

Abstract
Cortical GABAergic dysfunction may underlie the pathophysiology of psychiatric disorders, including schizophrenia. Here, we characterized a mouse strain in which the essential NR1 subunit of the NMDA receptor (NMDAR) was selectively eliminated in 40–50% of cortical and hippocampal interneurons in early postnatal development. Consistent with the NMDAR hypofunction theory of schizophrenia, distinct schizophrenia-related symptoms emerged after adolescence, including novelty-induced hyperlocomotion, mating and nest-building deficits, as well as anhedonia-like and anxiety-like behaviors. Many of these behaviors were exacerbated by social isolation stress. Social memory, spatial working memory and prepulse inhibition were also impaired. Reduced expression of glutamic acid decarboxylase 67 and parvalbumin was accompanied by disinhibition of cortical excitatory neurons and reduced neuronal synchrony. Postadolescent deletion of NR1 did not result in such abnormalities. These findings suggest that early postnatal inhibition of NMDAR activity in corticolimbic GABAergic interneurons contributes to the pathophysiology of schizophrenia-related disorders.
The discovery1 that the psychotomimetic drug phencyclidine noncompetitively blocks the NMDAR led to the theory that NMDAR hypofunction is important for the etiology and pathophysiology of schizophrenia2–6. This theory is supported by evidence that various NMDAR antagonists, including MK-801 and ketamine, induce a psychotic reaction in human subjects that resembles schizophrenia symptoms. These agents also reinstate pre-existing symptoms in stabilized schizophrenia patients7, suggesting that they utilize the same mechanisms that are already compromised in this disorder. Several genetically engineered mouse strains carrying mutations in the genes encoding NMDAR subunit proteins also displayed phenotypes that were similar to schizophrenia8,9. However, it remains to be determined whether a particular neuron type or a particular critical period of sensitivity underlies schizophrenia-related behaviors resulting from NMDAR hypofunction.
Postmortem brains of schizophrenia subjects suggest that dysfunction of GABAergic interneurons, particularly those containing the calcium-binding protein parvalbumin, may be a core feature of schizophrenia10,11. Supporting this notion, reduced expression of the GABA-synthesizing enzyme glutamic acid decarboxylase 67 (GAD67)12 and parvalbumin13 have been found in cortical interneurons of individuals with schizophrenia. The possibility that corticolimbic GABAergic interneurons are a prime target for NMDAR hypofunction14,15 is supported by three lines of evidence. First, acute systemic administration of NMDAR antagonists results in hyperactivity of cortical pyramidal neurons16 and spillover of cortical acetylcholine17 and glutamate18. These paradoxical cellular changes concur with brain-imaging data showing net cortical excitation after NMDA antagonist treatment in human subjects19,20. Second, GABAergic interneurons are disproportionally more sensitive to NMDAR antagonists than pyramidal neurons15,21, although the precise mechanisms for this difference is unresolved. NMDAR antagonist–induced cortical excitation may be a result of a preferential reduction in the firing of fast-spiking interneurons and resultant disinhibition of cortical excitatory neurons. Third, repeated administration of NMDAR antagonists decreases GAD67 and parvalbumin expression in cortical GABAergic neurons22–26, linking NMDAR hypofunction to dysfunction of GABAergic neurons.
To directly test the idea that NMDAR hypofunction in GABAergic interneurons produces elements of schizophrenia pathophysiology, we used the cre-loxP system to create a conditional knockout mouse strain in which early postnatal ablation of NR1 (also termed GluN1 or Grin1), an indispensable subunit of NMDAR, was targeted to cortical and hippocampal GABAergic neurons, a majority of which contained parvalbumin. We also generated a conditional NR1 knockout mutant in which NMDAR deletion occurs after adolescence in the same neuron population to assess whether adult onset of NMDAR deletion is critical for the emergence of schizophrenia pathophysiology.
Results
Cre expression confined to corticolimbic GABAergic neurons
We generated a Cre recombinase transgenic line in which Cre expression was driven by the Ppp1r2 gene promoter (protein phosphatase 1, regulatory (inhibitor) subunit 2) (Fig. 1). Endogenous Ppp1r2 is expressed in GABAergic neurons in the striatum, cortex, hippocampus and, to a lesser degree, pyramidal neurons in hippocampal CA1 (Fig. 1a and Supplementary Fig. 1). When crossed with a loxP-flanked Rosa26-lacZ reporter mouse, our new line Cre #4127 (Ppp1r2-cre+/−; referred to here as Ppp1r2-cre or simply cre) resulted in a scattered distribution of lacZ-positive neurons throughout the prefrontal cortex, neocortex and hippocampus (Fig. 1c–e and Supplementary Fig. 1). The dependence of Cre recombinase expression on the Ppp1r2 promoter was confirmed by coexpression of lacZ and Ppp1r2 (Fig. 1b). Over 92% of lacZ-positive neurons were GABAergic, as demonstrated by their coexpression of GAD67 (Fig. 1f and Supplementary Fig. 2), and consisted of 40–50% of the cortical GABAergic neurons across several cortical regions. No lacZ-positive neurons colocalized with the excitatory neuron markers TBR1 (Fig. 1g) or αCaMKII (Supplementary Fig. 2), suggesting that Cre recombination occurred exclusively in GABAergic neurons.
Figure 1.
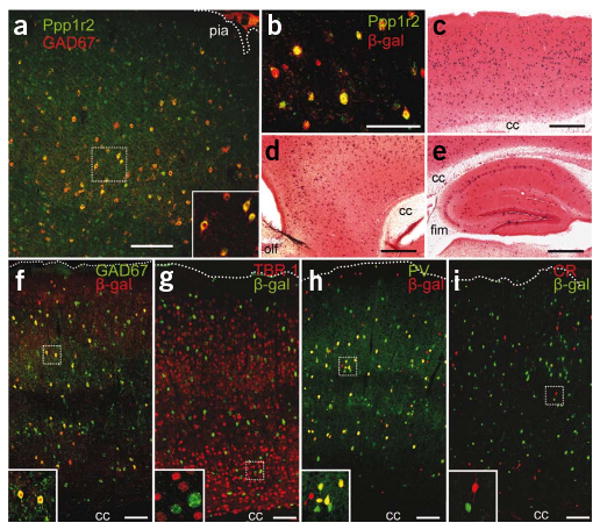
Generation of a corticolimbic GABAergic neuron-restricted Cre line. (a) Endogenous Ppp1r2 immunoreactivity in primary somatosensory cortex (S1) of a 4-week-old wild-type C57BL/6N mouse (Alexa 488, green) colocalized with antibody to GAD67 (Cy3, red). (b) Ppp1r2-cre mice (8-week-old) crossed with the loxP-flanked Rosa26-lacZ reporter. Cre recombination (β-galactosidase (β-gal)–Cy3, red) occurred in Ppp1r2-positive neurons (Alexa 488, green) in S1. (c–e) Spatial distribution of Cre recombinase activity in parasagittal sections from an 8-week-old Ppp1r2-cre; Rosa26-lacZ double-transgenic mouse stained with X-Gal (blue) and Safranin O (red). Sections are from S1 cortex (c), prefrontal cortex (d) and hippocampus (e). cc, corpus callosum, fim, fimbria; olf, olfactory bulb. (f– i) Immunofluorescence of coronal sections from Ppp1r2-Cre; Rosa26-lacZ mice. High-magnification confocal images were used to quantify colocalization. More than 92% of Cre-targeted neurons (β-gal positive) expressed GAD67 (f) but not TBR1 (g). Approximately 75% of GAD67-positive neurons were parvalbumin (PV) positive (h) and none were calretinin (CR) positive (i). Lower left insets, higher magnification of boxed regions. Scale bars represent 100 μm (a,b,f–i) and 500 μm (c–e).
