

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal motor neuron disease inherited in a small subset of patients. The SOD1(G93A) transgenic mouse models this subset of patients, and studies of this strain have suggested that endoplasmic reticulum (ER) stress and deficits in ER chaperone function are contributors to ALS pathophysiology. Here, we demonstrate that the reticulon family of proteins is a novel regulator of the ER chaperone protein disulfide isomerase (PDI), and that through PDI, reticulon-4A (Nogo-A) can protect mice against the neurodegeneration that characterizes ALS. We show that overexpressing reticulon protein induces a punctate redistribution of PDI intracellularly, both in vitro and in vivo. Conversely, reduction of endogenous NogoA expression causes a more homogeneous expression pattern in vivo. These effects occur without induction of the unfolded protein response. To examine the effect of PDI redistribution on ALS disease progression, we conducted survival and behavior studies of SOD1(G93A) mice. Deletion of a single copy of the NogoA,B gene accelerates disease onset and progression, while deletion of both copies further worsens disease. We conclude that NogoA contributes to the proper function of the ER resident chaperone PDI, and is protective against ALS-like neurodegeneration. Our results provide a novel intracellular role for reticulon proteins and support the hypothesis that modulation of PDI function is a potential therapeutic approach to ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by loss of cortical, brainstem, and spinal motor neurons (Pasinelli and Brown, 2006). Five to ten percent of ALS cases are inherited and known as familial ALS (fALS); the remaining 90% have no hereditary component and are known as sporadic ALS (Bruijn et al., 2004). Approximately 20% of fALS cases are caused by autosomal dominant mutations in superoxide dismutase 1 (SOD1), a ubiquitously expressed cytoplasmic enzyme (Pasinelli and Brown, 2006). Several lines of rodents that transgenically overexpress mutant human SOD1 (mSOD) exhibit many of the histological and clinical signs of neurodegeneration seen in the human disease (Gurney et al., 1994; Ripps et al., 1995; Bruijn et al., 1998; Howland et al., 2002; Matsumoto et al., 2006). Studies on these animals have provided some insight into the pathological processes of ALS, implicating aberrant protein folding and aggregation, excitotoxic injury, and dysfunctional axon transport (Bruijn et al., 2004; Kunst, 2004; Pasinelli and Brown, 2006). Despite numerous studies, however, our understanding of the pathophysiology of ALS remains incomplete.
Aberrant protein folding and aggregation are common to many neurodegenerative diseases, including ALS, Alzheimer's, Huntington's, Parkinson's, and prion disease (Kim et al., 2008). In mutant SOD1-fALS, misfolding and subsequent aggregation of mSOD protein overwhelms the protein folding capacity of the endoplasmic reticulum (ER), inducing ER stress and eventually forcing the cell to undergo apoptosis or autophagy (Ilieva et al., 2007; Nagata et al., 2007; Kanekura et al., 2009). Protein disulfide isomerase (PDI), an ER resident chaperone, may protect against this process. PDI is a major component of the ER stress pathway and catalyzes the formation and dissolution of disulfide bonds, thereby facilitating correct disulfide bond formation (Gilbert, 1998; Ni and Lee, 2007). PDI is upregulated in the symptomatic mSOD transgenic lumbar spinal cord, interacts directly with mSOD, and can prevent mSOD-mediated toxicity in vitro (Atkin et al., 2006, 2008).
The reticulon family of proteins is highly expressed in the endoplasmic reticulum, and certain isoforms of Reticulon 4 (Rtn4, also known as Nogo) are upregulated in murine and human ALS (Dupuis et al., 2002; Oertle et al., 2003; Jokic et al., 2005; Yan et al., 2006; Ferraiuolo et al., 2007; Yang and Strittmatter, 2007). Nogo-A (Rtn4A) has been studied extensively with regard to its specialized function in oligodendrocytes, where on the cell surface it interacts with Nogo receptor to function as a myelin-associated inhibitor of axon growth after brain and spinal cord injury. However, in the adult CNS, Nogo and other reticulons are also expressed within the neuronal ER. Evidence from non-neuronal cells in several model organisms indicates that ER-associated reticulons contribute to peripheral ER morphology (Caroni and Schwab, 1988; Bandtlow and Schwab, 2000; Fournier et al., 2001, 2002b; Kim et al., 2003; Cafferty and Strittmatter, 2006; Voeltz et al., 2006; Kiseleva et al., 2007; Hu et al., 2009).
Here, we tested the hypothesis that reticulons affect PDI function and thus influence the progression of mSOD-mediated amyotrophic lateral sclerosis. Using cell culture and the high-copy SOD1(G93A) transgenic mouse, we demonstrate that reticulons specifically alter intracellular distribution of PDI, and that mutation of Rtn4 in the mSOD murine model of ALS shortens lifespan in a gene dose-dependent manner. Our results suggest that reticulon proteins facilitate PDI function by correct localization to intracellular subdomains and thus help prolong survival in mSOD-mediated amyotrophic lateral sclerosis, offering a new molecular target for therapeutic intervention for ALS.
Materials and Methods
Cell culture.
COS-7 cells (ATCC) were cultured in DMEM with 10% FBS, 1 mm glutamine, and penicillin/streptomycin at 37°C in a humidified chamber under 5% CO2, in 4-well or 8-well chamber slides (Nunc) or in 96-well tissue culture plates (Falcon). COS-7 cells were passaged with 0.25% trypsin according to ATCC recommendations. Cells were transfected with Fugene 6.0 (Roche) or Lipofectamine 2000 (Invitrogen). Cells were transfected with constructs listed in the Results section.
Immunocytochemistry.
Cultured cells were fixed with 4% paraformaldehyde, then blocked in goat serum and incubated with primary antibodies and appropriate fluorophore-conjugated secondary antibodies. Stained cells were imaged on Zeiss Axiovert or ImageExpress (Molecular Devices) microscopes. Quantification was done with MetaExpress software (Molecular Devices). Antibodies recognizing myc (clone 9E10, Sigma), FLAG (Sigma), PDI (AssayDesigns), BiP (AssayDesigns), calnexin (AssayDesigns), calreticulin (Calbiochem), Grp94 (AssayDesigns), Erp57 (Sigma), and VAPB (AssayDesigns) were obtained commercially.
