

Abstract
Inflammation and angiogenesis are intimately linked, and their dysregulation leads to pathological angiogenesis in human diseases. 15-lipoxygenase (15-LOX) and lipoxin A4 receptors (ALX) constitute a LXA4 circuit that is a key feature of inflammatory resolution. LXA4 analogs have been shown to regulate vascular endothelial growth factor (VEGF)-A-induced angiogenic response in vitro. 15-LOX and ALX are highly expressed in the avascular and immune-privileged cornea. However, the role of this endogenous LXA4 circuit in pathological neovascularization has not been determined. We report that suture-induced chronic injury in the cornea triggered polymorphonuclear leukocytes (PMN) infiltration, pathological neovascularization, and up-regulation of mediators of inflammatory angiogenesis, namely VEGF-A and the VEGF-3 receptor (FLT4). Up-regulation of the VEGF circuit and neovascularization correlated with selective changes in both 15-LOX (Alox15) and ALX (Fpr-rs2) expression and a temporally defined increase in basal 15-LOX activity. More importantly, genetic deletion of 15-LOX or 5-LOX, key and obligatory enzymes in the formation of LXA4, respectively, led to exacerbated inflammatory neovascularization coincident with increased VEGF-A and FLT4 expression. Direct topical treatment with LXA4, but not its metabolic precursor 15-hydroxyeicosatetraenoic acid, reduced expression of VEGF-A and FLT4 and inflammatory angiogenesis and rescued 15-LOX knockout mice from exacerbated angiogenesis. In summary, our findings and the prominent expression of 15-LOX and ALX in epithelial cells and macrophages place the LXA4 circuit as an endogenous regulator of pathological angiogenesis.
Formation of a new functional microvasculature, neovascularization, is a fundamental response to ischemia and a salient feature of wound healing. The primary function of newly formed blood vessels is to increase tissue oxygen tension and delivery of essential nutrients and effector cells to restore normal function. However, aberrant neovascularization is a hallmark feature of chronic inflammation and is associated with numerous pathological conditions that include diabetic retinopathy, Crohn's Disease, atherosclerosis, and cancer.1–3
The growth of microvessels from existing vessels, angiogenesis, is tightly controlled by a range of angiogenic factors and inhibitors; a circuit that is highly evolved in avascular tissues such as the cornea. The vascular endothelial growth factor (VEGF) family of angiogenic factors and their receptors are key mediators of this process, which has led to the recent development and clinical use of anti-VEGF strategies for the treatment of pathological neovascularization in the retina, colon cancer, and lung cancer.2–5 Many insights into the endogenous role of the VEGF network have been gained by using the cornea,5,6 which maintains an immune-privileged and avascular state despite expression of VEGF-A and its immediate proximity to the vasculature.7 Specifically, recent findings have demonstrated that avascularity of the cornea requires expression of a soluble VEGF receptor-1 (sFLT1), which traps VEGF-A.8 In addition, the receptor for VEGF-C/VEGF-D, namely VEGF receptor-3 (VEGFR-3, FLT4), is a critical regulator of inflammatory neovascularization.9–11 FLT4 is of special of interest because its essential expression during development becomes restricted primarily to lymph vessels and activation of this endothelial receptor is a critical step in initiating lymphangiogenesis. However, FLT4 expression is also up-regulated in microvessels of tumors and wounds, and in macrophages and in addition is constitutively expressed in corneal epithelial cells.3,10–13 A recent report demonstrates that FLT4 is highly expressed in angiogenic sprouts and is a critical regulator of sustained heme-angiogenesis,12 which underscores the potential key role of this receptor in pathological neovascularization.
Inflammation is intimately associated with neovascularization especially during wound healing and ischemic injury. Lipid autacoids are some of the earliest signals that are released in response to injury or insult. In this regard, the 15-lipoxygenase pathway14,15 is of interest as it is one of the most inducible genes in macrophages and highly expressed in mucosal and corneal epithelial cells. Macrophages and epithelial cells are important regulators of angiogenesis, especially in avascular tissue such as the cornea.3,10,16 Macrophages have a central and well-documented role in angiogenesis, especially in tumors and inflammatory neovascularization. In the cornea, a well-established model tissue for studying inflammatory neovascularization, VEGF-A recruits macrophages, the major cell type to generate VEGF, which drives inflammatory heme and lymphangiogenesis. Corneal epithelial cells constitutively express the receptor for VEGF-C/VEGF-D (ie, FLT4), which has been proposed as a critical pathway for regulating inflammatory neovascularization.10
Human 15-LOX (ALOX15 and ALOX15B) generate 15S-hydroxyeicosatetraenoic acid (HETE) and mouse 12/15-LOX (Alox15) generates 15S-HETE and 12S-HETE from arachidonic acid. 15-HETE and 12-HETE have been shown to induce proliferation, migration, and tube formation in endothelial cells.17 More importantly, 15-HETE is a key intermediate in the formation of the well-studied anti-inflammatory mediator lipoxin A4 (LXA4) that is generated via the rate-limiting enzyme 5-lipoxygenase (5-LOX). A body of work18–24 has established that the anti-inflammatory actions, which are associated with the up-regulation of 15-LOX and/or 15S-HETE formation are mediated by LXA4 and its G-protein coupled receptor ALX. Recent reports have demonstrated that stable analogs of LXA4 inhibit VEGF induced angiogenic responses in endothelial cells.25–27 These metabolically stable analogs are mimetics of aspirin-triggered LXA4, an endogenous isomer whose synthesis can be triggered by aspirin-acetylated cyclooxygenase-2 rather than 15-LOX. This 15-epi-isomer of LXA4 resists metabolic inactivation and mediates it bioactions, like LXA4, via the ALX receptor. The intimate link between inflammation and angiogenesis and the ability of LXA428 and analogs of 15-epi LXA425–27 to inhibit VEGF-A induced angiogenic responses in vitro points toward a potential role of endogenous LXA4 circuits in pathological angiogenesis. However, the endogenous role of 15-LOX in the regulation of angiogenesis remains controversial. Reports have demonstrated that the enzyme or its products promote or inhibit angiogenic responses in in vitro studies.29–32 More importantly, the in vivo role of the 15-LOX pathway or the LXA4 circuit in pathological neovascularization remains to be clearly defined. To this end, we assessed the role the 15-LOX pathway and LXA4 circuit in chronic injury-induced inflammatory neovascularization.
Here, we report that inflammatory neovascularization and up-regulation of the VEGF circuit correlate with changes in both 15-LOX (Alox15) and LXA4 receptor (ALX) expression and temporally defined 15-LOX activity. More importantly, genetic deletion of 15-LOX or 5-LOX, key enzymes in the formation of LXA4, led to amplified neovascularization and expression of VEGF-A and FLT4 in the avascular cornea during chronic injury. LXA4, but not 15S-HETE, attenuated expression of VEGF-A and FLT4 and the angiogenic response, which provides evidence that selective autacoids from the prominent 15-LOX pathway have an endogenous role in limiting pathological neovascularization.
Materials and Methods
Animals
12/15-lipoxygenase (Alox15) and 5-lipoxygenase (Alox5) deficient mice (6 to 10 weeks, female) were purchased from Jackson Laboratory (Bar Harbor, ME). These Jax Gemm strains have a targeted mutation in 12/15-LOX or in 5-LOX that is in a background C57Bl/6J inbred strain. These mice do not express a functional “leukocyte-type” 12/15-LOX (Alox15) or 5-LOX (Alox5).33,34 Age and gender-matched congenic C57Bl/6J stock 000664 mice (6 to 10 weeks, female) were used as controls. Mice were maintained on a 12-hour day/night cycle and fed ad libitum a standard diet (Rat/Mouse diet LM-485, Harlan Tekland, Madison, WI).
