

Abstract
Plasmodium parasites lacking plasmepsin 4 (PM4), an aspartic protease that functions in the lysosomal compartment and contributes to hemoglobin digestion, have only a modest decrease in the asexual blood-stage growth rate; however, PM4 deficiency in the rodent malaria parasite Plasmodium berghei results in significantly less virulence than that for the parental parasite. P. berghei Δpm4 parasites failed to induce experimental cerebral malaria (ECM) in ECM-susceptible mice, and ECM-resistant mice were able to clear infections. Furthermore, after a single infection, all convalescent mice were protected against subsequent parasite challenge for at least 1 year. Real-time in vivo parasite imaging and splenectomy experiments demonstrated that protective immunity acted through antibody-mediated parasite clearance in the spleen. This work demonstrates, for the first time, that a single Plasmodium gene disruption can generate virulence-attenuated parasites that do not induce cerebral complications and, moreover, are able to stimulate strong protective immunity against subsequent challenge with wild-type parasites. Parasite blood-stage attenuation should help identify protective immune responses against malaria, unravel parasite-derived factors involved in malarial pathologies, such as cerebral malaria, and potentially pave the way for blood-stage whole organism vaccines.
The digested vacuole (DV) of malaria parasites performs hemoglobin degradation, which is a crucial process for parasite growth and survival within the host erythrocyte. In Plasmodium falciparum, the most important human malaria parasite, this is achieved with the contribution of several digestive vacuole proteases including three aspartic proteinases, the plasmepsins (PM) PfPM1, PfPM2, and PfPM4 and one histo-aspartic protease, PfHAP.1–5 The plasmepsins have long been studied as potential drug targets and subjected to functional and biochemical studies with the hope that inhibiting them would halt hemoglobin digestion and result in parasite death. Surprisingly, the systematic disruption of either individual or different combinations of the plasmepsin genes did not result in any striking growth defect. Presumably, this is due to redundant enzyme systems for digesting hemoglobin, which involve cysteine proteases, metalloproteases, and aminopeptidases, and to the presence of multiple pathways for the uptake of extracellular amino acids.6–8 The P. falciparum and Plasmodium reichnowi clades differ from other Plasmodium species in that they have four genes encoding DV plasmepsins. In P. falciparum only the disruption of all four plasmepsin genes, which eliminates all aspartic protease activity from the DV, resulted in delayed in vitro schizont maturation accompanied by reduced formation of hemozoin (an insoluble crystal produced during hemoglobin degradation) and less efficient processing of endosomal vesicles in the DV.4
We here investigated the impact of the loss of the various functions of the DV plasmepsins on parasite virulence by disrupting the single gene encoding the DV plasmepsin 4 (pm4) in the rodent malaria parasite Plasmodium berghei. This parasite is a well established and tractable model to study the function of Plasmodium genes in vivo and replicates several key features of human cerebral malaria.9,10 The phenotypic analysis of loss-of-function mutants has been used to gain an insight into a variety of host-parasite interactions.11 In this study, we confirm that the disruption of PM4, which results in loss of all aspartic proteinase activity targeted to its lysosomal compartments, has only a modest effect on the intraerythrocytic development of P. berghei parasites, but we observed dramatic differences in the virulence of these parasites compared with that of wild-type parasites. Specifically, we report the growth and multiplication characteristics of Δpm4 parasites in different mouse strains and demonstrate that these parasites neither induce experimental cerebral malaria (ECM) in ECM-susceptible mice nor kill the host by hemolytic anemia in ECM-resistant mice. In these latter mice, Δpm4 parasites induce a self-resolving infection, which generates spleen-dependent protective immune responses. This is the first report of a mutant P. berghei parasite that does not induce cerebral complications as the result of a single gene mutation.
Materials and Methods
Parasites
A number of mutant parasite lines carrying a disrupted P. berghei pm4 locus (PB000298.03.0) were independently generated in different laboratories. Additional parasite lines expressing the green fluorescent protein (GFP)-luciferase fusion protein were generated either on the wild-type or on the Δpm4 background. Parasites of the P. berghei ANKA clone 2.34 and clone cl15cy112 have been used as a control (wild-type) and for the generation of the Δpm4 mutant lines. In addition, these parasites have been used to generate the transgenic wild type (wt+) parasites (1037cl1 line) and wt++ parasites (gfp-luc/cl2 line) that express a fusion protein (GFP-Luc) encompassing the GFP (mutant3) and the luciferase (LUC-IAV) coding sequence.
Δpm4cl1 and Δpm4cl6 are two Δpm4 parasite lines deficient in expressing PM4. The pm4 gene has been disrupted by introducing the construct pRSpm4 into the genome of the P. berghei ANKA clone 2.34 as described below.