Transgenic mice.
Congenic C57/B6 mutant SOD-transgenic male mice [B6.Cg-Tg(SOD1*G93A)1Gur/J] were obtained from Jackson Laboratories and mated with congenic C57/B6 NogoA,B−/− (Kim et al., 2003) mice for the mSOD, NogoA,B experiment. Mice strains were called congenic once they had been backcrossed to the C57/B6 strain background at least 10 times. Mice null for the Nogo receptor (NgR) were backcrossed approximately six times to C57/B6 mice (Kim et al., 2004), and the resulting mice were mated with B6xSJL F1 mutant SOD transgenic male mice from Jackson Laboratories [B6SJL-Tg(SOD1*G93A)1Gur/J] for the mSOD, NgR experiment. All animals were housed in compliance with IACUC and Yale Animal Regulatory Committee. Animals were maintained on a 12 h light cycle, with regular food and water ad libitum.
Immunohistochemistry.
Animals were perfused with normal saline and 4% PFA. For PDI and calnexin distribution in spinal cord motor neurons, NogoA,B−/− mice were 6 months of age, Hb9-NogoA transgenic mice were 6 d of age, and each group had littermate controls. Lumbar spinal cord was dissected, postfixed briefly, then embedded in gelatin and cut along the transverse plane on a Leica Microsystems VT1000 S vibrating blade microtome. Free-floating sections were incubated overnight at 4°C with antibodies against protein disulfide isomerase and calnexin (SPA-890, 1:500, and SPA-891, 1:1000, respectively) from AssayDesigns. Sections were washed three times in PBS, then incubated with fluorophore-conjugated secondary antibodies. Ventral horns of spinal cord sections were imaged by an observer blind to genotype on a Zeiss Axiovert upright fluorescent microscope with identical exposure settings and identical postacquisition processing for each image. Motor neurons were identified visually by ventrolateral location in spinal cord and by large size (longest axis >15 μm), and PDI and calnexin distribution was analyzed with the free open-source software program CellProfiler (Carpenter et al., 2006) by an observer blind to genotype using the parameter correlating to Haralick textural feature #7 (Haralick et al., 1973):
Western blots.
Cerebellum and lumbar spinal cord tissues were dissected from SOD transgenic and nontransgenic animals, lysed in buffer, sonicated, and centrifuged at 22,000 × g for 10 min. Supernatant was retained for experiments. Bradford assay for protein concentration was performed, and 15 μg of protein loaded per lane. Western blots were probed with anti-NogoA antibody [rabbit, 1:500–1:2000 (GrandPré and Strittmatter, 2001)], anti-actin antibody (Santa Cruz Biotechnology sc-1616, 1:500), human-specific anti-SOD antibody (Santa Cruz Biotechnology sc-8636), and then imaged and quantified using LiCor Odyssey.
For Western blots of COS-7 cell lysates, COS-7 cells were transfected with the empty vector pcDNA3.1mychis or constructs encoding eGFP, NogoA-myc, NogoC-myc, FLAG-Rtn1, FLAG-VAPB, or LRRTM1-myc for 36 h, or were treated with thapsigargin 1 μm or ethanol for 24 h. Cells were collected with a cell scraper, suspended in lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.1% SDS, 1% sodium deoxycholate, and 1% Triton X-100) and sonicated. Cell lysates were centrifuged at 28,000 × g for 10 min, and the supernatant was mixed with 2× Laemmli sample buffer with 4% β-mercaptoethanol. Cell lysates were loaded onto 4–20% gradient Tris-HCl mini-gels (Bio-Rad), electrophoresed, transferred to PVDF membrane, blocked in LiCor blocking buffer, probed with anti-CHOP (Santa Cruz Biotechnology, sc-575) and anti-actin antibodies (Santa Cruz Biotechnology, sc-1616), washed, probed with LiCor secondary antibodies, and imaged and quantified on the LiCor Odyssey (Lincoln, Nebraska). Transfection rate was 72% as measured by GFP transfection and fluorescence-activated cell sorting.
Histology.
For ventral root axon profile analysis, animals were killed at 18 weeks and perfused with ice-cold normal saline and 4% PFA. Ventral and dorsal roots from spinal levels L4–5 were dissected, postfixed in osmium tetroxide, and embedded in Epon, and semithin sections were cut and stained with toluidine blue. Intact axon profiles were counted by an observer blinded to genotype.
Survival and behavior analysis.
Mice were weighed weekly beginning from 6 weeks of age. Change in weight from 9 weeks of age was calculated. Mice were trained on the accelerating rotating rod (rotarod) for 3 d, then performance was assessed weekly thereafter. Rotarod values are group averages consisting of averages of the best three of four consecutive trials for each mouse. Survival assays were done as previously described (Gurney et al., 1994). For Kaplan–Meier survival curves, “death” was defined as the cessation of life between 24 h observation intervals or a moribund condition in which the animal was no longer able to right itself after 30 s on either side and was killed for humane purposes. Statistical analysis was done with SPSS 14.0 using the log-rank test for Kaplan–Meier curves and repeated-measures multivariate ANOVA for rotarod analysis.
Results
Rtn4 alters KDEL protein localization
The importance of ER chaperones and ER stress has been demonstrated in numerous neurodegenerative diseases, including Alzheimer's, Parkinson's, and prion diseases (Ni and Lee, 2007). To be retained in the ER, many lumenal ER chaperones contain a C-terminal lysine–aspartic acid–glutamic acid–leucine (KDEL) motif, which allows these ER residents (including PDI, BiP/Grp78, Grp94, and calreticulin) to be retrieved from distal components of the secretory pathway (Pelham, 1996). To assess the influence of Rtn4 on ER chaperones, we transfected COS-7 cells, which normally express no detectable Rtn4 protein, with Rtn4 cDNA-containing plasmids and probed with an antibody that recognizes the KDEL motif.