Corneal Neovascularization and Treatment
All animal studies have been approved by the University of California, Berkeley in accordance with the NIH Guide for the Care and Use of Laboratory Animals and in strict accord with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Mice were anesthetized with ketamine (50 mg/kg) and xylazine (20 mg/kg) intraperitoneally and a drop of tetracaine-HCL 0.5% was applied to the eye to deliver local corneal anesthesia before injury. A single sterile 8.0 silk suture was placed intrastromally extending over the corneal apex, without disrupting the iris. Selected mice were treated topically t.i.d. with LXA4 or 15S-HETE (Cayman Chemical, Ann Arbor, MI), or sterile saline alone (PBS, ph 7.4) for two or seven days. Ethanol from the LXA4 and 15S-HETE solutions were rapidly removed under gentle stream of nitrogen and autacoids immediately resuspended in sterile PBS and applied to the eye (5 μl drop, t.i.d.). Eyes were enucleated at the respective time points under a stereo-microscope and corneas carefully dissected on ice to remove the limbus area and all noncorneal tissue. Isolated corneas were either snap frozen for RNA or lipidomic analyses, or immediately processed for immunohistochemistry.
Assessment of Neovascularization and Inflammation
Isolated corneas were rinsed in PBS, fixed in acetone (100%) for 30 minutes, blocked in 2% bovine serum albumin/PBS solution and incubated in PBS containing fluorescein isothiocyanate-conjugated CD31/PECAM-1 overnight (Santa Cruz Biotechnology, Santa Cruz, CA; 1:100). Flatmounts were prepared by sectioning the cornea and fixing them to slides. The images were taken with a Zeiss Axiophot laser scanning confocal microscope and neovascularization was quantified by manually tracing the length of all vessels using Image Pro-Express software (Cyber Media) and was expressed as total pixels. Corneal flatmounts from 5-LOX KO mice and their matched congenic wild-type controls were taken with a Zeiss Axioplan 2 microscope equipped with a Ludl motorized stage and z focus. Mosaic images were taken with a AxioCam MR camera and compiled using MosaiX and Zeiss AxioVision 4.5 software.
Myeloperoxidase Activity
Myeloperoxidase (MPO) activity, an index of tissue leukocyte infiltration, was measured 48 hours and 7 days post injury.35,36 In brief, corneas (1 cornea/data point) were homogenized with a hand held tissue grinder in 450 μl of 50 mmol/L potassium phosphate buffer containing 0.5% hexadecyltrimethylammonium bromide (pH 6.0). This was followed by sonication, freeze-thaw three times and a second sonication. The homogenates were then centrifuged and supernatants collected. MPO activity in supernatants was measured by spectrophotometry using o-dianisidine dihydrochloride oxidation as a colorimetric indicator. Calibration curves for MPO activities were established with PMN collected from 12 hours zymosan A-induced peritonitis exudates in mice.
Gene and Protein Expression
RNA from mouse corneas was isolated using RNA Easy Mini Kit (Qiagen Sciences, Maryland). RNA integrity was verified using agarose gel electrophoresis and quantified by spectrophotometry. RNA was reverse transcribed using a High-Capacity cDNA Kit (Applied Biosystems, Foster City, CA). Heme oxygenase (HO)-1 was amplified using 5′-GATAGAGCGCAACAAGCAGAA-3′ and 3′-CAGTGAGGCCCATACCAGAA-5′; VEGFA 5′-TCACCAAAGCCAGCACATAGGAGA-3′ and 3′-TTCGTTTAACTCAAGCTGCCTCGC-5′; Fprl1 5′-CATTTGGTTGGTTCATGTGCAA-3′ and 3′-AATACAGCGGTCCAGTGCAAT-5′; Fpr-rs2 5′-GCCAGG ACTTTCGTGGAGAGAT-3′ and 3′-GATGAACTGGTGCTTGAATCACT-5′; VEGF-R3 (FLT-4) 5′-CTGGCAAATGGTTACTCCATGA-3′ and 3′-ACAACCCGTGTGTCTTCACTG-5′; 12/15 LOX (Alox15) 5′-GCGACGCTGCCCAATCCTAATC-3′ and 3′-ATATGGCCACGCTGTTTTCTACC-5′; and soluble flt1 5′-AGGTGAGCACTGCGGCA-3′ and 3′-ATGAGTCCTTTAATGTTTGAC-5′. Nucleotide primers were selected from the Harvard Primer Bank (pga.mgh.harvard.edu/primerbank/) and verified by the NIH GenBank database or have recently been reported.35–39 Real-time PCR was performed using Fast SYBR Green Master Mix (Applied Biosystems) with a Step One Plus QPCR system (Applied Biosystems). Amplifications were run in duplicates and efficiencies for each primer pair were established. Comparative quantification of gene expression was performed by Step One software (Applied Biosystems) using the ΔΔCT method. Expression of all genes is referenced to a positive mRNA control that was generated by pooling mRNA from C57Bl/6J mouse spleen and kidney.
Lipid Mediator Lipidomics
For endogenous lipid autacoid analysis, corneas were homogenized with a hand-held tissue grinder in 66% methanol (4°C). The methanol contained deuterated internal standards, prostaglandin (PG)E2-d4, 15(S)- HETE-d8, and leukotriene B4 (LTB4)-d4 (400 pg/each), to calculate the recovery of prostanoids or mono-hydroxy- and dihydroxy-containing fatty acids. Lipid autacoids were extracted by solid phase using Accubond ODS-C18 cartridges (Agilent Technologies, Santa Clara, CA)40 and the average extraction recovery of our class specific deuterated internal standard was 81% for PGE2-d4, 60% for LTB4-d4, and 63% for 15-HETE-d8. Eicosanoids were identified and quantified by liquid chromatography (LC)/mass spectrometry (MS)/MS-based lipidomics.37,41–45 In brief, extracted samples were analyzed by a triple quadruple linear ion trap LC/MS/MS system (MDS SCIEX 3200 QTRAP) equipped with a LUNA C18–2 minibore column using a mobile phase (methanol:water:acetate, 65:35:0.02, v:v:v) with a 0.50 ml/flow rate. MS/MS analyses were performed in negative ion mode and prominent fatty acid metabolites were quantified by multiple reaction monitoring (MRM mode) using established transitions37,41,42,44,45 for PGE2 (351→271, 351→189 m/z), thromboxane B2 (369 →169 m/z), PGF2α (353→193 m/z), PGD2 (351→271 m/z), 5-HETE (319→115 m/z), 15-HETE (319→175 m/z), 12-HETE (319→179 m/z), 5,12-DiHETE/LTB4 (335→195 m/z), LXA4 (351→115 m/z), PGE2-d4 (335→275 m/z), LTB4-d4 (339→197 m/z), and 15-HETE-d8 (327→182 m/z). Calibration curves (1 to 1000 pg) and specific LC retention times for each compound were established with synthetic standards (Cayman Chemical, Ann Arbor, MI). Structures were confirmed for selected autacoids by MS/MS analyses using enhanced product ion mode with appropriate selection of the parent ion in quadrupole 1.
Statistics
All data are expressed as mean ± SEM unless otherwise indicated. Student's t-test was used to evaluate the significance of differences between two groups and multiple comparisons were performed by regression analysis and one-way analysis of variance. P values of less than 0.05 were considered significant.
Results
The cornea in mice and humans is avascular and in general devoid of leukocytes; hence, it is an ideal tissue to study inflammatory neovascularization. Neovascularization is a fundamental response to severe corneal injury or infection such as microbial keratitis and chemical burns and a key feature of the pathogenesis. We selected the well-established corneal suture model as it induces robust and quantifiable neovascularization as a consequence of chronic injury and irritation.10,11,16 Consistent with the model, the silk suture induced robust formation of new blood vessels originating from the vascular border of the cornea (limbus) that become visible by day 4 and are pronounced by day 7 (Figure 1, A–B).
Figure 1.
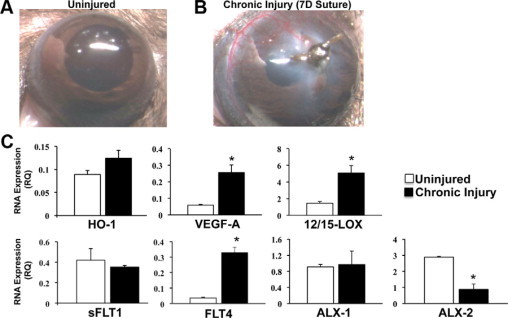
Inflammatory neovascularization differentially regulates expression of the LXA4 circuit. A: Representative images of an uninjured cornea. B: An 8.0 silk suture was placed intrastromally in female C57/BL6 mice. Image of cornea with neovascularization after 7 days of chronic injury. C: mRNA expression of selected genes was quantified by real-time PCR analyses in corneas after 7 days of chronic injury for heme oxygenase-1 (HO-1), vascular endothelial growth factor A (VEGF-A), soluble VEGF receptor-1 (sFLT1), VEGF receptor-3 (FLT4), 12 and /15 LOX (Alox15), and after 48 hours for ALXR-1 (Fprl1) and ALXR-2 (Fpr-rs2). mRNA expression was compared with matched untreated corneas and is expressed as relative quantity (RQ) compared with a mouse positive mRNA control (n = 4, *P < 0.05) using the ΔΔCT method and β-actin as a reference gene.
