

Abstract
Syndecan-1 (Sdc1) plays a major role in wound healing and modulates inflammatory responses. Sdc1 expression is reduced in lesions of patients with ulcerative colitis. The aim of this study was to investigate the role of Sdc1 in murine dextran sodium sulfate (DSS)-induced colitis. DSS colitis was induced in Sdc1-deficient (knockout (KO)) and wild-type mice by oral administration of 3% DSS. KO mice exhibited a significantly increased lethality as compared with wild-type controls (61 versus 5%, P < 0.05). Impaired mucosal healing and prolonged recruitment of inflammatory cells in KO mice were accompanied by significant up-regulation of tumor necrosis factor-α, CC chemokine ligand 3/macrophage inflammatory protein-1α, and vascular cell adhesion molecule-1, as determined by histological correlation between 0 and 15 days after colitis induction, TaqMan low-density array analysis, and quantitative real-time PCR. Treatment from days 7 through 14 with enoxaparin, a functional analogue of the Sdc1 heparan sulfate chains, significantly reduced lethality of KO mice due to DSS-induced colitis, which was correlated with improved mucosal healing. In vitro, Sdc1-deficient polymorphonuclear cells displayed increased adhesion to endothelial cells and intercellular adhesion molecule-1, and enoxaparin reverted adhesion to wild-type levels. Small interfering RNA-mediated knockdown of Sdc1 expression resulted in reduced basic fibroblast growth factor-mediated mitogen-activated protein kinase signaling and reduced Caco-2 cell proliferation. We conclude that Sdc1 has a protective effect during experimental colitis. The modification of missing Sdc1 function by heparin analogues may emerge as a promising anti-inflammatory approach.
Syndecan-1 (Sdc1) is the most important representative of the heparan sulfate proteoglycans (HSPGs) covering epithelial cell surfaces.1 It serves multiple biological roles, such as cell-matrix interactions, modulation of inflammatory responses, tumorigenesis, and wound healing.2–4 The highly conserved cytoplasmic domains of Sdc1 interact with scaffolding proteins and participate in integrin-mediated signaling events, thus providing a physical and functional link to the cytoskeleton. In addition, most of the extracellular-binding interactions are mediated by the heparan sulfate chains, which are structurally and functionally related to heparin, an extensively sulfated and epimerized derivative of heparan sulfate.1 Sdc1 serves as a coreceptor for several tyrosine kinase receptors. For example, it increases the activity of the complex of basic fibroblast growth factor (bFGF) and the FGF receptor and, therefore, contributes to improved wound healing via stimulation of keratinocyte proliferation.1,5 A role for Sdc1 in wound repair in vivo has been demonstrated in Sdc1-deficient (Sdc1-knockout (KO)) mice, which show delayed skin and corneal wound healing5 and functionally adverse repair following experimental myocardial infarction due to dysregulation of chemokine expression and matrix metalloproteinase-mediated tissue remodeling.6 Sdc1 forms chemotactic gradients due to binding of chemokines on heparan sulfate chains of the molecule. Therefore, Sdc1 is able to act as coreceptor for chemokine signaling.7,8 In addition, endothelial leukocyte recruitment and extravasation is modulated by Sdc1, possibly via interference with heparin-binding adhesion molecule function.9–12
Day et al13 described in 1999 a reduced expression of Sdc1 in patients with ulcerative colitis, which was linked to disrupted healing of colonic ulcers. In addition, this group demonstrated the benefit of the Sdc1 ectodomain for the FGF-induced proliferation of intestinal epithelial cell lines in vitro. The function of Sdc1 could be restored with heparin, representing a highly sulfated and epimerized form of heparan sulfate, the major functional constituent of the Sdc1 ectodomain.
Heparin sees widespread use as anticoagulant drug based on its antithrombin III-activating properties. Enoxaparin, is a low molecular weight heparin with similar features in vitro and in vivo like heparin; however, it exhibits a more favorable pharmacological side effect profile. Both low molecular weight heparins (enoxaparin) as well as heparin were recently found to possess anti-inflammatory properties.14
The hypothesis of Sdc1 being involved in the pathogenesis of ulcerative colitis is underlined by multiple clinical observations of patients who have been treated with heparins for different reasons.15 In a number of cases, this treatment has lead to an improved course of disease. A limited number of uncontrolled clinical trials with heparins in the treatment of low to medium active ulcerative colitis showed a variable outcome,16–18 which may be explained by variations in treatment regimes that may have failed to include the optimal dose, class of heparin, and mode of delivery. For example, most studies have involved either i.v. or s.c. delivery of heparin, whereas a more appropriate mode of delivery for stimulating mucosal healing might be the topical application or microsphere-mediated delivery of heparin.14,15 Furthermore, the outcome of heparin therapy may depend on the degree to which Sdc1 expression is reduced in inflammatory bowel disease (IBD) patients.19
Moreover, the expression of Sdc1 and the proinflammatory cytokine tumor necrosis factor-α (TNF-α) are inversely correlated in the colonic mucosa of patients with Crohn's disease,20 and a reduction of Sdc1 expression has been shown to result in increased TNF-α signaling in an in vitro model of protein-losing enteropathy,19,21 further suggesting a regulatory role for Sdc1 in proinflammatory cytokine signaling.
In this study, our goal was to characterize the impact of a Sdc1 deficiency on the dextran sodium sulfate (DSS)-induced colitis of the mouse. Furthermore, the efficacy of low molecular weight heparin to restore altered wound healing was studied in vivo. In addition, in vitro trials were performed to study the role of Sdc1 deficiency in the adhesion and transmigration of leukocytes under inflammatory conditions.
Materials and Methods
DSS Colitis and Enoxaparin Treatment
Sdc1-deficient (Sdc1 KO) mice on a C57BL/6 background5,12 and control wild-type mice were obtained from the clinical research laboratory of the Department of Gynecology, University of Muenster. Mice were bred, housed, and handled according to the guidelines of the local animal ethics committee (number A101/2005). Twelve-week-old male mice were placed in the Central Animal Facility of the University Hospital of Muenster with a 12-hour day/night light cycle and standard chow and water ad libitum. After 3 weeks of adaptation, the trials were performed as follows: starting on day 1 and continued to day 6 an oral administration of 3% DSS in drinking water was administered. On day 7, the water was switched to normal drinking water. Of 92 mice used in this study, 10 male wild-type mice and 10 Sdc1 KO mice were used to study the course of disease. The course of disease was monitored by daily weight measurement and a daily assessment of blood in stool with a commercially available stool test (Hemoccult; Beckman Coulter, Fullerton, CA).
Thirty-six wild-type mice and 36 Sdc1 KO mice were used for histological evaluation and for real-time PCR analysis of colon tissue divided in a treatment group of 18 mice and a control group of another 18 mice. The treatment group received an i.p. injection of enoxaparin (500 IU/kg/body weight) from days 7 to 14 (control group: 0.9% NaCl) once per day. Three animals out of each group were sacrificed for further histological examination on days 0, 3, 6, 9, 12, and 15.
Histological Examination
After euthanasia of animals, the proximal, medial, and distal colon was resected after laparotomy. The colon was flushed with PBS and covered with optimal cutting temperature Tissue-Tek (Sakura). The tissue was snap frozen in liquid nitrogen and stored at −80°C until use. Seven-micrometer frozen sections were cut, and standard H&E staining was performed as described previously.22 Severity of the colitis was assessed by an established scoring system as published before.23 For immunohistochemical analysis, the following primary antibodies were used: Sdc1 (rat anti-mouse Sdc1 mAb clone 281-2, diluted 1/100 in PBS with 1% BSA; BD Pharmingen, Heidelberg, Germany), Sdc2 (rabbit-anti-mouse, diluted 1/50; Santa Cruz Biotechnology, Santa Cruz, CA), Sdc3 (rat-anti-mouse mAb clone 312607, diluted 1/50; R&D Systems, Wiesbaden, Germany), Sdc4 (rat-anti-mouse clone KY/8.2, diluted 1/50; BD Pharmingen), F4/80 (rat-anti-mouse, clone BM8; eBioscience, San Diego, CA), CD4, CD8, and B220 (rat anti-mouse, clone C3T4 (CD4), clone 53-6.7 (CD8), and clone RA3-6B2 (B220), diluted 1/100; BD Pharmingen). M cells were stained with the UEA1 lectin method (Sigma-Aldrich, Deisenheim, Germany) as described elsewhere.23
Briefly, the sections were blocked with 10% goat serum or 1% BSA (Dako, Glostrup, Denmark) in PBS for 30 minutes. Incubation with the primary antibody was performed in a humid chamber overnight at 4°C. After three washes with PBS (5 minutes), the secondary antibody (dilution 1/1000 in 10% goat serum/PBS) was incubated for 1 hour at room temperature. For immunofluorescence microscopy, Alexa Fluor-labeled secondary antibodies were used (Invitrogen, Karlsruhe, Germany), while for conventional immunohistochemistry, the Vectastain ABC system (Vector Laboratories, Burlingame, CA) or the Dako EnVision system (Dako) were used, depending on the primary antibody used. For immunofluorescence microscopy, sections were washed again in PBS for 5 minutes, air-dried, and treated with prolong antifade kit (Invitrogen). For conventional immunohistochemistry, 3-amino-9- ethylcarbazole substrate (Dako) was used for color development, followed by Mayer's Hemalum counterstaining (Merck, Darmstadt, Germany). Pictures were taken with a Leica or Zeiss Axioscope microscope, and digital cameras directly connected to a PC system. Images were processed with Adobe Photoshop version 7.0 and Zeiss Axiovision software.
