

Abstract
Sodium arsenite (NaAs)-induced autophagic cell death (ACD) of a mouse renal tubular epithelial cell line (mProx24), which expresses enhanced levels of interleukin-6 (IL-6), was reduced by the suppression of autophagy by 3-methyladenine or Atg7 knockdown. The inhibition of the IL-6/signal transducer and activator of transcription 3 (STAT3) signal pathway by anti-IL-6 antibody or a Jak2 inhibitor (AG490) exaggerated ACD of mProx24 cells after NaAs challenge, attenuating STAT3 activation and reciprocally enhancing extracellular signal-regulated kinase (ERK) phosphorylation. In contrast, an ERK inhibitor, PD98059, reduced NaAs-induced ACD in mProx24 cells. Subcutaneous injection of NaAs (12.5 mg/kg) into BALB/c (wild-type) mice enhanced intrarenal expression of IL-6, mainly produced by tubular cells, and caused severe renal injury characterized by hemorrhages, acute tubular necrosis, cast formation, and brush border disappearance, with increases in serum urea nitrogen (blood urea nitrogen) and creatinine levels. In addition, IL-6-deficient (IL-6−/−) mice exhibited exaggerated histopathological changes with higher blood urea nitrogen and creatinine levels. Moreover, in IL-6−/− mice treated with NaAs, ACD in renal tubular cells was significantly augmented, along with diminished STAT3 activation and reciprocal enhancement of ERK signaling, compared with wild-type mice. Finally, the administration of exogenous IL-6 into wild-type mice significantly reduced NaAs-induced ACD along with diminished ERK activation and eventually alleviated acute renal dysfunction. Thus, IL-6/STAT3 signal pathway could inhibit ERK activation, a crucial step for ACD, eventually attenuating NaAs-induced renal dysfunction.
Arsenic is ubiquitously distributed in the natural environment such as soil, water, and air and is commonly associated with the ores of metals like copper, lead, and gold.1 Acute arsenic exposure can cause a profound injury to various organs, including kidney, liver, intestine, and brain, and can result in high mortality and morbidity,2 and massive renal tubular necrosis is a characteristic pathological feature of renal injury caused by acute arsenic exposure.3,4 Moreover, the environmental pollution of arsenic sometimes causes serious health problems in several developing countries, because chronic exposure to arsenic results in the dysfunctions in renal and nervous systems5,6 and often acts as carcinogen of skin, lung, bladder, liver, and kidney.7,8 On the other hand, arsenic trioxide (As2O3) has recently been shown to be effective for acute promyelocytic leukemia without causing bone marrow (BM) suppression,9–11 and its anticancer efficiency is being extended to several types of solid tumors.12
Programmed cell death is indispensable for various physiological processes, including development, maintenance of homeostasis, and regulation of immune system.13 Programmed cell death system can be classified into two major types, apoptosis and autophagic cell death. Apoptosis is designated as type I programmed cell death and is characterized by membrane blebbing, DNA fragmentation, and the preservation of organelles.14 In contrast, autophagic cell death, designated as type II programed cell death, exhibits the appearance of vacuoles engulfing bulk cytoplasm and cytoplasmic organelles such as mitochondria and endoplasmic reticulum.15,16 Some anticancer drugs are presumed to exert their actions by inducing autophagic cell death17 as well as apoptosis. Indeed, a potent anticancer agent, As2O3, can induce autophagic cell death in several malignant cells.18,19 Moreover, autophagic cell death was crucially involved in several diseases such as atherosclerosis, hypoxic neuronal death, and cardiomyopathy.20–24
Several cytokines can regulate the pathway involved in autophagic cell death. Th2 cytokines such as interleukin (IL)-4 and IL-13 can suppress autophagy by activating phosphatidylinositol 3-kinase.25 In sharp contrast, pro-inflammatory cytokines, tumor necrosis factor-α24 and interferon-γ,26 can promote autophagy in macrophage and vascular smooth muscle cells, respectively. IL-6 is produced by various types of cells and exhibits various similar activities as tumor necrosis factor-α on a wide variety of cells including lymphocytes, hepatocytes, and neuronal cells.27 However, it remains to be investigated on the effects of IL-6 on autophagic cell death.
We observed that sodium arsenite (NaAs) exposure caused autophagic cell death as well as IL-6 production in a murine renal tubular epithelial cell line, mProx24. Moreover, tubular cell necrosis due to autophagic cell death was observed in acute NaAs-induced renal injury. These observations prompted us to investigate the roles of IL-6 and its downstream signaling molecules in NaAs-induced autophagic death of renal tubular cells. We demonstrated that NaAs-induced autophagic cell death of mProx24 cells was augmented by anti-IL-6 antibodies (Abs) and inhibitors of Janus kinase 2 (JAK2) and that the inhibition of extracellular signal-regulated kinase (ERK)1/2 suppressed NaAs-induced autophagic cell death of mProx24 cells. These molecules that are localized downstream the IL-6/signal transducer and activator of transcription 3 (STAT3) pathway. Furthermore, genetic ablation of IL-6 gene and IL-6 administration enhanced and alleviated NaAs-induced acute renal tubular cell necrosis with autophagic cell death, respectively. Thus, IL-6-mediated signals can counteract NaAs-induced acute renal injury characterized by autophagic cell death of renal tubular epithelial cells.
Materials and Methods
Reagents and Abs
NaAs was purchased from Wako Chemical Industries (Osaka, Japan). 3-Methyladenine (3-MA, an inhibitor of autophagy), AG490 (a JAK2 inhibitor), and PD98059 (an ERK1/2 kinase inhibitor) were obtained from Sigma- Aldrich (Tokyo, Japan). E64d and pepstatin A (lysosomal protease inhibitors) were obtained from Calbiochem (San Diego, CA). The following polyclonal Abs (pAbs) were used in this study; goat anti-mouse IL-6 pAbs (R&D Systems, Minneapolis, MN), rabbit anti-LC3 pAbs (Sigma-Aldrich), rabbit anti-STAT3 pAbs, rabbit anti-phospholyrated-STAT3 (p-STAT3) at Tyr705 pAbs, rabbit anti-ERK1/2 pAbs, and rabbit anti-phospholyrated-ERK1/2 (p-ERK1/2) at Thr202/Tyr204 pAbs (Cell Signaling Technology, Danvers, MA). Murine recombinant IL-6 was prepared as described previously.28
Mice
Pathogen-free 8- to 10-week old male BALB/c mice were obtained from Sankyo Laboratories (Tokyo, Japan) and were designated as wild-type mice in the following experiments. Age- and sex-matched IL-6−/− mice, backcrossed to BALB/c mice for more than 10 generations, were used in this study as described previously.29 All mice were housed individually in cages under the specific pathogen-free conditions during the experiments. All of the experimental procedures were approved by Animal Research Committee of Wakayama Medical University (Wakayama, Japan).
NaAs-Induced Renal Injury
NaAs-induced renal injury was induced as described previously.3,4 Briefly, mice were administered s.c. with NaAs (12.5 mg/kg). In other series of experiments, mice received s.c. injection of recombinant IL-6 (0.5 μg/mouse) or vehicle (phosphate-buffered saline) 1 hour after NaAs challenge (13.5 mg/kg) or received i.p. injection of PD98059 (5 mg/kg in 10% DMSO) or vehicle (10% DMSO) immediately after NaAs challenge.30
Generation of BM Chimeric Mice
BM chimeric mice were prepared as described previously.31 Briefly, IL-6−/− BM to wild-type mice, wild-type BM to IL-6−/− mice, wild-type BM to wild-type mice, and IL-6−/− BM to IL-6−/− mice. BM cells were collected from the femurs of donor mice by aspiration and flushing. Recipient mice were irradiated to 15 Gy using an RX-650 irradiator (Faxitron X-ray, Wheeling, IL). Then, the animals i.v. received 5 × 106 BM cells from donor mice in a volume of 200 μl of sterile PBS under the anesthesia. Thereafter, the mice were housed in sterilized microisolator cages and were fed normal chow and autoclaved hyperchlorinated water for 60 days. Successful engraftment and reconstruction of the BM in the transplanted mice were confirmed by PCR analysis for wild-type or mutant IL-6 gene of peripheral blood of each chimeric mouse 30 days after BMT. After durable BM engraftment was confirmed, mice were treated with NaAs as described above.