Due to the small tissue size of the mouse cornea, we selected real-time PCR to quantify mRNA expression of key mediator of angiogenesis and pathways that limit the sequelae of corneal injury (Figure 1C). Chronic injury led to a significant 4.3-fold increase in mRNA levels of VEGF-A (injured = 0.256 ± 0.046 relative quantity [RQ] versus uninjured = 0.059 ± 0.005 RQ, n = 4) and a significant 9.2 fold increase in the receptor FLT4 (injured 0.330 ± 0.034 RQ versus uninjured = 0.036 ± 0.004 RQ, n = 4). In contrast, expression of the soluble FLT1 receptor (sFLT1), which traps VEGF-A in the cornea, did not change in response to injury. Consistent with the temporally defined induction of HO-1,46 an early response and cytoprotective gene, levels of HO-1 were no longer significantly elevated after 7 days of chronic injury compared with basal expression. Chronic injury differentially altered the expression of the resident LXA4 biosynthetic pathway and its receptor (Figure 1C), which are expressed in both recruited leukocyte and resident corneal epithelial cells.15,36 Specifically, both ALX receptors (Fprl1 and Fprs2) were expressed in the uninjured cornea of C57Bl/6J. Expression of ALX1 (Fprl1) did not change after 2 days of chronic injury. However, expression of ALX2 (Fprs2) decreased by 70% (injured = 2.89 ± 0.61 RQ versus uninjured = 0.873 ± 0.559 RQ, n = 4) despite the fact that 2 days is the initial phase of PMN infiltration, a cell type that expresses both ALX1 and ALX2.47 In sharp contrast mRNA levels for 12/15-LOX mRNA increased by 3.5 fold (injured = 5.07 ± 0.90 RQ versus uninjured = 1.46 ± 0.19 RQ, n = 4) as a consequence of 7-day chronic injury when directly compared with healthy corneas.
To assess if pathological angiogenesis was associated with temporally defined changes in endogenous lipid autacoid formation, we used LC/MS/MS-based lipidomics analyses, which demonstrated selective formation of LOX and cyclooxygenase-derived autacoids. Consistent with the time course of neovascularization and up-regulation of 12/15-LOX, endogenous levels of 15-HETE demonstrated a temporally defined increase from 33 pg/cornea in the uninjured cornea to a peak of 94 pg/cornea by day 4 (Figure 2; uninjured = 33 ± 6 pg/cornea versus injured = 94 ± 10 pg/cornea, n = 4) coinciding with the first appearance of functional blood vessels. 15-HETE is a metabolic intermediate for the formation of LXA4 in the cornea and thus a marker for the biosynthetic pathway.15 We were not able to consistently detect endogenous LXA4 formation in isolated single corneas of C57Bl/6J female mice. However, endogenous LXA4 formation in the cornea has been reported in a different strain of mice (BALB/c, males) in a model of self-resolving abrasion injury36; these differences in corneal LXA4 levels likely reflect differences in mouse strain, gender and injury model. Moreover, our lipidomic analysis did not measure tissue levels of 15-oxo-LXA4,45 the primary endogenous metabolite of LXA4, which may reflect a significant and established route of metabolic inactivation. Thus our measurements likely underestimated the corneal levels of LXA4, especially during chronic inflammation.
Figure 2.
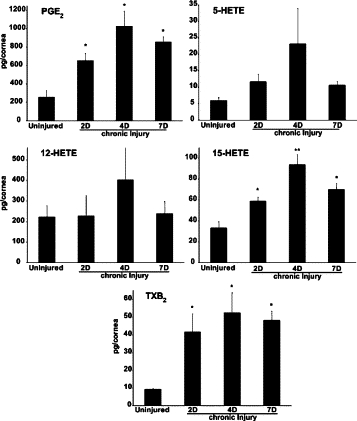
Inflammatory neovascularization is associated with the dynamic formation of select lipid autacoids. Endogenous lipid autacoids were quantified in corneas without injury and after 2, 4, and 7 days of chronic injury by MS based lipidomic analyses using triple quadrupole LC/MS/MS system (MDS SCIEX QTRAP 3200). Specific transition ions and multiple reaction monitoring was used to measure levels of >20 eicosanoids in a single mouse corneas. Significant levels of prostaglandin E2 (PGE2), 15-hydroxyeicosatetraenoic acid (15-HETE), thromboxane B2, 5-hydroxyeicosatetraenoic acid (5-HETE), and 12S- hydroxyeicosatetraenoic acid (12-HETE) were detected in injured and uninjured corneas (n = 4, *P < 0.05 versus uninjured, **P < 0.05 versus 2 D).
12-HETE levels were higher in both the uninjured (222 ± 55 pg/cornea, n = 4) and injured cornea by day 7 (237 ± 61 pg/cornea, n = 4) compared with 15-HETE levels, which reflects the expression of several distinct 12-LOX isozymes in the mouse cornea.15,36 Unlike 15-HETE, levels of 12-HETE did not increase in response to chronic injury or parallel the time course of neovascularization. Consistent with previous reports, both the uninjured and injured cornea demonstrated 5-LOX activity,36 a key and rate-limiting enzyme in the metabolism of 15-HETE to LXA4.47 5-LOX activity (ie, 5-HETE formations) did not increase during the time course of chronic injury and consistent with a previous report36 LTB4 formation was not detected in injured or uninjured corneas. In addition to 15-HETE, formation of the cyclooxygenase metabolites PGE2 and thromboxane B2 demonstrated a time dependent increase in response to chronic injury and their formation paralleled that of 15-HETE and neovascularization (Figure 2). PGE2 was the most prominent eicosanoid in the inflamed and vascularized cornea, peaking at 1.1 ng/cornea (1030 pg ± 167, n = 4) by day 4, which is consistent with its role as key mediator of inflammation and angiogenesis.33,48
To define the endogenous role of the 15-LOX in inflammatory neovascularization, we used the well-defined 12/15-LOX (Alox15) KO mice and matched congenic controls. In vivo deletion of 12/15-LOX, which is highly expressed in corneal epithelial cells and recruited leukocytes,36 correlated with a 2.8-fold (12/15-LOX KO = 5125 ± 500 PMN versus wild-type = 1770 ± 1277 PMN, n = 4) increase in the number of PMN after 7 days of chronic injury (Figure 3A). These findings are consistent with the notion that 15-LOX is a central pathway for the formation of pro-resolving and anti-inflammatory autacoids,18,21,49 such as LXA4, in the cornea.18,21,36 15-LOX activity (ie, 15-HETE formation) in the injured cornea of 12/15-LOX deficient mice was decreased by 60% (Figure 3B, wild-type = 59 ± 4 pg/cornea versus 12/15-LOX KO = 23 ± 9, pg/cornea, n = 4). The lack of complete ablation of 15-LOX activity in the Alox15 KO mice is in agreement with the fact that mice express multiple 12-LOX isozymes in the cornea,15 which can contribute toward the overall endogenous 15-HETE formation. We have previously demonstrated that topical LXA4, but not its metabolic intermediate 15-HETE, inhibits exacerbated inflammation in the cornea.35 In the current chronic injury model topical LXA4 (100 ng, t.i.d.) consistently inhibited PMN infiltration after 48 hours of chronic injury by 49%, when directly compared with saline treatment alone (Figure 3C, LXA4 = 9523 ± 3226 PMN/cornea versus saline = 18,750 ± 4417 PMN/cornea).
Figure 3.
