

Abstract
Understanding the origin of myofibroblasts in kidney is of great interest because these cells are responsible for scar formation in fibrotic kidney disease. Recent studies suggest epithelial cells are an important source of myofibroblasts through a process described as the epithelial-to-mesenchymal transition; however, confirmatory studies in vivo are lacking. To quantitatively assess the contribution of renal epithelial cells to myofibroblasts, we used Cre/Lox techniques to genetically label and fate map renal epithelia in models of kidney fibrosis. Genetically labeled primary proximal epithelial cells cultured in vitro from these mice readily induce markers of myofibroblasts after transforming growth factor β1 treatment. However, using either red fluorescent protein or β-galactosidase as fate markers, we found no evidence that epithelial cells migrate outside of the tubular basement membrane and differentiate into interstitial myofibroblasts in vivo. Thus, although renal epithelial cells can acquire mesenchymal markers in vitro, they do not directly contribute to interstitial myofibroblast cells in vivo. Lineage analysis shows that during nephrogenesis, FoxD1-positive(+) mesenchymal cells give rise to adult CD73+, platelet derived growth factor receptor β+, smooth muscle actin-negative interstitial pericytes, and these FoxD1-derivative interstitial cells expand and differentiate into smooth muscle actin+ myofibroblasts during fibrosis, accounting for a large majority of myofibroblasts. These data indicate that therapeutic strategies directly targeting pericyte differentiation in vivo may productively impact fibrotic kidney disease.
Understanding the origin and differentiation pathways of myofibroblasts in vivo is critical for identifying new therapeutic strategies for fibrosing disease. Myofibroblasts, contractile cells that deposit pathological extracellular matrix, were first believed to derive from a specialized perivascular cell known as the hepatic stellate cell when studied in the liver. In health these cells store retinoic acid in intracellular vesicles and cultured stellate cells possess all of the hallmarks of myofibroblasts in vitro.1 In other organ systems, similar perivascular cells have been postulated to be the source of myofibroblasts, but have been hard to define.2,3 Mesoderm-derived cells, when cultured in vitro, differentiate into cells with hallmarks of myofibroblasts, including most notably mesenchymal stem cells from bone marrow, as well as mesangial cells of the kidney, and cultured monocyte-derived macrophages.4–6 Whether mesoderm-derived pericytes (also called perivascular fibroblasts) give rise to kidney myofibroblasts remains controversial, partly because primary epithelial cells when cultured in vitro can be induced to express some genes that are also expressed in myofibroblasts.7–9 During carcinogenesis phenotypic alterations termed epithelial-to-mesenchymal transition (EMT) have been well characterized and promote cell migration, invasion, and metastasis.10 Further, a recent report suggests that other terminally differentiated cells such as endothelial cells can develop a myofibroblast phenotype in vitro and in vivo.11,12
It has been postulated that during kidney injury in vivo, epithelial cells undergo a phenotypic transition or can transdifferentiate into interstitial myofibroblasts by this same process of EMT.13,14 Subsequent studies both in vivo and in vitro support this hypothesis.15,16 The implication from these observations is that if the molecular mechanisms by which epithelial cells traverse the basement membrane and differentiate into myofibroblasts can be understood, novel antifibrotic strategies will be identified.
Epithelial cells are known to respond to injury in several ways. They undergo morphological changes, lose polarity, acquire stress fibers, and migrate along the basement membrane.17 They up-regulate inflammatory genes and genes that enhance their ability to survive in a hostile environment.18,19 In addition, they express some genes shared by embryonic mesenchymal cells transitioning to epithelium during nephrogenesis.20–22 Thus it has been suggested that in response to injury epithelial cells undergo EMT, recapitulating primitive mesenchymal cells of the intermediate mesoderm.9 This, however, is misleading since intermediate mesoderm cells do not express inflammatory and cell-survival genes that injured adult epithelial cells up-regulate, and expression of a limited number of genes shared by embryonic mesenchyme such as α smooth muscle actin (SMA), by itself, does not define injured epithelial cells as mesenchymal.23–25 Neoplastic epithelial cells have the capacity to metastasize, share some characteristics with myofibroblasts, and express or down-regulate key regulators of metastasis such as mts1 (S100A4 or FSP-1), Twist, Snail, and β-catenin, genes whose expression can also be activated in cultured epithelial cells.26–28 Proponents of the hypothesis that myofibroblasts in inflammation and scarring derive from epithelial cells have drawn on these observations to extend the term EMT to mean epithelial-to-myofibroblast transition.
Interstitial myofibroblasts are the principle source of interstitial collagens, including fibrillar collagens I and III. They are widely held to be the primary cell in the injured kidney that lays down the interstitial matrix that becomes fibrotic (For review see29). Many myofibroblasts express the actin fiber, αSMA that correlates with contractile and activated morphology, and recent studies confirmed that in the fibrotic kidney more than 80% of these produce fibrillary collagen.30 Although this is not specific to interstitial myofibroblasts (αSMA is also expressed by vascular smooth muscle cells), αSMA has long been used as a marker of myofibroblasts.
Although it is widely accepted that primary epithelial cells cultured in vitro up-regulate genes that result in a myofibroblast phenotype,9,25 and generate fibrillar collagens, the evidence that this occurs in vivo is less well-established. There are some published examples of epithelial cells transgressing intact or disrupted basement membrane or cells co-expressing established epithelial and fibroblast markers in vivo,31–33 but histological snapshots do not prove a lineage relationship, and cells may express a variety of antigens during injury. In our own extensive studies of injured epithelial cells in kidney repair, we concluded that non-epithelial cells do not migrate from interstitium into the tubule.34 Similarly, we have never observed a cell outside of the confines of the epithelial basement membrane that was positive for markers of epithelial injury. Explanations for a failure to make these observations in fixed tissues include the hypothesis that a cell exiting the confines of the basement membrane rapidly loses epithelial markers and only subsequently gains myofibroblast markers.35 However, in vitro, epithelial cells can express both fibroblast markers and epithelial markers simultaneously.36
Because efforts to design new antifibrotic therapies require a rigorous understanding of the cellular origin of myofibroblasts in vivo, we have performed lineage analysis of both renal epithelial cells and interstitial stromal cells during fibrosis in vivo. Transgenic or knock-in mice with lineage-restricted expression of bacterial Cre recombinase were used for genetic tracking of three cell populations. The HoxB7-Cre driver is expressed exclusively in the mesonephric duct and its derivatives, resulting in labeling of collecting duct epithelium and ureteral epithelium of adult kidney.37 In the Six2-Cre transgenic mouse, expression of Cre occurs in cap-mesenchyme and labels all non-ureteric, bud-derived, nephron epithelia, including podocytes, proximal tubule, loop of Henle, and connecting segment, but it does not label any interstitial cell population.34,38 FoxD1 is a well characterized marker of renal stromal cells, but not epithelia, during development, and we used FoxD1-Cre knock-in mice to genetically label renal stroma.39 We crossed these three Cre drivers against two different reporter lines to permanently and heritably label all epithelial cells of the entire nephron in adult mouse kidney or all stromal cells.34,38 We demonstrate that, contrary to the prevailing model, kidney epithelial cells do not become myofibroblasts in vivo during fibrotic disease. Rather, we show by genetic tracing that myofibroblasts derive from interstitial pericytes/perivascular fibroblasts.
Materials and Methods
Generation of Mice with Genetically Labeled Renal Epithelia and Stroma
The Six2-GC and HoxB7-Cre mice are described.38 FoxD1-GFP-Cre (FoxD1-GC) and FoxD1-GFP-Cre-ER(t2) (FoxD1-GCE) knock-in mice were generated using standard techniques. Full details of lines, their generation and use in identifying kidney lineage will be documented elsewhere (A.K. and A.P.M., unpublished data). Z/Red, NLSCre-Red, R26-LacZ mice and R26R mice were obtained from The Jackson Laboratory (Bar Harbor, ME). The studies were performed on a mixed background consisting of 129/B6. FoxD1-GCE+; R26R or FoxD1-GCE−; R26R pregnant females were given 6 mg of tamoxifen, i.p. in corn oil at embryonic day (e)10.5 or corn oil alone.
Fibrosis Models in Mice
All surgeries were performed according to protocols overseen by Animal Resources and Comparative Medicine at Harvard University. The complete unilateral ureteric obstruction (UUO) procedure is described elsewhere.30 Unilateral ischemia-reperfusion injury was performed as described, except mice underwent 35 minutes of clamping instead of 25 minutes.40 In brief, ureteral obstruction was performed through a left flank incision under general anesthesia. The ureter was identified and tied at the level of the lower pole of the kidney with two separate silk ties. Ischemia reperfusion injury to the left kidney only was performed through a similar incision under general anesthesia, at core body temperature of 36.7–37.2°C. A surgical clamp was placed over the artery and vein for 35 minutes and then removed. Return of blood flow was confirmed.