Cre recombination was detected in the cortex and hippocampus first at postnatal day 7 and the restriction of lacZ expression to GABAergic neurons was maintained for up to 18 weeks (Supplementary Fig. 3). Immunofluorescence staining in primary somatosensory cortex (S1) at 8 weeks revealed that >70% of the lacZ-positive neurons were parvalbumin positive (Fig. 1h) and 24% of the remaining population was labeled by antibody to Reelin (Supplementary Fig. 2). In complementary analysis, Cre-targeted neurons represented 75% of parvalbumin-positive interneurons and 30% of the Reelin-positive interneurons. In contrast, less than 3% of Cre-targeted neurons in the cortex were colabeled with antibody to somatostatin (Supplementary Fig. 2) and 15% of Cre-targeted neurons were labeled by antibody to neuropeptide Y (Supplementary Fig. 2). No colocalization was observed with antibody to calretinin in any brain area (Fig. 1i). Thus cre-loxP recombination in the Ppp1r2-cre line occurred early in postnatal development in 40–50% of cortical and hippocampal GABAergic interneurons, the majority of which were parvalbumin positive.
Ablation of NMDARs in corticolimbic interneurons
We crossed the Ppp1r2-cre mice with the loxP-flanked NR1 line A (NR1loxP/loxP-line A or simply Flox-A)27 to restrict NMDAR ablation to Cre-targeted GABAergic neurons. Homozygous NR1loxP/loxP; Ppp1r2-cre progeny (Ppp1r2-cre+/−; NR1loxP/loxP-line A, hereafter postnatal knockout mutant or simply mutant) were viable and fertile, with no gross morphological abnormalities revealed by Nissl staining. The spatial and temporal pattern of NR1 mRNA was analyzed by in situ hybridization (Fig. 2a–c). Expression of NR1 mRNA in control mice (Cre and Flox-A) was detected in most neurons expressing Gad67 mRNA. In contrast, NR1 mRNA expression was absent in 40–50% of cortical and hippocampal GAD67-positive neurons in 4-week-old mutant mice (Fig. 2c), suggesting that NR1 was knocked out in Cre-targeted GABAergic neurons. This pattern was maintained in the mutant mice up to 20 weeks, with no noticable decrease in NR1 mRNA being detected in other areas, including striatum, basolateral amygdala, thalamic reticular nucleus, cerebellum and the nucleus tractus solitarius (Fig. 2c and Supplementary Fig. 4). No difference was found for the ratio of Gad67 mRNA–positive neurons to the total number of neurons between genotypes, demonstrating an absence of neurodegeneration following NR1 ablation (medial prefrontal cortex, 20.1 ± 2.3% for control (Ppp1r2-cre and Flox-A, n = 4 in total) versus 19.6 ± 3.4% for mutant (n = 3), t test, P = 0.91; S1 cortex, 21.5 ± 2.3% for control versus 20.3 ± 3.5% for mutant, t test, P = 0.76). Moreover, there were no differences among genotypes in the number of NR1 mRNA–positive, Gad67 mRNA–negative neurons in the Cre-targeted areas, including the CA1 pyramidal neuron layer of the hippocampus at 20 weeks of age, indicating that there was no reduction of NR1 mRNA in excitatory neurons (Supplementary Fig. 5).
Figure 2.
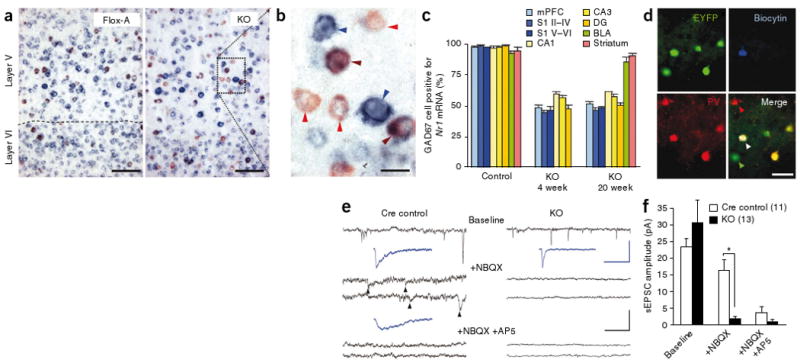
Restricted NR1 deletion in GABAergic neurons in cortex and hippocampus. (a) Representative photomicrographs of nonradioisotope double in situ hybridization of parasagittal sections of S1 from 4-week-old Flox-A (NR1loxP/loxP-line A) and mutant (Ppp1r2-cre+/−; NR1loxP/loxP-line A, KO) mice (GAD67, red; NR1 blue; colocalization, brown). Scale bars represent 50 μm. (b) High magnification of boxed area in the mutant section depicting GABAergic neurons with (brown arrows) and without (red arrows) detectable levels of NR1 mRNA. Non-GABAergic neurons expressing NR1 mRNA are indicated by blue arrows. Scale bar represents 20 μm. (c) Regional and temporal quantification of NR1 mRNA in GAD67-positive neurons. BLA, basolateral amygdala; DG, dentate gyrus; mPFC, medial prefrontal cortex. n = 3 for each mutant age, controls n = 4 at 4–20 weeks old. (d) Immunohistochemical characterization of a neuron filled with biocytin (blue) during whole-cell patch-clamp recording in S1 slices from Ppp1r2-cre mice crossed with the loxP-flanked Rosa26-EYFP mice. Cre-targeted neurons were identified by EYFP expression during recording and later stained for GFP (green). The white arrow indicates triple colocalization of EYFP, biocytin and parvalbumin (red). (e) Representative traces of whole-cell patch-clamp recordings from Cre-targeted neurons in S1 slices of Cre control (Ppp1r2-cre+/−; Rosa26-EYFPloxP/+) and mutant (Ppp1r2-cre+/−; NR1loxP/loxP-line A; Rosa26-EYFPloxP/+, KO) mice under different pharmacological conditions (black traces; black scale bars indicate 20 pA and 500 ms). Spontaneous EPSCs (arrowheads) recorded in the presence of NBQX in the Cre controls, but not in mutant mice, were reduced by the NMDA channel blocker AP5. Blue traces indicate the average of 20 sEPSCs; blue scale bars represent 20 pA and 50 ms. The recording of Cre control is from the neuron with the white arrow in d. (f) sEPSC amplitudes of Cre-targeted S1 neurons under baseline conditions, AMPA blockade (+NBQX), and AMPA/NMDA blockade (+NBQX/+APV). We observed no NMDA components in the mutant mice. Mann-Whitney U Test, * P < 0.05 versus Cre control. Data are mean ± s.e.m.; number of cells is indicated in parentheses from KO (n = 11) and Cre control (n = 10).