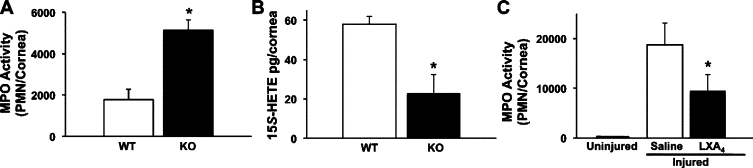
Deletion of a LXA4 biosynthetic pathway or topical treatment with LXA4 regulates PMN recruitment to the injured cornea. A: PMN content of corneas from 12/15-LOX KO mice and matched congenic wild-type (wt) controls was assessed by measuring tissue MPO activity after 7 days of chronic injury (n = 4, P < 0.02). A MPO calibration curve was established with inflammatory exudate peritoneal PMN and used to calculate relative tissue PMN numbers. B: 15-HETE formation, a metabolic precursor for LXA4 formation, was determined by LC/MS/MS-based lipidomic analyses after 4 days of chronic injury (n = 4; *P < 0.05). C: Topical action of LXA4 on PMN recruitment to the injured cornea was determined at 48 hours. Relative PMN numbers in uninjured corneas and injured corneas treated with either saline alone or LXA4 (100 ng, t.i.d.) were assessed by measuring total tissue MPO activity (n = 4, *P < 0.02).
Pathological neovascularization as a consequence of chronic injury (7 days) was markedly exacerbated in 12/15-LOX KO mice, which had a 180% increase in the total number of blood vessels, as compared with the matched congenic wild-type mice (Figure 4A, 12/15-LOX KO = 52899 ± 2477 pixels versus wild-type = 19016 ± 1490 pixels, n = 4). Topical LXA4 (100 ng, 7 days, t.i.d.) consistently attenuated pathological neovascularization by 38% (11775 ± 909 pixels, n = 3) and was able to markedly reduce the exacerbated neovascularization by 45% in 12/15-LOX KO mice (28979 ± 1448 pixels, n = 4), which have an impaired LXA4 biosynthetic pathway.36 Since 15S-HETE is a metabolic intermediate in the formation of LXA4, we assessed if 15S-HETE shared the potent bioactions of topical LXA4. Treatment with topical 15S-HETE (100 ng, t.i.d., 7 days), contrary to LXA4, markedly amplified pathological neovascularization by 87% (Figure 4B, 15S-HETE = 21516 ± 1048 pixels versus saline alone = 11497 ± 1142 pixels, n = 3).
Figure 4.
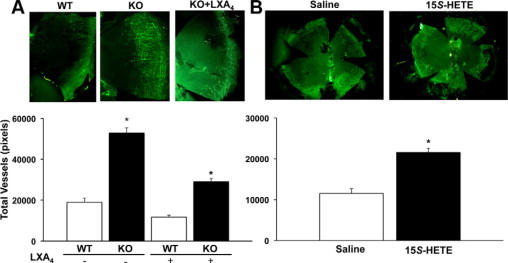
Topical LXA4 attenuates inflammatory neovascularization and inhibition of its biosynthetic pathway exacerbates pathological angiogenesis. A: Heme-angiogenesis in corneas with 7 days of chronic injury was assessed by immunohistochemistry using CD31 as a specific endothelial antigen. Corneas from 12/15-LOX KO or matched congenic wild-type (wt) mice treated with or without topical LXA4 (100 ng, t.i.d., 7 days) were collected and incubated in PBS containing fluorescein isothiocyanate-conjugated CD31/PECAM-1 monoclonal antibody and analyzed using a Zeiss Axiophot laser scanning confocal microscope. Images show neovascularization of representative corneal quadrants. All CD31+ vessels were traced manually and are expressed as total pixels (Image Pro Express 6.0) (n = 3 to 4; *P < 0.05). B: Topical 15-HETE exacerbates inflammatory neovascularization. Injured corneas were treated topically with either 15S-HETE (100 ng, t.i.d.) or sterile saline for 7 days. Images show representative whole corneal flat mounts (left panel, saline treatment alone; right panel 15S-HETE treatment). All CD31+ vessels were traced manually and are expressed as total pixels (n = 4; *P < 0.05).
To obtain proof of concept for a role of endogenous LXA4 in pathological neovascularization, we used 5-LOX KO mice, which are LXA4 knockout mice49–51 since the mouse genome only contains one 5-LOX enzyme, the obligatory and rate-limiting enzyme for LXA4 formation.
5-LOX is a well-studied and prominent enzyme in most leukocytes, especially PMN and macrophages.34 Recent reports have established expression and activity of this enzyme in uninjured and inflamed mouse corneas36 and RNA expression in bovine corneas and human corneal epithelium.52 Deletion of the LXA4 biosynthetic pathway in 5-LOX KO mice was associated with a 148% increase in pathological neovascularization (Figure 5) when directly compared with matched congenic wild-type mice (5-LOX KO = 164180 ± 9072 pixels versus wild-type = 66193 ± 6122 pixels, n = 4). Taken together, in vivo deletion of two key biosynthetic pathways for formation of endogenous LXA4 in the cornea (ie, 15-LOX and 5-LOX) provides strong evidence for a key role of endogenous LXA4 circuit as a key regulator of inflammatory neovascularization.
Figure 5.
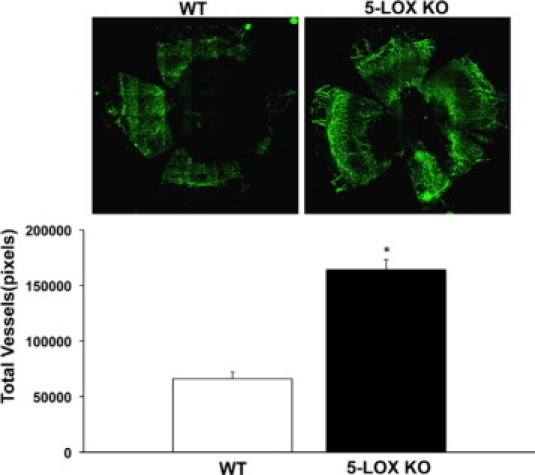
5-LOX/LXA4 KO mice have a phenotype of exacerbated neovascularization in response to chronic injury. 5-LOX KO mice, which cannot generate LXA4, and their matched congenic wild-type (wt) controls were subjected to chronic suture injury for 7 days. Corneas were removed and pathological angiogenesis quantified by immunohistochemistry using a Zeiss Axioplan 2 microscope. Mosaic images were taken with a AxioCam MR camera and compiled using MosaiX and Zeiss AxioVision 4.5 software. Inset shows representative whole corneal flatmounts (left panel, wild-type; right panel, 5-LOX KO). Vessel density was documented and expressed as total pixels (Image Pro Express 6.0) (n = 4; *P < 0.05).
Real-time PCR analysis was used to assess the impact of impaired 12/15-LOX activity on endogenous pathways that are markers of inflammatory neovascularization and stress responses during the initial stage of the angiogenic response (48 hours). mRNA expression of key mediators of inflammatory angiogenesis was markedly amplified in 12/15-LOX KO mice when directly compared with matched congenic wild-type mice (Figure 6). In accordance with exacerbated neovascularization VEGF-A (KO = 8.80 ± 1.32-fold versus wild-type = 4.60 ± 0.64-fold, n = 4), sFLT1 (KO = 7.79 ± 1.64-fold versus wild-type = 1.51 ± 0.272 fold, n = 4), and FLT4 (KO = 31.7 ± 9.27 versus wild-type = 10.4 ± 1.17-fold, n = 4) mRNA levels increased 91%, 413% and 210% in 12/15-LOX KO mice, respectively. Up-regulation of HO-1 and sFLT1 suggest a compensatory mechanism in this avascular and immune-privileged tissue to counter exacerbated neovascularization in the 12/15-LOX KO mice.
Figure 6.
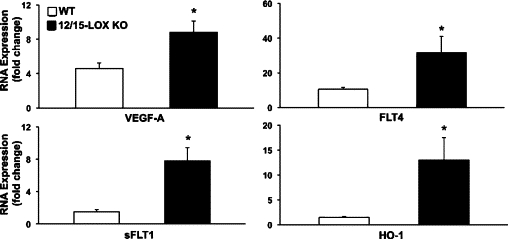
Deletion of 12/15 LOX up-regulates key regulators of inflammatory neovascularization. Corneas after 48 hours of chronic injury were collected from 12/15-LOX KO and matched congenic wild-type mice (n = 4). mRNA expression was quantified for HO-1, VEGF-A, sFLT1, and FLT4 using a Step One Plus QPCR system (Applied Biosystems). RNA expression for each gene was calculated using relative quantity (RQ) compared with a mouse positive mRNA control with the ΔΔCT method and β-actin as a reference gene. Data are expressed as fold change compared with the mouse positive mRNA control (n = 4, *P < 0.05).