Tissue Preparation and Histology
Mice were anesthetized, sacrificed, and immediately perfused via the left ventricle with ice-cold PBS for 2 minutes. Kidneys were hemi-sectioned and portions were snap frozen in liquid nitrogen. Other kidneys were fixed in 10% neutral buffered formalin at 4°C for 12 hours, processed, embedded in paraffin wax, sectioned, and stained with periodic acid-Schiff, picrosirius red or Jones’ Silver stain using standard procedures. Quantitative morphometry was performed as previously reported.41 Tubule injury was assessed by a semiquantitative scoring system as previously described.40 In brief injury was graded in periodic acid Schiff-stained sections on a scale from 0 to 3 (0, no changes; 1, changes affecting <25%; 2, changes affecting 25% to 50%; 3, changes affecting 50% to 100% of the section). To preserve native red fluorescent protein (RFP) epifluorescence which we found was quenched by prolonged fixation, some kidneys were fixed in PLP fixative (4% paraformaldehyde [PFA], 75 mmol/L l-lysine, 10 mmol/L sodium periodate) for 1 hour or 2 hours at 4°C, cryopreserved in 18% sucrose, and frozen, and 5-μm cryosections were prepared for direct visualization or immunofluorescence as described.40 Other kidneys were perfusion fixed with 4% PFA for 2 minutes, followed by sagittal hemisection and further fixation for 1 hour in 4% PFA, then cryopreserved in 18% sucrose before sectioning. Bromodeoxyuridine labeling was performed as previously described.42 Primary antibodies against the following proteins were used: DsRed (Rabbit, 1:500, Cat. No. PM005; MBL, Nagoya, Japan), E-cadherin (Rabbit, 1:200, Cat. No. 610404, BD Biosciences, San Jose, CA), αSMA (mouse fluorescein isothiocyanate-conjugated, 1:400, #F3777, mouse Cy3-conjugated, 1:400 #C6198, Sigma, St. Louis MO), platelet-derived growth factor receptor (PDGFR)β (1:500, Gift William Stallcup, Burnham Inst.), S100A4 (1:400 DAKO A5114 and 1:400 Abcam Ab27957 [both antibodies were tested and gave comparable results]). Primary antibodies against β-galactosidase (β-gal) were tested (Sigma #B0271, #G6282; Cappel #55976, Promega #Z378A) to detect the LacZ gene product, β-gal, by immunofluorescence. All four antibodies against β-gal were tested in frozen and formalin-fixed, paraffin-embedded sections but none of these gave a reliable signal by immunofluorescence or immunostain in adult kidneys of the R26-LacZ positive control mouse or Six2GC; R26R mice. It may be that β-gal enzymatic activity is the most reliable readout of LacZ expression in the fibrotic adult kidney. Affinity purified secondary antibodies were obtained from DAKO, Carpinteria CA, or Jackson Immunoresearch. All labeling with directly conjugated anti-αSMA antibodies was performed in 10% mouse serum to block nonspecific binding. In examples involving double-labeling with anti-αSMA antibodies, labeling with anti-DsRed antibodies followed by donkey anti-rabbit-Cy3 secondary was performed initially in 10% donkey serum, then after washing, directly conjugated anti-αSMA antibodies were applied in 10% mouse serum as previously described.30 Sections were mounted in Vectashield or Gel Mount or antifade Gold containing 4′, 6-diamino-2-phenylindole (VectorLabs, Burlingame CA). Epifluorescent images were taken with a Nikon TE2000 microscope, CoolSnap camera (Roper Scientific, Germany) and processed using IP lab software (BD Biosciences, San Jose, CA). Antibody enhancement of RFP signal was performed as previously described and gave identical results in vivo to native RFP fluorescence (data not shown).34 Confocal images were generated using a Nikon C1 D-Eclipse confocal microscope as previously described.40 Projection images were generated from 10 Z-stack images that were acquired at 0.1-μm steps. To allow for comparison between sections, all confocal settings including exposure time were kept constant between sections.
LacZ activity was measured by standard 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-gal) staining protocol on PFA-fixed frozen kidney sections.40 Care was taken to perform X-gal staining at neutral pH, and was performed for 16 hours at 37°C. After washing, sections were either counterstained with nuclear fast red or were postfixed (4% PFA 2 minutes), then were immunolabeled as detailed above before aqueous mounting. Morphometry of LacZ stained area in tissues sections was assessed using Fovea Pro Software as previously described.40
Tubule Cell Culture
Kidney proximal tubule cells were cultured exactly as described.43 After 7 days of culture the epithelial cells were confluent, then treated with transforming growth factor (TGF)β1 (10 ng/ml) or no additional treatment. Four days later, cells were fixed with 4% PFA 5 minutes, washed ×3 with PBS, permeabilized with 0.1% TX100, 5 minutes, washed ×3 with PBS, then labeled with antibodies as described in above.
Results
Our Model of Kidney Fibrosis Results in Epithelial Injury and Fibrosis
To study the process of EMT in the kidney we used a well-established mouse model of fibrosis, UUO. The model results in tubule cell injury due to obstruction of flow through the nephron and back-pressure.44 We studied control kidneys and two time points during the progression of the disease model, day 10 and day 14 since the population of myofibroblasts and fibrosis are well established and there is evidence of disruption of the tubular basement membrane, sites proposed to be a source of migrating epithelial cells (Supplementary Figure S1, A–C at http://ajp.amjpathol.org). The extent of renal injury and fibrosis achieved in this model was comparable with that seen in other studies.13,44
Six2-GC; R26R and HoxB7-Cre; R26R Genetically Label All Nephron Epithelia
We crossed reporter mice with a transgenic Cre driver line that expresses a fusion protein consisting of enhanced green fluorescent protein and bacterial Cre recombinase under regulation of the Six2 promoter (Six2-GC).34,38 Six2 is a transcription factor expressed transiently during development in cells of the metanephric mesenchyme fated to become renal epithelia, but not in cells fated to become kidney interstitial cells. Its renal expression is restricted solely to the cap mesenchyme during nephrogenesis and is not expressed in the adult kidney in health or induced after injury.34
Six2-GC+ mice were bred with ROSA26 reporter mice (Gtrosa26tm1Sor, hereafter referred to as R26R) (Figure 1A). As expected, kidneys from bigenic Six2-GC; R26R mice exhibited strong epithelial LacZ expression, as assessed by X-gal staining, from the glomerulus, where podocytes are labeled, through the entire nephron until the collecting duct (Figure 1B). Since collecting ducts of the adult kidney derive from the ureteric bud, this portion of the adult nephron is not labeled in Six2-GC; R26R kidneys. To label collecting duct epithelia, we crossed the HoxB7-Cre transgenic mouse, which directs expression of Cre recombinase in ureteric bud, during development to the R26R.37 HoxB7-Cre; R26R bigenic mice exhibited strong LacZ staining solely in collecting ducts (Figure 1). Negative and positive kidneys included control R26R mice that did not carry either the Six2-GC or HoxB7-Cre transgene and the ROSA26-LacZ (R26-LacZ) mice, respectively.
Figure 1.
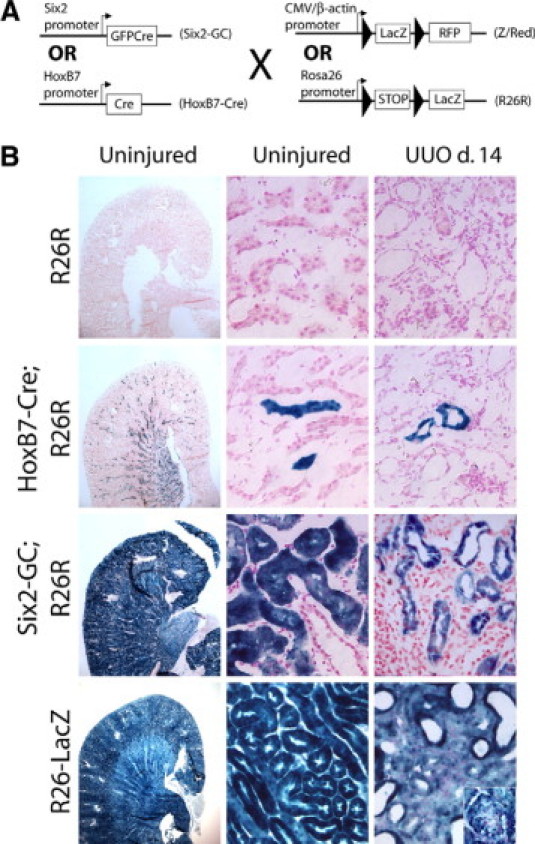
Control and day 14 unilateral ureteral obstruction kidneys from Six2-GC; R26R and HoxB7-Cre; R26R mice show no evidence for LacZ genetically labeled epithelial cells migrating into the interstitium. A: Bigenic Six2-GC; Z/Red mice activate GFPCre expression in renal progenitor cells present in metanephric mesenchyme as they differentiate into epithelial cells. Differentiation results in removal of the LoxP-LacZ-STOP-LoxP sequence in epithelial cells, leading to permanent, heritable expression of RFP in epithelial cells. HoxB7-Cre; Z/Red mice activate Cre in the ureteric bud during embryonic development. Cre activation removes the LoxP-LacZ-STOP-LoxP sequence in collecting duct cells, leading to permanent, heritable expression of RFP in collecting duct epithelial cells. For bigenic Six2-GC; R26R mice, or HoxB7-Cre; R26R mice, removal of the LoxP-STOP-LoxP sequence leads to permanent, heritable expression of the LacZ gene in epithelial cells. B: Representative light microscopy images of X-gal stain indicative of LacZ expression in control or day 14 UUO fibrotic kidneys of R26R mice, HoxB7-Cre; R26R mice Six2-GC; R26R mice and R26-LacZ positive control mice. Low power view (left panels) of whole kidneys stained identically with X gal solution indicates the high level of recombination and the high signal-to-noise ratio generated by LacZ staining. High power view (center panels) of control kidneys shows specificity of labeling and lack of interstitial cells deriving from epithelial cells. Note all interstitial and vascular cells as well as epithelial cells label with blue stain in the R26-LacZ kidneys (inset shows blue-stained glomerulus). High power view (right panels) of day 14 UUO kidneys. Note no staining in the interstitium of HoxB7Cre; R26R or Six2-GC; R26R mice but homogenous blue stain throughout the interstitium of positive control R26-LacZ kidneys.