To evaluate whether NMDAR function was absent in the Cre-targeted neurons, we induced coexpression of enhanced yellow fluorescent protein (EYFP) by Cre recombination (Fig. 2d) by crossing mutants or Pppr12-cre mice with a loxP-flanked Rosa26-EYFP line. Whole-cell patch-clamp recordings of EYFP-expressing neurons in S1 and hippocampal CA1 were performed in mutants (Ppp1r2-cre+/−; NR1loxP/loxP-line A; Rosa26-EYFPloxP/+) and Cre controls (Ppp1r2-cre+/−; Rosa26-EYFPloxP/+) at 6–26 weeks of age. In the presence of the AMPA-type glutamate receptor channel blocker 6-nitro-7-sulfamoylbenzo(f)quinoxaline-2,3-dione (NBQX), spontaneous excitatory postsynaptic currents (sEPSC) were observed in all the neurons examined in Cre-control mice, including fast-spiking neurons (Fig. 2e,f and Supplementary Fig. 6). sEPSCs were mediated by NMDAR channels, as they were subsequently blocked by the addition of the NMDA channel blocker d(−)-2-amino-5-phosphonovaleric acid (d-AP5). In contrast, in the presence of NBQX, no sEPSCs were observed in the S1 of the mutant mice and no further blockade was observed following the addition of d-AP5. EYFP-positive neurons in the hippocampal CA1 subfields of mutant mice yielded similar results (Supplementary Fig. 6). Kinetic analysis under baseline conditions also showed that the mean decay time of sEPSCs in Cre-targeted neurons in the mutant mice was significantly shorter than in Cre-controls (Mann-Whitney U test, P < 0.01, Supplementary Fig. 6), suggesting the absence of the slow NMDAR component of the sEPSC in the mutants. Immunocytochemical analysis confirmed that all of the biocytin filled–neurons during whole-cell patch-clamp recording of EYFP-positive neurons colocalized with GAD67. Many neurons also colocalized with parvalbumin (Fig. 2d) and some displayed fast-spiking, nonadapting firing patterns (Supplementary Fig. 6). Together, this suggests that NMDAR was functionally eliminated from the Cre-targeted GABAergic neurons in the mutant mice.
Psychiatric symptoms precipitated by social isolation stress
To assess the behavioral consequence of knocking out NMDAR in corticolimbic GABAergic neurons, we subjected mutant (Ppp1r2-cre+/−; NR1loxP/loxP-line A) males and age-matched control (Flox-A line, NR1loxP/loxP-line A or Cre, Ppp1r2-cre+/−) males (8–18 weeks old) to a battery of behavioral tests. Mice were single-housed for >1 week before testing unless otherwise noted. The mutant mice had no significant impairment in basic functions, including body growth rate, thermoregulation, basic auditory, visual and olfactory function, pain sensitivity and motor coordination (Supplementary Fig. 7). Spatial acuity, assessed by visually evoked potentials, was also normal. No obvious locomotor abnormality was apparent at 9–10 weeks of age (Supplementary Fig. 8). However, novelty-induced hyperlocomotion was prominent in the mutant mice during the first 3 min of spontaneous exploration in an open-field test (Fig. 3a), and they also spent substantially less time than controls exploring the unprotected center area of the open field (Fig. 3b), consistent with an anxiety-like phenotype. To explicitly test for anxiety-like behavior in the mutant mice, we subjected naive mice to an elevated plus-maze task. Group-housed mutant mice (3–5 per cage) exhibited clear anxiety-like behavior at 16 weeks of age, but not at 8 weeks (Fig.3c and Supplementary Fig. 8). This phenotype was precipitated by social isolation–induced stress and became evident in 8-week-old mutants that were single-housed for a week (Fig. 3c).
Figure 3.
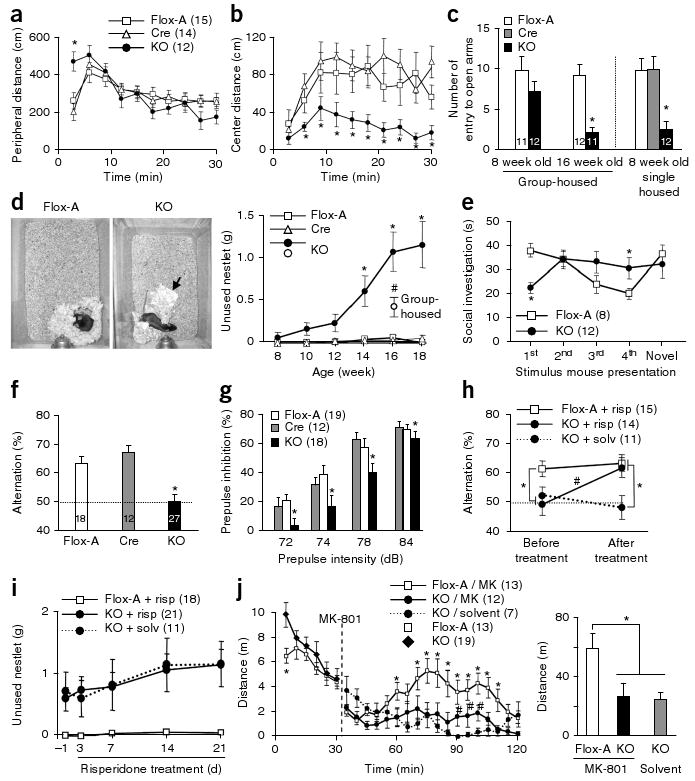
Early postnatal NR1 deletion leads to schizophrenia-related behaviors. (a,b) Increased peripheral locomotion following exposure to a novel open field and decreased exploration of the anxiogenic central area in mutant mice versus controls (Cre and Flox-A) (repeated measures ANOVA/LSD post hoc test, * P < 0.05 versus Flox-A and Cre). (c) Social isolation induced anxiety-like behavior in the elevated plus maze, as shown by the decreased number of entries into unprotected open arms by mutant mice compared with controls (ANOVA/LSD post hoc test, * P < 0.05 versus controls). (d) Left, representative pictures of the nests of Flox-A and mutant mice (arrow, unused material). Right, single-housed mutant mice displayed a significant deficit in nest construction starting at 14 weeks of age, as determined by weighing the unused nestlet material after overnight housing in a new cage (two-way ANOVA/LSD post hoc test, * P < 0.05 versus Flox-A and Cre). Group-housed mutant mice had a smaller deficit compared with age-matched single-housed mutants (16 weeks old, ANOVA/LSD post hoc test, # P < 0.05 versus controls and single-housed mutant mice). (e) The social-recognition test revealed impairment in social short-term memory in the mutant. Flox-A control mice decreased social investigation time following repeated exposures (1 min each) to a stimulus mouse. A fifth dishabituation trial elicited an increased response to a novel mouse, showing individual recognition. In contrast, mutant mice did not show habituation after repeated presentations of the stimulus mouse (second to fourth presentation, repeated measures ANOVA/LSD post hoc test, * P < 0.05 versus Flox-A), suggesting social amnesia, and reduced investigation time during the first presentation. (f) In the Y-maze spontaneous alternation task, the mutant mice alternated between the arms at the chance level (dotted line; ANOVA/LSD post hoc test, * P < 0.005 versus Flox-A and Cre). (g) Mutant mice at 10–12 weeks of age were impaired in prepulse inhibition of the auditory startle reflex across prepulse intensities (two-way ANOVA/LSD post hoc test, * P < 0.005 for genotype factor). (h) Chronic treatment with risperidone (2.5 mg per kg of body weight per d for 3 weeks in drinking water) ameliorated the working memory deficit of the mutant mice in the Y-maze spontaneous alternation task (repeated-measures ANOVA/LSD post hoc test, * P < 0.05, versus. Flox-A, # P < 0.001 mutant mice treated with risperidone versus solvent). Dotted line indicates chance level. (i) Chronic risperidone treatment did not rescue the deficit in social nest building (repeated-measures ANOVA/LSD post hoc test, P = 0.56 mutant mice treated with risperidone versus solvent, P < 0.01 mutant mice treated with risperidone versus Flox-A treated with risperidone). (j) Noncompetitive NMDAR antagonist–induced locomotion was diminished in mutant mice. Left, time course of MK-801–induced locomotor activity (broken line, MK-801 injection, 0.2 mg per kg intraperitoneal; repeated-measures ANOVA/LSD post hoc test, * P < 0.05 versus MK801-treated mutant mice (MK), # P < 0.05 versus mutant mice treated with solvent). Right, cumulative distance traveled after MK-801 treatment (30–120 min; one-way ANOVA/LSD post hoc test, * P < 0.02). Data are mean ± s.e.m.; n is indicated in parentheses or plot bars. Cre, Ppp1r2-cre+/−; Flox-A, NR1loxP/loxP-line A; KO, Ppp1r2-cre+/−; NR1loxP/loxP-line A.
A two-bottle saccharine-preference test was conducted to explore the effect of social isolation–induced stress on hedonistic-like/reward-seeking behavior. Mutant mice that received 8 weeks of social isolation beginning at 8 weeks of age had a decreased preference for sweet solutions (Supplementary Fig. 9) but showed no change in total fluid intake (Supplementary Fig. 9), indicating a mild anhedonia-like state. We did not observe this phenotype in 12-week-old mutant mice that received social isolation for 4 weeks (data not shown), suggesting that there was a quiescent period before the emergence of anhedonia-like symptoms. Anhedonia is commonly associated with depressive-like behavior. However, single-housed 12–15-week-old mutant mice performed normally in the forced swim test, a standard measure of behavioral despair for rodents that is often used to evaluate the efficacy of antidepressants (Supplementary Fig. 9).
Nest-building behavior was observed to assess social activity. Control mice consistently formed a clean and identifiable nest in a distinct location of a new cage using all of the cotton nestlet provided as starting material the evening before (Fig. 3d). In contrast, mutant mice did not build a clear nest by the morning. Quantitative analysis revealed a clear nest-building deficit, which was prominent in mutants older than 12 weeks of age. Notably, mutant mice that were group-housed until 16 weeks of age had a milder nest-building deficit, suggesting that this phenotype was also precipitated by the social isolation–induced stress.
Decreased breeding efficiency was also observed in the mutant mice compared with control mice (data not shown). To determine whether the inefficient breeding was a result of an impairment in mating behavior, mutant males were exposed to superovulated naive wild-type C57BL/6N females in the evening and copulatory success was assessed 12 h later. The number of females with copulatory plugs was significantly lower when paired with mutants (>12 week old) that had been socially isolated from 7 weeks of age. This deficit in mating behavior was not a result of reproductive organ dysfunction, as pairs that successfully bred produced normal-sized litters (Supplementary Fig. 9).
Behavioral features reminiscent of human schizophrenia
To extend our analysis of the mutant to social behaviors, we subjected 12-week-old mutant mice and control littermates to a social-recognition test in a novel cage (Fig. 3e). Control mice demonstrated social investigation (intense sniffing) toward a 4-week-old ‘stimulus male’ during the first 1-min presentation but spent less time investigating the same mouse in subsequent presentations, reflecting an intact social short-term memory. During the last trial, subjects were presented with a new stimulus male and controls again demonstrated social investigation (increased sniffing), suggesting normal social recognition and intact social short-term memory. In contrast, the mutant mice did not investigate the stimulus mouse during the first 1-min presentation, which may be a result of enhanced novelty-induced anxiety (Supplementary Fig. 9). However, mutant mice showed increased social investigation toward the stimulus mouse after the second trial, which was maintained in subsequent trials without habituation (Fig. 3e). If the stimulus male was present continuously for 10 min in a subsequent test, both the mutant and control mice displayed decreased social investigation (Supplementary Fig. 9). This suggests that the impaired habituation of social investigation by the mutants may be a result of a deficit in short-term memory. To test for a short-term memory deficit, we conducted a test of spontaneous Y-maze alternations, a spatial working memory task based on the natural tendency of mice to alternate the choice of maze arms. Although reliable alternation was observed in control mice, mutant mice displayed a reduction in alternation to chance levels (Fig. 3f), suggesting the presence of a spatial working memory deficit, which is well documented in schizophrenia28.
Prepulse inhibition (PPI) of the startle reflex, a measure of sensorimotor gating, is also decreased in human schizophrenia29. We observed impaired PPI of the auditory startle reflex in 10–12 week-old single-housed mutant mice (Fig. 3g), although they had normal reflex amplitudes (Supplementary Fig. 7). Age-matched, group-housed mutant mice showed a similar impairment in PPI (Supplementary Fig. 9), suggesting that this deficit is unaffected by social isolation–induced stress.