Rtn4 transfection causes a dramatic redistribution of KDEL-motif proteins (Fig. 1, top row, arrows). KDEL proteins in cells transfected with Rtn4 have a distinct punctate expression pattern, compared with a homogeneous pattern in untransfected cells. These puncta do not colocalize with Golgi or endosomal markers (data not shown), or with Rtn4 itself. In contrast, Rtn4 transfection does not cause redistribution of the KDEL-negative ER transmembrane protein, calnexin (Fig. 1, middle row). A control for overexpression of an ER membrane protein in these cells, VAPB/ALS8, causes no KDEL redistribution (Fig. 1, bottom row).
Figure 1.
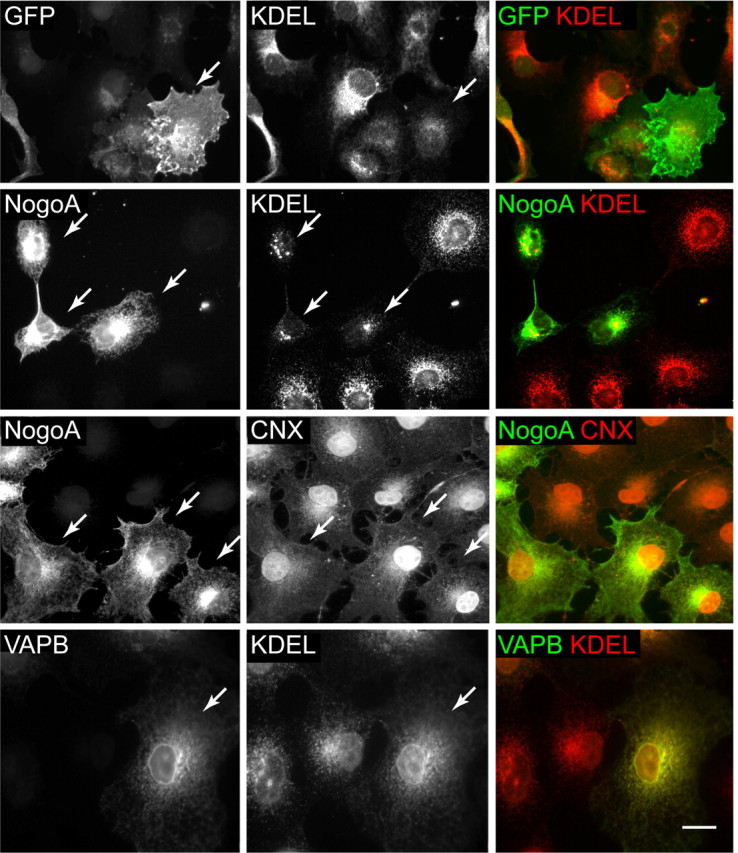
NogoA expression alters distribution of KDEL-motif proteins. COS-7 cells were transfected with NogoA (Rtn4A), GFP, and VAPB (left column) and probed with antibodies against ER proteins (center column). Top row, GFP transfection of COS-7 cells has no effect on KDEL protein expression. Second row, NogoA transfection causes redistribution of KDEL proteins into puncta (arrows). This redistribution is absent in nontransfected cells in same panel. Third row, Transfection with NogoA has no effect on distribution of calnexin, a non-KDEL ER protein. Bottom row, Transfection with VAPB, an ER transmembrane protein, has no effect on KDEL protein distribution; KDEL antibody immunoreactivity remains in a diffuse pattern and expression levels are not increased. Merged images are in right column. Arrows point to transfected cells. Scale bar, 20 μm.
Rtn4 and protein disulfide isomerase
Each chaperone has a different role in the ER. PDI is a member of the thioredoxin family and catalyzes formation and dissolution of disulfide bonds (Appenzeller-Herzog and Ellgaard, 2008); calreticulin is part of the calnexin/calreticulin system (Michalak et al., 2002); BiP and Grp94 are ER homologues of Hsp70 and Hsp90, respectively (Ni and Lee, 2007). Erp57 is a PDI homolog that shares enzymatic functions with PDI, but unlike PDI, forms direct interactions with the ER lectins calnexin and calreticulin (Oliver et al., 1999). Using immunocytochemistry, we examined individual members of the KDEL family to look for differential effects of Rtn4 on their redistribution. Rtn4 expression has little to no effect on Grp78/Bip, a moderate effect on Grp94 and calreticulin, and the greatest effect on PDI distribution (Fig. 2). Reticulon expression can cause redistribution of the PDI-related protein Erp57 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), but the effect is minimal compared with PDI. Our results demonstrate that Nogo affects a subset of ER chaperones, and may affect protein folding and disulfide bond formation specifically via its greater influence on PDI than other KDEL and thioredoxin family members.
Figure 2.

NogoA differentially affects individual members of the KDEL protein family. COS-7 cells transfected with NogoA (left column, arrows) were probed with antibodies recognizing individual members of the KDEL protein family (center column). PDI exhibits the greatest punctate redistribution in NogoA-expressing cells, Grp94 and calreticulin (CLRT) exhibit less redistribution, and BiP is not redistributed by NogoA expression, when compared with untransfected cells in the same panels. Merged images are in right column. Arrows point to transfected cells. Scale bar, 20 μm.
The reticulons and protein disulfide isomerase
In mammals, the reticulon protein family has four members, Rtn1–4, each with a number of isoforms [e.g., Rtn1A–C (Oertle and Schwab, 2003; Yang and Strittmatter, 2007)]. All family members contain a C-terminal reticulon homology domain (RHD), which consists of two hydrophobic regions linked by an ∼66-amino acid region (GrandPré et al., 2000). The N-terminal regions of each reticulon share little homology, however, and their functions within the ER remain largely unclear. If Rtn4's impact on PDI expression is mediated by the RHD, then other reticulon family members might have a similar effect.
To address the specificity of Rtn4's effect on PDI, COS-7 cells were transfected with plasmids containing the coding sequence for each of the reticulon family members: NogoA (Rtn4A), NogoC (Rtn4C), Rtn1C, Rtn2A, and Rtn3, in addition to VAPB and GFP. We found that multiple reticulon family members alter PDI distribution in a statistically significant manner (Fig. 3A; for quantification, see Fig. 3B) (*p < 0.05, **p < 0.01, n = 3 experiments in all cases, 124–224 cells per antigen). VAPB and GFP, in contrast, have no effect on PDI distribution. Importantly, transfection does not increase the total intensity of PDI expression compared with control transfection (Fig. 4C), indicating that reticulon expression affects the intracellular spatial arrangement of endogenous PDI without altering total expression levels.