Flow Cytometry
To study the composition of intestinal lymphoid subpopulations, flow cytometry was performed.24 Briefly, Peyer's patches (PPs) and lamina propria lymphocytes from the colon were extracted and analyzed by flow cytometry. All PPs were counted and resected from the intestine. PPs were homogenized mechanically with a Falcon cell mesh (pore size, 70 μm; BD Pharmingen) and mixed with ice-cold RPMI 1640 medium and 10% fetal calf serum. After centrifugation at 10,000 × g for 5 minutes, the supernatant was discarded, and the pellet was washed with ice-cold PBS. This washing/centrifugation step was performed 3 times. The pellet was resuspended in ice-cold PBS and added to Ficoll (Genaxxon Bioscience, Biberach, Germany). After centrifugation at 5000 × g for 15 minutes, the layer containing the leukocytes was retrieved and placed into a fluorescence-activated cell sorting tube. After a washing step with PBS and centrifugation, the pellet was incubated with an antibody for surface markers CD4 (direct fluorescein isothiocyanate-conjugated mAb, clone C3T4; BD Pharmingen), CD8 (direct phycoerythrin-conjugated mAb, clone 53-6.7; BD Pharmingen), and B220 (direct allophycocyanin-conjugated mAb, clone RA3-6B2; BD Pharmingen). Furthermore, in lamina propria lymphocyte populations, the markers CD3 (clone 145-2C11), CD11c (clone HC3), CD25 (clone 7D4), CD127 (clone A7R34), and Gr-1 (clone RB6-8C5) (all antibodies purchased from BD Pharmingen) were examined. Fluorescence-activated cell sorting analysis was performed with a FACSCalibur flow cytometer from BD Biosciences (Heidelberg, Germany).
Analysis of Cell Proliferation and bFGF-Dependent Signal Transduction in Sdc1 Silenced Caco-2 Cells
The human colon carcinoma cell line Caco-2 (German Collection of Microorganisms and Cell Cultures, Department of Human and Animal Cell Cultures, Braunschweig, Germany) was cultured in minimal essential medium (Invitrogen) containing 10% fetal calf serum, 1% penicillin/streptomycin and 1 mm of l-glutamine. Small interfering RNA (siRNA)-mediated knockdown of Sdc1 expression was performed exactly as previously described,25 using siRNA numbers 12527 and 12432 (Ambion, Cambridgeshire, UK) targeting the coding region of Sdc1 and a negative control siRNA (number 301698; Qiagen, Hilden, Germany) at 40 nmol/L and Dharmafect reagent (Dharmacon, Lafayette, CO). Cell proliferation was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide assay essentially as previously described,26 using 5000 Caco-2 cells/well plated 2 days after Sdc1 silencing for an incubation period of 3 days. bFGF-mediated mitogen-activated protein kinase (MAPK) activation was studied 72 hours after Sdc1 knockdown. Silenced serum-starved Caco-2 cells were stimulated with 20 nmol/L bFGF (R&D Systems, Wiesbaden, Germany) for 0, 10, or 20 minutes, respectively, and cell extracts were subjected to SDS-PAGE and Western blotting for phosphorylated and total p44/42 MAPK using rabbit polyclonal antibodies (Cell Signaling Technology, Beverly, MA) exactly as described previously.25 All experiments were performed at least three times.
Static Leukocyte-Endothelial Cell Adhesion Assay
Polymorphonuclear (PMN) cells were purified from the bone marrow of Sdc1-deficient and wild-type mice by Histopaque (Sigma-Aldrich) gradient centrifugation as previously described,11 and leukocyte adhesion to the murine bEnd.3 endothelial cell line, derived from viral oncogene-immortalized brain endothelial cells27 was measured under static conditions. Briefly, PMN cells were fluorescently labeled with 2,7 -bis-(2 carboxyethyl)-5-carboxyfluorescein acetoxymethyl ester (Invitrogen), washed, and incubated in sextuplets (2 × 106 cells/ml, 50 μl/well) with confluent bEnd.3 monolayers in 96-well plates (10 minutes, 37°C). For inhibition experiments, PMN cells were preincubated for 30 minutes with 10 or 25 μg/ml enoxaparin, followed by incubation in the presence of the drug as described above. After three washes with PBS, adhering cells were lysed, and the fluorescence signal was quantified in a Spectramax fluorimeter (excitation, 485 nm; emission, 535 nm). Statistical analysis was performed using Student's t-test. A value of P < 0.05 was considered statistically significant.
Intercellular Adhesion Molecule-1 Adhesion Assay
Adhesion of Sdc1-deficient and wild-type mouse PMN cells to recombinant Intercellular adhesion molecule-1 (ICAM-1) was performed essentially as described.28 Briefly, 10 μg/ml recombinant murine ICAM-1 protein in PBS/1% fetal calf serum was coated onto 96-well flat-bottom plates (Maxisorp; Nunc, Wiesbaden, Germany). Murine PMN cells, prepared as described above, were added and incubated at a concentration of 3 × 105 cells/well for 45 minutes at 4°C to allow for cell-ICAM-1 interactions. For inhibition experiments, PMN cells were preincubated for 30 minutes with 10 or 25 μg/ml enoxaparin, followed by incubation in the presence of the drug as described above. Plates were washed four times with PBS, and adherent cells were fixed using 1% paraformaldehyde (w/v) in PBS. Adhering cells were observed at 32-fold magnification using a Zeiss Axioskop microscope (Zeiss, Göttingen, Germany) equipped with a charge-coupled device camera. Cell numbers were quantified and expressed as cells/mm2 with the assistance of Zeiss Axiovision imaging software. Statistical analysis was performed using Student's t-test. A value of P < 0.05 was considered statistically significant.
Selectin-Binding Assay
Granulocytes were incubated with 10 μg/ml E- and P-selectin-Fc proteins consisting of the extracellular parts of the murine selectins and the Fc portion of human IgG essentially as described previously.29,30 Human IgG1 (Sigma-Aldrich) served as negative control. Before protein incubation, unspecific binding was blocked by incubation with 10 μg/ml Fc block 2.4G2 (BD Pharmingen). Phycoerythrin-conjugated donkey-anti-human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) served to visualize binding using a FACSCanto flow cytometer and FACSDiva software (BD Biosciences).