Recent reports have demonstrated that stable analogs of LXA4 inhibit VEGF-A-induced angiogenic responses in isolated endothelial cells25,26,28 and the murine airpouch model27 by inhibiting downstream signals of the VEGF-2 receptor (FLK1). In view of intimate and dynamic interactions of inflammatory and angiogenic pathways in physiological responses to injury, we assessed the in vivo actions of native LXA4 on selected pathways that regulate the angiogenic and inflammatory responses to chronic injury in the cornea (Figure 7). A fundamental feature of exacerbated/aberrant inflammation, especially in the immune privileged cornea, is the up-regulation of protective pathways such as HO-1 and 12/15-LOX.36,38,46,53 Direct topical treatment with LXA4 (100 ng, t.i.d., Figure 7) significantly reduced levels of HO-1 and 12/15-LOX by 4.4 fold (LXA4 = 0.010 ± 0.002 RQ versus saline = 0.044 ± 0.042 RQ, n = 4) and 3.3 fold (LXA4 = 0.015 ± 0.02 RQ versus saline = 0.050 ± 0.02 RQ, n = 4), respectively. In accordance with the observation that exogenous amplification of the LXA4 circuit reduced expression of resident protective circuits in the injured cornea, topical LXA4 also significantly reduced mRNA levels of VEGF-A and FLT4 by 66% (LXA4 = 0.120 ± 0.024 RQ versus saline = 0.358 ± 0.001 RQ, n = 4) and 80% (0.015 ± 0.005 RQ versus 0.003 ± 0.001 RQ), respectively. LXA4 mediates its established bioactions via the ALX receptor, which is expressed predominantly in the epithelium of healthy corneas and recruited leukocytes.36,47 Direct corneal treatment with LXA4 abrogated expression of both LXA4 receptors (ALX-1, LXA4 = 0.040 ± 0.018 RQ versus saline = 1.05 ± 0.447 RQ; ALX-2 LXA4 = 0.274 ± 0.097 RQ versus saline = 1.710 ± 0.678 RQ, n = 3) in the injured cornea suggesting a ligand-dependent negative feedback loop in this tissue. This response is distinct from the innate immune responses in the peritoneal cavity,54 where transgene expression of human ALX in a positive feed forward loop increases endogenous LXA4 formation by the inflammatory exudate.
Figure 7.
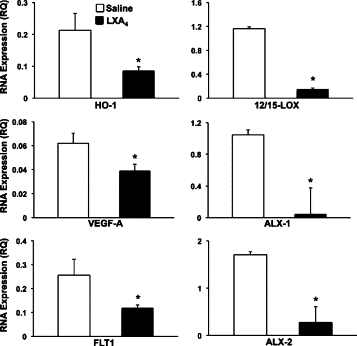
Direct topical treatment with LXA4 down-regulates expression of key regulators of inflammatory angiogenesis. After suture injury corneas were treated with either saline alone or LXA4 (100 ng, t.i.d.) for 48 hours. mRNA expression was quantified for HO-1, VEGF-A, FLT4, and both LXA4 receptors (ALX-1, ALX-2) using a real-time PCR system. Relative expression was compared with a mouse positive mRNA control (n = 4, *P < 0.05).
Discussion
A coordinated and synergistic array of pro- and anti-angiogenic signals regulates the multicellular process of angiogenesis that leads to the formation of functional blood and/or lymph vessel.3 Inflammation and angiogenesis are triggered, especially in response to injury and insult, by the same molecular events and in a reciprocal fashion these two fundamental responses sustain each other. Indeed, there is strong evidence for a close association or co-dependence between chronic inflammation and angiogenesis in several human diseases.2,3
An essential feature of acute inflammation is the resolution of leukocytes and restoration of normal tissue function.55 It is well established that dynamic lipid circuits, such as the LXA4 circuit, have a pivotal role in the active resolution of normal and vital inflammatory responses.49,55–57 Hence, it stands to reason that resolution of heme- or lymphangiogenesis, like inflammation, is an active and highly coordinated process that is regulated by common or shared pathways. 12/15-lipoxygenase is a central biosynthetic pathway in mucosal tissues and the cornea for the formation of LXA4,15 an eicosanoid that is formed in both humans and mice.49,55–57 In addition, 12/15-LOX is a key enzyme for the formation of ω−3 polyunsaturated fatty acid-derived mediators, namely protectins, resolvins, and maresins,36,58–61 which have demonstrated ant-inflammatory and anti-apoptotic and/or pro-resolving bioactions. These 15-LOX products, namely protectins and resolvins, are formed in the eye.36,58,60,62 Hence, in addition to LXA4, 15-LOX can generate additional classes of potent anti-inflammatory mediators, which can contribute to the protective function of this enzyme in the eye. It is of particular interest that chronic injury of the cornea and pathological angiogenesis are associated with increased expression and functional activity of 12/15-LOX (Figure 1 and 2). More importantly, in vivo deletion of 12/15-LOX (Alox15) resulted in marked amplification of inflammatory neovascularization (Figures 3 and 4), which provides direct evidence that this prominent ocular enzyme15,58,60,63,64 has a key role in attenuating inflammatory neovascularization. The notion that 15-LOX is a protective pathway in acute inflammatory responses is supported by studies that demonstrate that in vivo transfection with human 15-LOX (ALOX15) provides functional protection against immune-mediated renal injury.65 Moreover, transgenic rabbits,54,66 which selectively express human ALOX15 in monocytes/macrophages and PMN, demonstrate functional protection against neutrophil-mediated tissue injury, microbial inflammation, and atherosclerosis.
The endogenous role of 15-LOX in angiogenesis is not well defined and has not been explored in models of inflammatory neovascularization. In vitro studies that either add 15S-HETE to microvascular endothelial cell lines30,31 or express human 15-LOX (ALOX15) in rabbit skeletal muscle32 have led to conflicting conclusions, namely, that the 15-LOX pathway either induces angiogenic responses or inhibits VEGF-A induced angiogenesis, respectively. 15-lipoxygenase can initiate the potential formation of several distinct classes of lipid autacoids that have pleiotropic actions. Endogenous formation of two 15-LOX-derived products are firmly established in several animal models of inflammation and human tissue, namely 15S-HETE and LXA4.14,47,49,57 A body of work49,55,57,67,68 has established the endogenous formation of LXA4 and its role as a key mediator of inflammatory resolution.55,57 More importantly, unlike other 15-LOX metabolites, data from receptor transgenic mice and detailed structure-activity studies have established specific ALX receptors in humans (FPRL1) and mice (Fprl1 and Fpr-rs2) as a molecular mechanism for LXA4's protective in vivo actions.47
In accordance with our previous studies in male BALB/c mice36 and the original identification of ALX in human corneas,69 we detected basal expression of ALX-1 (Fprl1) in uninjured corneas of female C57Bl/6J mice and were also able to detect expression of ALX-2 (Fpr-rs2). More importantly, our findings demonstrate that ALX-2 expression is markedly down-regulated as a consequence of chronic injury, which is coincident with pathological neovascularization. If reduced expression of ALX2 attenuates the protective actions of endogenous LXA4 and the mechanism for the dynamic regulation of the corneal ALX2 receptor remains to be determined and are of great interest. Our findings are in line with a recent report39 that confirmed expression of a LXA4 receptor (Fprs-rs2) in the cornea of BALB/c mice. Also note, unlike this report,39 we detected formation of both LXA4 receptors in uninjured corneas and observed dynamic regulation of ALX-2, which may reflect differences in the sensitivity of analytical methods, mouse strain, and/or gender differences.
The rationale for focusing on the in vivo actions of LXA4 and activation of the corneal ALX receptor36,70 as a likely 12/15-LOX induced circuit that attenuates inflammatory neovascularization was based on several independent lines of evidence: 1) the key observation that inflammatory neovascularization was associated with a specific decrease in one of the mouse LXA4 receptors (Fpr-rs2, Figure 1); 2) previous findings that demonstrate that corneal formation of LXA4 is attenuated in 12/15-LOX KO mice;36 and 3) reports that have demonstrated that stable mimetics of LXA4 inhibit VEGF-induced angiogenesis.25–27,39
More importantly, in vivo deletion of 5-LOX, which is the obligatory and rate-limiting enzyme for LXA4 formation47,49 led to pronounced amplification of neovascularization (Figure 6).