Figure 3.

Each reticulon family member causes redistribution of PDI. COS-7 cells were transfected with constructs encoding each of the reticulon protein family members [NogoA (Rtn4A), NogoC (Rtn4C), Rtn1C, Rtn2A, and Rtn3, top row] and VAPB (control), and probed for protein disulfide isomerase (PDI, middle row). A, Each individual reticulon causes a dramatic redistribution of intracellular PDI into puncta when compared with untransfected cells and VAPB, another ER membrane transmembrane protein (arrows). B, Quantitation of number of protein puncta per cell is presented for each protein transfected and detected. NogoA, NogoC, Rtn1, and Rtn2 all strongly induce puncta of PDI but not BiP or Calnexin (Clnx). C, NogoA-induced PDI puncta (arrows) do not colocalize with LC3II-positive vesicles induced by starvation, indicating that these PDI puncta are unlikely to be autophagosomes. Scale bar, 20 μm in A and C. Means ± SEM are graphed. *p < 0.05, **p < 0.01, using Student's t test. n = 3 experiments in all cases, 124–224 cells per antigen.
Figure 4.

Transfection with reticulons does not alter UPR induction. A–C, COS-7 cells were transfected with individual members of the reticulon family and with VAPB (control, top row), and probed for BiP, a common marker for the unfolded protein response (UPR, middle row). A, Compared with control, reticulon transfection does not increase BiP expression. B, Quantification of integrated BiP intensity per transfected cell. C, Quantification of integrated PDI intensity per transfected cell. BiP and PDI expression are not induced with transfection alone. Merged images are in bottom row. Arrows indicate transfected cells. Scale bar, 20 μm. Means ± SEM are graphed. Student's t test. n = 3 experiments in all cases, 124–224 cells per antigen. D, Immunoblot of COS-7 cell lysates probed with antibody against CHOP after overexpression of the indicated proteins or treatment with thapsigargin. E, Quantification of immunoblot signal demonstrating induction of CHOP expression with thapsigargin 1 μm treatment, but lack of induction of CHOP expression after expression of reticulons or VAPB.
Interestingly, reticulon proteins can form aggregates when transfected into COS-7 cells, especially Rtn2. Reticulon aggregate-like structures are never seen to colocalize with PDI or KDEL structures, however.
The punctate structures that PDI forms resemble autophagosomes, and it seemed possible that the puncta formed by PDI could be vesicles in the autophagy pathway. To address this possibility, COS-7 cells transfected with reticulons were starved and probed with an anti-LC3II antibody. LC3II, a microtubule-associated protein, is cleaved from LC3I to LC3II upon activation of the autophagy pathway, and associates with autophagosomes (Klionsky et al., 2008). Although starvation-induced puncta positive for LC3II immunoreactivity are observed, these puncta are not positive for PDI immunoreactivity. Furthermore, PDI puncta induced by transfection with NogoA are not positive for LC3II immunoreactivity (Fig. 3C). Therefore, reticulon-induced PDI puncta are unlikely to be autophagosomes.
Reticulon-induced PDI puncta are not associated with the unfolded protein response
One possible explanation for these observations was that transfection with reticulons induces the unfolded protein response (UPR). The UPR is a major cellular response to stress and one of its downstream effects is to increase expression of ER chaperone proteins, including PDI (Malhotra and Kaufman, 2007). Simply overexpressing a foreign protein in a cell via transient transfection has also been shown to induce the UPR. To address these possibilities, we examined BiP expression in cells transfected with each of the reticulon family members or VAPB; BiP is upregulated upon induction of the UPR (Malhotra and Kaufman, 2007). We found that BiP expression is unaltered by transfection of reticulon family members compared with control transfection (Fig. 4A; for quantification, see Figs. 1A, 4B). To further confirm the absence of UPR induction, we examined expression levels of CHOP/GADD153, a major downstream effector of the UPR, in COS-7 cell lysates after overexpression of reticulons, VAPB or LRRTM1, or after treatment with thapsigargin, an inhibitor of the endoplasmic reticulum Ca2+ ATPase known to induce the UPR (Fig. 4D,E). CHOP levels increase dramatically with thapsigargin treatment, and increase to a lesser extent with LRRTM1 expression, consistent with our unpublished observations that LRRTM1 causes a mild UPR induction. In contrast, reticulon expression does not elevate CHOP expression above the levels induced by transfection with the empty vector pcDNA3.1 or a GFP plasmid (Fig. 4E). We conclude therefore that reticulons do not induce the UPR, and the reticulon effect on PDI distribution is a process independent of the UPR.
PDI distribution in spinal cord motor neurons
We wondered whether reticulons have the same pronounced effect on PDI expression in vivo, and specifically in neurons. Therefore, we examined whether and how genetic alteration of Nogo expression affects murine PDI distribution in spinal motor neurons. In the adult mouse, NogoA is expressed in spinal motor neurons and oligodendrocytes (Fig. 5A) (Huber et al., 2002). Mutation of Nogo via gene trapping completely abrogates expression of NogoA and NogoB isoforms in mouse spinal cord (Kim et al., 2003). PDI expression in spinal motor neurons of wild-type mice is visibly punctate (Fig. 5B, top left), but in NogoA,B−/− mice, PDI expression is considerably more homogeneous (Fig. 5B, bottom left). Using the CellProfiler software package to quantify homogeneity of PDI expression (see Materials and Methods), we found a statistically significant increase in PDI homogeneity in the NogoA,B−/− motor neurons compared with wild-type motor neurons (Fig. 5C) (p < 0.01). Calnexin distribution, in contrast, is unchanged in the absence of Nogo expression, consistent with our findings in COS-7 cell culture (Fig. 5B, right; for quantification, see Fig. 5C).
Figure 5.