Quantitative Real-Time PCR and Low-Density TaqMan Microarray Analysis
Animals were sacrificed, and the medial to distal colon was resected. Colon tissue was immediately placed in RNAlater solution (Ambion, Huntington, UK), followed by preparation of total RNA using the RNAeasy kit (Qiagen). Five hundred nanograms of total RNA was transcribed using the high-capacity cDNA synthesis kit (Applied Biosystems). cDNA corresponding to 0.5 ng of total RNA was used as a template in the PCR consisting of ABI MasterMix (Applied Biosystems, Darmstadt, Germany) and predesigned TaqMan gene expression systems (Applied Biosystems) or the TaqMan Low-Density Mouse Immune Panel (ABI number 4367786), according to the manufacturers instructions. PCR was performed using 7900HT Fast and 7300 Real-Time PCR Systems (Applied Biosystems). For detection of Sdc1 to Sdc4, P-Selectin, TNF-α, vascular cell adhesion molecule-1 (VCAM-1), ICAM-1, CC chemokine ligand (CCL)3/macrophage inflammatory protein-1α (MIP-1α), CCL2/ monocyte chemoattractant protein-1, interleukin-6 mRNA, primers Mm00448918_m1 (Sdc1 exon 2–3), Mm00484718_m1 (Sdc2 exon 4–5), Mm01179831_m1 (Sdc3 exon 2–3), Mm00488527_m1 (Sdc4 exon 4–5), Mm 00441295_m1 (P-Selectin exon 8–9), Mm00443258_m1 (TNF α exon 1–2), Mm00449197_m1 (VCAM-1 exon 5–6), Mm00516023_m1 (ICAM-1 exon 2–3), Mm00441242_m1 (CCL2 exon 1–2), Mm00441258_m1 (CCL3 exon 1–2), and Mm00446190_m1 (interleukin-6 exon 2–3) were used and normalized to the expression of mammalian 18S rRNA (Hs99999901_s1, all primers from Applied Biosystems). Quantitative real-time PCR was performed using the ABI PRISM 7300 Sequence Detection System (Applied Biosystems) by using the default thermal cycling conditions (10 minutes at 95°C and then 40 cycles of 15 seconds at 95°C plus 1 minute at 60°C). Relative quantification was performed using the comparative cycle threshold method. For low-density array analysis 2 (Sdc1 KO DSS), or 3 (Sdc1 KO control, wild-type control, DSS) biological replicates were used. For quantitative real-time PCR, 5–9 biological replicates were used. For statistical analysis of ΔCt values, Student's t-test was used. A value of P < 0.05 was considered statistically significant.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling Assay
The terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay was used on frozen sections of the colon according to the manufacturers manual (TdT-fragEL; Calbiochem, San Diego, CA). The apoptotic cells were visualized by using the horseradish peroxidase reaction with diaminobenzidine. Hematoxylin was used as counterstaining. For negative controls, distilled H2O was substituted for the TdT during the labeling step.
In Vivo BrdU labeling
5-Bromo-2′-deoxyuridine (BdrU; Sigma-Aldrich, Munich, Germany) was used to visualize DNA replication in epithelial cells. BrdU (50 mg/kg body weight) was given i.p. 24 hours before resection of the colon. The resected colon was embedded in Tissue-Tek optimal cutting temperature compound (Sakura-Fintek, Torrance, CA) and immediately frozen in liquid nitrogen. BdrU was visualized on cryosections by a monoclonal antibody against BdrU conjugated with biotin, followed by incubation with streptavidin-horseradish peroxidase and 3-amino-9- ethylcarbazole substrate (BrdU in situ detection kit; BD Pharmingen). Sections were counterstained with hematoxylin before microscopy. Quantitative analysis of BrdU labeling was performed by determining the number of BrdU-positive cells per well-defined crypt, excluding cross-sectioned crypts from the analysis. A minimum of 200 crypts from three independent experimental series were analyzed per treatment group.
Statistical Analysis
For statistical analysis of body weight, average body weight and SD were determined for each day. To test for significance of the results, Student's t-test was applied when data were normally distributed; otherwise, the Mann-Whitney U-test was applied using Sigma Stat 3.1 statistical software. Survival of animals was evaluated with the log-rank test. Values of P < 0.05 were considered significant.
Results
Effects of Enoxaparin in DSS Colitis of Sdc1-Deficient Mice
Murine colitis was induced by application of 3% DSS in drinking water for 6 days. The average body weight of the animals was 22 g (+/−1 g) at the beginning of the experiment before starting DSS. Weight loss was observed, starting on days 4 to 5 of DSS application. Enoxaparin or 0.9% NaCl as placebo was used in a therapeutic setting starting on day 7 when DSS treatment was stopped.
We compared four groups of animals in terms of weight loss, survival, as well as blood loss via the stool as parameters of disease progression. At first we evaluated the effect of enoxaparin treatment on the course of colitis in wild-type mice. Both groups showed similar weight loss without significant changes in the weight loss curves between enoxaparin-treated and placebo-treated wild-type groups (Figure 1A). The onset of hematochezia was seen on day 3 and persisted until day 10 after colitis induction with DSS in wild-type mice. Enoxaparin-treated wild-type mice suffered from hematochezia until day 12 after DSS application. The onset of hematochezia in Sdc1 KO mice DSS colitis started on day 3 and lasted until day 12 after DSS induction. Enoxaparin-treated Sdc1 KO mice did not show a prolonged rectal bleeding past day 12 (data not shown).
Figure 1.
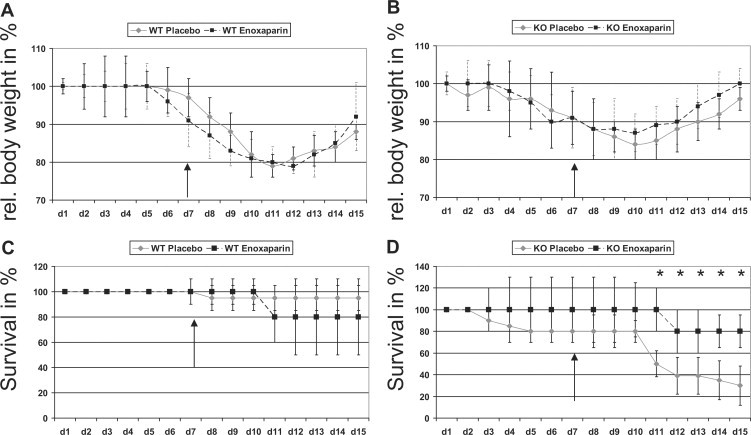
A: Course of DSS colitis indexed as weight loss in wild-type (WT) mice receiving either 0.9% saline or enoxaparin (500 IE/kg/body weight/once per day). B: Course of DSS colitis in Sdc1 KO mice receiving either 0.9% saline or enoxaparin (500IE/kg/body weight/once per day). C: Survival rates of wild-type mice with DSS colitis. D: Survival rates of KO mice with DSS colitis. ∗P < 0.05. Arrows indicate the start of enoxaparin application.
On day 15, 95% of wild-type mice treated with 0.9% NaCl survived colitis, whereas 80% of wild-type mice receiving enoxaparin survived the course of disease (Figure 1C). In summary, there was no statistical significance regarding survival rates between these two groups, but a tendency toward a poorer outcome of enoxaparin-treated animals.
Second, we induced DSS colitis in Sdc1 KO mice in the same way as it was performed in wild-type mice. Weight loss started around 24 hours earlier as compared with wild-type mice. Because of the bias of higher lethality, the weight curve is similar to the weight curve of wild-type mice without significant changes (Figure 1B). Interestingly, the onset of regaining weight was seen earlier in enoxaparin-treated Sdc1 KO mice compared with placebo-treated Sdc1 KO mice. Statistically significant benefit for Sdc1 KO mice treated with enoxaparin was seen for survival rates (Figure 1D). Sdc1 KO mice treated with 0.9% NaCl had a poor outcome with only 40% survival on day 15, whereas Sdc1 KO animals treated with enoxaparin had a survival rate of ∼80% (P < 0.05 by log-rank test). Sdc1 KO mice weight data interfered with the accuracy of the weight data due to the high lethality of Sdc1 KO mice and the increased survival of wild-type mice during DSS colitis. In summary, enoxaparin treatment significantly improves the outcome of DSS colitis in Sdc1 KO mice mainly due to an improved recovery phase with little difference in the induction phase.
Enoxaparin Treatment Improves Intestinal Wound Healing in Sdc1-Deficient Mice during DSS-Induced Colitis
To evaluate the mechanism of the observed effect of enoxaparin in Sdc1 KO mice, a histology-based analysis of the colon was performed. Animals from all four groups were analyzed after DSS exposure and sacrificed on days 3, 6, 9, 12, and 15, respectively. The colon was removed, and specimens from the proximal, middle, and distal colon were examined.
To characterize the inflammatory process we used a histology scoring system in a blinded fashion.23 Briefly, inflammatory activity was characterized by the severity of inflammatory damage and destruction of cytoarchitecture of the gut (loss of epithelial cells (grade 1) versus large ulcerations penetrating deep muscular layers (grade 4)) as well as the amount of leukocyte infiltration (eg, epithelial infiltration (grade 1) versus transmural infiltration (grade 4)).