Kidney Epithelial Cells Do Not Become Myofibroblasts in Vivo
Six2GC; R26R bigenic, HoxB7-Cre; R26R bigenic, R26-LacZ, or control R26R mice were analyzed at day 10 or 14 after UUO or sham surgery. Sections were stained for LacZ activity alone (Figure 1, right column) or Xgal-stained and co-labeled with directly conjugated antibodies against αSMA (see Supplementary Figure S2 at http://ajp.amjpathol.org). We viewed 10 high power fields per sagittal section, from three different levels in the cortex from three different kidneys. We did not identify any X-gal-positive cells in the interstitium of HoxB7-Cre; R26R or Six2-GC; R26R mice. Importantly, all interstitial cells were strongly positive for LacZ activity in the positive control R26-LacZ mice, indicating that the fate marker can be expressed in the myofibroblast lineage.
To verify that interstitial myofibroblasts were generated in our injury model, kidney sections were labeled with directly conjugated antibodies against the myofibroblast marker αSMA after X-gal staining. Despite robust interstitial αSMA immunoreactivity, we did not identify a single αSMA-positive cell that stained for β-gal in normal kidneys or kidneys from day 10 or day 14 following UUO surgery from either the Six2-GC; R26R or HoxB7-Cre; R26R mice. Control R26R mice showed no evidence of β-gal staining under the same conditions whereas in R26-LacZ mice αSMA+ cells localized with β-gal staining, confirming that the LacZ fate marker is capable of being expressed in the myofibroblast lineage (see Supplementary Figure S2, A−C at http://ajp.amjpathol.org).
Six2-GC; Z/Red and HoxB7-Cre; Z/Red Mice Allow High Resolution Cell Tracking after UUO
Because we were unable to define conditions that allowed for unambiguous immunodetection of LacZ using four different anti-LacZ antibodies (See Materials and Methods, data not shown), we repeated our analysis in reporter mice using a fluorescent protein fate marker. The ACTB-Bgeo,-DsRed.MST (hereafter referred to as Z/Red) reporter line expresses dsRed.MST (hereafter referred to as red fluorescent protein RFP) after Cre-mediated recombination (Figures 1–3 and Supplementary Figure S3 at http://ajp.amjpathol.org). Z/Red mice crossed with Six2-GC or HoxB7-Cre mice strongly expressed RFP in either metanephric mesenchyme-derived nephron epithelia or collecting duct, respectively (Figures 2 and 3).34,38 The proportion of noncollecting duct epithelial tubule cells expressing detectable levels of RFP during fibrosis in control kidneys was >95% and was not changed following ureteral obstruction (data not shown).
Figure 2.
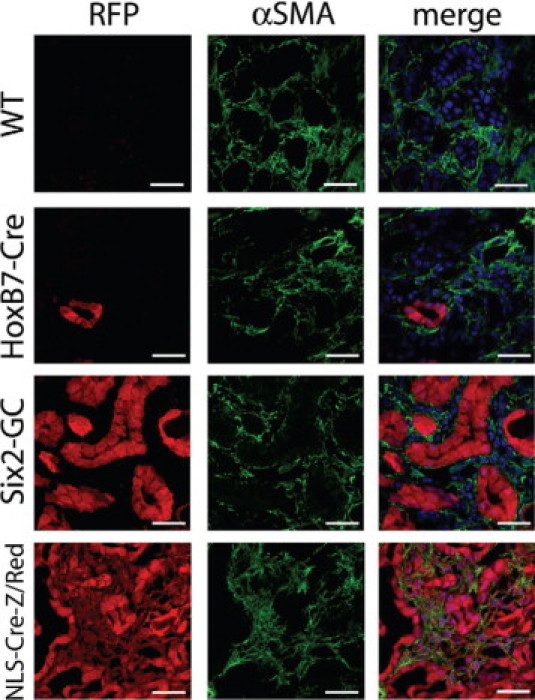
Day 10 unilateral ureteral obstruction kidneys from Six2-GC; Z/Red and HoxB7-Cre; Z/Red mice show no evidence for RFP+ genetically labeled epithelial cells, becoming interstitial cells expressing αSMA+. Representative confocal images of native RFP epifluorescence (left panels) or fluorescein isothiocyanate-conjugated antibody detection of αSMA (green, center panels) of day 10 UUO fibrotic kidneys of Z/Red mice, HoxB7-Cre; Z/Red mice Six2-GC; Z/Red mice and NLS-Cre-Z/Red mice. Composite images with nuclei in blue are seen (right panels). Note that all interstitial cells expressing αSMA co-express RFP in the NLS-Cre-Z/Red positive control mice, but no αSMA+ cells co-express RFP in the other mice. Scale bar = 25 μm.
Figure 3.
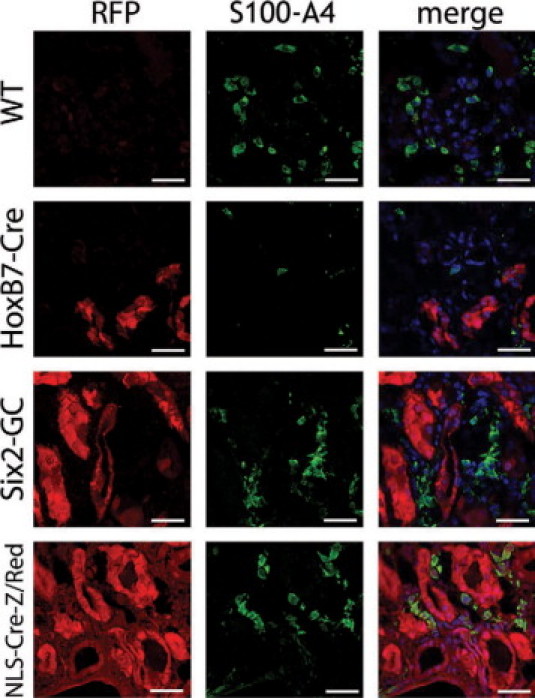
Day 10 unilateral ureteral obstruction kidneys from Six2-GC; Z/Red and HoxB7-Cre; Z/Red mice show no evidence for RFP+ genetically labeled epithelial cells, becoming interstitial cells or acquiring the interstitial cell marker S100A4. Representative confocal images of native RFP epifluorescence (left panels) or fluorescein isothiocyanate-conjugated antibody detection of S100A4 (green, center panels) of day 10 UUO fibrotic kidneys of Z/Red mice, HoxB7-Cre; Z/Red mice Six2-GC; Z/Red mice and NLS-Cre-Z/Red mice. Composite images with nuclei in blue are seen (right panels). Note that all interstitial cells expressing S100A4 co-express RFP in the NLS-Cre-Z/Red positive control mice, but no S100A4+ cells co-express RFP in the other mice. Scale bar = 25 μm.
As a positive control, we used a Z/Red transgenic mouse line with deletion of the floxed stop in the germ line (hereafter referred to as NLSCre-Z/Red).45,46 NLSCre-Z/Red mice harbor the same transgene insertion site as the Z/Red mice, ruling out epigenetic differences in transgene expression between the two lines. In kidneys of NLSCre-Z/Red mice, all epithelial, glomerular, and interstitial cells expressed RFP (Figures 2 and 3, and Supplementary Figure S3 at http://ajp.amjpathol.org) and in fibrotic kidney all cells including interstitial myofibroblasts express RFP (Figures 2 and 3, and Supplementary Figure S3 at http://ajp.amjpathol.org), confirming that the genetic lineage marker RFP can be expressed and easily detected in the interstitial compartment.
High Resolution Analysis of Epithelial Fate in Six2GC; Z/Red and HoxB7-Cre; Z/Red Kidneys
We examined 10 complete kidney sagittal sections from both Six2GC; Z/Red and HoxB7-Cre; Z/Red bigenic mice (n = 3 each genotype) from different tissue levels and from control or 10- and 14-day obstructed kidneys (Figures 2 and 3, and Supplementary Figure S3 at http://ajp.amjpathol.org). In negative control, uninjured Z/Red kidneys no interstitial cells were positive for RFP and only vascular smooth muscle cells were positive for αSMA, and these cells did not co-express RFP (not shown). In HoxB7-Cre; Z/Red mice, collecting ducts strongly expressed RFP, but no other tubule segments or interstitial cells were labeled. In day 10 and day 14 UUO kidneys the collecting ducts remained robustly RFP-positive, but no RFP-positive interstitial cells were identified, despite clear immunodetection of both αSMA or S100A4 positive interstitial cells (Figures 2 and 3, and Supplementary Figure S3 at http://ajp.amjpathol.org).
Similar results were obtained from the Six2-GC; Z/Red mice, where proximal, distal and Loop of Henle epithelial cells of healthy mice strongly expressed RFP without any interstitial RFP expression. After UUO, the epithelial cells remained robustly RFP positive but no RFP-positive interstitial cells were identified (Figures 2 and 3, and Supplementary Figure S3 at http://ajp.amjpathol.org). Although many interstitial cells expressed the myofibroblast marker αSMA, and S100A4, none of these cells were epithelial cells that expressed RFP (Figures 2 and 3).