Determination of Blood Urea Nitrogen and Creatinine in the Serum
At the indicated time intervals after NaAs challenge, sera were collected to determine blood urea nitrogen (BUN) and creatinine (CRE) levels with a Fuji DRI-CHEM 5500V (Fuji Medical System, Tokyo, Japan), according to the manufacturer's instructions.
Histopathological and Immunohistochemical Analyzes
Kidney tissues were obtained at the indicated time intervals after NaAs challenge. Paraffin-embedded sections (6 μm thick) were prepared and subjected to H&E staining or PAS staining. The degree of renal damage was scored with examination of hemorrhages, the disappearance of PAS-positive brush border, and cast formation in 10 randomly chosen regions in each sample at a magnification of ×100. Renal morphological alterations were graded on a scale of 0 to 3+: 0, normal; 1+, slight; 2+, moderate; and 3+, severe. The mean of all of the fields was taken as the damage score in each sample. Immunohistochemical analyses for IL-6 or LC3 were performed as described previously.3,4 Histopathological and immunohistochemical evaluation was conducted by an examiner without a prior knowledge on the experimental procedures.
Quantification of Intrarenal Arsenic Contents
Intrarenal arsenic contents were analyzed with the use of certified arsenic standard solution (As 100: Lot. RWN9791; Wako Chemical Industries).3,4 Briefly, a portion of the kidney (∼100 mg) was digested in nitric acid (100 mg/ml). Total arsenic was determined using graphite furnace atomic absorption spectrometry ACF-6800 (Shimadzu, Kyoto, Japan). The data were expressed as arsenic (μg)/sample weight (g).
Cell Culture
A murine renal proximal tubular epithelial cell line (mProx24), derived from C57BL/6J adult mouse kidney,32 was maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum at 37°C in 5% CO2. Then, the cells were incubated for the indicated time intervals in the presence of various combinations of NaAs (20 μmol/L), goat anti-IL-6 pAbs (2 μg/ml), 3-MA (2 mmol/L), AG490 (50 μmol/L), PD98059 (20 μmol/L), E64d (10 μg/ml), and pepstatin A (10 μg/ml). Cell viability was assessed with a Cell Counting Kit (Dojin Laboratories, Kumamoto, Japan) to count living cells with the combination of 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium and 1-methoxyphenazine methosulfate.
Transient Transfection of the GFP-LC3 into mProx24 Cells
mProx24 cells at 70% confluence were transfected with EGFP-LC3 plasmid33 (a gift from Dr. T. Yoshimori, Osaka University, Japan) using Lipofectamine reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. After 24 hours, cells were treated with NaAs, and the images were observed using a LSM5Pascal Exciter (Carl Zeiss Japan, Tokyo, Japan) laser scanning confocal microscope.
Atg7 Knockdown by Small Interfering RNA
Small interfering RNA (siRNA) (Accell SMART pool siRNA Reagent) for Atg7 were purchased from Thermo Fisher Scientific (Yokohama, Japan). Nontargeting siRNA (Accell Non-targting siRNA number 1; Thermo Fisher Scientific) was used as a control. mProx24 cells at ∼30% confluence were transfected with siRNA (1 μmol/L) in Accell delivery media (Thermo Fisher Scientific). After 72 hours, cells were treated with NaAs, followed by cell viability assay or Western blotting analyses.
Electron Microscopy
Transmission electron microscopy was performed with some modifications, as described previously.34 Briefly, mProx24 cells were harvested by trypsinization and fixed with 1% glutaraldehyde in 100 mmol/L phosphate buffer (pH 7.4) at 4°C for 1 hour. After washing in 100 mmol/L PBS, the cells were postfixed in 2% osmium tetroxide at 4°C for 1 hour, dehydrated in graded ethanol, and embedded in Quetol-812 resin (Nisshin EM, Tokyo, Japan). Ultrathin sections (70 nm thick) of the cells were stained with uranyl acetate and lead citrate and observed with a transmission electron microscope (JEM-1220; JEOL, Tokyo, Japan).
Semiquantitative RT-PCR
A semiquantitative RT-PCR was conducted as described previously.35 Total RNAs were extracted from kidney samples or mProx24 cells using ISOGEN (Nippon Gene, Toyama, Japan). Five micrograms of total RNA was reverse-transcribed at 42°C for 1 hour in 20 μl of reaction mixture containing mouse Moloney leukemia virus reverse transcriptase (Toyobo, Osaka, Japan) and oligo(dT) primers (Amersham Biosciences Japan, Tokyo, Japan). Thereafter, cDNA was amplified together with Taq polymerase (Nippon Gene) using specific primers (Table 1). The PCR products were fractionated on a 2% agarose gel and visualized by ethidium bromide staining. The band intensities were measured using NIH Image Analysis Software version 1.61 (National Institutes of Health, Bethesda, MD) and were calculated as the ratios to β-actin.
Table 1.
Sequences of the Primers Used for RT-PCR
| Transcript | Sequence | Annealing temperature (°C) | Cycle | Product size (bp) |
|---|---|---|---|---|
| IL-6 | (F) 5′-TGGCCAAAGTTCCTGACTTGTTTG-3′ | 64 | 36 | 489 |
| (R) 5′-CAGGCTATTTAACCAAGTGGTGCT-3′ | ||||
| MRP1 | (F) 5′-GTTCCCTCCGCATGAACTTG-3′ | 55 | 30 | 550 |
| (R) 5′-CTGGCTCATGCCTGGACTCTG-3′ | ||||
| MRP2 | (F) 5′-TGAACTGGCTAGTGAGGA-3′ | 55 | 30 | 379 |
| (R) 5′-GTCAGTCTCCCTCGAAGG-3′ | ||||
| β-Actin | (F) 5′-TTCTACAATGAGCTGCGTGTGGC-3′ | 62 | 28 | 456 |
| (R) 5′-CTCATAGCTCTTCTCCAGGGAGGA-3′ |
F, forward primer; R, reverse primer.
Western Blotting Analysis of Mouse Kidney Proteins
At the indicating time intervals after NaAs challenge, kidney tissues were obtained and homogenized with a lysis buffer (10 mmol/L PBS (pH 7.4) containing 0.01% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) containing complete protease inhibitor mixture, and phosphatase inhibitor mixtures for serine/threonine protein phosphatases and tyrosine protein phosphatases (P2850 and P5726; Sigma-Aldrich) and were centrifuged to obtain lysates. Thereafter, equal amounts of protein were separated on 10 or 15% SDS-polyacrylamide gel electrophoresis and transferred to an Immobilon-P transfer membrane (Millipore, Billerica, MA). After the incubation of the membrane with anti-LC3 pAbs, anti-STAT3 pAbs, anti-p-STAT3 pAbs, anti-ERK1/2 pAbs, p-ERK1/2 pAbs, or anti-β-actin, immune complexes were detected using an enhanced chemiluminescence reagent (Millipore), according to the manufacturer's instructions. In another series of experiments, mProx24 cells were scraped and centrifuged. The cell pellets were dissolved with SDS sample buffer. Thereafter, Western blotting analysis was performed to detect STAT3, p-STAT3, ERK1/2, p-ERK1/2, and anti-β-actin, as mentioned above.
Statistical Analysis
The means and SEMs were calculated for all parameters determined in this study. Statistical significance was evaluated using analysis of variance (analysis of variance) or Mann-Whitney U-test. A value of P < 0.05 was accepted as statistically significant.