On day 9, placebo-treated Sdc1 KO mice showed a strong epithelial disintegration with ulcerations, edema, muscular thickening, and wide areas of epithelial denudation; nearly no intact mucosa was seen (Figure 2A). Colonic tissue from placebo and enoxaparin-treated wild-type mice had less severe signs of epithelial damage with ulcerations and edema; epithelial healing on day 9 was detectable. On day 15, histological changes in placebo-treated Sdc1 KO mice persisted, whereas wild-type mice (both placebo- and enoxaparin-treated wild-type mice) and Sdc1 KO mice treated with enoxaparin almost completely recovered (Figure 2B). We also specifically evaluated the amount of leukocyte infiltration (Figure 2C); however, no significant changes were seen among the four groups with one exception: Sdc1 KO mice treated with placebo showed a persistent leukocyte infiltration on day 15. Data suggest that enoxaparin might substitute for the loss of some of the functions induced by Sdc1 KO regarding the epithelial integrity and leukocyte infiltration.
Figure 2.
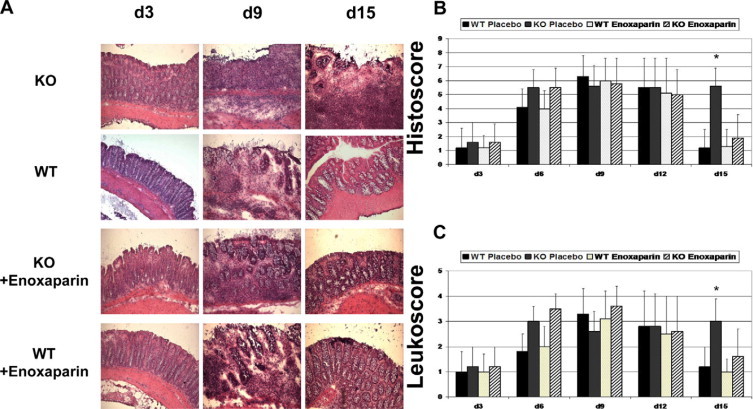
A: Representative histology of KO mice with placebo (first row) and enoxaparin treatment (third row) compared with normal wild-type (WT) DSS colits (second row) and wild-type treated with enoxaparin (fourth row). H&E staining, magnification, ×200. B: Inflammatory changes indexed as histology score. C: Leukocyte infiltration score. ∗P < 0.05.
BrdU labeling of Sdc1 KO mice undergoing DSS colitis confirmed a lack of regeneration on day 6 compared with wild-type mice undergoing DSS colitis (Figure 3A). In contrast, epithelial repair was considerably delayed in Sdc1 KO mice, taking place at day 15, when repair was complete in wild-type mice (Figure 3B) The terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling assay (Figure 3C) showed that apoptosis in the area of the epithelium persisted in Sdc1 KO mice on day 15 compared with wild-type mice during DSS colitis. No differences were seen between both groups on days 1 and 6.
Figure 3.
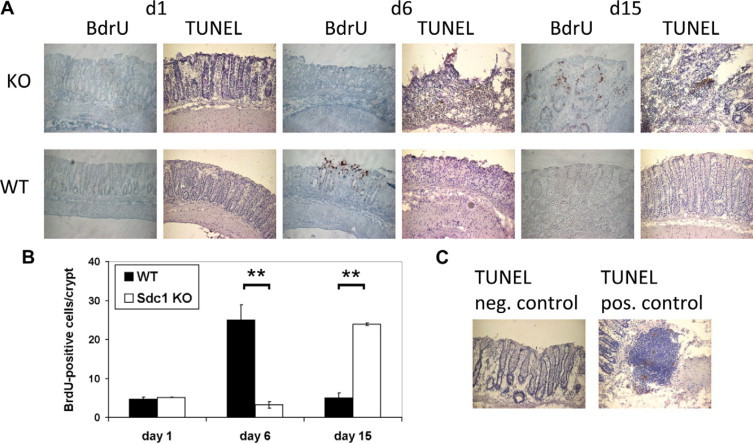
Altered cell proliferation and apoptosis in Sdc1 KO mice subjected to DSS colitis. A: BrdU and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay. A delay in epithelial repair is observed in Sdc1 KO mice versus wild-type (WT) mice. Prolonged epithelial apoptosis is detectable in KO mice until day 15. Representative immunohistochemical stainings of BrdU-positive cells at day 1, day 6, and day 15, ×400. B: Quantitative analysis of BrdU-positive cells per well-defined colonic crypt. ∗∗P < 0.01. C: Positive and negative controls of the TUNEL staining.
Expression Analysis of Sdc1 and Inflammation-Related Genes in DSS Colitis
We next investigated the expression of Sdc1 mRNA in colon samples of wild-type control and DSS-treated mice. Quantitative real-time PCR analysis revealed a slight reduction of Sdc1 expression during DSS colitis (not significant, P = 0.61) (Figure 4, A and B; also Supplemental Table 1, see http://ajp.amjpathol.org). At the mRNA level, Sdc1 was the most abundantly expressed member of the syndecan family in unchallenged wild-type colon tissue (Figure 4A). Immunohistochemistry confirmed a primarily epithelial staining of Sdc1 in wild-type mice during DSS colitis (Supplemental Figure S1, see http://ajp.amjpathol.org). No increase in Sdc2 to Sdc4 immunostaining and mRNA expression was observed in Sdc1 KO compared with wild-type mice, suggesting the absence of a potential compensatory up-regulation of other syndecan family members. To further characterize the influence of Sdc1 on intestinal inflammation, we also studied mRNA expression of 93 inflammation-related genes using TaqMan microfluidic card low-density PCR array technology. About 25% of the investigated genes were either not expressed, or at the limit of detection, whereas ∼50% of the genes were not consistently differentially expressed between Sdc1 KO and wild-type mice. However, a number of relevant proinflammatory genes were up-regulated in colon tissue of DSS-treated Sdc1-deficient compared with wild-type mice (Figure 5, A–D; also Supplemental Table 1, see http://ajp.amjpathol.org). Among these genes the expression of six identified relevant targets (P-selectin, TNF-α, VCAM-1, macrophage inflammatory protein-1α (CCL3/MIP-1α), monocyte chemoattractant protein-1 (CCL2/MCP-1), and interleukin-6) was further investigated in a larger sample collective (n = 9) by real-time PCR. Compared with wild-type controls the expression of the proinflammatory cytokine TNF-α, the chemokine CCL3/MIP-1α, and of the leukocyte adhesion molecules P-selectin and VCAM-1 was up-regulated 3- to 60-fold in DSS-treated Sdc1-deficient colon tissue compared with wild-type controls (P < 0.05) suggesting that syndecan might also influence intestinal leukocyte trafficking. A differential regulation of monocyte chemoattractant protein-1, IL-6, and of the adhesion molecule ICAM-1 could not be independently confirmed by quantitative PCR (Supplemental Table 1, see http://ajp.amjpathol.org, and results not shown).
Figure 4.
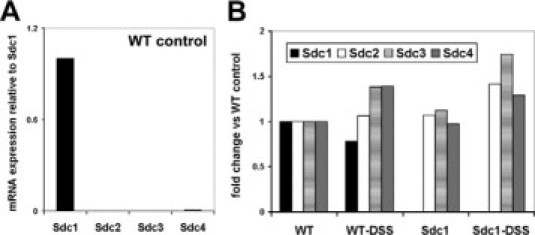
Expression of Sdc1 to Sdc4 in wild-type (WT) and Sdc1 KO mice subjected to DSS colitis. A and B: Quantitative real-time PCR analysis of Sdc1 to Sdc4. Sdc1 KO and WT mice were treated with saline (control) or DSS for 7 days, respectively, mRNA was prepared from colon samples of five to nine mice, transcribed into cDNA, and subjected to quantitative real-time PCR analysis. The fold change of the mean Ct values, as calculated by the 2−(ΔΔCt) method, is shown (Supplemental Table SI, see http://ajp.amjpathol.org for Ct values and SD). No significant differences in the expression of Sdc2 to Sdc4 were observed between WT and Sdc1 KO mice (Supplemental Figure S1, see http://ajp.amjpathol.org for immunohistochemical investigation of Sdc1 to Sdc4).
Figure 5.