In the positive control, NLS-Cre-Z/Red mice all kidney cells expressed RFP at baseline (not shown). In day-10 and day-14 UUO kidneys all cells remained robustly RFP-positive by epifluorescence confocal microscopy. RFP-positive interstitial cells co-expressed the myofibroblast markers αSMA and S100A4, indicating that the RFP transgene is capable of being expressed in these cell types (Figures 2 and 3). To confirm that we had not missed any interstitial cells in Six2-GC; Z/Red or HoxB7-Cre; Z/Red expressing RFP at levels that we could not detect by epifluorescence, we labeled sections from all experimental mice with antibodies specific for RFP and detected these with Cy3 conjugated antibodies. There was very high specificity using these antibodies, but we still did not detect any interstitial cells expressing RFP in the negative control mice or Six2-GC; Z/Red or HoxB7-Cre; Z/Red mice and in the NLS-Cre-Z/Red mice the antibody enhancement did not lead to detection of any additional interstitial cells (data not shown). Thus in these studies despite renal fibrosis with dense accumulation of αSMA+ cells and S100A4+ cells in the interstitium none of these cells derived from epithelial cells of the nephron. Throughout these studies we did not detect α-SMA or S100A4 expression in injured epithelial cells.
Cultured Six2GC+; Z/Red Kidney Epithelial Cells Up-Regulate αSMA and S100A4 in Vitro
We have shown in vivo that injured epithelial cells do not migrate into the interstitium and do not express the proteins αSMA and S100A4, despite severe injury. Many reports have documented up-regulation of these two genes in epithelial cells cultured in vitro in the presence of cytokines such as TGFβ1.25 To determine whether primary kidney epithelial cells from Six2GC; Z/Red mice, cultured ex vivo, had the capacity to express these proteins in vitro, which some investigators have used to define an EMT process, we cultured mouse cortical tubule cells using established methods that maintain tubule epithelial characteristics.43 The epithelial origin of cells within confluent monolayers was confirmed by examining RFP epifluorescence. Cells were cultured in culture medium alone, with TGFβ1, or with FCS for 4 days, then fixed and assessed for expression of E-cadherin, αSMA, and S100A4 (Figure 4, A–H). Primary cultured cells did not express αSMA or S100A4, but, in response to TGFβ1 treatment, both proteins were readily detected. A proportion of cells also lost expression of E-cadherin (Figure 4, G and H).
Figure 4.
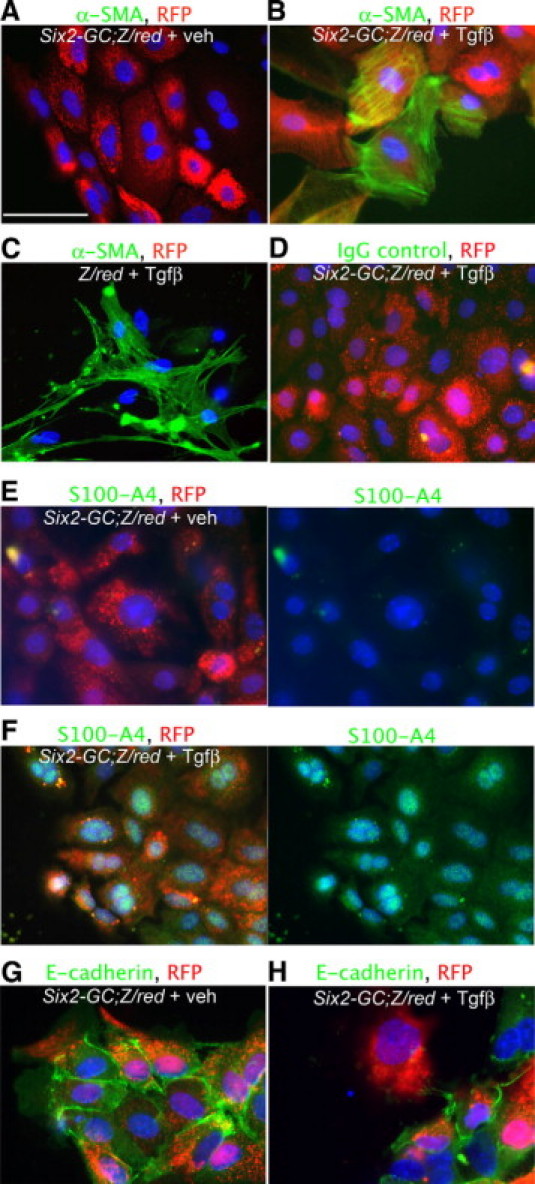
Cultured tubule cells from kidneys of Six2-GC; Z/Red mice, co-express αSMA and S100A4 cultured in the presence of transforming growth factor-β1. A: Day 10 cultured Six2-GC; Z/Red tubule cells fluoresce red from RFP expression, but do not label for αSMA. Scale bar = 50 μm. B: Day 10 cultured Six2-GC; Z/Red tubule cells grown in the presence of TGFβ1 for 4 days fluoresce red with RFP and co-label for αSMA (green). C: Day 10 cultured Z/Red tubule cells grown in the presence of TGFβ1 for 4 days show no evidence of RFP expression but co-label with αSMA (green). D: Day 10 cultured Six2-GC; Z/Red tubule cells grown in the presence of TGF1 for 4 days fluoresce red with RFP show no evidence of non-specific binding with irrelevant IgG control primary antibodies. E: Split panel images (right shows green channel only) Day 10 cultured Six2-GC; Z/Red tubule cells fluoresce red with RFP and do not label for S100A4. F: Split panel images showing Day 10 cultured Six2-GC; Z/Red tubule cells grown in the presence of TGFβ1 for 4 days fluoresce red with RFP and co-label for S100A4 (green) in the cytoplasm and nucleus. G: Day 10 cultured Six2-GC; Z/Red tubule cells fluoresce red with RFP and label for E-cadherin (green). H: Day 10 cultured Six2-GC; Z/Red tubule cells grown in the presence of TGFβ1 for 4 days also fluoresce red with RFP and co-label for E cadherin (green), however some cells lose E-cadherin expression.
Analysis of Pericyte Fate in FoxD1-GC; R26R and FoxD1-GCE; R26R Adult Mouse Kidneys Following Ureteral Obstruction
We have identified PDGFRβ positive interstitial pericytes as candidate interstitial cells that can differentiate into myofibroblasts.30,47 Since we found no contribution to the myofibroblast pool from epithelium, we performed lineage analysis to test the hypothesis that pericytes/perivascular fibroblasts are the primary source of interstitial myofibroblasts in the UUO fibrosis model. We genetically labeled pericytes with a Cre driver line in which a GFP-Cre (GC) fusion protein is expressed under control of the FoxD1 locus (hereafter referred to as FoxD1-GC) (Figure 5A). FoxD1 is a forkhead/winged helix transcription factor expressed during kidney development in metanephric mesenchyme only in cells fated to become stromal cells of the kidney, including mesangial cells, vascular smooth muscle cells and pericytes, but not endothelial cells.48 In FoxD1-GC; R26R adult mouse kidneys, mesangial cells, kidney vascular smooth muscle cells and many interstitial cells were identified by LacZ stain (Figure 5B). These interstitial cells expressed the pericyte marker PDGFRβ and this marker was restricted to cells that had expressed FoxD1 developmentally (Figure 5, C–D), in keeping with our previous observations,30,47 and indicating that these cells are both developmentally homogeneous and that all adult pericytes derive from FoxD1+ progenitors. FoxD1 derived cells also expressed CD73, a marker which has been used to describe perivascular fibroblast cells elsewhere.49 However since there was no αSMA expression in interstitial cells of normal kidney, none of the LacZ labeled interstitial cells expressed αSMA (not shown). Following 14 days of UUO injury to adult kidneys of FoxD1-GC; R26R mice there was marked expansion LacZ stained cells exclusively in the interstitium of the kidney (Figure 5E), and these cells labeled almost exclusively with the myofibroblast marker αSMA (Figure 5, F–G). Furthermore αSMA+ interstitial cells were almost exclusively positive for LacZ indicating that there was no significant population of cells becoming myofibroblasts that had not previously expressed FoxD1. Although FoxD1 was not detected in normal adult kidney, following UUO injury in adult kidney FoxD1 transcripts were detected by PCR (Figure 5H). It was therefore not possible to test definitively whether myofibroblasts derive from pericytes using FoxD1-GC; R26R mice since injury-induced activation of FoxD1 in myofibroblasts would label myofibroblasts with the heritable marker at the time of injury. To circumvent this problem, we developed a knock-in mouse with GFP-CreER(T2) protein expressed under control of the FoxD1 locus (FoxD1-GCE). CreER(T2) is a tamoxifen-inducible form of Cre recombinase. To fate map pericytes during fibrosis we pulsed FoxD1-GCE; R26R litters (Figure 6A) at embryonic day 10.5 post conception (e10.5) with a single dose of tamoxifen given to the mother (6 mg, i.p.).
Figure 5.