To ask whether the behavioral deficits of our mutant could be reversed by antipsychotics, we evaluated the behavior of the mutants following chronic treatment with the second generation antipsychotic risperidone. Chronic oral administration of risperidone for 3 weeks alleviated the deficit in spatial working memory (Fig. 3h) without affecting general locomotor activity in the Y maze (Supplementary Fig. 9). The PPI deficits in the mutant mice (16 weeks old on average) were also partially ameliorated by risperidone (Supplementary Fig. 9). However, not all the behavioral deficits (such as the nest building deficit; Fig. 3i) were reversed by risperidone.
Psychomotor effects elicited by systemic administration of the noncompetitive NMDAR antagonist MK-801 provide an animal model for psychotic symptoms of schizophrenia30. Locomotor hyperactivity induced by intraperitoneal administration of MK-801, which was observed in the Flox-A controls, was largely diminished in the mutant mice (Fig. 3j). The result suggested that NMDAR deletion from 40–50% of corticolimbic GABAergic neurons during early postnatal period may be sufficient to elicit a brain state similar to that of the MK-801–sensitized mice, thereby resulting in the occlusion of MK-801–induced hyperlocomotion.
Reduced GAD67 and parvalbumin with cortical disinhibition
We quantified the levels of GAD67 and parvalbumin proteins in GABAergic neurons under confocal microscopy. GAD67 and parvalbumin expression was decreased in Cre-targeted GABAergic neurons of mutants, but not in the nontargeted neurons (Fig. 4a–c and Supplementary Fig. 10). Cre recombinase expression alone did not reduce the level of GAD67 or parvalbumin, suggesting that dysregulation of GABAergic neuron maturation was a specific consequence of NMDAR deletion in postnatal development.
Figure 4.
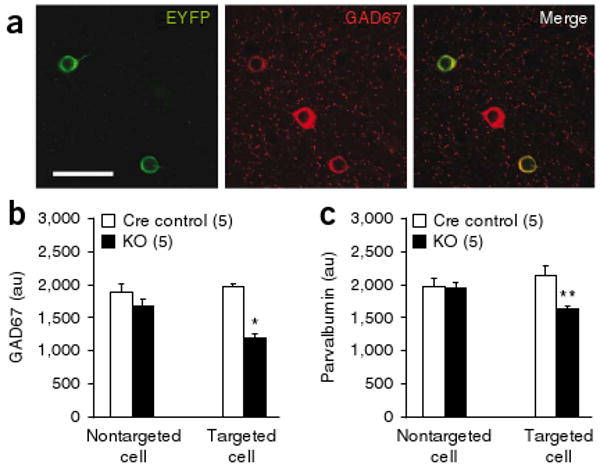
Decreased expression of GAD67 and parvalbumin in NR1-deleted neurons. (a) Confocal photomicrographs of coronal sections (layer II/III S1) immunostained with antibodies to GFP (green, Alexa488) and GAD67 (red, Cy3) to identify Cre-targeted neurons from mutant or Ppp1r2-cre mice crossed with loxP-flanked Rosa26-EYFP mice (12 weeks old). Scale bar represents 40 μm. (b,c) Quantitative analysis of GAD67 (b) and parvalbumin (c) immunoreactivity in Cre-targeted and nontargeted neurons in S1 coronal sections from Cre controls (Ppp1r2-cre+/−; Rosa26-EYFPloxP/+) and mutant mice (Ppp1r2-cre+/−; NR1loxP/loxP-line A; Rosa26-EYFPloxP/+) at 12–14 weeks old). Bars depict mean ± s.e.m., animal number is indicated in parantheses, two-way ANOVA/LSD post hoc test, * P < 0.005, ** P < 0.01.
The decrease in GAD67 could lead to reduced GABA synthesis and release and a subsequent state of disinhibition in cortical excitatory neurons16. To assess this possibility, we conducted in vivo tetrode recordings from mouse S1 during exploration of unfamiliar linear tracks (Fig. 5). Putative pyramidal neurons were defined by a relatively broad spike waveform (peak to trough width >380 μs; Fig. 5b) with a negative curvilinear afterhyperpolarization (Fig. 5a). Putative pyramidal neurons from 13–14-week-old mutant mice showed increased mean firing rates compared with age-matched controls (Fig. 5c) and decreased cross-correlation with nearby neurons (Fig. 5d,e). There was no difference in exploratory behavior between genotypes, including running speed on the linear track (3.2 ± 0.7 cm s−1 for control versus 3.5 ± 1 cm s−1 for mutant mice, t test, P = 0.85). These results indicated that the mutant mice had augmented, but unsynchronized, activity in excitatory neurons.
Figure 5.
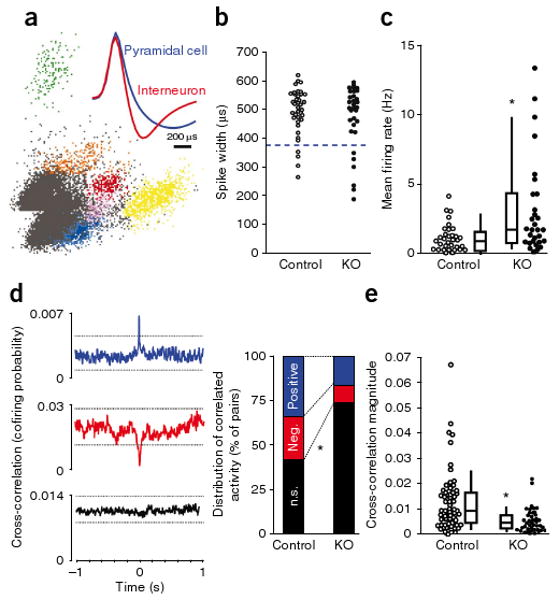
Increased firing of cortical excitatory neurons accompanied by reduced neuronal synchrony. (a) A representative cluster analysis of spike waveforms, sorted into individual units on the basis of spike amplitude distribution. Inset, average spike shape for putative pyramidal neuron (blue) and interneuron (red). Note the characteristic difference in the slope of the afterhyperpolarization phase. (b) No difference in spike-width distributions in neurons recorded from S1 in 13–14 week-old freely moving mutant mice (Ppp1r2-cre+/−; NR1loxP/loxP-line A, n = 4, KO) and controls (n = 2 for Cre (Ppp1r2-cre+/−), n = 2 for Flox-A (NR1loxP/loxP-line A)) during exploration of an unfamiliar linear track. Units with spike widths >380 μs were considered to be putative pyramidal neurons. (c) Mean firing rates of putative S1 pyramidal neurons were significantly higher in mutant mice compared with controls. Each dot is the mean firing rate for an individual neuron (n = 34 control, n = 29 mutant mice). Box plots depict medians (box centers), interquartile ranges (box boundaries) and 10–90th percentiles (whiskers). Mann-Whitney U test, * P < 0.005. (d) Synchronized activity of pairs of nearby neurons from the recording shown in c was assessed by computing the cross-correlation for all pairs of pyramidal neurons recorded from a single tetrode. Representative cross-correlograms from controls exemplifying three different correlation responses: significantly positive (blue), significantly negative (red) and nonsignificant cross-correlation (black). Dotted lines indicate 99% confidence interval. Mutants showed a higher incidence of nonsignificantly correlated pairs of neurons (controls, 26 of 62 pairs (42%); mutant mice s, 31 of 42 pairs (74%); χ2 test, * P < 0.005]. (e) Decrease in the magnitude of cross-correlations across all pairs in mutant mice regardless of the polarity. Each dot represents an individual pair. Mann-Whitney U test, * P < 0.0005.