Quantitative real-time PCR analysis of P-selectin, TNF-α, MIP-1α/CCL3, and VCAM-1 expression. Sdc1 KO and wild-type (WT) mice were treated with saline (control) or DSS for 7 days, respectively, mRNA was prepared from colon samples of five to nine mice, transcribed into cDNA, and subjected to quantitative real-time PCR analysis. A: Expression of P-selectin. B: Expression of the proinflammatory cytokine TNF-α. C: Expression of the chemokine MIP-1α/CCL3. D: Expression of the vascular adhesion molecule VCAM-1. The fold change of the mean Ct values, as calculated by the 2−(ΔΔCt) method, is shown (Supplemental Table 1, see http://ajp.amjpathol.org for Ct values and SD). ∗P < 0.05 versus WT control.
Enoxaparin Inhibits Increased Adhesion of Sdc1-Deficient PMN Cells to Endothelial Cells and to the Adhesion Molecule ICAM-1
Since leukocyte infiltration during DSS colitis was increased in Sdc1-deficient compared with wild-type mice, we next investigated leukocyte-endothelial interactions of PMN cells from these mice in a static adhesion assay. Adhesion of Sdc1-deficient PMN cells to the murine bEnd.3 endothelial cell line was increased by 25% (P < 0.05) compared with wild-type control PMNs (Figure 6A). Although treatment of wild-type PMN cells with enoxaparin (10 to 25 μg/ml) did not alter leukocyte-endothelial interactions (Figure 6B), enoxaparin treatment reduced endothelial adhesion of Sdc1 KO PMN cells by ∼35% (P < 0.05), indicating a role for the heparan sulfate chains of Sdc1 in the adhesion process (Figure 6C). We next aimed at identifying the target molecule that may mediate increased ligand binding and leukocyte adhesion in Sdc1-deficient PMN cells. Previous data from our laboratory suggested that ICAM-1 may be a prime candidate in this process,2,9,12 and pharmacological inhibition of ICAM-1 has been shown to ameliorate DSS colitis.31 Thus, we performed in vitro adhesion assays of Sdc1 KO and wild-type PMN cells to recombinant murine ICAM-1 (Figure 6D). Compared with controls, Sdc1 KO PMN cells displayed ∼45% increased adhesion to ICAM-1. Of note, similar to our PMN-endothelial adhesion assays, enoxaparin was able to decrease ICAM-1 adhesion of Sdc1-deficient PMN cells to wild-type levels (Figure 6D). Because the altered ability of granulocytes to bind to endothelium may also be caused by changes in interactions with endothelial selectins, we analyzed binding of soluble E- and P-selectin proteins to granulocytes. Wild-type and Sdc1-deficient granulocytes bound to E-selectin and P-selectin with similar efficiency (Figure 6E). Moreover, fluorescence-activated cell sorting analysis revealed no difference in the expression of the integrin subunits CD11a, CD11b, and CD49, and of the selectin ligand P-selectin glycoprotein ligand-1 between Sdc1 KO and wild-type neutrophils (results not shown).
Figure 6.

In vitro leukocyte adhesion assays and Selectin binding assays. A–D: PMNs were purified from the bone marrow of Sdc1-deficient and wild-type (WT) mice and subjected to static adhesion assays to bEnd3 endothelial cells (A–C) or immobilized ICAM-1 (D) as described in Materials and Methods. Sdc1-deficient PMNs display increased adhesion to endothelial cells and ICAM-1, which can be decreased to WT levels by enoxaparin. Enx = enoxaparin treatment. E: Selectin-binding assay. WT and Sdc1-deficient (Sdc1 KO) granulocytes were incubated with E- and P-selectin Fc chimeric proteins, respectively. Fluorescence obtained with a secondary anti-Fc antibody was measured on a flow cytometer. ∗P < 0.05 (A–C).
Normal Gut-Associated Lymphatic Tissue Composition in Sdc1-Deficient Mice
To test whether Sdc1-deficient mice exhibit an overall disturbed gut-associated lymphatic tissue, we examined the complete small bowel, colon, PPs, and spleen by H&E staining of frozen sections. No obvious changes could be detected. Moreover, we analyzed lymphocyte subsets within the lamina propria and within PPs as well as within the spleen of wild-type and Sdc1 KO mice. Flow cytometric analysis showed similar amounts of B cells, CD4 T cells, and CD8 T cells (Supplemental Figure S2, see http://ajp.amjpathol.org). Furthermore, no differences for Gr1-positive granulocytes, CD11c dendritic cells and CD25 or CD127-positive T cells were found. In addition, we were not able to find differences in the architecture of PPs between Sdc1 KO and wild-type animals. For example, the localization of B cells and T cells within the PP was not affected. The appearance of M cells in the follicle-associated epithelium of Sdc1 KO mice was comparable with that in wild-type animals. Immunohistochemical staining of macrophages in both Sdc1 KO and wild-type colon did not show differences (Supplemental Figure S2, see http://ajp.amjpathol.org).
siRNA-Mediated Knockdown of Sdc1 in Caco-2 Cells Results in Decreased bFGF-Mediated MAPK Signaling and Reduced Proliferation
Since the coreceptor function of Sdc1 for receptor tyrosine kinases1,25 may have contributed to the epithelial repair phenotype of Sdc1 KO mice, we investigated the role of Sdc1 in bFGF-mediated MAPK signal transduction and in colonic epithelial cell proliferation in vitro. siRNA-mediated silencing of Sdc1 in Caco-2 cells resulted in an >50% reduction of Sdc1 mRNA expression (Figure 7A). Low Sdc1 expression was associated with a significant reduction of cell proliferation (Figure 7B). Of note, bFGF-mediated activation of p44/42 MAPK signaling was induced in control siRNA-treated Caco-2 cells already 10 minutes after bFGF stimulation, whereas no activation was observed in Sdc1-silenced cells, confirming a coreceptor role for Sdc1 in bFGF-mediated activation of colonic epithelial cell proliferation.
Figure 7.
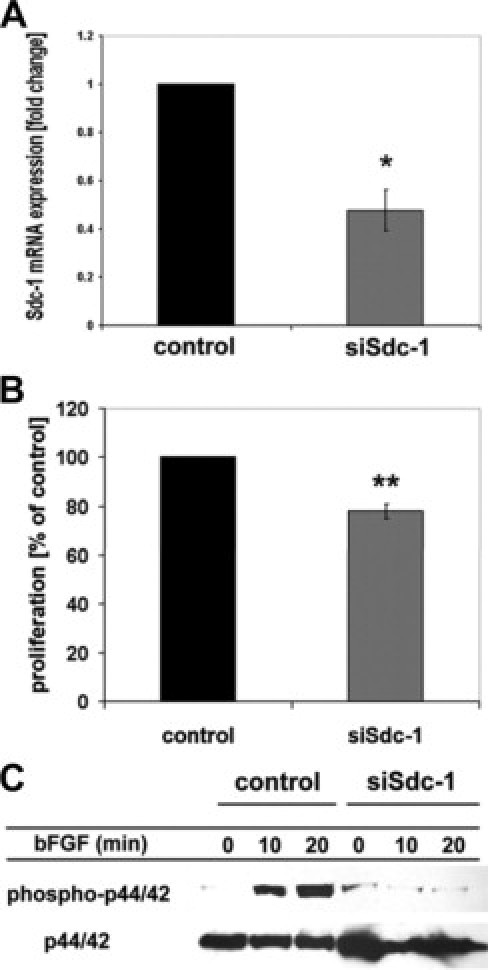
siRNA knockdown results in decreased Caco-2 cell proliferation and reduced bFGF-dependent MAPK signaling. Caco-2 cells were subjected to siRNA mediated knockdown of syndecan-1 mRNA expression as described in the Methods section. A: Quantitative real-time PCR analysis confirms successful knockdown of Sdc1 expression by >50% (n = 3, P < 0.01). B: Cell proliferation is significantly reduced in Sdc1-silenced Caco-2 cells (n = 3, P < 0.001). C: Western blot analysis of bFGF-mediated p44/42 MAPK activation in Sdc1-silenced Caco-2 cells. Caco-2 cells were transfected with a control siRNA or a Sdc1 siRNA construct and serum-starved for 24 hours after transfection. Cells were treated ±20 nmol/L bFGF for 0, 10, or 20 minutes, lysed, and analyzed by Western blotting for phospho-p44/42 MAPK and total p44/42 MAPK.