Results
Induction of Autophagic Cell Death in mProx24 Cells after NaAs Challenge
NaAs-induced acute renal injury is characterized by a massive renal tubular cell necrosis.3,4 Because arsenic trioxide can induce autophagic cell death in several tumor cells,18,19 we first examined whether NaAs-induced renal tubular cell necrosis was due to autophagic cell death. LC3 protein induces autophagosome formation through its conversion of LC3-I to LC3-II,33,36,37 and therefore, the amount of LC3-II is increased as autophagic process proceeds. Hence, we examined LC3 protein in NaAs-treated mProx24 cells as a maker of autophagic cell death. NaAs challenge increased LC3-II amounts (Figure 1, A and B) and decreased the viability of mProx24 cells (Figure 1C). Ultrastructurally, NaAs-treated cells exhibited typical features of autophagic cell death, with the appearance of vacuoles engulfing bulk cytoplasm and cytoplasmic organelles (Figure 1D). An autophagy inhibitor, 3-MA, abrogated NaAs-induced increases in LC3-II and decreases in cell viability (Figure 1, A–C). Similarly, the block of autophagy by the knockdown of Atg7, a key molecule for autophagy, increased the cell viability after NaAs treatment, compared with control siRNA-treated cells (Figure 1, E and F) but augmented the amount of activated caspase 3 (data not shown). Thus, these observations would indicate that NaAs mainly caused autophagic cell death in mProx24 cells.
Figure 1.
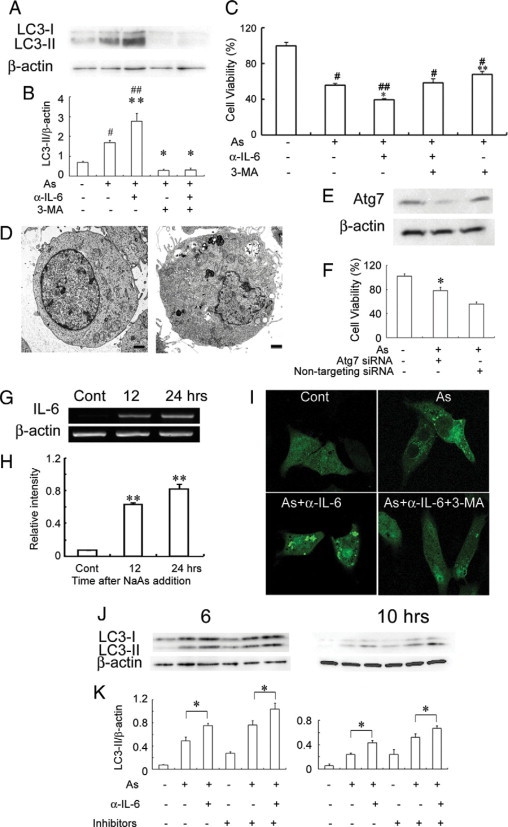
A and B: Effects of anti-IL-6 pAbs and 3-MA on NaAs-induced conversion of LC3-I to LC3-II in mProx24 cells after 12 hours of culture. Western blotting analyses were performed as described in Materials and Methods. Representative results from six independent experiments are shown in A. B: LC3-II/β-actin levels were densitometrically determined. All values represent means ± SEM (six independent experiments). **P < 0.01; *P < 0.05, versus mProx24 cells cultured with only NaAs. #P < 0.01; ##P < 0.05, versus untreated mProx24 cells. C: Effects of anti-IL-6 pAbs and 3-MA on the viability of mProx24 cells cultured with NaAs for 12 hours. All values represent means ± SEM (n = 16 wells). ##P < 0.01; #P < 0.05, versus control. **P < 0.01; *P < 0.05, versus cultured with only NaAs. D: The ultrastructure of untreated and NaAs-treated mProx24 cells (left: untreated; right: NaAs-treated). Numerous autophagic vacuoles were detected in NaAs-treated mProx 24 cells. Representative results from three independent experiments is shown here (bars = 1 μm). E: The effects of siRNA treatment on Atg7 expression. mProx24 cells were transfected with Atg7 siRNA or nontargeting control siRNA. Whole-cell lystes were subjected to Western blotting with anti-Atg7 pAbs. F: Effects of Atg7 knockdown on viability of mProx24 cell cultured with NaAs for 12 hours. All values represent means ± SEM (n = 6 wells). *P < 0.05, versus nontargeting siRNA trasfected mProx24 cells. G and H: RT-PCR analysis for IL-6 gene expression in mProx24 cells cultured in the presence of NaAs. Representative results from six independent experiments are shown in G. The ratios of IL-6 to β-actin are calculated and shown in H. All values represent means ± SEM (n = 6 independent experiments). **P < 0.01, versus control. I: Fluorescence microscopic determination of GFP-LC3 distributions in mProx24 cells treated with NaAs, NaAs+anti-IL-6 pAbs, or NaAs+anti-IL-6 pAbs + 3-MA for 12 hours. Representative results from six independent experiments are shown here. J and K: Effects of E64d and pepstatin A on NaAs-induced conversion of LC3-I to LC3-II in mProx24 cells. NaAs-induced LC3-II formation in mProx24 cells cultured in the presence or absence of E64d and pepstatin A were analyzed as described in Materials and Methods. Representative results from six independent experiments are shown in J. LC3-II/β-actin ratios were densitometrically determined and shown in K. All values represent means ± SEM (six independent experiments). *P < 0.05.
Enhanced NaAs-Induced Autophagic Cell Death of mProx24 Cells by IL-6 Neutralization
Concomitantly, NaAs exposure progressively augmented IL-6 gene expression in mProx24 cells (Figure 1, G and H), which expressed constitutively IL-6 receptor and gp130 mRNA (data not shown). These observations prompted us to examine the role of endogenous IL-6 in NaAs-induced renal tubular cell death. Anti-IL-6 pAbs further augmented NaAs-induced increases in LC3-II and decreases in cell viability (Figure 1, A–C). Consistently, anti-IL-6 pAbs further enhanced NaAs-induced LC3 puncta formation in GFP-LC3-transfected mProx24 cells, whereas 3-MA apparently reversed the effects of anti-IL-6 pAbs (Figure 1I). Either autophagosome accumulation or enhanced autophagic pathway can increase LC3-II amount.38 Lysosomal protease inhibitors augment the increase in LC3-II arising from enhanced autophagic pathway but not autophagosome accumulation. Thus, we examined the effects of lysosomal protease inhibitors on LC-3II amounts in mProx24 cells treated with NaAs and/or anti-IL-6 pAbs. At the indicated time points, two distinct lysosomal protease inhibitors, E64d and pepstatin A, further increased the amount of LC3-II in mProx24 cells treated with NaAs or NaAs plus anti-IL-6 pAbs (Figure 1, J and K). Thus, these observations would indicate that endogenously-produced IL-6 could suppress NaAs-induced autophagic cell death of renal tubular epithelial cells.