Early postnatal NR1 deletion crucial for phenotype development
NR1 mRNA is abundantly expressed in cortical and hippocampal interneurons throughout life (Fig. 2c). We next asked whether knocking out NR1 in corticolimbic interneurons at a later developmental time point would produce behavioral phenotypes similar to those observed when NR1 was knocked out early in postnatal development. To achieve knockout of the NR1 gene in adults in the same Cre-targeted populations, we used a second loxP-flanked NR1 line (NR1loxP/loxP-line B, referred to here as Flox-B)31 with two loxP sites located ∼12 kb apart, resulting in substantially delayed recombination. NR1 ablation in corticolimbic GABAergic neurons in the resultant knockout mutant (Ppp1r2-cre+/−; NR1loxP/loxP-line B, referred to here as adult knockout mutant) obtained by crossing Ppp1r2-cre mice with the Flox-B line began at 8 weeks and was completed by 20 weeks (Fig. 6a).
Figure 6.
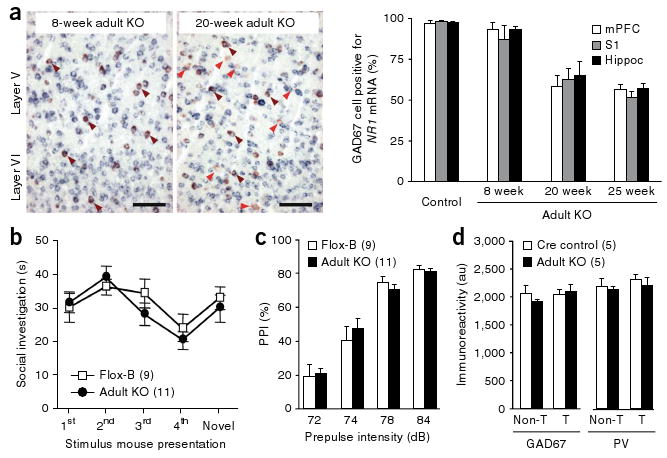
No schizophrenia-related phenotypes were observed following adult NR1 deletion. (a) Left, representative photomicrographs from cortex of the adult knockout mutant (Ppp1r2-cre+/−; NR1loxP/loxP-line B) prior to onset of recombination (8 weeks old) and after recombination was completed (20 weeks old). Brown arrows indicate colocalization of Gad67 and NR1 mRNAs, and red arrows indicate GAD67-positive neurons lacking NR1 mRNA. Right, quantification of double in situ hybridization detection of NR1 and Gad67 mRNA in the adult knockout mutants (n = 3 for each age). (b,c) Single-housed adult knockout mutants tested at 25–29 weeks of age were not impaired in a social-recognition test (b) or PPI (c). (d) After crossing with the loxP-flanked Rosa26-EYFP mice, adult knockout mutants (Ppp1r2-cre+/−; NR1loxP/loxP-line B; Rosa26-EYFPloxP/+) showed no significant change in GAD67 or parvalbumin immunoreactivity in Cre-targeted neurons of layer II/III S1 compared to Cre controls (Ppp1r2-cre+/−; Rosa26-EYFPloxP/+) at 26–27 weeks old. T, Cre-targeted neurons; Non-T, nontargeted neurons. Data are mean ± s.e.m.; n is indicated in parentheses.
Behavioral analysis of single-housed adult knockout mutant males and Flox-B control littermates were conducted 5 weeks after the completion of NR1 knockout (at 25 weeks) to parallel the time course used in the postnatal knockout experiments. Adult knockout mutants did not show the behavioral deficits observed in the postnatal knockout mutant mice and displayed normal social recognition (Fig. 6b), normal PPI of the auditory startle reflex (Fig. 6c) and normal behavior in the elevated plus maze (4.87 ± 1.2 entries to open arms for Flox-B control versus 3.82 ± 1.4 for adult knockout mutant, t test, P = 0.58) and in the Y-maze spontaneous alternation task (alternation index (%); 65.8 ± 6.1 for Flox-B control versus 65.2 ± 4.6 for adult knockout mutant, t test, P = 0.93). Even at >32 weeks of age, adult knockout mutants did not have a nest-building impairment (0.02 ± 0.01 g of unused nestlet for Flox-B control versus 0.016 ± 0.02 g for adult knockout mutant) or anxiety-like behaviors (data not shown), indicating that the absence of deficits in adult knockout mutants was not a result of a delayed onset of schizophrenia-related symptoms. There was also no reduction in GAD67 or parvalbumin immunoreactivity in the Cre-targeted neurons of the adult knockout mutants immediately after the completion of NR1 deletion (20 weeks old; data not shown) or at 26–27 weeks (Fig. 6d), suggesting that GABAergic dysfunction in adult knockout mutants was negligible. Moreover, in vivo electrophysiological recordings revealed no differences in mean firing rates or cross-correlation values between nearby pyramidal neurons in adult knockout mutants versus controls (Fig. 7a–c). Collectively, these results suggest that early postnatal NMDAR hypofunction in corticolimbic GABAergic neurons is crucial for the development of schizophrenia-related symptoms in mice.
Figure 7.
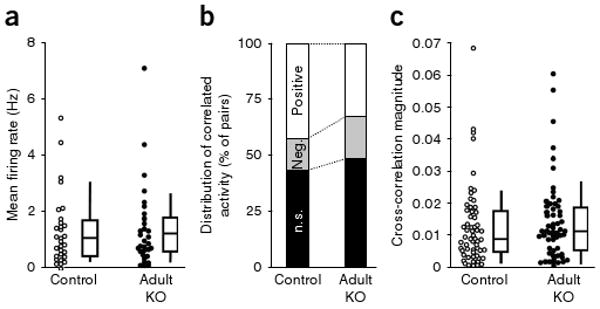
Adult NR1 deletion did not alter firing rate or synchronous firing of cortical excitatory neurons. (a) We found no difference in the mean firing rates of putative pyramidal neurons from S1 between 25–28-week-old controls (n = 2 for Cre (Ppp1r2-cre+/−), n = 2 for Flox-B (NR1loxP/loxP-line B)) and adult knockout mutants (Ppp1r2-cre+/−; NR1loxP/loxP-line B, (n = 4) during exploration of an unfamiliar linear track. Each dot represents the mean firing rate of an individual neuron (n = 31 for control, n = 33 for mutant). Mann-Whitney U test, P = 0.87. (b) Synchronized activity of pairs of nearby neurons from the recording shown in a. No difference was observed in the distribution of cross-correlation patterns between adult knockout mutants and age-matched controls (χ2 test, P = 0.24). (c) There was no difference in the magnitude of cross-correlations across all pairs between groups (Mann-Whitney U test, P = 0.32). Box plots depict medians (box centers), interquartile ranges (box boundaries) and 10–90th percentiles (whiskers). Each dot represents an individual pair.