Discussion
Sdc1 and DSS Colitis
The DSS colitis of the mouse is the most established and understood model of intestinal inflammation.32 In this well-studied model, a chemical disruption of the epithelial barrier leads to alterations of the crypts, ulcerations and neutrophil infiltration in a highly reproducible setting. TNF-α plays a crucial role in this model as demonstrated by elevated levels of TNF-α33 and by the benefit of TNF-α blockage in animal and human disease models.34,35 Furthermore, disruption of the epithelial barrier leads to the production of cytokines and migration of inflammatory cells via activated endothelial cells. ICAM-1 plays a vital role in this inflammatory setting.36 Our data demonstrate differentially increased TNF-α expression in Sdc1 KO mice during DSS colitis. Stimulation of epithelial cells with the proinflammatory cytokine TNF-α induces Sdc1 down-regulation and results in its shedding after cleavage by matrix metalloproteinases, thereby enhancing cellular migration.37 Reduced Sdc1 expression is correlating with increased TNF-α levels and activity.19–21 Increased recruitment of TNF-α-producing leukocytes in Sdc1 KO mice emerges as a major mechanistic aspect of this phenotype, since Sdc1 modulates leukocyte recruitment and the inflammatory response in experimental models: in Sdc1 KO mice subjected to contact allergies, an increased expression of ICAM-1 relative to wild-type mice was found.2,12 In addition, a differentially increased leukocyte-endothelial interaction was observed in vitro and in vivo following i.p. TNF-α stimulation.9,10 Our data suggest that persistent leukocyte infiltration may have been induced by increased adhesion of Sdc1-deficient PMNs to endothelial cells and ICAM-1, concomitant with an alteration of chemokine, proinflammatory cytokine, and vascular adhesion molecule expression, such as TNF-α, P-selectin, VCAM-1, and CCL3. The migration of leukocyte subsets within the intestine was not significantly affected under noninflammatory conditions. CCL3 has been shown to be up-regulated in the acute inflammation phase of DSS colitis in C57BL/6 mice38 and in the colon tissue of IBD patients.39 Thus, differential up-regulation of CCL3 expression in Sdc1 KO mice as a secondary consequence of increased leukocyte recruitment may have promoted exaggerated inflammation. VCAM-1 was differentially up-regulated in Sdc1 KO mice during DSS colitis. Moreover, we could identify increased interaction of Sdc1-deficient neutrophils with ICAM-1 as a molecular mechanism of increased leukocyte influx leading to an overshooting inflammatory reaction as well as to a failure to resolve the leukocyte infiltrate and potentially to increased lethality of Sdc1-deficient mice in the DSS colitis model. Recent results suggest that Sdc1 modulates the direct interaction of ICAM-1 with its integrin counterreceptor, because CD18-blocking antibodies are able to reduce increased binding of Sdc1 KO PMN cells to ICAM-1.12 In contrast to ICAM-1, no difference in E-selectin and P-selectin binding was observed between Sdc1 KO and wild-type neutrophils. These findings are in accordance with previous intravital microscopy data, suggesting that integrin-mediated tight leukocyte adhesion, rather than selectin-dependent leukocyte rolling, is primarily affected in Sdc1 KO mice.10 Although heparin and heparan sulfate can modulate endothelial leukocyte recruitment in vivo at the level of selectin-mediated rolling,40 a later step of leukocyte recruitment appears to be affected in Sdc1 KO mice.
Sdc1 and Intestinal Inflammation
In this report, we were able to show that Sdc1 is essentially involved in controlling intestinal inflammation, because Sdc1-deficient mice exhibit a significantly worsened course of DSS colitis with a high lethality, failure to resolve leukocyte infiltration, and a disturbed, prolonged epithelial healing. Sdc1 is the most important HSPG of the epithelial cell surfaces,1,13 serving multiple roles in proliferation and cell repair, and as a coreceptor for cytokine and chemokine signaling.2,5,6,10,11 First evidence for the role of Sdc1 in the field of IBD came from Day et al15 in 1999, who showed immunohistochemically that Sdc1 is less expressed in ulcers of ulcerative colitis patients. In vitro, shedding of the extracellular domain of syndecan in intestinal epithelial cells disrupted fibroblast growth factor-dependent proliferation, which could be restored by the adding of the soluble ectodomain containing mainly heparan sulfate side chains as well as by adding heparin, a chemically similar molecule of the heparan sulfate side chains of the syndecans. These observations lead to the hypothesis that this altered biology of syndecans and their heparan sulfate side chains plays a role during the pathogenesis of IBD, opening the view for a new possible mechanism for the intended use of heparin in ulcerative colitis patients. In a recent study, investigating the role of syndecan for the integrity of the epithelial barrier, Bode et al41 showed that syndecan plays a critical role during protein-losing enteropathy and that the loss of Sdc1 increases basal protein leakage in a dose-dependent fashion. Furthermore, Sdc1 deficiency promoted cytokine associated protein leakage. TNF-α and Sdc1 acted synergistically in this process. Since the epithelial barrier function is essential for the pathogenesis of IBD,42 and given these data, it can be speculated that challenging the animals with DSS, triggering TNF-α production (especially in a system already lacking the protective factor of Sdc1 for the gut) promoted lethality in our model.
Sdc1 Modulates Intestinal Epithelial Cell Proliferation
Apart from increased leukocyte recruitment, the lack of Sdc1 lead to decreased cell proliferation, prolonged apoptosis and delayed tissue repair. These findings are in accordance with previous publications on the coreceptor role of Sdc1 in mitogenic growth factor signaling1,2,10,11,13 and on delayed skin and corneal wound repair in Sdc1 KO mice, which is partially due to decreased epithelial cell proliferation rates.5 Our in vitro data in Sdc1 silenced Caco-2 cells confirm and extend these findings, because Sdc1 mRNA knockdown resulted in reduced bFGF-mediated MAPK activation and reduced cell proliferation. Therefore, reduced epithelial cell proliferation due to a lack of the coreceptor function of Sdc1 for tyrosine kinase receptor emerges as an additional possible mechanism of the increased severity of DSS colitis in Sdc1 KO mice, which promotes delayed wound repair and reepithelialization.
Enoxaparin as a Substitute for Sdc1 Function
Unfortunately, the pharmacotherapy of IBD comes along with significant side effects.43 Accordingly, investigation in alternative fields of new anti-inflammatory drugs or mechanisms is mandatory. The worsened outcome of Sdc1 KO mice after DSS challenge could be reversed by treatment with the low molecular weight heparin, enoxaparin. Wild-type animals treated with enoxaparin showed a nonsignificant tendency for increased mortality compared with placebo-treated animals.
Because of its similar ligand binding properties,1,3,4 it can be expected that heparin can substitute for a variety of biological functions of HSPG. For example, both heparin and the Sdc1 ectodomain can be a coreceptor of the fibroblast growth factor in epithelial cell lines.15,25 It was suggested that heparan sulfate might be secreted by epithelial cells to retain an intact intestinal barrier after shedding of dead epithelial cells.44 A lack of this mechanism might contribute to the worse outcome of Sdc1 KO mice, and substitution by heparin might restore this mechanism. Different work groups were able to reproduce the positive effect of heparan sulfate glycosaminoglycans, especially heparins and heparin derivatives in colitis models, eg, in trinitrobenzene sulfonic acid or oxazolone induced colitis.14,45 However, since soluble heparin can act as a mimic of both cis- (cell surface) and trans- (shed) conformations of Sdc1, we cannot fully differentiate between possible differential roles of soluble and membrane-bound forms of Sdc1 in the DSS colitis model. In addition, it has to be considered that heparin may also mimic the effects of other HSPGs. However, very little is known about the role of these HSPGs in the context of colonic inflammation. Because of the less-extended conformation of the glypican core proteins and the closer localization of their HS chains to the plasma membrane,1 the HS chains of Sdc1 are more likely to form the first line of binding sites for extracellular ligands than glypican HS. Basement membrane HSPG such as perlecan may play a role in sustaining epithelial integrity and barrier function; however, they should be similarly affected in Sdc1 KO and wild-type mice during DSS colitis.
Mechanistically, additional aspects of heparin or heparin-derivative effects, such as TNF-α neutralization,46 inhibition of P-selectin-mediated leukocyte migration,47 or CD14 dependent macrophage activation,48 need to be considered.