The Enhancement of NaAs-Induced Intrarenal ERK1/2 Signaling by the Absence of IL-6/STAT3 Signal Pathway
STAT3, a main signal transducer of IL-6,21,39 and ERK1/2 were presumed to be involved in the suppression and promotion of autophagy, respectively.40 Possible suppression of ERK signaling by the JAK2/STAT3 axis,41 prompted us to examine the effects of AG490 (a JAK2 inhibitor) and PD98059 (an ERK kinase inhibitor) on NaAs-induced autophagic cell death and the viability in mProx24 cells. The treatment with anti-IL-6 pAbs or AG490 further augmented autophagy as evidenced by increases in LC3-II amounts, together with reduced STAT3 activation and reciprocally enhanced ERK phosphorylation, and eventually decreased the cell viability (Figure 2). On the contrary, PD98059 suppressed ERK phosphorylation with few effects on NaAs-induced STAT3 phosphorylation and attenuated autophagic cell death of mProx24 cells as evidenced by reduced LC3-II amount and increased cell viability (Figure 2, B and E). Moreover, when 3-MA was added to the culture medium, cell viability was significantly increased in mProx 24 cells treated with NaAs+AG490 (NaAs+AG490 versus NaAs+AG490+ 3MA: 31.1 ± 1.8% vs. 54.8 ± 6.3%, P < 0.05), with reduced LC3-II formation (data not shown). In contrast, anti-IL-6 pAbs failed to modulate the activities of other signaling molecules involved in autophagic processes, such as p38 mitogen-activated protein kinase,42,43 eukaryotic translation inhibition factor 2α,44 Akt,45 mammalian target of rapamycin,39,45 and Bcl246 in mProx24 cells cultured with NaAs (data not shown). These observations suggested that the IL-6/STAT3/JAK2 axis can suppress NaAs-induced autophagic cell death of mouse renal tubular epithelial cells by dampening ERK signal pathway and eventually play a protective role in NaAs-induced cell injury.
Figure 2.
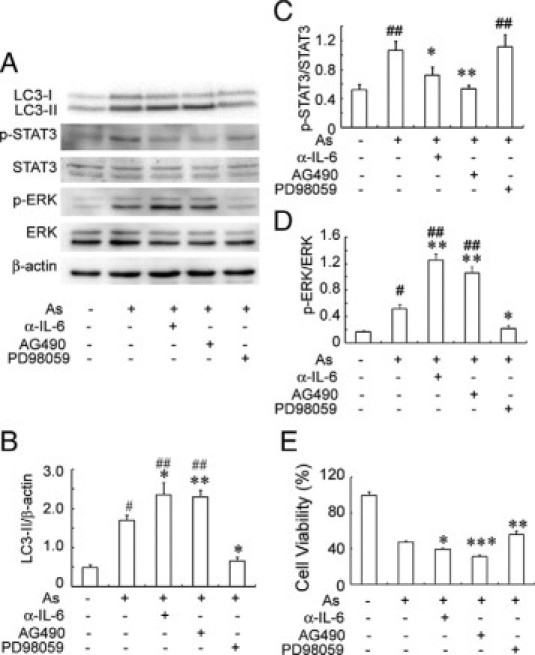
A: Effects of anti-IL-6 pAbs, AG490, and PD98059 on NaAs-induced autophagic cell death and the phosphorylation of STAT3 and ERK in mProx24 cells. Western blotting analyses were performed as described in Materials and Methods. Western blotting analysis using anti-β-actin Ab confirmed that an equal amount of protein was loaded onto each lane. Representative results from six independent experiments are shown in A. LC3-II/β-actin ratios (B), p-STAT3/STAT3 ratios (C), and p-ERK/ERK ratios (D) were densitometrically determined and are shown here. All values represent means ± SEM (six independent experiments). **P < 0.01; *P < 0.05, versus mProx24 cells cultured with only NaAs. #P < 0.01; ##P < 0.05, versus untreated mProx24 cells. E: Effects of anti-IL-6 pAbs, AG490, and PD98059 on the viability of mProx24 cells cultured with NaAs for 12 hours. All values represent means ± SEM (n = 16 wells). ***P < 0.005; **P < 0.01; *P < 0.05, versus cultured with only NaAs.
Enhanced NaAs-Induced Renal Dysfunction in the Absence of IL-6
We next examined whether NaAs exposure could enhance intrarenal IL-6 expression. The intrarenal IL-6 mRNA expression was transiently increased at 3 and 6 hours after s.c. injection of NaAs and returned to nearly basal level later than 10 hours after the challenge (Figure 3, A and B). Consistently, immunoreactive IL-6 proteins were detected in a small proportion of tubular epithelial cells in untreated mice and NaAs treatment markedly increased IL-6-positive epithelial cell numbers (Figure 3C). To explore the pathophysiological roles of endogenous IL-6 in vivo, we administered NaAs to wild-type and IL-6−/− mice. There were no significant differences in serum BUN and CRE levels between untreated wild-type and IL-6−/− mice, but NaAs challenge increased serum BUN and CRE levels to larger extents in IL-6−/− mice than wild-type ones (Figure 4, A and B). Concomitantly, IL-6−/− mice exhibited more severe histopathological alterations such as hemorrhages, acute tubular necrosis, cast formation, and the disappearance of PAS-positive brush border, compared with wild-type mice (Figure 4, C–G). However, in wild-type and IL-6−/− mice, the intrarenal gene expression of multidrug resistance-associated protein (MRP)1 and MRP2, which were essentially involved in arsenite efflux,3,4,47,48 were induced, to a similar extent, after NaAs challenge (Figure 5, A and B). Moreover, there was no significant difference in intrarenal arsenic contents after NaAs challenge between both strains (Figure 5C). Furthermore, to clarify the roles of BM-derived cells in NaAs-induced renal injury, we generated BM chimeric mice between wild-type and IL-6−/− mice. Irrespective of the origin of BM, NaAs-induced renal injury was significantly augmented in IL-6−/− mice, compared with wild-type mice, as evidenced by serum BUN and CRE levels (Figure 6). These observations implied that NaAs-iduced renal injury could be counterbalanced by IL-6 produced by non-BM-derived cells, possibly renal tubular cells.
Figure 3.
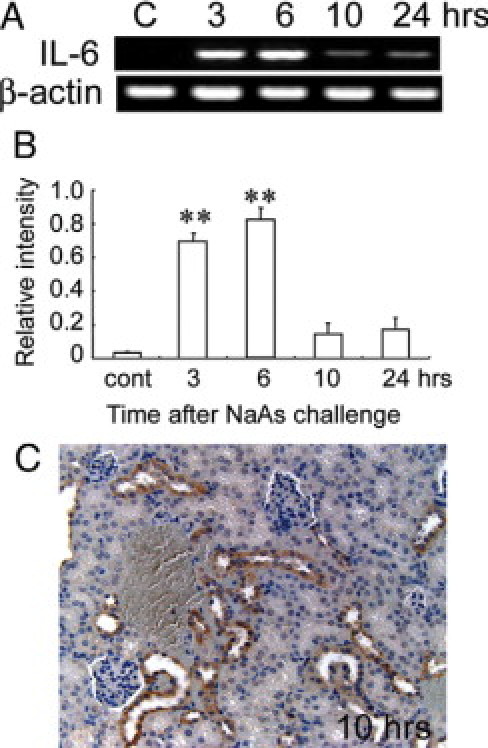
A and B: RT-PCR analysis for IL-6 mRNA expression in the kidneys of wild-type mice at the indicated time intervals after NaAs challenge. Representative results from six independent experiments are shown in A. B: The ratio of IL-6 to β-actin was calculated. All values represent means ± SEM (n = 6 animals). **P < 0.01, NaAs-treated wild-type mice versus control mice. C: Immunohistochemical detection of IL-6 protein in the kidneys of wild-type mice at 10 hours after NaAs challenge. Representative results from six individual animals are shown here. At 10 hours after NaAs challenge, IL-6 protein was immunohistochemically detected in most renal tubular cells. Original magnification, ×200.
Figure 4.