Discussion
We found, to the best of our knowledge, for the first time that the integrity of NMDAR in cortical and hippocampal interneurons in early postnatal development was critical for maintaining normal GABAergic function. Furthermore, a restricted deletion of NMDAR in early postnatal corticolimbic interneurons was sufficient to trigger several behavioral and pathophysiological features in mice that resemble human schizophrenia. Together, our results provide strong experimental support for the long-standing hypothesis that corticolimbic NMDAR hypofunction is a primary site of schizophrenia pathogenesis14,15.
Cortical interneuron NMDAR hypofunction and schizophrenia
Our postnatal NR1 knockout mutant mice share several pathophysiological features that are typical of major psychiatric disorders, especially, the constellation of symptoms found in schizophrenia. In particular, the mutant mice exhibited both positive symptoms, such as psychomotor agitation, and negative symptoms, such as a reduced preference for sweet solution and deficits in nesting/mating, which mirror anhedonia and social withdrawal. In addition, the mutant mice had cognitive symptoms such as deficits in spatial working memory and short-term social memory. Impaired sensorimotor gating, as indicated by decreased PPI of the startle reflex in our mutant mice, is also often observed in individuals with schizophrenia29. Similarly, the NR1-deleted cortical GABAergic neurons had reduced GAD67 and parvalbumin levels, concurring with reduced expression of these markers12,13 in the postmortem cortex of individuals with schizophrenia. The disinhibition of cortical excitatory neurons and reduced neuronal synchrony that we observed in the postnatal knockout mutants is also consistent with hyperactivity of the dorsolateral prefrontal cortex during a working-memory task, which is seen in individuals with schizophrenia32.
Notably, several mutant phenotypes, including deficits in nest building and mating, anhedonic, and anxiety-like behaviors were first observed after 12 weeks, suggesting that there is a latency period between NR1 knockout and the emergence of these phenotypes. This latency period resembles the premorbid stage that precedes the emergence of symptoms that is characteristic of several major psychiatric disorders, particularly schizophrenia33. Social isolation–induced stress exacerbated the expression of these phenotypes in the mutant mice, similar to the stress-induced precipitation typical of psychiatric illnesses in human, which is particularly well-characterized in schizophrenia34.
Utility of conditional NR1 mutants for schizophrenia research
The phenotypes observed in our mutants reflect many, but not all, of the clinical features of human schizophrenia. For instance, the finding that spatial working memory deficits were ameliorated by chronic treatment with risperidone is consistent with evidence that risperidone alleviates phencyclidine-induced behavioral abnormalities in rodents, including working-memory deficits35. However, there are conflicting reports about the efficacy of risperidone to ameliorate deficits in verbal and spatial working memory in schizophrenia subjects36,37.
Although a number of schizophrenia susceptibility genes have been implicated in the modulation of NMDAR activity38, genetic linkage studies have not directly associated polymorphisms in the NR1 subunit with schizophrenia39. It is also unlikely that genetic predispositions of human schizophrenia are manifested solely in corticolimbic GABAergic neurons. Nevertheless, we expect that our strategy to spatially and temporally restrict NMDAR hypofunction will provide insights into the mechanisms of cellular and behavioral manifestation of schizophrenia-related phenotypes. Indeed, restricting the genetic deletion of NR1 to corticolimbic GABAergic neurons during early postnatal development was sufficient to trigger the development of schizophrenia-related phenotypes after adolescence in mice.
Evidence for neurodevelopmental origin of pathophysiology
Schizophrenia-related behaviors in mice were absent when genetic NR1 ablation in GABAergic neurons occurred after adolescence. It appears that the dysregulation of GABAergic neurons, as revealed by the reduction of GAD67 and parvalbumin levels, is not simply attributed to NR1 deletion from GABAergic neurons. Instead, the NR1 deletion impaired the postnatal maturation of GABAergic neurons, and, in the absence of proper GABAergic inhibition, the refinement of cortical circuitry may be impaired40. Accordingly, postnatal NR1 knockout in the corticolimbic GABAergic neurons contributed to an increase in excitatory neuronal activity and reduced neuronal synchrony; no such phenotypes were observed in adult NR1 knockout mutant mice. The mechanisms by which NMDA hypofunction impairs cortical maturation and underlies the pathological phenotype warrants future research.
This idea of abnormal maturation of cortical circuits is consistent with the neurodevelopmental hypothesis of schizophrenia41. Notably, early postnatal NR1 deletion in our mice corresponds to a period encompassing late gestation up to 2 years in human infants42. We speculate that a disturbance of NMDAR function in cortical interneurons during this early developmental period, resulting from genetic or epigenetic alteration of NMDARs or downstream signaling molecules, would lead to abnormal cortical maturation and increase susceptibility to psychiatric illness after adolescence. Our postnatal NR1 knockout mutants promise to provide insights into approaches for understanding the development of human psychiatric illnesses such as schizophrenia.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/natureneuroscience/.
Supplementary Material
Acknowledgments
We thank N. Heintz for pLD53.SCAEB plasmid and the BAC homologous recombination protocol, F. Costantini for the loxP-flanked Rosa26-EYFP mouse strain, D.L. Brautigan for antibody to Ppp1r2, B. Condie and J. Rubenstein for Gad67 cDNA, J. Pickel for oocyte injections, J.N. Crawley for advice on behavioral testing, S. Zhang, J. Okolonta and M. Taylor for technical and animal care assistance, H. Matsunami for in situ hybridization protocol, Y. Kubota for immunostaining protocol and J. Yamamoto for Neuralynx/Xclust2 conversion software. We thank D.R. Weinberger, M.M. Behrens, G. Kunos, I. Henter, K.M. Christian, H. Giesen and H.A. Nash for critical comments on the manuscript. We also acknowledge the CURE/Digestive diseases research center at the University of California Los Angeles and the US National Institute of Mental Health Chemical Synthesis and Drug Supply Program for antibodies and risperidone, respectively. This work was supported by the Intramural Research Program of the US National Institute of Mental Health and of the US National Institute on Alcohol Abuse and Addiction.
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
Author Contributions: J.E.B. and K.N. designed most of the experiments and wrote the paper. V.Z. was responsible for the slice physiology data. E.R.S. and Z.J. conducted some of the behavioral testing. G.Y. and E.M.Q. were responsible for the visually evoked potentials. Y.L. provided animal resources. All other experiments were data-collected by J.E.B. All authors discussed the results and commented on the manuscript.