It is important to note that except for LXA4, other 5-LOX products such as leukotrienes are potent and well-established pro-inflammatory mediators in both humans and mice.33 Inflammation is an essential feature of injury-induced neovascularization and amplifies the angiogenic response in the cornea.16,71 Hence, the finding that 5-LOX deletion leads to amplified neovascularization indicates an unexpected protective role for this pathway in the immune privileged cornea, which strongly implicates LXA4 as the metabolic product. Taken together, our data from 5-LOX KO mice, which are LXA4 knockouts,50,51 and from 12/15-LOX KO mice provide strong evidence for endogenous LXA4 as a key regulator of inflammatory neovascularization.
Topical treatment with LXA4 initiated immediately after inducing chronic injury not only attenuated formation of blood vessels, but also critical mediators of inflammatory neovascularization, namely VEGF-A and FLT4. In addition, topical LXA4 reduced expression of HO-1, a stress gene and a marker of inflammation, whose degree of expression directly correlates with the degree of the inflammatory response37,72 and is a key feature of non-resolving inflammation in the cornea.38 Contrary to LXA4, topical 15-HETE exacerbated inflammatory neovascularization (Figure 4), which supports the notion that the protective actions of 15-LOX require metabolism of 15-HETE to LXA4 by 5-LOX, which is expressed in leukocytes49 and the cornea.36
The findings that topical LXA4 inhibits and in vivo deletion of LXA4 formation exacerbates VEGF-A and FLT4 expression provides a compelling argument for regulation of angiogenic responses by the resident LXA4 circuit. It is important to recognize that the VEGF-A circuit, in addition to its role as a key angiogenic factor, is also involved in macrophage chemotaxis, a cell type that not only is a major source but also a target for the bioactions of VEGF-A. VEGF-A induces release of several prominent inflammatory mediators in primary endothelial cells such as interleukins 6 and 8, and tumor necrosis factorα28 and was originally identified as a potent vascular permeability factor.73 Moreover, local intravitreal injection of VEGF-A leads to local adhesion of leukocytes and monocyte chemotaxis,3 while VEGF-A traps significantly inhibit leukocyte infiltration in the suture injury model.16 These findings underscore the important role of VEGF-A as a modulator of inflammatory function. Hence, LXA4's ability to inhibit VEGF-A up-regulation in response to injury indicates that LXA4 may regulate the early phase of both angiogenic and inflammatory responses. In this context it also important to note that our model is inflammatory based neovascularization. Hence, it remains to be determined if our findings regarding the protective actions of the resident LXA4 circuit extend to hypoxia-driven angiogenesis.
FLT4 is the endothelial receptor for VEGF-C and VEGF-D and mediates lymphangiogenesis. Recent evidence suggests that FLT4 has a key role in sprouting angiogenesis and thus may drive angiogenesis.12 The observation that FLT4 is required for spontaneous heme-angiogenesis in the cornea of corn1 mice11 support the notion for a pivotal role in pathological neovascularization. It is of particular interest that a functional FLT4 receptor is expressed in corneal epithelial cells.10 How FLT4 ligation and proven kinase activity10 regulates epithelial function remains to be defined. What is clear is that chronic injury as early as 48 hours, which is initial phase of angiogenesis, strikingly up-regulates expression of FLT4. Moreover, deletion of protective LXA4 biosynthetic pathways correlates with amplified expression FLT4 while topical treatment with LXA4 inhibits it expression (Figure 6, 7). Detailed studies are required to define the cellular targets for LXA4 in the cornea (ie, FLT4 expressing endothelial cells, epithelial cells and/or macrophages) and if activation of VEGF circuits precedes inflammation or is a consequence of injury-induced inflammation.
Our findings regarding the endogenous role LXA4 in the model of suture induced angiogenesis are in general agreement with a recent report39 that assessed the therapeutic actions (subconjunctival injections) of a stable analog of LXA4 and two ω−3 PUFA-derived anti-inflammatory autacoids. However, in regards to FLT4 expression our findings with 12/15-LOX KO mice and direct topical treatment with LXA4 both indicate that FLT4 expression is regulated by exogenous and endogenous LXA4 in the cornea, while the report by Lin and colleagues39 shows a strong trend but not significant inhibition in male BALB/c mice. These differences are likely due to differences in the treatment protocols (ie, direct daily t.i.d. corneal treatment versus indirect treatment by subconjunctival injection every 48 hours), the use of a stable LXA4 analog versus native LXA4 and mouse strain differences (C57Bl/6J versus BALB/c).
ALX receptors are expressed in leukocytes, epithelial cells and vascular endothelial cells, hence, LXA4 can regulate the function of all cell types critical to corneal neovascularization. LXA4's well-established anti-inflammatory actions with PMN, macrophages, and epithelial cells are consistent with our finding that therapeutic amplification attenuates while in vivo inhibition of endogenous LXA4 formation exacerbates chronic inflammation. However, it is important to recognize that LXA4 and stable analogs of LXA4 have been shown to directly regulate signaling of the VEGF-2 (FLK1) receptor and its expression in endothelial cells.25,26,28 Thus, the ability of endogenous and therapeutic LXA4 to inhibit inflammatory neovascularization is likely extends to direct regulation of angiogenesis. In this regard, it is important to recognize that angiogenic responses as a consequence of corneal injury do not require leukocyte infiltration since complete depletion of PMN and monocytes attenuates but does not prevent angiogenesis.71 Moreover, the initiating signals or cell types for injury-induced angiogenic response have not been elucidated in the cornea. In view of the fact that the healthy cornea, especially the central cornea, contains no leukocytes the initial targets for LXA4 are likely epithelial cells, since these cells are a predominant cell source of ALX in human and mouse corneas.15,36
In summary, our current results demonstrate that resident LXA4 circuits are key determinants for the degree of inflammatory neovascularization in response to chronic injury. Genetic deletion of LXA4 biosynthetic pathways or amplification with exogenous LXA4 provides strong evidence that this protective circuit controls key regulators of the angiogenic and inflammatory response in the cornea. In view of the prominent expression of 15-LOX and the ALX receptor in epithelial cells and macrophages, the endogenous role the LXA4 circuit in pathological angiogenesis clearly warrants further studies.
Acknowledgements
We thank Dr. Lars Bellner at New York Medical College for skillful and expert help with immunohistochemistry and image analyses of corneal neovascularization in the 15-LOX KO mice and congenic controls.
Footnotes
Supported in part by grants from the National Eye Institute EY016136 and P30EY003176.
