, Takaishi H. Synthetic microbial chemistry. XXII. Synthesis of monocerin, an antifungal, insecticidal and phytotoxic heptaketide metabolite of Exserohilum monoceras. Tetrahedron. 1989;45:1639–1646. [Google Scholar]
- (16).Mitsunobu O, Yamada M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967;40:2380–2382. [Google Scholar]
- (17).Botstein D, Chervitz SA, Cherry JM. Yeast as a model organism. Science. 1997;277:1259–1260. doi: 10.1126/science.277.5330.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Almeida B, Silva A, Mesquita A, Sampaio-Marques B, Rodrigues F, Ludovico P. Drug-induced apoptosis in yeast. Biochim. Biophys. Acta. 2008;1783:1436–1448. doi: 10.1016/j.bbamcr.2008.01.005. [DOI] [PubMed] [Google Scholar]
- (19).Zabrocki P, Bastiaens I, Delay C, Bammens T, Ghillebert R, Pellens K, De Virgilio C, Van Leuven F, Winderick J. Phosphorylation, lipid raft interaction and traffic of alpha-synuclein in a yeast model for Parkinson's disease. Biochim. Biophys. Acta. 2008;1783:1767–1780. doi: 10.1016/j.bbamcr.2008.06.010. [DOI] [PubMed] [Google Scholar]
- (20).Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nguyen TL, McGrath C, Hermone AR, Burnett JC, Zaharevitz DW, Day BW, Wipf P, Hamel E, Gussio R. A common pharmacophore for a diverse set of colchicine site inhibitors using a structure-based approach. J. Med. Chem. 2005;48:6107–6116. doi: 10.1021/jm050502t. [DOI] [PubMed] [Google Scholar]
- (22).Dupeyre G, Chabot GG, Thoret S, Cachet X, Seguin J, Guenard D, Tillequin F, Scherman D, Koch M, Michel S. Synthesis and biological evaluation of (3,4,5-trimethoxyphenyl)indol-3-ylmethane derivatives as potential antivascular agents. Bioorg. Med. Chem. 2006;14:4410–4426. doi: 10.1016/j.bmc.2006.02.037. [DOI] [PubMed] [Google Scholar]
- (23).Liou J-P, Wu C-Y, Hsieh H-P, Chang C-Y, Chen C-M, Kuo C-C, Chang J-Y. 4- and 5-Aroylindoles as novel classes of potent antitubulin agents. J. Med. Chem. 2007;50:4548–4552. doi: 10.1021/jm070557q. [DOI] [PubMed] [Google Scholar]
- (24).Jia Y, Zhu J. Palladium-catalyzed, modular synthesis of highly functionalized indoles and tryptophans by direct annulation of substituted o-haloanilines and aldehydes. J. Org. Chem. 2006;71:7826–7834. doi: 10.1021/jo061471s. [DOI] [PubMed] [Google Scholar]
- (25).Tullberg E, Schacher F, Peters D, Frejd T. Solvent-free Heck–Jeffery reactions under ball-milling conditions applied to the synthesis of unnatural amino acids precursors and indoles. Synthesis. 2006;7:1183–1189. [Google Scholar]
- (26).Davis DP, Kirk KL, Cohen LA. New synthesis of 2-nitroimidazoles. J. Heterocycl. Chem. 1982;19:253–256. [Google Scholar]
- (27).Molecular Operating Environment (MOE 2007.09) Chemical Computing Group, Inc.; Montreal, Quebec, Canada: http://www. chemcomp.com. [Google Scholar]
- (28).FlexX 3.0. BioSolveIT GmbH; Sankt Augustin, Germany: http://www.biosolveit.de. Accessed on 11/12/2008. [Google Scholar]
- (29).Code “fragment_rmsd.svl” obtained from SVL exchange website: SVL Exchange: An SVL Code Exchange Site for the MOE User Community. Chemical Computing Group, Inc.; Montreal, Canada: 2004. http://svl.chemcomp.com. Accessed on 11/12/2008. [Google Scholar]
- (30).Zodiac 0.5b. http://www.zeden.org. Accessed on 11/12/2008.
- (31).Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003;38:1–21. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- (32).Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure–activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.