Finally, the beneficial effect of heparin on TNF-α induced protein-losing enteropathy,19 may have contributed to the improved outcome in Sdc1 KO mice, because Sdc1 deficiency exacerbates protein-losing enteropathy. In our experimental model, enoxaparin treatment not only helped to decrease inflammation in the colon but also effectively abolished increased leukocyte-endothelial interactions and ICAM-mediated leukocyte adhesion in vitro. Enoxaparin could inhibit leukocyte-endothelial interactions as efficiently as conventional heparin.9 The differential modulation of leukocyte recruitment by heparin in Sdc1 KO and wild-type mice may therefore represent an important mechanistic aspect of the beneficial effect of heparin in this model.
A few clinical trials with heparin treatment in steroid-resistant ulcerative colitis were performed. In some cases, this therapeutic approach was successful.16,17 However, a meta-analysis18 could not prove efficacy in the treatment of patients with ulcerative colitis.
With respect to the underlying causes of decreased Sdc1 expression in ulcerative colitis patients,13 it is not yet clear if the reduced Sdc1 expression is a secondary consequence of the active colitis or if a pre-existing reduction in Sdc1 expression makes a subgroup of patients more susceptible for ulcerative colitis. In the latter case, a differential diagnosis of these patients may be required, which may constitute a potential limitation for a general therapeutic application of heparinoids in IBD. At the same time, the existence of such a subgroup could serve to explain controversial results on the efficacy of heparin therapy in IBD. Therefore, further histopathological studies on these subjects would be desirable.
Conclusions
The observation of a worse course of DSS colitis in Sdc1-deficient mice supports the hypothesis that altered expression of the main HSPG, Sdc1, plays a substantial role during the process of colon inflammation. Heparin derivatives and formulations with increased potency of targeting heparan-sulfate associated anti-inflammatory mechanisms have to be evaluated, because different mechanisms connected to Sdc1 function are beneficial in this process. Our findings show that HSPGs such as Sdc1 contribute to the mucosal integrity by affecting epithelial cell function, cell proliferation, expression of chemokines, and cytokines, as well as by influencing the function of cell adhesion molecules. Mechanistically, the lack of Sdc1 promoted increased leukocyte recruitment under inflammatory conditions, possibly by removal of a steric hindrance of ICAM-1-counterreceptor interactions. Increased leukocyte recruitment in vivo promoted differentially increased expression of cytokines and chemokines, among which TNF-α is known to be a major player in the pathogenesis of DSS colitis. The lack of Sdc1 further hampered efficient wound repair, because it is required for tyrosine kinase receptor-mediated mitogenic signaling. Enoxaparin is able to reduce lethality in the course of DSS colitis in Sdc1 KO mice. In addition, enoxaparin modifies the complex biological dysfunction of missing Sdc1 during leukocyte recruitment in vitro. Therefore, targeting HSPG interactions might provide novel therapeutic approaches for the treatment of IBD, with a potential benefit for patient collectives displaying low colonic Sdc1 expression.
Acknowledgements
We thank Drs. Dietmar Vestweber and Georg Varga for discussions, Ruth Seelige for performing the P-selectin ligand binding assay, and Mrs. Birgit Pers, Sonja Dufentester, Monika Offers, and Ruth Goez for expert technical assistance.
Footnotes
Supported by Münster University Hospital grant “Innovative Medizinische Forschung” IMF GÖ 120415 (to M.G. and T.K.).
M.F. and M.G. contributed equally to this study.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
TaqMan®low density array and-real time PCR analysis of inflammation-related gene expression in colon samples from control and DSS-treated mice.
References
- 1.Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Götte M. Syndecans in inflammation. FASEB J. 2003;6:575–5913. doi: 10.1096/fj.02-0739rev. [DOI] [PubMed] [Google Scholar]
- 3.Yip GW, Smollich M, Götte M. Therapeutic value of glycosaminoglycans in cancer. Mol Cancer Ther. 2006;5:2139–2148. doi: 10.1158/1535-7163.MCT-06-0082. [DOI] [PubMed] [Google Scholar]
- 4.Bishop JR, Schuksz M, Esko JD. Heparan sulfate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 5.Stepp MA, Gibson HE, Gala PH, Iglesia DD, Pajoohesh-Ganji A, Pal-Ghosh S, Brown M, Aquino C, Schwartz AM, Goldberger O, Hinkes MT, Bernfield M. Defects in keratinocyte activation during wound healing in syndecan-1 deficient mouse. J Cell Sci. 2002;115:4517–4531. doi: 10.1242/jcs.00128. [DOI] [PubMed] [Google Scholar]
- 6.Vanhoutte D, Schellings MW, Götte M, Swinnen M, Herias V, Wild MK, Vestweber D, Chorianopoulos E, Cortés V, Rigotti A, Stepp MA, Van de Werf F, Carmeliet P, Pinto YM, Heymans S. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation. 2007;115:475–482. doi: 10.1161/CIRCULATIONAHA.106.644609. [DOI] [PubMed] [Google Scholar]
- 7.Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- 8.Götte M. Syndecans as modulators of chemokine function. In: Linkes WP, editor. Progress in Chemokine Research. Nova Science Publishers; Hauppauge, NY: 2007. p. 85. [Google Scholar]
- 9.Götte M, Bernfield M, Joussen AM. Increased leukocyte-endothelial interactions in syndecan-1 deficient mice involve heparan-sulfate dependent and independent steps. Curr Eye Res. 2005;6:417–422. doi: 10.1080/02713680590956289. [DOI] [PubMed] [Google Scholar]
- 10.Götte M, Joussen AM, Klein C, Andre P, Wagner DD, Hinkes MT, Kirchhof B, Adamis AP, Bernfield M. Role of syndecan-1 in leukocyte-endothelial interactions in the ocular vasculature. Invest Ophthalmol Vis Sci. 2002;43:1135–1141. [PubMed] [Google Scholar]
- 11.Parish CR. Heparan sulfate and inflammation. Nat Immunol. 2005;6:861–862. doi: 10.1038/ni0905-861. [DOI] [PubMed] [Google Scholar]
- 12.Kharabi Masouleh B, Ten Dam GB, Wild MK, Seelige R, van der Vlag J, Rops AL, Echtermeyer FG, Vestweber D, van Kuppevelt TH, Kiesel L, Götte M. Role of the heparan sulfate proteoglycan syndecan-1 (CD138) in delayed-type hypersensitivity. J Immunol. 2009;182:4985–4993. doi: 10.4049/jimmunol.0800574. [DOI] [PubMed] [Google Scholar]
- 13.Day R, Ilyas M, Daszak P, Talbot I, Forbes A. Expression of syndecan-1 in inflammatory bowel disease and a possible mechanism of heparin therapy. Dig Dis Sci. 1999;44:2508–2515. doi: 10.1023/a:1026647308089. [DOI] [PubMed] [Google Scholar]
- 14.Pellequer Y, Meissner Y, Ubrich N, Lamprecht A. Epithelial heparin delivery via microspheres mitigates experimental colitis in mice. J Pharmacol Exp Ther. 2007;321:726–733. doi: 10.1124/jpet.106.117226. [DOI] [PubMed] [Google Scholar]
- 15.Day R, Forbes A. Heparin, cell adhesion, and pathogenesis of inflammatory bowel disease. Lancet. 1999;354:62–6514. doi: 10.1016/S0140-6736(98)09267-8. [DOI] [PubMed] [Google Scholar]
- 16.Evans RC, Wong VS, Morris AI, Rhodes JM. Treatment of corticosteroid-resistant ulcerative colitis with heparin—a report of 16 cases. Aliment Pharm Ther. 1997;1:1037–1040. doi: 10.1046/j.1365-2036.1997.00252.x. [DOI] [PubMed] [Google Scholar]
- 17.Gaffney P, Caffney Doyle A, Hogan J, Hayes D, Annis P. Paradoxical response to heparin in 10 patients with ulcerative colits. Am J Gastroenterol. 1995;90:220–223. [PubMed] [Google Scholar]
- 18.Malhotra S, Kondal A, Shafiq N, Sidhu S, Bhasin DK, Pandhi P. A comparison of observational and controlled trials of heparin in ulcerative colitis. Int J Clin Pharmacol Ther. 2004;42:690–694. doi: 10.5414/cpp42690. [DOI] [PubMed] [Google Scholar]
- 19.Bode L, Eklund EA, Murch S, Freeze HH. Heparan sulfate depletion amplifies TNF-α-induced protein leakage in an in vitro model of protein-losing enteropathy. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1015–G1023. doi: 10.1152/ajpgi.00461.2004. [DOI] [PubMed] [Google Scholar]