NogoA causes punctate distribution of PDI in mouse spinal cord. A, NogoA is expressed in ventral horn motor neurons (arrowheads) and myelinating oligodendrocytes (arrow) in adult murine spinal cord. B, PDI puncta are easily visible in spinal motor neurons of wild-type mice, while PDI expression is diffuse in NogoA,B−/− motor neurons; calnexin (CNX) expression is similar in NogoA,B−/− and wild-type motor neurons. Scale bar, 20 μm. C, Quantification of PDI and calnexin intracellular distribution pattern in motor neurons using CellProfiler. PDI expression is much more diffuse in Nogo−/− motor neurons than wild type, but there is no difference in calnexin expression across genotypes. D, Schematic showing construct used to make Hb9-NogoA-myc transgenic mouse. E, At 6 d of age, NogoA is strongly and selectively expressed in ventral horn large motor neurons visualized with anti-c-myc antibody staining. Scale bar, 30 μm. F, PDI expression is more punctate and less homogeneous in the Hb9-NogoA-myc transgenic mouse large motor neurons compared with the wild-type mouse at P6. Calnexin subcellular distribution is indistinguishable between transgenic and nontransgenic animals. Scale bar, 20 μm. G, Quantification of PDI and calnexin expression pattern (CellProfiler). Means ± SEM are graphed. n = 4–6 animals per group, 45–58 cells per group. **p < 0.01 using Student's t test.
To address whether increasing Nogo expression above normal expression levels would affect PDI distribution in vivo, we generated a transgenic mouse line expressing myc-tagged NogoA driven by the postmitotic motor neuron promoter Hb9 (Fig. 5D) (Wichterle et al., 2002). In Hb9-NogoA transgenic mice, NogoA is strongly expressed above wild-type levels in ventral horn motor neurons at an early age (P6), as detected with anti-c-myc staining (Fig. 5E). In support of our in vitro results, PDI expression is more discrete and punctate in transgenic motor neurons with increased levels of NogoA compared with motor neurons with wild-type amounts of NogoA expression (Fig. 5F, left). Quantification of homogeneity demonstrated a statistically significant effect of Nogo on homogeneity of PDI expression (Fig. 5G) (p < 0.01), while calnexin homogeneity remained unaltered. The degree to which PDI is inhomogeneous and punctate in wild-type mice increases during postnatal development, being greater in adult mice (Fig. 5B) than at P6 (Fig. 5E). The developmental timing of this transition is not defined here. Combined, our results suggest that decreasing Nogo expression below baseline causes PDI expression to be more diffuse while increasing Nogo expression above baseline causes PDI to localize to more punctate, discrete intracellular regions of the cell. Calnexin, another ER resident protein, is not affected by Nogo expression. Whether the intracellular areas of PDI localization are membrane-bound, associated with other cellular structures, or passing through a specific trafficking compartment remains to be determined.
NogoA mutation accelerates motor neuron degeneration in the SODG93A murine mouse model of amyotrophic lateral sclerosis
Recent publications have demonstrated an important role for the endoplasmic reticulum in many neurodegenerative diseases, including ALS (Kim et al., 2008; Kanekura et al., 2009). PDI is one of the most abundant ER chaperones, is upregulated in spinal cord of the mSOD transgenic mouse, and has also been shown to interact directly with SOD and prevent mSOD toxicity in vitro (Atkin et al., 2008). Due to its effect on PDI, we asked whether Nogo plays a role in the motor neuron-specific degeneration in ALS and in the most common murine mouse model of ALS, the SOD(G93A) transgenic (mSOD) mouse. Spinal roots of mSOD, NogoA,B−/− mice were examined at a stage where animals were visibly weak, and thus were likely to have degeneration of motor axons. Dorsal (sensory) root axons are unaffected by mSOD expression or the NogoA,B mutation (data not shown), but in ventral (motor) roots, axon degeneration and disorganization occurs to a significantly greater extent in mSOD mice with the NogoA,B mutation (Fig. 6A). Quantification of intact motor neuron axons demonstrated a dramatically accelerated disease process in the mSOD, NogoA,B−/− mouse compared with mSOD alone (Fig. 6B) (p < 0.05). No degeneration is seen in ventral root axons of wild-type mice.
Figure 6.

Loss of NogoA,B expression accelerates degeneration of ventral root axons in the SOD1G93A murine model of ALS. A, Semithin sections of L4–5 ventral roots from mSOD and wild-type mice with or without homozygous mutation of NogoA,B were stained with toluidine blue. Mutation of NogoA,B does not alter number of intact healthy motor axons, but mutation of NogoA,B in the context of the mSOD transgene causes increased disorganization and degeneration. B, Quantification of number of intact, healthy axons (Intact Ventral Root Fibers) per ventral root. Scale bar, 20 μm. Means ± SEM are graphed. n = 3 per condition. *p < 0.05, **p < 0.01, ***p < 0.01 using Student's t test.
We wondered whether Nogo might affect ALS-like degeneration simply by increasing expression levels of mSOD protein, as it is known that changes in SOD transgene copy number directly alter degeneration and survival time of mSOD mice (Alexander et al., 2004). High amounts of mSOD protein levels were detected in mSOD transgenic mouse brain via Western blot, but mSOD protein expression is not observed in nontransgenic mice (Fig. 7A). mSOD protein expression is unaltered by mutation of NogoA,B in mSOD transgenic mouse cerebellum, indicating that the effect of Nogo on mSOD-mediated degeneration is not due to changes in absolute levels of mSOD protein itself (Fig. 7B,C).
Figure 7.

Mutation of NogoA,B does not increase or decrease mSOD protein expression levels. A, Mutant human SOD (hSOD) is clearly detected with a human-specific anti-SOD antibody; none is detected in spinal cord lysate from wild-type mice. B, Western blot of hSOD protein expression levels for mSOD-positive mice lacking zero, one, and two copies of NogoA,B. C, Quantification of hSOD protein expression, normalized for actin levels, shows no difference among NogoA,B genotypes. Means ± SEM are graphed. No statistical difference found using Student's t test. n = 2 per genotype.