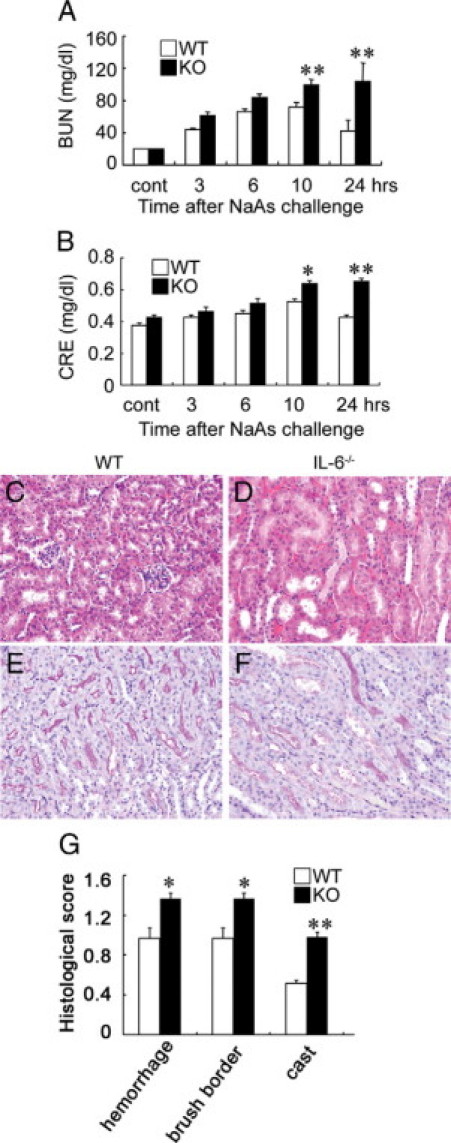
A and B: Determination of serum BUN (A) and serum CRE (B) levels in wild-type (WT) and IL-6−/− mice at the indicated time intervals after NaAs challenge. All values represent means ± SEM (n = 15). *P < 0.05; **P < 0.01, WT versus IL-6−/− mice. C–F: Histopathological observations of the kidneys from WT (C and E) and IL-6−/− mice (D and F). Representative results from six individual animals are shown here. The specimens were obtained from mice at 10 hours after NaAs challenge and were stained with H&E (C and D) or PAS staining (E and F). In IL-6−/− mice, massive tubular necrosis with cast formation and severe hemorrhages (D), and the disappearance of PAS-positive brush border were observed (F). In contrast, the histopathological changes were less evident in WT mice, compared with IL-6−/− mice (C and E). Original magnification, ×200. G: The histopathological score of the kidneys was determined as described in Materials and Methods. All values represent mean ± SEM (n = 15). *P < 0.05; **P < 0.01, WT versus IL-6−/− mice.
Figure 5.
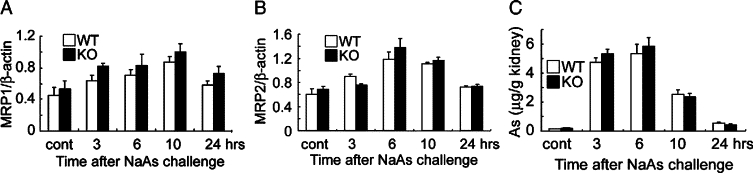
A and B: RT-PCR analysis for gene expression for MRP1 and MRP2 was performed at 3, 6, 10, and 24 hours after NaAs challenge, as described in the Materials and Methods. The ratios of MRP1 (A) and MRP2 (B) to β-actin were calculated and are shown here. All values represent means ± SEM (n = 6 animals). C: Intrarenal arsenic contents in wild-type and IL-6−/− mice were determined by atomic absorption spectrometry at the indicated time intervals after NaAs challenge, and the data were expressed as the arsenic amount (μg)/the kidney weight (g). All values represent means ± SEM (n = 6 animals).
Figure 6.
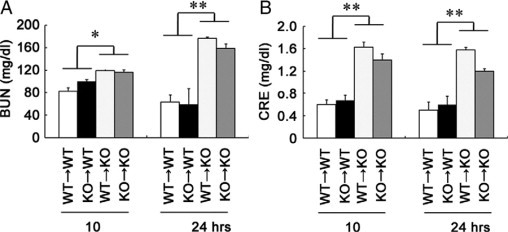
The effects of BM transplantation on NaAs-induced renal injury. Recipient mice were transplanted with BM cells from IL-6−/− or wild-type (WT) donors as described in Materials and Methods. BM chimeric mice were injected with NaAs at 60 days after BM transplantation. Serum BUN (A) and serum CRE (B) levels in BM chimeric mice at 10 and 24 hours after NaAs challenge. Each value represents mean ± SEM (n = 6). *P < 0.05; **P < 0.01, WT recipient versus knockout (KO) recipient mice.
The Effects of IL-6/STAT3 Signal Pathways on NaAs-Induced Autophagic Cell Death of Renal Tubular Cells
Moreover, in line with in vitro study, LC3 protein was detected in the renal tubular epithelial cells of wild-type mice, and the immunoreactivities were more evident in the kidneys of IL-6−/− mice, compared with wild-type mice (Figure 7A). An electron microscopical analyses further demonstrated that NaAs treatment caused autophagic vacuole accumulation in tubular epithelial cells to a larger extent in IL-6−/− mice than in wild-type mice (data not shown). Consistently, Western blotting analysis demonstrated that NaAs challenge increased intrarenal LC3-II amounts in IL-6−/− mice to a larger extent, compared with wild-type mice (Figure 7, B and C). Moreover, NaAs challenge increased intrarenal amount of p-STAT3 to a larger extent in wild-type than IL-6−/− mice (Figure 7, B and D). On the contrary, the same treatment increased intrarenal amount of phosphorylated ERK1/2 to a larger extent in IL-6−/− mice than wild-type mice (Figure 7, B and E). In sharp contrast, the s.c. injection of recombinant IL-6 (0.5 μg/mouse) to wild-type mice significantly reduced the increase in BUN levels induced by a higher dose of NaAs (13.5 mg/kg), compared with vehicle treatment (Figure 8A). Consistently, histopathological damages were apparently attenuated in IL-6-treated wild-type mice, compared with PBS-treated wild-type mice (data not shown). Moreover, the activation of ERK1/2 and the amounts of LC3-II were significantly reduced in wild-type mice injected with recombinant IL-6 (Figure 8B). Finally, PD98059 treatment significantly reduced BUN level (PD98059 versus vehicle: 54.7 ± 4.9 vs. 73.7 ± 6.4 mg/dl, P < 0.05; n = 5 in each group) at 10 hours after NaAs challenge in wild-type mice, suggesting that IL-6 could protect renal tissue through inhibition of ERK activation. These observations would indicate that exogenous IL-6 can be therapeutically effective for NaAs-induced renal injury, probably by suppressing autophagic cell death of renal tubular cells.
Figure 7.
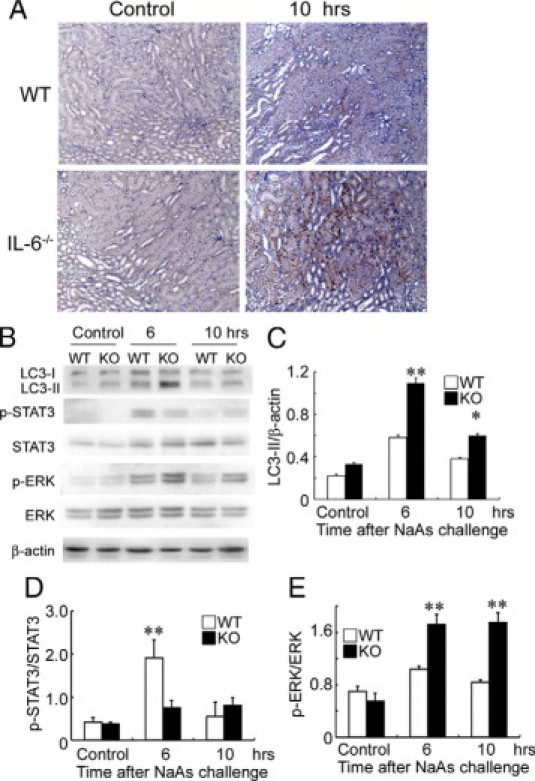
A: Immunohistochemical detection of LC3, a marker of autophagy, in the kidneys of wild-type (WT) and IL-6−/− mice after NaAs challenge. Representative results from six individual animals are shown here. At 10 hours after NaAs challenge, LC3 immunoreactivities were apparently enhanced in renal tubular cells in IL-6−/− mice, compared with wild-type mice. Original magnification, ×200. B: Western blotting analysis of the conversion of LC3-I to LC3-II, p-STAT3, STAT3, p-ERK, ERK, and β-actin in the kidney from WT and IL-6−/− mice at 6 and 10 hours after NaAs challenge. Representative results from six independent experiments are shown here. C–E: LC3-II/β-actin ratios (C), p-STAT3/STAT3 ratios (D), and p-ERK/ERK ratios (E) obtained by densitometry are shown here. All values represent means ± SEM (n = 6 animals). **P < 0.01; *P < 0.05, wild-type versus IL-6−/− mice.