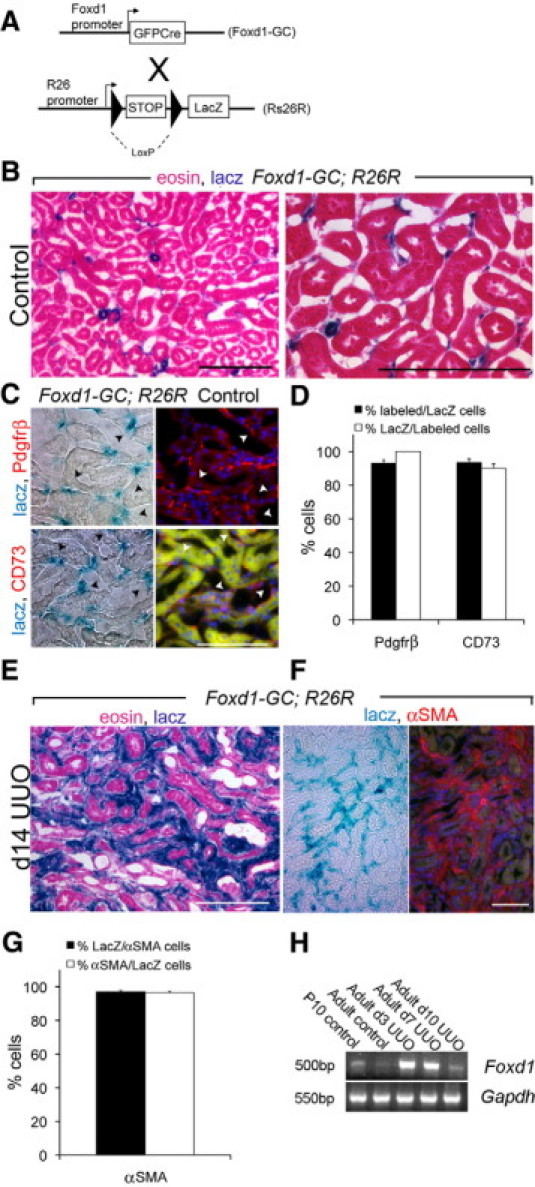
Characterization of FoxD1 metanephric mesenchyme-derived cells in the adult kidney of FoxD1-GC; R26R mice. A: Bigenic FoxD1-GC; R26R mice activate GFPCre expression in renal progenitor cells that are fated to remain as stromal cells, removing the LoxP-STOP-LoxP sequence in stromal cells, leading to permanent, heritable expression of LacZ in stromal cells. B: Low power and higher power views of adult kidney cortex showing LacZ+, blue stained arteriolar smooth muscle and many perivascular cells surrounding peritubular capillaries. C: Images (split panel light and fluorescence microscopy) of normal adult kidney showing co-expression of LacZ with Pdgfrβ or CD73. D: Graph showing the proportion of Prgfrβ+, or CD73+ labeled interstitial cells co-expressing LacZ and the proportion of LacZ interstitial cells co-expressing either Pdgfrβ or CD73. E: Low-magnification image of FoxD1-GC; R26R mouse kidney following 14 days of ureteral obstruction. Note extensive expansion of interstitial LacZ+, blue-stained cells, but no staining of the epithelial compartment. F: Low magnification split light and fluorescence image showing co-expression of LacZ+ blue cells with αSMA (red) after UUO day 14. G: Graph showing quantification of the proportion of αSMA cells that co-express LacZ and the proportion of LacZ cells that co-express αSMA after kidney UUO day 14. H: RT-PCR for endogenous Foxd1 transcripts (40 cycles) compared with Gapdh (32 cycles), in day 10 pup kidney, normal adult kidney and adult kidney at times after UUO injury. Note absence of Foxd1 in normal adult kidney but gene expression following injury (n = 5/group). Scale bar = 100 μm.
Figure 6.
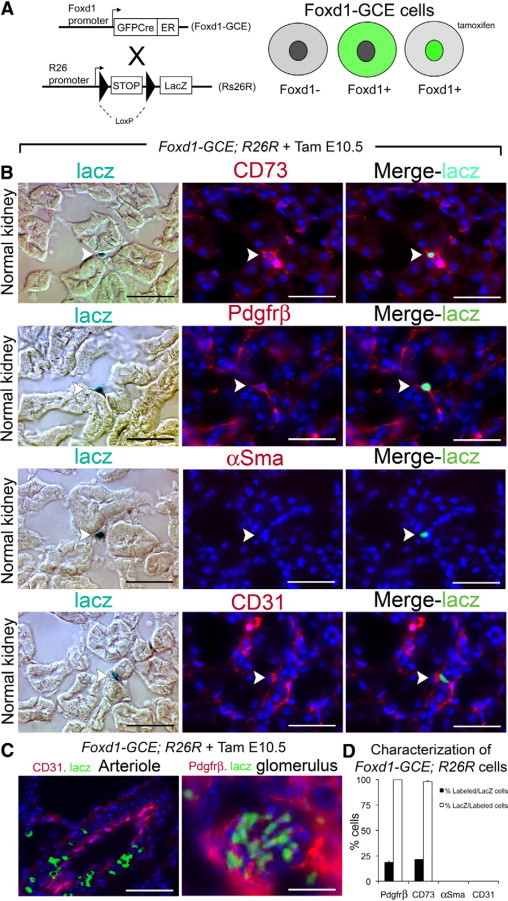
Characterization of FoxD1 metanephric mesenchyme derived interstitial pericytes in the adult kidney of FoxD1-GCE; R26R mice. A: Bigenic FoxD1-GCE; R26R mice express GFP CreERT2 fusion protein (green) under control of the endogenous FoxD1 regulatory sequences in metanephric mesenchyme fated to differentiate into stromal cells. In the absence of tamoxifen, the fusion protein is excluded from the nucleus and does not mediate recombination between loxP sites. During a tamoxifen (Tam) pulse, GFPCreERT2 translocates to the nucleus activating fate marker expression, leading to permanent, heritable expression of LacZ in a cohort kidney stromal cells. B: Light and fluorescence photomicrographs showing LacZ expressing cells in the normal kidney of FoxD1-GCE; R26R mice treated with Tam on E10.5 only. Note positive cells (arrows) are exclusively in the interstitium and express the markers CD73, Pdgfrβ, but do not express αSMA or CD31. C: FoxD1 fated mesenchyme cells also become kidney arteriolar VSMCs and glomerular mesangial cells (left and center panels). Scale bar = 50 μm. D: Graph showing the proportion of Prgfrβ+, CD73+, αSma+, CD31+ labeled interstitial cells co-expressing LacZ and the proportion of LacZ interstitial cells co-expressing Pdgfrβ, CD73, αSma, CD31 in normal kidneys of FoxD1-GCE; Rs26R mice.
Kidneys from adult FoxD1-GCE; R26R mice that had received tamoxifen during nephrogenesis had blue-stained (LacZ+) cells in the interstitium without any LacZ+ cells in tubular epithelium. These interstitial cells were CD73+, PDGFRβ+, αSMA−, and CD31− identifying them as pericytes/perivascular fibroblasts (Figure 6, B and D),29,30 similar to the cells identified in FoxD1-GC; R26R mice. We confirmed that these LacZ+ cells were not resident macrophages, as they were F4/80 negative in kidney and no cells were labeled in spleen or peripheral blood leukocytes (not shown). In medium sized arteries of uninjured kidneys αSMA+ smooth muscle was LacZ+, and mesangium of glomeruli was also LacZ+ (Figure 6C). The single pulse of tamoxifen labeled approximately 20% of pericytes as determined by the proportion of LacZ+, PDGFRβ+ double-positive cells, compared with the total number of PDGFRβ+ perivascular cells. LacZ+, CD73+ Alternatively, double-position cells as compared with total CD73+ interstitial cells (Figure 6, B and D). In an attempt to increase the number of pericytes that were labeled, additional doses of tamoxifen were given to pregnant mice but this triggered fetal demise. Since nephrogenesis proceeds from e10.5 onward but tamoxifen lasts only about 24 hours, incomplete labeling of pericytes was due to submaximal tamoxifen administration and was unavoidable.
To definitively establish whether pericytes undergo proliferative expansion and differentiate into myofibroblasts during fibrosis, we subjected FoxD1-GCE; R26R mice to UUO that had received tamoxifen or vehicle at e10.5. At day 4 and day 10 following UUO surgery there was a marked increase in perivascular cells labeling with LacZ (Figure 7A–D). FoxD1-GCE; R26R kidneys that received vehicle and not tamoxifen had no LacZ expression either in uninjured or fibrotic kidneys (Figure 7, A and D). In tamoxifen-treated mice, all of the LacZ+ cells had acquired expression of αSMA at day 10, and all continued to express PDGFRβ+ (Figure 7, B, C, E, and F), indicating that the LacZ+ cells had differentiated into myofibroblasts and that they derived from pericytes. None of the LacZ+ stained cells expressed F4/80 (data not shown). To assess whether FoxD1-derived pericytes alone accounted for the appearance of myofibroblasts we determined whether there had been any change in the ratio of LacZ labeled cells to total pericyte/myofibroblasts during progression of UUO disease (Figure 7F). To do this we used interstitial cell PDGFRβ expression, since all pericytes and all myofibroblasts (Figure 5D, co-label of day 10 UUO kidney indicates 100% αSMA+ cells co-express PDGFRβ [not shown] and previous studies30) consistently express PDGFRβ. In FoxD1-GCE; R26R mice treated with tamoxifen at e10.5, the proportion of PDGFRβ+ cells that co-expressed LacZ was consistently about ∼20% (Figure 7F) in both normal and diseased kidney despite the tremendous expansion of LacZ cells (Figure 7D). The finding of only ∼20% of cells labeled was expected since only this proportion of original pericytes was labeled by tamoxifen injection during nephrogenesis. The absence of dilution of the proportion of PDGFRβ+ interstitial cells that expressed the fate marker LacZ, before and after disease indicates, however, that only cells deriving from FoxD1 precursors contribute to the myofibroblast pool and therefore the predominant cell type responsible for the accumulation of interstitial myofibroblasts is the pericyte/perivascular fibroblast.
Figure 7.