Loss of Rtn-4A,B shortens mouse lifespan in the mSOD transgenic model of ALS
To determine whether NogoA's effect on PDI distribution might influence survival in the murine model of ALS, NogoA,B−/− mice from an F5 C57/B6 background were bred with mSOD mice on a B6/SJL background. All mice were on the same noncongenic background and survival assays were performed (Fig. 8A). mSOD mice lacking one or two copies of NogoA,B died 13 and 20 d earlier than mSOD control mice, respectively (p = 0.015, p < 0.001, mean ages of death ± SEM were mSOD, 142.2 ± 2.5 d, n = 9; mSOD, NogoA,B+/−, 128.7 ± 2.1 d, n = 29; mSOD, NogoA,B−/−, 122.0 ± 2.0 d, n = 19).
Figure 8.
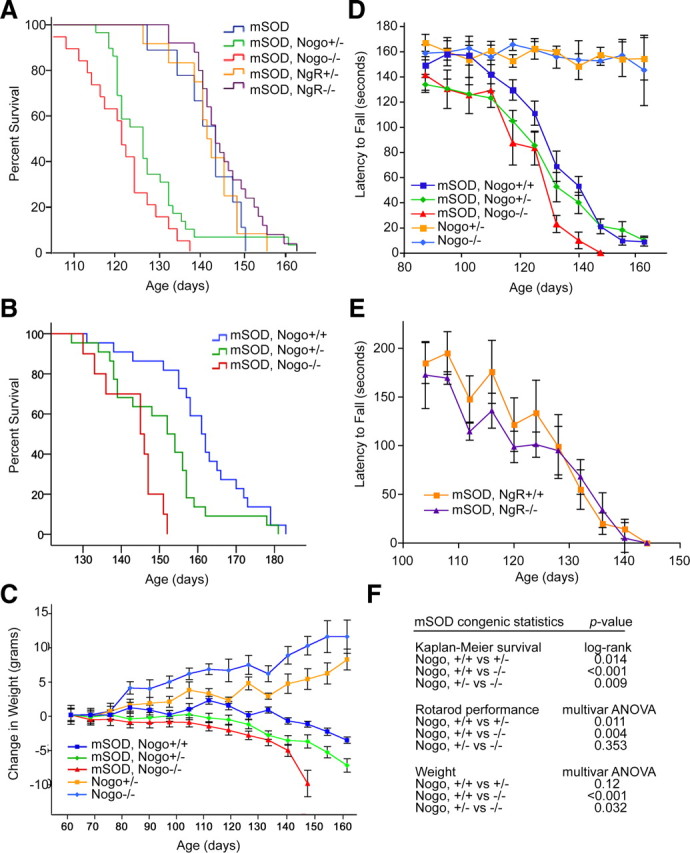
NogoA,B mutation shortens lifespan of the SOD1G93A murine model of ALS, but Nogo receptor mutation has no effect. A, Kaplan–Meier curve demonstrating protective effect of NogoA,B on mSOD mouse survival and lack of effect of Nogo receptor (NgR) on mSOD mouse survival. All genotypes are on the same noncongenic background. Mean ages of death with SE were mSOD, 142.2 ± 2.5 d, n = 9; mSOD, NogoA,B+/−, 128.7 ± 2.1 d, n = 29; mSOD, NogoA,B−/−, 122.0 ± 2.0 d, n = 19; mSOD, NgR+/−, 142.7 ± 2.2 d, n = 12; mSOD, NgR−/−, 146.1 ± 1.5 d, n = 25. Log-rank test: mSOD versus mSOD, NogoA,B+/−, p = 0.015; mSOD versus mSOD, NogoA,B−/−, p < 0.001, mSOD, NogoA,B+/− versus mSOD, NogoA,B−/−, p = 0.067. There was no statistical significance among mSOD NgR−/− mice. B, Kaplan–Meier curve demonstrating gene dose-dependent protective effect of NogoA,B on mSOD mouse survival on congenic background. Mean ages of death with SE were mSOD, Nogo+/+, 161.0 ± 2.7 d, n = 22; mSOD NogoA,B+/−, 150.8 ± 2.8 d, n = 22; mSOD NogoA,B−/−, 143.2 ± 2.4 d, n = 10. See 8F for log-rank comparisons. C, Change in weight from baseline for congenic mSOD, NogoA,B mice. D, Performance on rotating rod of NogoA,B−/− mice compared with wild-type and mSOD mice. E, Performance on rotating rod of mSOD, NgR-null mice compared with mSOD, wild-type mice shows no difference between mSOD, NgR−/−, and mSOD mice. Multivariate ANOVA analysis, p = 0.993. F, Statistics for congenic experiments.
Strain background has been shown to significantly influence mSOD mouse lifespan (Heiman-Patterson et al., 2005). To determine whether strain background was playing a role in the influence of NogoA,B on mSOD mouse survival, congenic B6 NogoA,B−/− mice were bred with congenic B6 mSOD transgenic mice, and F2 progeny were monitored for lifespan, weight, and motor performance (Fig. 8B–D). Consistent with published reports, congenic mSOD animals lived longer than noncongenic mSOD mice by ∼18 d. NogoA,B mutation in mSOD mice shortens lifespan of these mice from a mean survival of 161.0 ± 2.7 d in mSOD mice (n = 22) to 150.8 ± 2.8 d in mSOD, NogoA,B+/− mice (n = 22) and 143.2 ± 2.4 d in mSOD, NogoA,B−/− mice (n = 10) (Fig. 8B). Weight loss also begins earlier and is accelerated in mSOD, NogoA,B−/− mice compared with mSOD, NogoA,B wild-type animals (Fig. 8C). NogoA,B−/− mice without the mSOD transgene gain more weight than their heterozygous littermates and live to 18–24 months of age without signs of neurodegeneration (Fig. 8C and data not shown). Motor function as measured by performance on the accelerating rotating rod (rotarod) is also decreased in mSOD, NogoA,B−/− mice compared with mSOD, NogoA,B wild-type animals (Fig. 8D). Thus, Nogo mutation alone has no influence over motor performance as animals age, and in fact may cause increased weight gain over time, but in the setting of the mSOD transgene, Nogo mutation accelerates disease onset and progression. We conclude that Nogo expression has a protective effect in the murine model of mSOD-mediated ALS.