References
- 1.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 3.Costa C, Incio J, Soares R. Angiogenesis and chronic inflammation: cause or consequence? Angiogenesis. 2007;10:149–166. doi: 10.1007/s10456-007-9074-0. [DOI] [PubMed] [Google Scholar]
- 4.Noel A, Jost M, Lambert V, Lecomte J, Rakic JM. Anti-angiogenic therapy of exudative age-related macular degeneration: current progress and emerging concepts. Trends Mol Med. 2007;13:345–352. doi: 10.1016/j.molmed.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Cao Y, Langer R. A review of Judah Folkman's remarkable achievements in biomedicine. Proc Natl Acad Sci USA. 2008;105:13203–13205. doi: 10.1073/pnas.0806582105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gimbrone MA, Jr, Cotran RS, Leapman SB, Folkman J. Tumor growth and neovascularization: an experimental model using the rabbit cornea. J Natl Cancer Inst. 1974;52:413–427. doi: 10.1093/jnci/52.2.413. [DOI] [PubMed] [Google Scholar]
- 7.Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol. 2003;3:879–889. doi: 10.1038/nri1224. [DOI] [PubMed] [Google Scholar]
- 8.Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, Kleinman ME, Caldwell RB, Lin Q, Ogura Y, Orecchia A, Samuelson DA, Agnew DW, St Leger J, Green WR, Mahasreshti PJ, Curiel DT, Kwan D, Marsh H, Ikeda S, Leiper LJ, Collinson JM, Bogdanovich S, Khurana TS, Shibuya M, Baldwin ME, Ferrara N, Gerber HP, De Falco S, Witta J, Baffi JZ, Raisler BJ, Ambati J. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Hamrah P, Cursiefen C, Zhang Q, Pytowski B, Streilein JW, Dana MR. Vascular endothelial growth factor receptor-3 mediates induction of corneal alloimmunity. 2004. Ocul Immunol Inflamm. 2007;15:275–278. doi: 10.1080/09273940701382317. [DOI] [PubMed] [Google Scholar]
- 10.Cursiefen C, Chen L, Saint-Geniez M, Hamrah P, Jin Y, Rashid S, Pytowski B, Persaud K, Wu Y, Streilein JW, Dana R. Nonvascular VEGF receptor 3 expression by corneal epithelium maintains avascularity and vision. Proc Natl Acad Sci USA. 2006;103:11405–11410. doi: 10.1073/pnas.0506112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cursiefen C, Ikeda S, Nishina PM, Smith RS, Ikeda A, Jackson D, Mo JS, Chen L, Dana MR, Pytowski B, Kruse FE, Streilein JW. Spontaneous corneal hem- and lymphangiogenesis in mice with destrin-mutation depend on VEGFR3 signaling. Am J Pathol. 2005;166:1367–1377. doi: 10.1016/S0002-9440(10)62355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Yla-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454:656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 13.Paavonen K, Mandelin J, Partanen T, Jussila L, Li TF, Ristimaki A, Alitalo K, Konttinen YT. Vascular endothelial growth factors C and D and their VEGFR-2 and 3 receptors in blood and lymphatic vessels in healthy and arthritic synovium. J Rheumatol. 2002;29:39–45. [PubMed] [Google Scholar]
- 14.Kuhn H, O'Donnell VB. Inflammation and immune regulation by 12/15-lipoxygenases. Prog Lipid Res. 2006;45:334–356. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Gronert K. Lipoxins in the eye and their role in wound healing. Prostaglandins Leukot Essent Fatty Acids. 2005;73:221–229. doi: 10.1016/j.plefa.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, D'Amore PA, Dana MR, Wiegand SJ, Streilein JW. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. 2004;113:1040–1050. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nie D, Tang K, Diglio C, Honn KV. Eicosanoid regulation of angiogenesis: role of endothelial arachidonate 12-lipoxygenase. Hemost Thromb Vasc Biol. 2000;95:2304–2311. [PubMed] [Google Scholar]
- 18.Bannenberg GL, Aliberti J, Hong S, Sher A, Serhan C. Exogenous pathogen and plant 15-lipoxygenase initiate endogenous lipoxin a4 biosynthesis. J Exp Med. 2004;199:515–523. doi: 10.1084/jem.20031325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brezinski ME, Serhan CN. Selective incorporation of (15S)-hydroxyeicosatetraenoic acid in phosphatidylinositol of human neutrophils: agonist-induced deacylation and transformation of stored hydroxyeicosanoids. Proc Natl Acad Sci USA. 1990;87:6248–6252. doi: 10.1073/pnas.87.16.6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy BD, Romano M, Chapman HA, Reilly JJ, Drazen J, Serhan CN. Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J Clin Invest. 1993;92:1572–1579. doi: 10.1172/JCI116738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, Colgan SP, Stahl GL, Merched A, Petasis NA, Chan L, Van Dyke TE. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171:6856–6865. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- 22.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 23.Nassar GM, Morrow JD, Roberts LJ, II, Lakkis FG, Badr KF. Induction of 15-lipoxygenase by interleukin-13 in human blood monocytes. J Biol Chem. 1994;269:27631–27634. [PubMed] [Google Scholar]
- 24.Chavis C, Godard P, Crastes de Paulet A, Damon M. Formation of lipoxins and leukotrienes by human alveolar macrophages incubated with 15(S)-HETE: a model for cellular cooperation between macrophages and airway epithelial cells. Eicosanoids. 1992;5:203–211. [PubMed] [Google Scholar]
- 25.Cezar-de-Mello PF, Nascimento-Silva V, Villela CG, Fierro IM. Aspirin-triggered Lipoxin A4 inhibition of VEGF-induced endothelial cell migration involves actin polymerization and focal adhesion assembly. Oncogene. 2006;25:122–129. doi: 10.1038/sj.onc.1209002. [DOI] [PubMed] [Google Scholar]
- 26.Cezar-de-Mello PF, Vieira AM, Nascimento-Silva V, Villela CG, Barja-Fidalgo C, Fierro IM. ATL-1, an analogue of aspirin-triggered lipoxin A4, is a potent inhibitor of several steps in angiogenesis induced by vascular endothelial growth factor. Br J Pharmacol. 2008;153:956–965. doi: 10.1038/sj.bjp.0707650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fierro IM, Kutok JL, Serhan CN. Novel lipid mediator regulators of endothelial cell proliferation and migration: aspirin-triggered-15R-lipoxin A(4) and lipoxin A(4) J Pharmacol Exp Ther. 2002;300:385–392. doi: 10.1124/jpet.300.2.385. [DOI] [PubMed] [Google Scholar]
- 28.Baker N, O'Meara SJ, Scannell M, Maderna P, Godson C. Lipoxin A4: anti-inflammatory and anti-angiogenic impact on endothelial cells. J Immunol. 2009;182:3819–3826. doi: 10.4049/jimmunol.0803175. [DOI] [PubMed] [Google Scholar]
- 29.Mochizuki N, Kwon YG. 15-lipoxygenase-1 in the vasculature: expanding roles in angiogenesis. Circ Res. 2008;102:143–145. doi: 10.1161/CIRCRESAHA.107.170191. [DOI] [PubMed] [Google Scholar]
- 30.Bajpai AK, Blaskova E, Pakala SB, Zhao T, Glasgow WC, Penn JS, Johnson DA, Rao GN. 15(S)-HETE production in human retinal microvascular endothelial cells by hypoxia: novel role for MEK1 in 15(S)-HETE induced angiogenesis. Invest Ophthalmol Vis Sci. 2007;48:4930–4938. doi: 10.1167/iovs.07-0617. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava K, Kundumani-Sridharan V, Zhang B, Bajpai AK, Rao GN. 15(S)-hydroxyeicosatetraenoic acid-induced angiogenesis requires STAT3-dependent expression of VEGF. Cancer Res. 2007;67:4328–4336. doi: 10.1158/0008-5472.CAN-06-3594. [DOI] [PubMed] [Google Scholar]
- 32.Viita H, Markkanen J, Eriksson E, Nurminen M, Kinnunen K, Babu M, Heikura T, Turpeinen S, Laidinen S, Takalo T, Yla-Herttuala S. 15-lipoxygenase-1 prevents vascular endothelial growth factor A- and placental growth factor-induced angiogenic effects in rabbit skeletal muscles via reduction in growth factor mRNA levels. NO bioactivity, and downregulation of VEGF receptor 2 expression. Circ Res. 2008;102:177–184. doi: 10.1161/CIRCRESAHA.107.155556. [DOI] [PubMed] [Google Scholar]
- 33.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 34.Funk CD, Chen XS, Johnson EN, Zhao L. Lipoxygenase genes and their targeted disruption. Prostaglandins Other Lipid Mediat. 2002;68–69:303–312. doi: 10.1016/s0090-6980(02)00036-9. [DOI] [PubMed] [Google Scholar]
- 35.Biteman B, Hassan IR, Walker E, Leedom AJ, Dunn M, Seta F, Laniado-Schwartzman M, Gronert K. Interdependence of lipoxin A4 and heme-oxygenase in counter-regulating inflammation during corneal wound healing. FASEB J. 2007;21:2257–2266. doi: 10.1096/fj.06-7918com. [DOI] [PubMed] [Google Scholar]