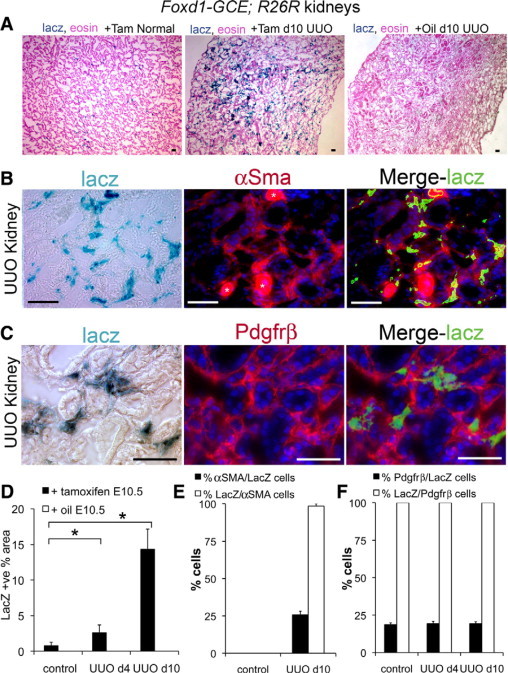
FoxD1-derived kidney pericytes/perivascular fibroblasts are myofibroblast progenitors following unilateral ureteral obstruction. A: Low magnification images of LacZ stained normal (left) and day 10 UUO (center) kidney FoxD1-GCE; R26R kidney treated with tamoxifen in utero on e10.5. Note marked expansion of interstitial blue stain following UUO. In day 10 UUO FoxD1-GCE; R26R kidney treated with vehicle (oil) on e10.5 there is no interstitial blue staining. (B, C) Representative split image light and fluorescence micrographs of day 10 UUO kidney structures from FoxD1-GCE; R26R mice treated on e10.5 with tamoxifen, showing anti-αSMA-Cy3 immunolabeling and anti-Pdgfrβ-Cy3 labeling. Note many αSMA+ or Pdgfrβ+ interstitial cells co-express LacZ (*denotes arteriole). D: Graph of area of LacZ staining in control (day 0) kidneys, or kidneys day 4 and day 10 following UUO from FoxD1-GCE; R26R mice treated on e10.5 with tamoxifen (n = 6/group). Note that the total area of LacZ+ cells increases markedly in the interstitium in response to UUO, but that without tamoxifen injection on e10.5 there are no LacZ cells in control of diseased kidneys. E: Graph showing the percentage of LacZ+ cells that co-express αSMAon day 10 of UUO disease and the percentage of αSMA+ cells that co-express LacZ. F: Graph showing the percentage of LacZ+ cells that co-express Pdgfrβ with time of disease and the proportion of Pdgfrβ+ cells that co-express LacZ with time of disease (n = 6/group). Scale bar = 50 μm. *P < 0.05.
Fate Mapping Epithelia and Pericytes in a Second Fibrosis Model Confirms the Pericyte and Not Epithelial Origin of Myofibroblasts
Having identified pericytes as myofibroblast precursors in the UUO fibrosis model, we next asked whether this lineage relationship was a generalized phenomenon by repeating the studies in a different fibrosis model. We performed ischemia-reperfusion injury using a 35-minute warm ischemia time (longer than the typical 25-minute interval) to induce fibrosis rather than repair. At day 15 after ischemia-reperfusion injury in Six2-GC; Z/Red mice, we observed the appearance of abundant interstitial myofibroblasts, but none of these cells expressed RFP (Supplemental Figure S4A at http://ajp.amjpathol.org). This established that epithelial cells do not traverse the basement membrane and contribute to the myofibroblast pool in the ischemia-reperfusion injury model of fibrosis, confirming the conclusions drawn using the UUO model. We next subjected FoxD1-GCE; R26R mice labeled with tamoxifen at e10.5 to the same ischemia-reperfusion injury protocol. A similar expansion of LacZ+ interstitial pericytes was seen in the injured kidney, and as expected, all of the LacZ+ cells were also PDGFRβ+ (Supplemental Figure S4, B and C at http://ajp.amjpathol.org). These observations indicate that pericyte proliferation and differentiation into interstitial myofibroblasts is a stereotyped response to fibrotic stimuli.
Discussion
Efforts to develop new antifibrotic therapies for chronic kidney diseases require an understanding of the cellular origin of myofibroblasts. Here, we present several lines of evidence against a model of fibrogenesis in which injured epithelial cells exit the tubule and become interstitial myofibroblasts in vivo. While epithelial cells cultured in vitro and exposed to cytokines for sustained periods acquire some myofibroblast markers and characteristics, we could not find evidence that epithelial cells directly contribute to the interstitial myofibroblast population in vivo, suggesting that if this does occur, it is infrequent and not a major mechanism of fibrosis in the models studied. While we have no clear explanation for these results discordant with others, we note that different strategies for transgene detection (anti-LacZ antibody versus LacZ enzymatic activity) is a prominent methodologic difference from prior studies.13 We cannot rule out a mouse strain-specific effect either. Regardless of the reasons for these different findings, we provide for the first time definitive lineage tracing evidence that interstitial pericytes/perivascular fibroblasts differentiate into myofibroblasts and undergo proliferative expansion during fibrosis. Additionally, our analysis indicates that the pericyte-derived myofibroblast population is sufficient to account for the large majority of myofibroblasts present in fibrotic kidney.
These studies included important controls and design considerations that lend weight to the conclusions. We achieved efficient genetic labeling of all of the epithelial cells of the nephron, including those derived from the ureteric bud, so we can be confident that we did not miss a small fraction of unmarked epithelial cells that might have transitioned into interstitial myofibroblasts. Importantly, we could readily detect either RFP+ or LacZ+ interstitial myofibroblasts in the respective positive control mice, so if epithelial cells from Six2GC or HoxB7Cre labeled kidneys did become interstitial myofibroblasts our methods would have identified them. Robust and direct methods to detect fate markers are critical for interpretation of genetic lineage analysis. To detect RFP and LacZ we used neither antigen retrieval nor antibody enhancement. The native fluorescence from RFP was sufficient in all tissues of the NLS-Cre-Z/Red mouse positive control and in the Six2GC; Z/Red mouse, combined with the lack of significant autofluorescence in our negative control Six2GC-; Z/Red mice indicates that no artifacts obscured the interpretation of our findings. Similarly, we used the enzymatic activity of the LacZ gene product to generate stain as a direct robust marker of LacZ expression rather than anti-LacZ immunostaining, which we have repeatedly found to be unreliable in the adult kidney (see Materials and Methods). The Xgal stain for LacZ expression in tissue also gave a high signal-to-noise ratio.
The pericyte identity of FoxD1-GC and FoxD1-GCE labeled interstitial cells was confirmed by positive expression of PDGFRβ and CD73, and the absence of expression of the myofibroblast marker αSMA, the endothelial cell marker CD31, and the macrophage marker F4/80. We verified the presence of myofibroblasts in the interstitium by co-labeling tissues with antibodies against αSMA, S100A4, or PDGFRβ.30 We labeled fixed–frozen sections without antigen retrieval, and detected very clear signal for αSMA using directly conjugated antibodies. We also detected very clear signal for S100A4 in interstitial cells. We did not identify any interstitial cells in Six2GC; R26R, Six2GC; Z/Red, HoxB7; R26R or HoxB7; Z/Red kidneys co-expressing either αSMA or S100A4 and the epithelial fate markers RFP or LacZ. In recent studies using a Collagen1α1-GFP reporter mouse, we validated that in the UUO kidney fibrosis model, the antigens αSMA and PDGFRβ are faithful markers of collagen-I producing cells (ie, myofibroblasts), from day 2 through day 14 of this UUO model. Our studies therefore comprehensively label any cells that might be called fibroblasts or myofibroblasts.30 We have used two different polyclonal antibodies against the calcium sensing metastasis associated protein S100A4, both of which gave comparable results showing expected expression in the cytoplasm and also in the nucleus and are therefore confident about the specificity of S100A4 detection.30 Nevertheless, despite the lack of epithelial cells contributing to myofibroblasts in vivo, in vitro studies using genetic labeled, RFP+, cultured primary tubule epithelial cells, from Six2GC+; Z/Red mice, activated expression of both αSMA and S100A4, confirming that primary epithelial cells are capable of expressing some mesenchymal genes in vitro, but not that they undergo a complete phenotypic conversion to a myofibroblast cell in vivo, as has been argued.
For some time, a prevailing view has held that by unraveling the molecular mechanisms of EMT, new therapies to prevent fibrosis will be developed. Our studies serve to realign the study of fibrosis toward understanding myofibroblast activation and proliferation, since we can find no evidence for direct transition of renal epithelia into interstitial myofibroblasts in our models. Although EMT is well characterized in other contexts such as cancer metastasis and development, we note that lineage analysis techniques have led to a reappraisal of the importance of EMT in other tissues such as pancreas, for example.50 Furthermore, our studies challenge the notion that epithelial expression of the intermediate protein αSMA or the calcium binding protein S100A4 defines a cell as mesenchymal.
In separate studies using kinetic mathematical modeling we have suggested pericytes, supportive and pro-angiogenic cells of peritubular capillaries, are a major source of myofibroblasts in the UUO model of fibrosis.30 Evidence that myofibroblasts in the diseased kidney derive from a highly proliferative mesenchymal interstitial precursor has been suggested by several other investigators.49,51–54 The fact that FoxD1+ offspring express both PDGFRβ and CD73 in the normal kidney indicates that the interstitial precursor described by others is the same pericyte/perivascular fibroblast precursor we describe in these studies. This is the first study, however, to employ genetic fate marking of both the epithelial and interstitial compartments to identify myofibroblast origins in fibrosis. Recombination in the absence of tamoxifen was undetectable in FoxD1-GCE kidneys, an important characteristic of this model since ‘leaky’ recombination in the absence of tamoxifen is reported in some CreER mouse lines. Because FoxD1 is induced in kidney mesenchmye fated to become stromal cells of the kidney,34,39 it represents a useful genetic tool to modify stromal cells in health and disease. We show here that stromal cells of FoxD1-CreERt2 mice pulse-labeled by tamoxifen generate CD73+, PDGFRβ+, SMA− pericytes/perivascular fibroblasts, as well as mesangial cells and vascular smooth muscle cells. Following either UUO or ischemia-reperfusion injury we observed a marked expansion of interstitial cells that were pulse labeled at e10.5. The fact that the expanded LacZ labeled cell population expresses PDGFRβ and αSMA confirms that the LacZ labeled cells are myofibroblasts. Together with data from Collagen1α1-GFP mice,30 we conclude that pericytes and not epithelial cells give rise to the vast majority of interstitial myofibroblasts.