Figure 8.
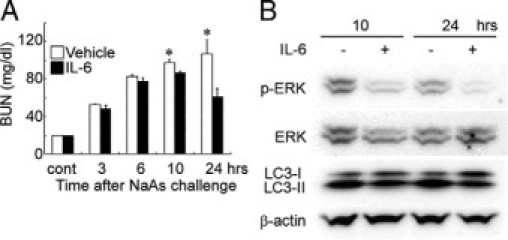
Effects of exogenous IL-6 on renal injury in NaAs-treated wild-type mice. A: Determination of serum BUN levels in wild-type mice treated with IL-6 at 1 hour after NaAs challenge. All values represent means ± SEM (n = 6). *P < 0.05, IL-6 versus vehicle. B: The effects of IL-6 treatment on Erk, LC3-I, and LC3-II. Proteins were extracted from the kidney of vehicle- or IL-6-treated mice at 10 and 24 hours after NaAs challenge and were subjected to Western blotting analysis of p-ERK, ERK, LC3-I, LC3-II, and β-actin. Representative results from six independent experiments are shown here.
Discussion
IL-6 is a pleiotropic cytokine produced by inflammatory and resident cells and has a crucial role in the pathogenesis of various diseases.27 IL-6 exacerbated renal injury caused by ischemia-reperfusion.49–51 In line with these observations, metallothionein I/II-null mice exhibited enhanced susceptibility to NaAs toxicity, together with enhanced serum IL-6 levels.2 Likewise, NaAs challenge enhanced intrarenal IL-6 mRNA expression and IL-6 contents in renal tubular cells. Thus, we hypothesized that the absence of IL-6 could attenuate NaAs-induced renal injury. Contrary to our expectation, the genetic disruption of IL-6 augmented NaAs-induced renal injury, implying that IL-6 could be protective for NaAs-induced renal dysfunction.
ATP-binding cassette transporter proteins such as MRP1 and MRP2 are presumed to be responsible for arsenite efflux, and therefore, their different expression levels among mouse strains can cause differences in intrarenal arsenic contents, thereby determining the susceptibility to NaAs-induced nephrotoxicity.3,4,47,48 Actually, impaired arsenic excretion from the kidneys by reduced intrarenal MRP1 expression can cause the accumulation of arsenic contents and eventually enhance NaAs-induced renal injury3,4 as observed in interferon-γ-deficient mice.3 However, NaAs challenge induced intrarenal expression of MRP1 and MRP2, to similar extents, in wild-type and IL-6−/− mice, and there was no significant difference in intrarenal arsenic contents between wild-type and IL-6−/− mice. Supportingly, intrarenal interferon-γ was expressed to similar extents in wild-type and IL-6−/− mice (data not shown). Thus, the different susceptibility to NaAs nephrotoxicity between these two strains was not ascribed to intrarenal MRP expression levels and intrarenal arsenic concentrations.
Autophagic cell death was presumed to be involved in various diseases20–24 including ischemia-reperfusion renal injury.23 Because As2O3 acted as an anticancer agent by inducing autophagic cell death in malignant cells,18,19 arsenite can induce autophagic cell death in renal cells. Indeed, NaAs challenge induced autophagic cell death in renal cells, particularly tubular epithelial cells. Several cytokines can regulate autophagic cell death.24,26 Of interest is that the activation of STAT3, a main signal transducer of IL-6, could suppress autophagic cell death of several types of cells including glioma cells and cardiomyocytes.21,39 Likewise, anti-IL-6 pAbs or STAT3 inhibition by AG490 significantly enhanced NaAs-induced autophagic cell death in a mouse tubular epithelial cell line, mProx24. Consistently, autophagic cell death of renal tubular epithelial cells was more evident in IL-6−/− mice than wild-type mice. Moreover, the analysis on BM chimeric mice unraveled that NaAs-iduced renal injury could be counterbalanced by IL-6 produced by non-BM-derived cells, possibly renal tubular cells. Collectively, these observations suggest that IL-6/STAT3 signaling pathway can act as a negative regulator of autophagic cell death in renal tubular epithelial cells induced by NaAs injection.
It still remains elusive whether autophagy is protective or injurious in renal injury. We demonstrated that inhibition of autophagy with 3-MA or Atg7 gene ablation increased cell viability in NaAs-treated mProx24 cells, indicating that autophagy was detrimental in NaAs-induced renal injury. Likewise, Suzuki and colleagues demonstrated that inhibition of autophagy protected tubular epithelial cells from H2O2-induced cell death.23 On the contrary, several lines of evidence demonstrated that inhibition of autophagy by 3-MA treatment or the gene knockdown of Beclin-1 increased the cell death in renal tubular cells treated with cisplatin, a nephrotoxicant implying that autophagy would be cytoprotective during cisplatin-induced renal cell injury.52,53 Thus, the biological consequences of autophagy may depend on the types of renal injury.
We observed that NaAs induced ERK1/2 phosphorylation in mouse kidney tissue as well as a mouse renal tubular epithelial cell line. NaAs-induced ERK activation is presumed to mediated by Src-dependent activation of epidermal growth factor receptor (EGFR) in various normal and tumor cells in vitro.54,55 Activated ERK1/2 can phosphorylate Gα-interacting protein, which can accelerate the rate of GTP hydrolysis by the α subunit of the trimeric Gi3 protein.56 Because acceleration of GTP hydrolysis can control lysosomal-autophagic catabolism, ERK1/2 is presumed to promote autophagic cell death in several types of tumor cells.56–58 The blockade of IL-6 augmented NaAs-induced autophagic cell death, along with enhanced ERK1/2 activation in vivo and in vitro without any effects on other signaling pathway involved in autophagic cell death58 (data not shown). Moreover, an ERK1/2 inhibitor attenuated autophagic cell death in a murine renal tubular epithelial cell line treated with NaAs, and reduced NaAs-induced renal injury as well as intrarenal autophagic cell death in vivo. Thus, the activation of ERK signal pathway was also essentially involved in the promotion of NaAs-induced autophagic cell death and subsequent renal injury.
Evidence is accumulating to indicate the presence of the cross-talk between STAT3 and ERK signal pathways in several cell lines.41,59 Arany et al41 demonstrated that, under oxidative stress, STAT3 activation could inhibit EGFR-mediated ERK signaling in murine renal proximal tubular cells (TKPTS). Likewise, we observed that anti-IL-6 pAbs or a Jak2 inhibitor, AG490, significantly suppressed NaAs-induced STAT3 activation and reciprocally enhanced NaAs-induced ERK activation in mProx24 cells and that the net result was the promotion of their autophagic cell death. In the case of STAT3-induced inhibition of EGFR-mediated ERK1/2 activation, STAT3-induced SOCS3 can degrade EGFR in a ubiquitination-dependent manner,60 or activated STAT3 competitively can inhibit the binding of Grb2 to SH2 domain of EGFR.61 Thus, it is likely that STAT3 can be upstream ERK1/2. This can explain why ERK1/2 activation persisted in renal tissues at 10 hours after NaAs treatment when p-STAT3 was barely detected (Figure 7B). However, it still remains an open question which mechanism underlies the cross-talk between STAT3 and ERK1/2 pathways in NaAs- induced renal injury.