Nogo has been studied extensively as a myelin-associated inhibitor of axon regeneration after injury through its surface expression on oligodendrocytes and interaction with axonal Nogo receptor (NgR) (GrandPré et al., 2000; Fournier et al., 2001, 2002a; Venkatesh et al., 2007). This occurs separately from any intracellular trafficking of PDI and other ER proteins. Thus, we hypothesized that NgR genotype would not alter ALS disease. To test this, mSOD transgenic mice were bred with NgR−/− mice on a noncongenic background, and behavior and survival were evaluated. NgR mutation has no effect on survival or motor performance of mSOD transgenic mice (Fig. 8A,E). Mean ages of survival were mSOD, 142.2 ± 2.5 d, n = 9; mSOD, NgR+/−, 142.7 ± 2.2 d, n = 12; mSOD, NgR−/−, 146.1 ± 1.5 d, n = 25; there was no statistical significance among groups. This finding supports the hypothesis that Nogo's genetic effect on survival is a function of intracellular, ER-associated Nogo and its influence on PDI distribution, rather than oligodendrocyte surface Nogo-A interaction with axonal Nogo receptor.
Discussion
In this study, we demonstrate a novel link between the reticulon family of proteins and ER resident chaperones, in particular PDI. Reticulons dramatically alter the intracellular distribution of PDI without perturbing the overall structure of the ER or distribution of other ER residents such as calnexin and VAPB. Both in vitro and in vivo, higher levels of reticulon expression cause an increase in the punctate distribution of PDI, whereas lower levels cause PDI expression to be more diffuse. To date, little is known about the function of intracellular reticulon protein, though it has been proposed that reticulons could influence the curvature of the ER and nuclear membrane, possibly through the family of GTPases known as atlastins (Voeltz et al., 2006; Kiseleva et al., 2007; Hu et al., 2009). The interaction with ER chaperones seen here, however, allowed us to surmise that reticulons may be involved in a neurodegenerative disease such as ALS, and indeed, mutation of Nogo, a member of the reticulon family, accelerates disease onset and progression in the SOD1(G93A) transgenic murine model of ALS. This finding demonstrates a novel protective role for NogoA and possibly the entire family of reticulon proteins in motor neuron disease.
This novel protective role for reticulons and specifically for Nogo is distinct from Nogo's role as a myelin-associated inhibitor of axon outgrowth, which is detrimental for recovery following spinal cord injury (SCI). The fact that Nogo has the opposite effect in ALS degeneration from its effect on SCI recovery further supports our hypothesis that it is intracellular, ER-associated reticulon that influences the neurodegeneration occurring in these ALS-like mice and not the Nogo that is found on the surface of oligodendrocytes and interacts with NgR. It is important to note that a report by Jokic et al. (2006) observed that NogoA mutation increased the lifespan of SOD1(G93A) transgenic mice, but the NogoA mutation in that study is known to increase expression levels of NogoB (Simonen et al., 2003). Therefore, it is likely that the overall expression level of the reticulon homology domain, the region of Nogo common to NogoA–C, is increased in that NogoA knock-out mouse, and it is the increase in total amounts of Nogo that has a protective effect in their paper. This is further supported by the fact that the punctate redistribution of PDI can be caused by all reticulon family members. It is likely to be the C-terminal reticulon homology domain mediating the redistribution of PDI rather than each nonhomologous N-terminal region having the same effect.
Several issues remain at the end of this study. While PDI expression is affected by reticulons, a range of markers do not colocalize with PDI puncta; therefore, reticulon-induced puncta may define an unidentified cellular compartment. Such puncta support the protective function of PDI in ALS-like neurodegeneration, yet the mechanistic basis for the link between distribution and function is not clear. At this stage, we cannot be sure whether the puncta are vesicular in nature, protein aggregates, or a distinct intracellular compartment. Interestingly, neither reticulon nor other ER markers are present in these structures. Further elucidation of their identity and character could help our understanding of how reticulons influence this particular ER chaperone population. Second, it remains an open question, both in our work and in that of others, how PDI protects against mSOD-dependent neurodegeneration. Our experiments show that Nogo does not simply titrate induction of the unfolded protein response. Previous papers have shown a decrease in mSOD aggregate formation via PDI, but whether such SOD1 aggregates represent a toxic insult in ALS or a distinct degradative pathway is not known. Future studies will define the nature and mechanism whereby PDI puncta assist cellular handling of mutant SOD1, and therefore shed light on how these puncta might slow the progressive degeneration found in the mSOD model of ALS.
We demonstrate here that reticulons induce a punctate distribution of the ER chaperone PDI, and that these PDI puncta are protective against the degeneration that takes place in the mSOD mouse model of ALS, lengthening lifespan and slowing disease progression. Agents that increase PDI puncta, therefore, whether reticulons or others, should be of therapeutic use in ALS.
Footnotes
This work was supported by a Medical Scientist Training Program training grant (TG 2T32GM07205) from the National Institutes of Health (NIH) to Y.S.Y., by a K08 award (NS056212) from the National Institute of Neurological Disorders and Stroke to N.Y.H., and by grants to S.M.S. from The ALS Association, the NIH, and the Falk Medical Research Trust.