An important implication of these findings is that endothelial perturbation may be an early initiating event in the development of fibrosis, since endothelial cells and pericytes contact each other in the renal interstitium. Injury-induced alteration in endothelial cell delivered factors such as PDGF, for example, may represent the first step in pericyte differentiation into a myofibroblast. Since pericytes are known in other tissues to supply endothelial cell survival factors such as angiopoietins, subsequent differentiation of these cells into myofibroblasts with migration away from vasculature may also lead to endothelial cell death and capillary rarefaction.55 Chronic kidney disease is characterized by a reduction in the density of peritubular capillaries and targeting pericytes to prevent vascular injury may be a novel therapeutic approach for the treatment of progressive kidney diseases.56 Future studies should focus on identification of the factors regulating differentiation and migration of the pericyte away from the capillary. Whether a subset of the pericytes identified herein might represent a kidney-specific mesenchymal stem cell population deserves further investigation as well. Similarities between MSC and pericytes include shared expression of PDGFRβ, NG2 (anti-chondroitin sulfate proteoglycan-4), and CD73, a perivascular niche for both cell types in vivo and ability to secrete large quantities of bioactive trophic molecules that regulate angiogenesis and wound healing.57,58
In conclusion, we have used comprehensive genetic lineage analysis to clarify the role of epithelial cells and interstitial cells in the generation of myofibroblasts during kidney injury and fibrosis. We provide definitive lineage tracing evidence that kidney epithelial cells do not become myofibroblasts in vivo, but show that interstitial pericytes are myofibroblast progenitors in fibrotic kidney disease.
Acknowledgements
We thank Dr. Adam Lacy-Hulbert (Massachusetts General Hospital) for useful discussions, and Dr. William Stallcup, Burnham Institute, for anti-PDGFRβ antibodies, and Dr Bing Li (Harvard Medical School) for assistance with laminin localization and generating images detecting laminin in the kidneys.
Footnotes
Supported by NIH grants DK073628 (B.D.H.), DK073299, DK084077, DK87389 (J.S.D.), DK054364 (A.P.M.) National Science Council, Taiwan NSC-095-2314-B-002-310 and NSC-095-SAF-I-564-601-TMS (S.L.L.), support from the Harvard Stem Cell Institute (B.D.H.) and the Gottschalk award, American Society of Nephrology (J.S.D.).
These studies were presented in part at the American Society of Nephrology, 2008 and Keystone Conference on Fibrosis, 2009.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
Unilateral ureteral obstruction in Six2-GC+; Z/Red mice results in severe tubule injury fibrosis and disruption of basement membranes. (A) In normal kidney αSMA (green) is only in vascular smooth muscle cells, but there is induction of numerous α-SMA labeled interstitial myofibroblasts in UUO d10. Scale bar = 50μm. (B) Picrosirius red staining of paraffin sections from d0 control and d14 UUO kidney. Scale bars = 50μm. (C) Immunodetection of Laminin (red) and lotus lectin binding to detect proximal tubules in healthy kidney and d10 UUO kidney. Note multiple areas of disrupted tubule basement membrane in the UUO kidney (arrows). Scale bar = 200μm. (D) Graphs showing tubular dilatation score of d10 UUO kidneys compared with control (left), BrdU positive tubule nuclei in UUO kidneys (center) and morphometric assessment of fibrosis by area of picrosirius red staining in d14 UUO compared with control (right).
In d14 UUO kidney from Six2-GC; R26R mice, αSMA stained myofibroblasts do not express the epithelial marker LacZ. (A) Kidney sections from d0 control and d14 UUO Six2-GC; ROSA26R mice stained with X-gal for bacterial β-gal activity (green [false color]), and co-labeled with directly conjugated anti-αSMA antibodies (red). Left panels show identical images to right panels except that the red channel is absent. Note while there is strong staining for LacZ in tubules there is no co-expression of LacZ in cells expressing αSMA. (B) Kidney sections from d14 UUO Six2-GC mice co-stained with X-gal for bacterial β-gal activity (green), and αSMA (red). Note there is no LacZ activity in these kidneys stained under identical conditions. (C) Kidney sections from d0 control and d14 UUO ROSA26-LacZ mice (positive control) that express LacZ in all tissues stained with X-gal for LacZ activity (green), and co-labeled with directly conjugated anti-αSMA antibodies (red). Left panels show identical images to right panels except that the red channel is absent. Note that X-gal staining is present in all cells and LacZ activity in myofibroblasts is clearly visible in cells coexpressing αSMA (see higher magnification inset in lower panels). Scale bars = 50μm.
Day 10 unilateral ureteral obstruction kidneys from Six2-GC+; Z/Red and HoxB7-Cre; Z/Red mice show no evidence for RFP+ genetically labeled epithelial cells, becoming interstitial cells expressing αSMA+. Representative low magnification confocal images of native RFP epifluorescence (left panels) or FITC-conjugated antibody detection of αSMA (green, centre panels) of d10 UUO fibrotic kidneys of Z/Red mice, HoxB7-Cre; Z/Red mice Six2GC; Z/Red mice and NLS-Cre-Z/Red mice. Composite images with nuclei in blue are seen (right panels). Note that all interstitial cells expressing αSMA coexpress RFP in the NLS-Cre-Z/Red positive control mice but no αSMA+ cells coexpress RFP in the other mice. Scale bar = 50μm.
Following unilateral ischemia reperfusion injury, interstitial myofibroblasts derive from FoxD1 fated but not Six2 fated kidney mesenchyme. (A) Low magnification (upper) and high magnification (lower) confocal images of Six2GC; Z/red mouse kidneys 15d following IRI showing marked expansion of interstitial αSMA+ cells (green) but none co-express the epithelial fate marker RFP (red) which is clearly visible in all metanephric mesenchyme-derived tubular epithelial cells. (B) By contrast FoxD1-GCE; Rs26R adult kidneys pulsed with Tam on E10.5 to label a cohort of pericytes (blue) in utero, show marked expansion of LacZ stained cells 15 days following IRI injury due to expansion of the originally stained cells, and (C) the LacZ+ blue stained interstitial cells 15d following IRI co-express the marker Pdgfrβ (marker = 50μm), confirming their identity as myofirboblasts.
References
- 1.Friedman SL, Roll FJ, Boyles J, Arenson DM, Bissell DM. Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix. J Biol Chem. 1989;264:10756–10762. [PubMed] [Google Scholar]
- 2.Ivarsson M, Sundberg C, Farrokhnia N, Pertoft H, Rubin K, Gerdin B. Recruitment of type I collagen producing cells from the microvasculature in vitro. Exp Cell Res. 1996;229:336–349. doi: 10.1006/excr.1996.0379. [DOI] [PubMed] [Google Scholar]
- 3.Schlondorff D. The glomerular mesangial cell: an expanding role for a specialized pericyte. FASEB J. 1987;1:272–281. doi: 10.1096/fasebj.1.4.3308611. [DOI] [PubMed] [Google Scholar]
- 4.Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001;98:2396–2402. doi: 10.1182/blood.v98.8.2396. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RJ, Floege J, Yoshimura A, Iida H, Couser WG, Alpers CE. The activated mesangial cell: a glomerular “myofibroblast”? J Am Soc Nephrol. 1992;2:S190–S197. doi: 10.1681/ASN.V210s190. [DOI] [PubMed] [Google Scholar]
- 6.Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;171:5537–5546. doi: 10.4049/jimmunol.171.10.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyer B, Tucker GC, Valles AM, Gavrilovic J, Thiery JP. Reversible transition towards a fibroblastic phenotype in a rat carcinoma cell line. Int J Cancer Suppl. 1989;4:69–75. doi: 10.1002/ijc.2910440719. [DOI] [PubMed] [Google Scholar]
- 8.Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127:2021–2036. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okada H, Danoff TM, Kalluri R, Neilson EG. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol. 1997;273:F563–F574. doi: 10.1152/ajprenal.1997.273.4.F563. [DOI] [PubMed] [Google Scholar]
- 10.Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007;101:816–829. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- 11.Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 13.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest. 2003;112:503–516. doi: 10.1172/JCI17913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334:1448–1460. doi: 10.1056/NEJM199605303342207. [DOI] [PubMed] [Google Scholar]
- 18.Ouellette AJ, Malt RA, Sukhatme VP, Bonventre JV. Expression of two “immediate early” genes, Egr-1 and c-fos, in response to renal ischemia and during compensatory renal hypertrophy in mice. J Clin Invest. 1990;85:766–771. doi: 10.1172/JCI114502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida T, Tang SS, Hsiao LL, Jensen RV, Ingelfinger JR, Gullans SR. Global analysis of gene expression in renal ischemia-reperfusion in the mouse. Biochem Biophys Res Commun. 2002;291:787–794. doi: 10.1006/bbrc.2002.6535. [DOI] [PubMed] [Google Scholar]
- 20.Dressler GR, Woolf AS. Pax2 in development and renal disease. Int J Dev Biol. 1999;43:463–468. [PubMed] [Google Scholar]