Nevertheless, our present observations suggest that the balance between IL-6/STAT3- and ERK1/2-mediated signals can modulate NaAs-induced renal injury by affecting autophagic cell death in renal tubular epithelial cells. We demonstrated that IL-6 could reduce NaAs-induced renal injury, even when administered at 1 hour after NaAs challenge. Thus, IL-6 may be a novel therapeutic modality for acute NaAs intoxication cases in emergency rooms. Moreover, in patients treated with As2O3, the combined administration of As2O3 and recombinant IL-6 may give more therapeutic benefits by minimizing As2O3-induced adverse effects such as renal dysfunction.
Acknowledgements
We thank Dr. Tamotsu Yoshimori (Osaka University, Japan) for his gift of EGFP-LC3 plasmid. We thank Ms. Mariko Kawaguchi for her excellent assistant in the preparation of this paper.
Footnotes
Supported in part from grants-in-aids from the Ministry of Education, Culture, Science, and Technology of the Japanese Government (to T.K. and A.K.) and a research grant from Uehara Memorial Foundation (to T.K.).
References
- 1.Oremland RS, Stolz JF. The ecology of arsenic. Science. 2003;300:939–944. doi: 10.1126/science.1081903. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, Liu YP, Goyer RA, Chanzar W, Waalkes MP. Metallothionein-I/II null mice are more sensitive than wild-type mice to the hepatotoxic and nephrotoxic effects of oral or injected inorganic arsenicals. Toxicol Sci. 2000;55:460–467. doi: 10.1093/toxsci/55.2.460. [DOI] [PubMed] [Google Scholar]
- 3.Kimura A, Ishida Y, Hayashi T, Wada T, Yokoyama H, Sugaya T, Mukaida N, Kondo T. Interferon-γ plays protective roles in sodium arsenite-induced renal injury by up-regulating intrarenal multidrug resistance-associated protein 1 expression. Am J Pathol. 2006;169:1118–1128. doi: 10.2353/ajpath.2006.060024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimura A, Ishida Y, Wada T, Yokoyama H, Mukaida N, Kondo T. MRP-1 expression levels determine strain-specific susceptibility to sodium arsenic-induced renal injury between C57BL/6 and BALB/c mice. Toxicol Appl Pharmacol. 2005;203:53–61. doi: 10.1016/j.taap.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Snow E. Metal carcinogenesis: mechanistic implications. Pharmacol Ther. 1992;53:31–65. doi: 10.1016/0163-7258(92)90043-y. [DOI] [PubMed] [Google Scholar]
- 6.Thompson DJ. A chemical hypothesis for arsenic methylation in mammals. Chem Biol Interact. 1993;88:89–114. doi: 10.1016/0009-2797(93)90086-e. [DOI] [PubMed] [Google Scholar]
- 7.Goering PL, Aposhian HV, Mass MJ, Cebrian M, Beck BD, Waalkes MP. The enigma of arsenic carcinogenesis: role of metabolism. Toxicol Sci. 1999;49:5–14. doi: 10.1093/toxsci/49.1.5. [DOI] [PubMed] [Google Scholar]
- 8.Abernathy CO, Liu Y-P, Longfellow D, Aposhian HV, Beck B, Fowler B, Goyer R, Menzer R, Rossman T, Thompson C, Waalkes MP. Arsenic: health effects, mechanisms of actions and research issues. Environ Health Perspect. 1999;107:593–597. doi: 10.1289/ehp.99107593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, Han ZG, Ni JH, Shi GY, Jia PM, Liu MM, He KL, Niu C, Ma J, Zhang TD, Paul P, Naoe T, Kitamura K, Miller W, Waxman S, Wang ZY, Chen SJ, Chen Z. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukaemia (APL): As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- 10.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukaemia (APL). II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–3360. [PubMed] [Google Scholar]
- 11.Soignet SL, Maslak PZ, Wang G, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, Pandolfi PP, Warrell RP., Jr Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339:1341–1348. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]
- 12.Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007;33:542–564. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Uchiyama Y, Shibata M, Koike M, Yoshimura K, Sasaki M. Autophagy-physiology and pathophysiology. Histochem Cell Biol. 2008;129:407–420. doi: 10.1007/s00418-008-0406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ker JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bursch W, Ellinger A, Gerner C, Fröhwein U, Schulte-Hermann R. Programmed cell death (PCD): apoptosis, autophagic PCD, or others? Ann NY Acad Sci. 2000;926:1–12. doi: 10.1111/j.1749-6632.2000.tb05594.x. [DOI] [PubMed] [Google Scholar]
- 16.Zakeri Z, Bursch W, Tenniswood M, Lockshin RA. Cell death: programmed, apoptosis, necrosis, or other? Cell Death Differ. 1995;2:87–96. [PubMed] [Google Scholar]
- 17.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 18.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–2108. [PubMed] [Google Scholar]
- 19.Qian W, Liu J, Jin J, Ni W, Xu W. Arsenic trioxide induces not only apoptosis but also autophagic cell death in leukemia cell lines via up-regulation of Beclin-1. Leuk Res. 2007;31:329–339. doi: 10.1016/j.leukres.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 20.Takemura G, Miyata S, Kawase Y, Okada H, Maruyama R, Fujiwara H. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy. 2006;2:212–214. doi: 10.4161/auto.2608. [DOI] [PubMed] [Google Scholar]
- 21.Miyata S, Takemura G, Kawase Y, Li Y, Okada H, Maruyama R, Ushikoshi H, Esaki M, Kanamori H, Li L, Misao Y, Tezuka A, Toyo-Oka T, Minatoguchi S, Fujiwara T, Fujiwara H. Autophagic cardiomyocyte death in cardiomyopathic hamsters and its prevention by granulocyte colony-stimulating factor. Am J Pathol. 2006;168:386–397. doi: 10.2353/ajpath.2006.050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, Uchiyama Y, Takahara S, Imai E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun. 2008;368:100–106. doi: 10.1016/j.bbrc.2008.01.059. [DOI] [PubMed] [Google Scholar]
- 24.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. 2006;84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 25.Wright K, Ward SG, Kolios G, Westwick J. Activation of phosphatidylinositol 3-kinase by interleukin-13. An inhibitory signal for inducible nitric-oxide synthase expression in epithelial cell line HT-29. J Biol Chem. 1997;272:12626–12633. doi: 10.1074/jbc.272.19.12626. [DOI] [PubMed] [Google Scholar]
- 26.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 27.Akira S, Kishimoto T. IL-6 and NF-IL6 in acute-phase response and viral infection. Immunol Rev. 1992;127:25–50. doi: 10.1111/j.1600-065x.1992.tb01407.x. [DOI] [PubMed] [Google Scholar]
- 28.Yasumoto K, Mukaida N, Harada A, Kuno K, Akiyama M, Nakashima E, Fujioka N, Mai, Kasahara T, Fujimoto-Ouchi K, Mori K, Tanaka Y, Matsushima M. Molecular analysis of the cytokine network involved in cachexia in colon 26 adenocarcinoma-bearing mice. Cancer Res. 1995;55:921–927. [PubMed] [Google Scholar]
- 29.Lin ZQ, Kondo T, Ishida Y, Takayasu T, Mukaida N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J Leukoc Biol. 2003;73:713–721. doi: 10.1189/jlb.0802397. [DOI] [PubMed] [Google Scholar]