References
- 1.Lodge D, Anis NA. Effects of phencyclidine on excitatory amino acid activation of spinal interneurones in the cat. Eur J Pharmacol. 1982;77:203–204. doi: 10.1016/0014-2999(82)90022-x. [DOI] [PubMed] [Google Scholar]
- 2.Javitt DC. Negative schizophrenic symptomatology and the PCP (phencyclidine) model of schizophrenia. Hillside J Clin Psychiatry. 1987;9:12–35. [PubMed] [Google Scholar]
- 3.Olney JW. In: Excitatory Acid Acids in Health and Disease. Lodge D, editor. Wiley; London: 1988. pp. 337–351. [Google Scholar]
- 4.Deutsch SI, Mastropaolo J, Schwartz BL, Rosse RB, Morihisa JMA. “glutamatergic hypothesis” of schizophrenia. Rationale for pharmacotherapy with glycine. Clin Neuropharmacol. 1989;12:1–13. [PubMed] [Google Scholar]
- 5.Krystal JH, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 6.Coyle JT. The glutamatergic dysfunction hypothesis for schizophrenia. Harv Rev Psychiatry. 1996;3:241–253. doi: 10.3109/10673229609017192. [DOI] [PubMed] [Google Scholar]
- 7.Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- 8.Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends Neurosci. 2001;24:527–533. doi: 10.1016/s0166-2236(00)01886-5. [DOI] [PubMed] [Google Scholar]
- 9.Labrie V, Lipina T, Roder JC. Mice with reduced NMDA receptor glycine affinity model some of the negative and cognitive symptoms of schizophrenia. Psychopharmacology (Berl) 2008;200:217–230. doi: 10.1007/s00213-008-1196-6. [DOI] [PubMed] [Google Scholar]
- 10.Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 11.Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 12.Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52:293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Eyles DW, McGrath JJ, Reynolds GP. Neuronal calcium-binding proteins and schizophrenia. Schizophr Res. 2002;57:27–34. doi: 10.1016/s0920-9964(01)00299-7. [DOI] [PubMed] [Google Scholar]
- 14.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 15.Grunze HC, et al. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16:2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci USA. 2004;101:8467–8472. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasegawa M, et al. MK-801 increases endogenous acetylcholine release in the rat parietal cortex: a study using brain microdialysis. Neurosci Lett. 1993;150:53–56. doi: 10.1016/0304-3940(93)90106-u. [DOI] [PubMed] [Google Scholar]
- 18.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. 1997;154:805–811. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- 20.Vollenweider FX, et al. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG) Eur Neuropsychopharmacol. 1997;7:9–24. doi: 10.1016/s0924-977x(96)00039-9. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Clark S, Lewis DV, Wilson WA. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: a potential mechanism of neurotoxicity. J Neurosci. 2002;22:3070–3080. doi: 10.1523/JNEUROSCI.22-08-03070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cochran SM, et al. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- 23.Keilhoff G, Becker A, Grecksch G, Wolf G, Bernstein HG. Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience. 2004;126:591–598. doi: 10.1016/j.neuroscience.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 24.Rujescu D, et al. A pharmacological model for psychosis based on N-methyl-d-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry. 2006;59:721–729. doi: 10.1016/j.biopsych.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 25.Behrens MM, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- 26.Morrow BA, Elsworth JD, Roth RH. Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacology (Berl) 2007;192:283–290. doi: 10.1007/s00213-007-0708-0. [DOI] [PubMed] [Google Scholar]
- 27.Dang MT, et al. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci USA. 2006;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piskulic D, Olver JS, Norman TR, Maruff P. Behavioral studies of spatial working memory dysfunction in schizophrenia: a quantitative literature review. Psychiatry Res. 2007;150:111–121. doi: 10.1016/j.psychres.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 30.Bubeníková-Valesová V, Horácek J, Vrajová M, Höschl C. Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci Biobehav Rev. 2008;32:1014–1023. doi: 10.1016/j.neubiorev.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- 32.Whitfield-Gabrieli S, et al. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc Natl Acad Sci USA. 2009;106:1279–1284. doi: 10.1073/pnas.0809141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cannon M, et al. Premorbid social functioning in schizophrenia and bipolar disorder: similarities and differences. Am J Psychiatry. 1997;154:1544–1550. doi: 10.1176/ajp.154.11.1544. [DOI] [PubMed] [Google Scholar]
- 34.Agid O, Kohn Y, Lerer B. Environmental stress and psychiatric illness. Biomed Pharmacother. 2000;54:135–141. doi: 10.1016/S0753-3322(00)89046-0. [DOI] [PubMed] [Google Scholar]
- 35.Lim EP, Verma V, Nagarajah R, Dawe GS. Propranolol blocks chronic risperidone treatment-induced enhancement of spatial working memory performance of rats in a delayed matching-to-place water maze task. Psychopharmacology (Berl) 2007;191:297–310. doi: 10.1007/s00213-006-0664-0. [DOI] [PubMed] [Google Scholar]
- 36.Houthoofd SA, Morrens M, Sabbe BG. Cognitive and psychomotor effects of risperidone in schizophrenia and schizoaffective disorder. Clin Ther. 2008;30:1565–1589. doi: 10.1016/j.clinthera.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 37.Reilly JL, Harris MS, Keshavan MS, Sweeney JA. Adverse effects of risperidone on spatial working memory in first-episode schizophrenia. Arch Gen Psychiatry. 2006;63:1189–1197. doi: 10.1001/archpsyc.63.11.1189. [DOI] [PubMed] [Google Scholar]
- 38.Carter CJ. Schizophrenia susceptibility genes converge on interlinked pathways related to glutamatergic transmission and long-term potentiation, oxidative stress and oligodendrocyte viability. Schizophr Res. 2006;86:1–14. doi: 10.1016/j.schres.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 39.Martucci L, et al. N-methyl-d-aspartate receptor NR1 subunit gene (GRIN1) in schizophrenia: TDT and case-control analyses. Am J Med Genet B Neuropsychiatr Genet. 2003;119B:24–27. doi: 10.1002/ajmg.b.20014. [DOI] [PubMed] [Google Scholar]
- 40.Huang ZJ. Activity-dependent development of inhibitory synapses and innervation pattern: role of GABA signalling and beyond. J Physiol (Lond) 2009;587:1881–1888. doi: 10.1113/jphysiol.2008.168211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 42.Hagberg H, Ichord R, Palmer C, Yager JY, Vannucci SJ. Animal models of developmental brain injury: relevance to human disease. A summary of the panel discussion from the Third Hershey Conference on Developmental Cerebral Blood Flow and Metabolism. Dev Neurosci. 2002;24:364–366. doi: 10.1159/000069040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.