- 21.Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372:679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- 22.Witzgall R, Brown D, Schwarz C, Bonventre JV. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J Clin Invest. 1994;93:2175–2188. doi: 10.1172/JCI117214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118:1657–1668. doi: 10.1172/JCI34487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mazzucchelli L. Protein S100A4: too long overlooked by pathologists? Am J Pathol. 2002;160:7–13. doi: 10.1016/S0002-9440(10)64342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 26.Tulchinsky EM, Grigorian MS, Ebralidze AK, Milshina NI, Lukanidin EM. Structure of gene mts1, transcribed in metastatic mouse tumor cells. Gene. 1990;87:219–223. doi: 10.1016/0378-1119(90)90305-b. [DOI] [PubMed] [Google Scholar]
- 27.Vega S, Morales AV, Ocana OH, Valdes F, Fabregat I, Nieto MA. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004;18:1131–1143. doi: 10.1101/gad.294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008;130:247–262. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng S, Lovett DH. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am J Pathol. 2003;162:1937–1949. doi: 10.1016/S0002-9440(10)64327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ng YY, Huang TP, Yang WC, Chen ZP, Yang AH, Mu W, Nikolic-Paterson DJ, Atkins RC, Lan HY. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int. 1998;54:864–876. doi: 10.1046/j.1523-1755.1998.00076.x. [DOI] [PubMed] [Google Scholar]
- 33.Tyler JR, Robertson H, Booth TA, Burt AD, Kirby JA. Chronic allograft nephropathy: intraepithelial signals generated by transforming growth factor-beta and bone morphogenetic protein-7. Am J Transplant. 2006;6:1367–1376. doi: 10.1111/j.1600-6143.2006.01339.x. [DOI] [PubMed] [Google Scholar]
- 34.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 36.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- 37.Yu J, Carroll TJ, McMahon AP. Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development. 2002;129:5301–5312. doi: 10.1242/dev.129.22.5301. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3:169–181. doi: 10.1016/j.stem.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hatini V, Huh SO, Herzlinger D, Soares VC, Lai E. Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of Winged Helix transcription factor BF-2. Genes Dev. 1996;10:1467–1478. doi: 10.1101/gad.10.12.1467. [DOI] [PubMed] [Google Scholar]
- 40.Duffield JS, Park KM, Hsiao LL, Kelley VR, Scadden DT, Ichimura T, Bonventre JV. Restoration of tubular epithelial cells during repair of the postischemic kidney occurs independently of bone marrow-derived stem cells. J Clin Invest. 2005;115:1743–1755. doi: 10.1172/JCI22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, Bonventre JV, Hughes J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheridan AM, Schwartz JH, Kroshian VM, Tercyak AM, Laraia J, Masino S, Lieberthal W. Renal mouse proximal tubular cells are more susceptible than MDCK cells to chemical anoxia. Am J Physiol. 1993;265:F342–F350. doi: 10.1152/ajprenal.1993.265.3.F342. [DOI] [PubMed] [Google Scholar]
- 44.Diamond JR, Kees-Folts D, Ding G, Frye JE, Restrepo NC. Macrophages, monocyte chemoattractant peptide-1, and TGF-beta 1 in experimental hydronephrosis. Am J Physiol. 1994;266:F926–F933. doi: 10.1152/ajprenal.1994.266.6.F926. [DOI] [PubMed] [Google Scholar]
- 45.Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- 46.Vintersten K, Monetti C, Gertsenstein M, Zhang P, Laszlo L, Biechele S, Nagy A. Mouse in red: red fluorescent protein expression in mouse ES cells, embryos, and adult animals. Genesis. 2004;40:241–246. doi: 10.1002/gene.20095. [DOI] [PubMed] [Google Scholar]
- 47.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Palmer R, Gao X, Kreidberg J, Gerald W, Hsiao L, Jensen RV, Gullans SR, Haber DA. Transcriptional activation of placental growth factor by the forkhead/winged helix transcription factor FoxD1. Curr Biol. 2003;13:1625–1629. doi: 10.1016/j.cub.2003.08.054. [DOI] [PubMed] [Google Scholar]
- 49.Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol. 2008;130:141–155. doi: 10.1007/s00418-008-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morton RA, Geras-Raaka E, Wilson LM, Raaka BM, Gershengorn MC. Endocrine precursor cells from mouse islets are not generated by epithelial-to-mesenchymal transition of mature beta cells. Mol Cell Endocrinol. 2007;270:87–93. doi: 10.1016/j.mce.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le Hir M, Hegyi I, Cueni-Loffing D, Loffing J, Kaissling B. Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem Cell Biol. 2005;123:335–346. doi: 10.1007/s00418-005-0788-z. [DOI] [PubMed] [Google Scholar]
- 52.Fujigaki Y, Muranaka Y, Sun D, Goto T, Zhou H, Sakakima M, Fukasawa H, Yonemura K, Yamamoto T, Hishida A. Transient myofibroblast differentiation of interstitial fibroblastic cells relevant to tubular dilatation in uranyl acetate-induced acute renal failure in rats. Virchows Arch. 2005;446:164–176. doi: 10.1007/s00428-004-1155-5. [DOI] [PubMed] [Google Scholar]
- 53.Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res Ther. 2005;7:R1113–R1123. doi: 10.1186/ar1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Faulkner JL, Szcykalski LM, Springer F, Barnes JL. Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am J Pathol. 2005;167:1193–1205. doi: 10.1016/S0002-9440(10)61208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–523. doi: 10.1161/01.RES.0000182903.16652.d7. [DOI] [PubMed] [Google Scholar]
- 56.Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens. 2004;13:1–7. doi: 10.1097/00041552-200401000-00001. [DOI] [PubMed] [Google Scholar]
- 57.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, Norotte C, Teng PN, Traas J, Schugar R, Deasy BM, Badylak S, Buhring HJ, Giacobino JP, Lazzari L, Huard J, Peault B. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 58.Plotkin MD, Goligorsky MS. Mesenchymal cells from adult kidney support angiogenesis and differentiate into multiple interstitial cell types including erythropoietin-producing fibroblasts. Am J Physiol Renal Physiol. 2006;291:F902–F912. doi: 10.1152/ajprenal.00396.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Unilateral ureteral obstruction in Six2-GC+; Z/Red mice results in severe tubule injury fibrosis and disruption of basement membranes. (A) In normal kidney αSMA (green) is only in vascular smooth muscle cells, but there is induction of numerous α-SMA labeled interstitial myofibroblasts in UUO d10. Scale bar = 50μm. (B) Picrosirius red staining of paraffin sections from d0 control and d14 UUO kidney. Scale bars = 50μm. (C) Immunodetection of Laminin (red) and lotus lectin binding to detect proximal tubules in healthy kidney and d10 UUO kidney. Note multiple areas of disrupted tubule basement membrane in the UUO kidney (arrows). Scale bar = 200μm. (D) Graphs showing tubular dilatation score of d10 UUO kidneys compared with control (left), BrdU positive tubule nuclei in UUO kidneys (center) and morphometric assessment of fibrosis by area of picrosirius red staining in d14 UUO compared with control (right).
In d14 UUO kidney from Six2-GC; R26R mice, αSMA stained myofibroblasts do not express the epithelial marker LacZ. (A) Kidney sections from d0 control and d14 UUO Six2-GC; ROSA26R mice stained with X-gal for bacterial β-gal activity (green [false color]), and co-labeled with directly conjugated anti-αSMA antibodies (red). Left panels show identical images to right panels except that the red channel is absent. Note while there is strong staining for LacZ in tubules there is no co-expression of LacZ in cells expressing αSMA. (B) Kidney sections from d14 UUO Six2-GC mice co-stained with X-gal for bacterial β-gal activity (green), and αSMA (red). Note there is no LacZ activity in these kidneys stained under identical conditions. (C) Kidney sections from d0 control and d14 UUO ROSA26-LacZ mice (positive control) that express LacZ in all tissues stained with X-gal for LacZ activity (green), and co-labeled with directly conjugated anti-αSMA antibodies (red). Left panels show identical images to right panels except that the red channel is absent. Note that X-gal staining is present in all cells and LacZ activity in myofibroblasts is clearly visible in cells coexpressing αSMA (see higher magnification inset in lower panels). Scale bars = 50μm.
Day 10 unilateral ureteral obstruction kidneys from Six2-GC+; Z/Red and HoxB7-Cre; Z/Red mice show no evidence for RFP+ genetically labeled epithelial cells, becoming interstitial cells expressing αSMA+. Representative low magnification confocal images of native RFP epifluorescence (left panels) or FITC-conjugated antibody detection of αSMA (green, centre panels) of d10 UUO fibrotic kidneys of Z/Red mice, HoxB7-Cre; Z/Red mice Six2GC; Z/Red mice and NLS-Cre-Z/Red mice. Composite images with nuclei in blue are seen (right panels). Note that all interstitial cells expressing αSMA coexpress RFP in the NLS-Cre-Z/Red positive control mice but no αSMA+ cells coexpress RFP in the other mice. Scale bar = 50μm.
Following unilateral ischemia reperfusion injury, interstitial myofibroblasts derive from FoxD1 fated but not Six2 fated kidney mesenchyme. (A) Low magnification (upper) and high magnification (lower) confocal images of Six2GC; Z/red mouse kidneys 15d following IRI showing marked expansion of interstitial αSMA+ cells (green) but none co-express the epithelial fate marker RFP (red) which is clearly visible in all metanephric mesenchyme-derived tubular epithelial cells. (B) By contrast FoxD1-GCE; Rs26R adult kidneys pulsed with Tam on E10.5 to label a cohort of pericytes (blue) in utero, show marked expansion of LacZ stained cells 15 days following IRI injury due to expansion of the originally stained cells, and (C) the LacZ+ blue stained interstitial cells 15d following IRI co-express the marker Pdgfrβ (marker = 50μm), confirming their identity as myofirboblasts.