- 30.Lee PJ, Zhang X, Shan P, Ma B, Lee CG, Homer RJ, Zhu Z, Rincon M, Mossman BT, Elias JA. ERK1/2 mitogen-activated protein kinase selectively mediates IL-13-induced lung inflammation and remodeling in vivo. J Clin Invest. 2006;116:163–173. doi: 10.1172/JCI25711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishida Y, Kimura A, Kondo T, Hayashi T, Ueno M, Takakura N, Matsushima K, Mukaida N. Essential roles of the CC chemokine ligand 3-CC chemokine receptor 5 axis in bleomycin-induced pulmonary fibrosis through regulation of macrophage and fibrocyte infiltration. Am J Pathol. 2007;170:843–854. doi: 10.2353/ajpath.2007.051213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takaya K, Koya D, Isono M, Sugimoto T, Sugaya T, Kashiwagi A, Haneda M. Involvement of ERK pathway in albumin-induced MCP-1 expression in mouse proximal tubular cells. Am J Physiol. 2003;284:F1037–F1045. doi: 10.1152/ajprenal.00230.2002. [DOI] [PubMed] [Google Scholar]
- 33.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsui T, Kinoshita T, Morikawa Y, Tohya K, Katsuki M, Ito Y, Kamiya A, Miyajima A. K-Ras mediates cytokine-induced formation of E-cadherin-based adherens junctions during liver development. EMBO J. 2002;21:1021–1030. doi: 10.1093/emboj/21.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-γ in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J. 2002;16:1227–1236. doi: 10.1096/fj.02-0046com. [DOI] [PubMed] [Google Scholar]
- 36.Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem. 2004;279:36268–36276. doi: 10.1074/jbc.M401461200. [DOI] [PubMed] [Google Scholar]
- 37.Aoki H, Kondo Y, Aldape K, Yamamoto A, Iwado E, Yokoyama T, Hollingsworth EF, Kobayashi R, Hess K, Shinojima N, Shingu T, Tamada Y, Zhang L, Conrad C, Bogler O, Mills G, Sawaya R, Kondo S. Monitoring autophagy in glioblastoma with Ab against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy. 2008;4:1–9. doi: 10.4161/auto.5668. [DOI] [PubMed] [Google Scholar]
- 38.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 39.Aoki H, Iwado E, Eller MS, Kondo Y, Fujiwara K, Li GZ, Hess KR, Siwak DR, Sawaya R, Mills GB, Gilchrest BA, Kondo S. Telomere 3′ overhang-specific DNA oligonucleotides induce autophagy in malignant glioma cells. FASEB J. 2007;21:2918–2930. doi: 10.1096/fj.06-6941com. [DOI] [PubMed] [Google Scholar]
- 40.Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007;72:29–39. doi: 10.1124/mol.106.033167. [DOI] [PubMed] [Google Scholar]
- 41.Arany I, Megyesi JK, Nelkin BD, Safirstein RL. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int. 2006;70:669–674. doi: 10.1038/sj.ki.5001604. [DOI] [PubMed] [Google Scholar]
- 42.Cui Q, Tashiro S, Onodera S, Minami M, Ikejima T. Oridonin induced autophagy in human cervical carcinoma HeLa cells through Ras, JNK, and P38 regulation. J Pharmacol Sci. 2007;105:317–325. doi: 10.1254/jphs.fp0070336. [DOI] [PubMed] [Google Scholar]
- 43.Tang G, Yue Z, Talloczy Z, Hagemann T, Cho W, Messing A, Sulzer DL, Goldman JE. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17:1540–1555. doi: 10.1093/hmg/ddn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 45.Shinojima N, Yokoyama T, Kondo Y, Kondo S. Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin-induced autophagy. Autophagy. 2007;3:635–637. doi: 10.4161/auto.4916. [DOI] [PubMed] [Google Scholar]
- 46.Cárdenas-Aguayo Mdel C, Santa-Olalla J, Baizabal JM, Salgado LM, Covarrubias L. Growth factor deprivation induces an alternative nonapoptotic death mechanism that is inhibited by Bcl2 in cells derived from neural precursor cells. J Hematother Stem Cell Res. 2003;12:735–748. doi: 10.1089/15258160360732759. [DOI] [PubMed] [Google Scholar]
- 47.Rosen BP. Biochemistry of arsenic detoxification. FEBS Lett. 2002;529:86–92. doi: 10.1016/s0014-5793(02)03186-1. [DOI] [PubMed] [Google Scholar]
- 48.Miller DS, Shaw JR, Stanton CR, Barnaby R, Karlson KH, Hamilton JW, Stanton BA. MRP2 and acquired tolerance to inorganic arsenic in the kidney of killifish (Fundulus heteroclitus) Toxicol Sci. 2007;97:103–110. doi: 10.1093/toxsci/kfm030. [DOI] [PubMed] [Google Scholar]
- 49.Kielar ML, John R, Bennett M, Richardson JA, Shelton JM, Chen L, Jeyarajah DR, Zhou XJ, Zhou H, Chiquett B, Nagami GT, Lu CY. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol. 2005;16:3315–3325. doi: 10.1681/ASN.2003090757. [DOI] [PubMed] [Google Scholar]
- 50.Patel NS, Chatterjee PK, Di Paola R, Mazzon E, Britti D, De Sarro A, Cuzzocrea S, Thiemermann C. Endogenous interleukin-6 enhances the renal injury, dysfunction, and inflammation caused by ischemia/reperfusion. J Pharmacol Exp Ther. 2005;312:1170–1178. doi: 10.1124/jpet.104.078659. [DOI] [PubMed] [Google Scholar]
- 51.Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, Axelrod JH. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol. 2008;19:1106–1115. doi: 10.1681/ASN.2007070744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol. 2008;294:F777–F787. doi: 10.1152/ajprenal.00590.2007. [DOI] [PubMed] [Google Scholar]
- 53.Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–640. doi: 10.1038/ki.2008.214. [DOI] [PubMed] [Google Scholar]
- 54.Liu ZM, Huang HS. As2O3-induced c-Src/EGFR/ERK signaling is via Sp1 binding sites to stimulate p21WAF1/CIP1 expression in human epidermoid carcinoma A431 cells. Cell Signal. 2006;18:244–255. doi: 10.1016/j.cellsig.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka-Kagawa T, Hanioka N, Yoshida H, Jinno H, Ando M. Arsenite and arsenate activate extracellular signal-regulated kinases 1/2 by an epidermal growth factor receptor-mediated pathway in normal human keratinocytes. Br J Dermatol. 2003;149:1116–1127. doi: 10.1111/j.1365-2133.2003.05704.x. [DOI] [PubMed] [Google Scholar]
- 56.Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Gα-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem. 2000;275:39090–39095. doi: 10.1074/jbc.M006198200. [DOI] [PubMed] [Google Scholar]
- 57.Schiemann WP, Bartoe JL, Nathanson NM. Box3-independent signaling mechanisms are involved in leukemia inhibitory factor receptor α- and gp130-mediated stimulation of mitogen-activated protein kinase: evidence for participation of multiple signaling pathways which converge at Ras. J Biol Chem. 1997;272:16631–16636. doi: 10.1074/jbc.272.26.16631. [DOI] [PubMed] [Google Scholar]
- 58.Codogno P, Meijer A. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 59.Kritikou EA, Sharkey A, Abell K, Came PJ, Anderson E, Clarkson RW, Watson CJ. A dual, nonredundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003;130:3459–3468. doi: 10.1242/dev.00578. [DOI] [PubMed] [Google Scholar]
- 60.Xia L, Wang L, Chung AS, Ivanov SS, Ling MY, Dragoi AM, Platt A, Gilmer TM, Fu XY, Chin YE. Identification of both positive and negative domains within the epidermal growth factor receptor COOH-terminal region for signal transducer and activator of transcription (STAT) activation. J Biol Chem. 2002;277:30716–33023. doi: 10.1074/jbc.M202823200. [DOI] [PubMed] [Google Scholar]
- 61.Zhang T, Ma J, Cao X. Grb2 regulates Stat3 activation negatively in epidermal growth factor signalling. Biochem J. 2003;376:457–464. doi: 10.1042/BJ20030668. [DOI] [PMC free article] [PubMed] [Google Scholar]