References
- Alexander GM, Erwin KL, Byers N, Deitch JS, Augelli BJ, Blankenhorn EP, Heiman-Patterson TD. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Brain Res Mol Brain Res. 2004;130:7–15. doi: 10.1016/j.molbrainres.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783:535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, Horne MK. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Bandtlow CE, Schwab ME. NI-35/250/nogo-a: a neurite growth inhibitor restricting structural plasticity and regeneration of nerve fibers in the adult vertebrate CNS. Glia. 2000;29:175–181. doi: 10.1002/(sici)1098-1136(20000115)29:2<175::aid-glia11>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, Strittmatter SM. The Nogo-Nogo receptor pathway limits a spectrum of adult CNS axonal growth. J Neurosci. 2006;26:12242–12250. doi: 10.1523/JNEUROSCI.3827-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85–96. doi: 10.1016/0896-6273(88)90212-7. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis L, Gonzalez de Aguilar JL, di Scala F, Rene F, de Tapia M, Pradat PF, Lacomblez L, Seihlan D, Prinjha R, Walsh FS, Meininger V, Loeffler JP. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:358–365. doi: 10.1006/nbdi.2002.0522. [DOI] [PubMed] [Google Scholar]
- Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27:9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Gould GC, Liu BP, Strittmatter SM. Truncated soluble Nogo receptor binds Nogo-66 and blocks inhibition of axon growth by myelin. J Neurosci. 2002a;22:8876–8883. doi: 10.1523/JNEUROSCI.22-20-08876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPré T, Gould G, Wang X, Strittmatter SM. Nogo and the Nogo-66 receptor. Prog Brain Res. 2002b;137:361–369. doi: 10.1016/s0079-6123(02)37027-4. [DOI] [PubMed] [Google Scholar]
- Gilbert HF. Protein disulfide isomerase. Methods Enzymol. 1998;290:26–50. doi: 10.1016/s0076-6879(98)90005-2. [DOI] [PubMed] [Google Scholar]
- GrandPré T, Strittmatter SM. Nogo: a molecular determinant of axonal growth and regeneration. Neuroscientist. 2001;7:377–386. doi: 10.1177/107385840100700507. [DOI] [PubMed] [Google Scholar]
- GrandPré T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–444. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Haralick RM, Shanmugam K, Dinstein I. Textural features for image classification. IEEE Trans Syst Man Cybern. 1973;3:610–621. [Google Scholar]
- Heiman-Patterson TD, Deitch JS, Blankenhorn EP, Erwin KL, Perreault MJ, Alexander BK, Byers N, Toman I, Alexander GM. Background and gender effects on survival in the TgN(SOD1–G93A)1Gur mouse model of ALS. J Neurol Sci. 2005;236:1–7. doi: 10.1016/j.jns.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brösamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci. 2002;22:3553–3567. doi: 10.1523/JNEUROSCI.22-09-03553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva EV, Ayala V, Jové M, Dalfó E, Cacabelos D, Povedano M, Bellmunt MJ, Ferrer I, Pamplona R, Portero-Otín M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111–3123. doi: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Pradat PF, Dupuis L, Echaniz-Laguna A, Muller A, Dubourg O, Seilhean D, Hauw JJ, Loeffler JP, Meininger V. Nogo expression in muscle correlates with amyotrophic lateral sclerosis severity. Ann Neurol. 2005;57:553–556. doi: 10.1002/ana.20420. [DOI] [PubMed] [Google Scholar]
- Jokic N, Gonzalez de Aguilar JL, Dimou L, Lin S, Fergani A, Ruegg MA, Schwab ME, Dupuis L, Loeffler JP. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 2006;7:1162–1167. doi: 10.1038/sj.embor.7400826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura K, Suzuki H, Aiso S, Matsuoka M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol Neurobiol. 2009;39:81–89. doi: 10.1007/s12035-009-8054-3. [DOI] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPré T, Qiu D, Strittmatter SM. Axon regeneration in young adult mice lacking Nogo-A/B. Neuron. 2003;38:187–199. doi: 10.1016/s0896-6273(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Kiseleva E, Morozova KN, Voeltz GK, Allen TD, Goldberg MW. Reticulon 4a/NogoA locates to regions of high membrane curvature and may have a role in nuclear envelope growth. J Struct Biol. 2007;160:224–235. doi: 10.1016/j.jsb.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst CB. Complex genetics of amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:933–947. doi: 10.1086/426001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Okada Y, Nakamichi M, Nakamura M, Toyama Y, Sobue G, Nagai M, Aoki M, Itoyama Y, Okano H. Disease progression of human SOD1 (G93A) transgenic ALS model rats. J Neurosci Res. 2006;83:119–133. doi: 10.1002/jnr.20708. [DOI] [PubMed] [Google Scholar]
- Michalak M, Robert Parker JM, Opas M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium. 2002;32:269–278. doi: 10.1016/s0143416002001884. [DOI] [PubMed] [Google Scholar]
- Nagata T, Ilieva H, Murakami T, Shiote M, Narai H, Ohta Y, Hayashi T, Shoji M, Abe K. Increased ER stress during motor neuron degeneration in a transgenic mouse model of amyotrophic lateral sclerosis. Neurol Res. 2007;29:767–771. doi: 10.1179/016164107X229803. [DOI] [PubMed] [Google Scholar]
- Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertle T, Schwab ME. Nogo and its paRTNers. Trends Cell Biol. 2003;13:187–194. doi: 10.1016/s0962-8924(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Oertle T, Klinger M, Stuermer CA, Schwab ME. A reticular rhapsody: phylogenic evolution and nomenclature of the RTN/Nogo gene family. FASEB J. 2003;17:1238–1247. doi: 10.1096/fj.02-1166hyp. [DOI] [PubMed] [Google Scholar]
- Oliver JD, Roderick HL, Llewellyn DH, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell. 1999;10:2573–2582. doi: 10.1091/mbc.10.8.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pelham HR. The dynamic organisation of the secretory pathway. Cell Struct Funct. 1996;21:413–419. doi: 10.1247/csf.21.413. [DOI] [PubMed] [Google Scholar]
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME. Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron. 2003;38:201–211. doi: 10.1016/s0896-6273(03)00226-5. [DOI] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Sheu SS, Giger RJ. Molecular dissection of the myelin-associated glycoprotein receptor complex reveals cell type-specific mechanisms for neurite outgrowth inhibition. J Cell Biol. 2007;177:393–399. doi: 10.1083/jcb.200702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- Yan R, Shi Q, Hu X, Zhou X. Reticulon proteins: emerging players in neurodegenerative diseases. Cell Mol Life Sci. 2006;63:877–889. doi: 10.1007/s00018-005-5338-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YS, Strittmatter SM. The reticulons: a family of proteins with diverse functions. Genome Biol. 2007;8:234. doi: 10.1186/gb-2007-8-12-234. [DOI] [PMC free article] [PubMed] [Google Scholar]