- 20.Principi M, Day R, Marangi S, Burattini O, De Francesco V, Ingrosso M, Pisani A, Panella C, Forbes A, Di Leo A, Francavilla A, Ierardi E. Differential immunohistochemical expression of syndecan-1 and tumor necrosis factor α in colonic mucosa of patients with Crohn's disease. Immunopharmacol Immunotoxicol. 2006;28:185–195. doi: 10.1080/08923970600815048. [DOI] [PubMed] [Google Scholar]
- 21.Bode L, Salvestrini C, Park PW, Li JP, Esko JD, Yamaguchi Y, Murch S, Freeze HH. Heparan sulfate plays a central role in a dynamic in vitro model of protein-losing enteropathy. J Biol Chem. 2006;281:7809–7815. doi: 10.1074/jbc.M510722200. [DOI] [PubMed] [Google Scholar]
- 22.Anthoni C, Laukoetter MG, Rijcken E, Vowinkel T, Mennigen R, Müller S, Senninger N, Russell J, Jauch J, Bergmann J, Granger DN, Krieglstein CF. Mechanisms underlying the anti-inflammatory actions of boswellic acid derivates in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1131–G1137. doi: 10.1152/ajpgi.00562.2005. [DOI] [PubMed] [Google Scholar]
- 23.Hausmann M, Obermeier F, Paper DH, Balan K, Dunger N, Menzel K, Falk W, Schoelmerich J, Herfarth H, Rogler G. In vivo treatment with the herbal phenylethanoid acteoside ameliorates intestinal inflammation in dextran sulfate sodium-induced colitis. Clin Exp Immunology. 2007;148:373–381. doi: 10.1111/j.1365-2249.2007.03350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lügering A, Floer M, Westphal S, Maaser C, Spahn TW, Schmidt MA, Domschke W, Williams IR, Kucharzik T. Absence of CCR6 inhibits CD4+ regulatory T cell development and M cell formation in inside Peyer's patches. Am J Pathol. 2005;166:1647–1654. doi: 10.1016/S0002-9440(10)62475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolova V, Koo CY, Ibrahim SA, Wang Z, Spillmann D, Dreier R, Kelsch R, Fischgräbe J, Smollich M, Rossi LH, Sibrowski W, Wülfing P, Kiesel L, Yip GW, Götte M. Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis. 2009;30:397–407. doi: 10.1093/carcin/bgp001. [DOI] [PubMed] [Google Scholar]
- 26.Seidler DG, Faiyaz-Ul-Haque M, Hansen U, Yip GW, Zaidi SH, Teebi AS, Kiesel L, Götte M. Defective glycosylation of decorin and biglycan, altered collagen structure, and abnormal phenotype of the skin fibroblasts of an Ehlers-Danlos syndrome patient carrying the novel Arg270Cys substitution in galactosyltransferase I (β4GalT-7) J Mol Med. 2006;84:583–594. doi: 10.1007/s00109-006-0046-4. [DOI] [PubMed] [Google Scholar]
- 27.Montesano R, Pepper MS, Möhle-Steinlein U, Risau W, Wagner EF, Orci L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell. 1990;62:435–445. doi: 10.1016/0092-8674(90)90009-4. [DOI] [PubMed] [Google Scholar]
- 28.Varga G, Balkow S, Wild MK, Stadtbaeumer A, Krummen M, Rothoeft T, Higuchi T, Beissert S, Wethmar K, Scharffetter-Kochanek K, Vestweber D, Grabbe S. Active MAC-1 (CD11b/CD18) on DCs inhibits full T cell activation. Blood. 2007;109:661–669. doi: 10.1182/blood-2005-12-023044. [DOI] [PubMed] [Google Scholar]
- 29.Hahne M, Jäger U, Isenmann S, Hallmann R, Vestweber D. Five tumor necrosis factor-inducible cell adhesion mechanisms on the surface of mouse endothelioma cells mediate the binding of leukocytes. J Cell Biol. 1993;121:655–664. doi: 10.1083/jcb.121.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gotsch U, Borges E, Bosse R, Boggemeyer E, Simon M, Mossmann H, Vestweber D. VE-cadherin antibody accelerates neutrophil recruitment in vivo. J Cell Sci. 1997;110:583–588. doi: 10.1242/jcs.110.5.583. [DOI] [PubMed] [Google Scholar]
- 31.Panés J, Aceituno M, Gil F, Miquel R, Piqué JM, Salas A, McLean P. Efficacy of an inhibitor of adhesion molecule expression (GI270384X) in the treatment of experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G739–G748. doi: 10.1152/ajpgi.00059.2007. [DOI] [PubMed] [Google Scholar]
- 32.Cooper HS, Murthy SNS, Shah RS, Sedergam DJ. Clinicopathologic study of dextran sulphate sodium experimental murine colitis. Lab Invest. 1993;69:238. [PubMed] [Google Scholar]
- 33.Boismenu R, Chen Y, Chou K, El-Sheikh A, Buelow R. Orally administered RDP 58 reduces the severity of dextran sodium sulphate induced colitis. Ann Rheum Dis. 2002;61(Suppl 2):ii19–ii24. doi: 10.1136/ard.61.suppl_2.ii19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kojouharoff G, Hans W, Obermeier F, Männel DN, Andus T, Schölmerich J, Gross V, Falk W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin Exp Immunol. 1997;107:353–358. doi: 10.1111/j.1365-2249.1997.291-ce1184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandborn WJ, Hanauer SB. Antitumor necrosis factor therapy for inflammatory bowel disease: a review of agents, pharmacology, clinical results, and safty. Inflamm Bowel Dis. 1999;5:119–133. doi: 10.1097/00054725-199905000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Taniguchi T, Tsukada H, Nakamura H, Kodama M, fukada K, saito Y, Miyasaka M, Seino Y. Effects of the anti-ICAM-1 monoclonal antibody on dextran sodium sulphate-induced colitis in rats. J Gastroenterol Hepatol. 1998;13:945–949. doi: 10.1111/j.1440-1746.1998.tb00766.x. [DOI] [PubMed] [Google Scholar]
- 37.Day RM, Mitchell TJ, Knight SC, Forbes A. Regulation of epithelial syndecan-1 expression by inflammatory cytokines. Cytokine. 2003;21:224–233. doi: 10.1016/s1043-4666(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 38.Melgar S, Drmotova M, Rehnström E, Jansson L, Michaëlsson E. Local production of chemokines and prostaglandin E2 in the acute, chronic and recovery phase of murine experimental colitis. Cytokine. 2006;35:275–283. doi: 10.1016/j.cyto.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Banks C, Bateman A, Payne R, Johnson P, Sheron N. Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn's disease. J Pathol. 2003;199:28–35. doi: 10.1002/path.1245. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 41.Bode L, Salvestrini C, Park PW, Li JP, Esko JD, Yamaguchi Y, Murch S, Freeze HH. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest. 2008;118:229–238. doi: 10.1172/JCI32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wehkamp J, Koslowski M, Wang G, Stange EF. Barrier dysfunction due to distinct defensin deficiencies in small intestinal and colonic Crohn's disease. Mucosal Immunol. 2008;1(Suppl 1):67–74. doi: 10.1038/mi.2008.48. [DOI] [PubMed] [Google Scholar]
- 43.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 44.Watson AJ, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, McKenna M, Rose T, Montrose MH. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129:902–912. doi: 10.1053/j.gastro.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 45.Belmiro CL, Castelo-Branco MT, Melim LM, Schanaider A, Elia C, Madi K, Pavão MS, de Souza HS. Unfractionated heparin and new heparin analogues from ascidians (chordate-tunicate) ameliorate colitis in rats. J Biol Chem. 2009;284:11267–11278. doi: 10.1074/jbc.M807211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bendayan I, Casaldaliga J, Castello F, Miro Heparin therapy and reversal of protein-losing enteropathy in a case with congenital heart disease. Pediatr Cardiol. 2000;21:267–268. doi: 10.1007/s002460010055. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Broen J, Varki A, Esko J. Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J Clin Invest. 2002;110:127–136. doi: 10.1172/JCI14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dziarski R, Gupta D. Heparin, sulfated heparinoids, and lipoteichoic acids bind to the 70-kDa peptidoglycan/lipopolysaccharide receptor protein on lymphocytes. J Biol Chem. 1994;269:2100–2110. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TaqMan®low density array and-real time PCR analysis of inflammation-related gene expression in colon samples from control and DSS-treated mice.