- 36.Gronert K, Maheshwari N, Khan N, Hassan IR, Dunn M, Laniado Schwartzman M. A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J Biol Chem. 2005;280:15267–15278. doi: 10.1074/jbc.M410638200. [DOI] [PubMed] [Google Scholar]
- 37.Hassan IR, Gronert K. Acute changes in dietary omega-3 and omega-6 polyunsaturated fatty acids have a pronounced impact on survival following ischemic renal injury and formation of renoprotective docosahexaenoic acid-derived protectin D1. J Immunol. 2009;182:3223–3232. doi: 10.4049/jimmunol.0802064. [DOI] [PubMed] [Google Scholar]
- 38.Seta F, Bellner L, Rezzani R, Regan RF, Dunn MW, Abraham NG, Gronert K, Laniado-Schwartzman M. Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am J Pathol. 2006;169:1612–1623. doi: 10.2353/ajpath.2006.060555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin Y, Arita M, Zhang Q, Saban DR, Chauhan SK, Chiang N, Serhan CN, Dana R. Novel anti-inflammatory and pro-resolving lipid mediators block inflammatory angiogenesis. Invest Ophthalmol Vis Sci. 2009;50:4743–4752. doi: 10.1167/iovs.08-2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gronert K, Clish CB, Romano M, Serhan CN. Transcellular regulation of eicosanoid biosynthesis. Methods Mol Biol. 1999;120:119–144. doi: 10.1385/1-59259-263-5:119. [DOI] [PubMed] [Google Scholar]
- 41.Serhan CN, Lu Y, Hong S, Yang R. Mediator lipidomics: search algorithms for eicosanoids, resolvins, and protectins. Methods Enzymol. 2007;432:275–317. doi: 10.1016/S0076-6879(07)32012-0. [DOI] [PubMed] [Google Scholar]
- 42.Murphy RC, Barkley RM, Zemski Berry K, Hankin J, Harrison K, Johnson C, Krank J, McAnoy A, Uhlson C, Zarini S. Electrospray ionization and tandem mass spectrometry of eicosanoids. Anal Biochem. 2005;346:1–42. doi: 10.1016/j.ab.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez-Periz A, Horrillo R, Ferre N, Gronert K, Dong B, Moran-Salvador E, Titos E, Martinez-Clemente M, Lopez-Parra M, Arroyo V, Claria J. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 2009;23:1946–1957. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiang N, Takano T, Clish CB, Petasis NA, Tai H-H, Serhan CN. Aspirin-triggered 15-epi-lipoxin A4 (ATL) generation by human leukocytes and murine peritonitis exudates: development of a specific 15-epi-LXA4 ELISA. J Pharmacol Exp Ther. 1998;287:779–790. [PubMed] [Google Scholar]
- 45.Clish CB, Levy BD, Chiang N, Tai HH, Serhan CN. Oxidoreductases in lipoxin A4 metabolic inactivation: a novel role for 15-onoprostaglandin 13-reductase/leukotriene B4 12-hydroxydehydrogenase in inflammation. J Biol Chem. 2000;275:25372–25380. doi: 10.1074/jbc.M002863200. [DOI] [PubMed] [Google Scholar]
- 46.Bellner L, Vitto M, Patil KA, Dunn MW, Regan R, Laniado-Schwartzman M. Exacerbated corneal inflammation and neovascularization in the HO-2 null mice is ameliorated by biliverdin. Exp Eye Res. 2008;87:268–278. doi: 10.1016/j.exer.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chiang N, Serhan CN, Dahlen SE, Drazen JM, Hay DW, Rovati GE, Shimizu T, Yokomizo T, Brink C. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol Rev. 2006;58:463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 48.Nie D, Honn KV. Eicosanoid regulation of angiogenesis in tumors. Semin Thromb Hemost. 2004;30:119–125. doi: 10.1055/s-2004-822976. [DOI] [PubMed] [Google Scholar]
- 49.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat Immunol. 2002;3:76–82. doi: 10.1038/ni745. [DOI] [PubMed] [Google Scholar]
- 51.Aliberti J, Serhan C, Sher A. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii infection. J Exp Med. 2002;196:1253–1262. doi: 10.1084/jem.20021183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liminga M, Oliw EH. Studies of lipoxygenases in the epithelium of cultured bovine cornea using an air interface model. Exp Eye Res. 2000;71:57–67. doi: 10.1006/exer.2000.0852. [DOI] [PubMed] [Google Scholar]
- 53.Gronert K. Lipid autacoids in inflammation and injury responses: a matter of privilege. Mol Interv. 2008;8:28–35. doi: 10.1124/mi.8.1.7. [DOI] [PubMed] [Google Scholar]
- 54.Devchand PR, Arita M, Hong S, Bannenberg G, Moussignac RL, Gronert K, Serhan CN. Human ALX receptor regulates neutrophil recruitment in transgenic mice: roles in inflammation and host defense. FASEB J. 2003;17:652–659. doi: 10.1096/fj.02-0770com. [DOI] [PubMed] [Google Scholar]
- 55.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 2007;447:869–874. doi: 10.1038/nature05877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–416. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 58.Calandria JM, Marcheselli VL, Mukherjee PK, Uddin J, Winkler JW, Petasis NA, Bazan NG. Selective survival rescue in 15-lipoxygenase-1 deficient retinal pigment epithelial cells by the novel docosahexaenoic acid-derived mediator, neuroprotectin D1. J Biol Chem. 2009;284:17877–17882. doi: 10.1074/jbc.M109.003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, Oh SF, Spite M. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. 2009;206:15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin Q, Patil KA, Gronert K, Sharma SC. Neuroprotectin D1 inhibits retinal ganglion cell death following axotomy. Prostaglandins Leukot Essent Fatty Acids. 2008;79:201–207. doi: 10.1016/j.plefa.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 61.Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–2783. doi: 10.1172/JCI25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Connor KM, SanGiovanni JP, Lofqvist C, Aderman CM, Chen J, Higuchi A, Hong S, Pravda EA, Majchrzak S, Carper D, Hellstrom A, Kang JX, Chew EY, Salem N, Jr, Serhan CN, Smith LE. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat Med. 2007;13:868–873. doi: 10.1038/nm1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chang MS, Schneider C, Roberts RL, Shappell SB, Haselton FR, Boeglin WE, Brash AR. Detection and subcellular localization of two 15S-lipoxygenases in human cornea. Invest Ophthalmol Vis Sci. 2005;46:849–856. doi: 10.1167/iovs.04-1166. [DOI] [PubMed] [Google Scholar]
- 64.Liminga M, Fagerholm P, Oliw EH. Lipoxygenases in corneal epithelia of man and cynomolgus monkey. Exp Eye Res. 1994;59:313–321. doi: 10.1006/exer.1994.1113. [DOI] [PubMed] [Google Scholar]
- 65.Munger KA, Montero A, Fukunaga M, Uda S, Yura T, Imai E, Kaneda Y, Valdivielso JM, Badr KF. Transfection of rat kidney with human 15-lipoxygenase suppresses inflammation and preserves function in experimental glomerulonephritis. Proc Natl Acad Sci USA. 1999;96:13375–13380. doi: 10.1073/pnas.96.23.13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen J, Herderick E, Cornhill JF, Zsigmond E, Kim HS, Kuhn H, Guevara NV, Chan L. Macrophage-mediated 15-lipoxygenase expression protects against atherosclerosis development. J Clin Invest. 1996;98:2201–2208. doi: 10.1172/JCI119029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Godson C, Brady HR. Lipoxins: novel anti-inflammatory therapeutics? Curr Op Invest Drugs. 2000;1:380–385. [PubMed] [Google Scholar]
- 68.McMahon B, Mitchell S, Brady HR, Godson C. Lipoxins: revelations on resolution. Trends Pharmacol Sci. 2001;22:391–395. doi: 10.1016/s0165-6147(00)01771-5. [DOI] [PubMed] [Google Scholar]
- 69.Gronert K, Gewirtz A, Madara JL, Serhan CN. Identification of a human enterocyte lipoxin A4 receptor that is regulated by interleukin (IL)-13 and interferon gamma and inhibits tumor necrosis factor alpha-induced IL-8 release. J Exp Med. 1998;187:1285–1294. doi: 10.1084/jem.187.8.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang LC, Petkova TD, Reins RY, Proske RJ, McDermott AM. Multifunctional roles of human cathelicidin (LL-37) at the ocular surface. Invest Ophthalmol Vis Sci. 2006;47:2369–2380. doi: 10.1167/iovs.05-1649. [DOI] [PubMed] [Google Scholar]
- 71.Sholley MM, Gimbrone MA, Jr, Cotran RS. The effects of leukocyte depletion on corneal neovascularization. Lab Invest. 1978;38:32–40. [PubMed] [Google Scholar]
- 72.Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- 73.Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22:201–220. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]