688cl2 and 688cl3 are two Δpm4 parasite lines deficient in expressing PM4. The pm4 gene has been disrupted by introducing construct pL1095 into the genome of cl15cy1 parasites as described below.
wt+ (or 1037cl1) is a reference transgenic parasite line that expresses GFP-Luc under the control of the schizont-specific ama-1 promoter. The gfp-luc is inserted into the p230p locus (PB000214.00.0) on chromosome 3 of parasites of cl15cy1. This line does not contain a drug-selectable marker and has been selected by flow-sorting of GFP-expressing parasites directly after transfection as described below.
1092cl4 and 1092cl6 are two Δpm4+ parasite lines deficient in expressing PM4. The pm4 gene has been disrupted by introducing construct pL1095 into the genome of transgenic parasites of line 1037cl1. Both lines express GFP-luciferase under the control of the schizont specific ama-1 promoter.
wt++ is a reference transgenic parasite line that expresses GFP-Luc under the control of the schizont-specific ama-1 promoter. The transgene is inserted into the c-/d-rrna gene unit by single crossover recombination as described below. This line contains a tgdhfr/ts drug-selectable marker cassette.
Mice
Swiss-OF1 mice (OF1 ico, construct 242, age 6 weeks, Charles River Laboratories, Inc., Wilmington, MA), C57BL/6 (age 6 weeks, Charles River Laboratories, Inc.), CBA/J (age 6 to 8 weeks, Charles River Laboratories, Inc.), Swiss-CD1 (age 6 to 8 weeks; Harlan Sprague-Dawley, Indianapolis, IN), BALB/c (age 6 to 8 weeks; Harlan Sprague-Dawley, Indianapolis, IN), and NIH Swiss (age 6 weeks; Harlan Sprague-Dawley, Indianapolis, IN) were used. All studies in which animals are involved have been performed according to the regulations of the Dutch “Animal On Experimentation Act” and guidelines 86/609/EEG, the Italian regulation D.L 27 January 1992, n. 116, and the US Public Health Service Policy on Humane Care and Use of Laboratory Animals, as approved by the Institutional Animal Care and Use Committee of the University of Florida.
Generation of Δpm4 parasites
Parasite Lines Δpm4cl1 and Δpm4cl6
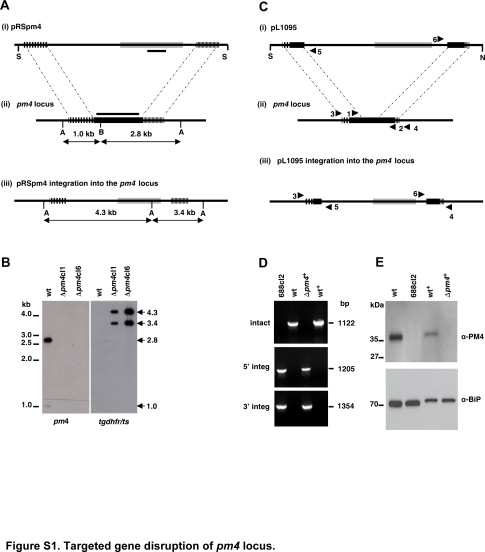
These parasite lines were generated using the DNA construct pRSpm4. This construct contains the following elements (Supplemental Figure S1A, see http://ajp.amjpathol.org): i) a5′-untranslated region (UTR) 954-bp PCR fragment of the pm4 (sense 5′-CCGGGCCCTACAAAATATTTTCATAAGTTGGC-3′ [ApaI site is underlined] and antisense 5′-CCATCGATTTCCATTTTGAACTAATTAAAG-3′ [ClaI site is underlined]); ii) a 3′-UTR 813-bp PCR fragment of the pm4 (sense 5′-GGGAATTCTTATATATGATATATTACACGTAC-3′ [EcoRI site is underlined] and antisense 5′-CGGGATCCATGGTTTTACGATTTAAACTTTC-3′ [BamHI site is underlined]); and iii) the tgdhfr/ts drug-selectable marker cassette (Supplemental Figure S1A, see http://ajp.amjpathol.org). The plasmid was linearized with SacII and PvuII and used for the generation of Δpm4 lines. For the generation of Δpm4cl1 and Δpm4cl6, blood stages of the P. berghei ANKA clone 2.34 parasites were transfected with 10 μg of DNA linear fragments of pRSpm4, and mutant parasites were obtained by the standard method of drug (pyrimethamine) selection in mice.13 Pyrimethamine-resistant parasites were subsequently cloned by limiting dilution. The disruption of the pm4 locus was confirmed by diagnostic PCR (data not shown) and by Southern blot analysis of BamHI (B)- and AvaII (A)-digested genomic DNA (Supplemental Figure S1B, see http://ajp.amjpathol.org). Loss of expression of PM4 was confirmed by Western blot analysis as described below (data not shown).
Parasite Lines 688cl2, 688cl3, 1092cl4, and 1092cl6
These parasite lines were generated using the DNA construct pL1095. To generate the construct, we first amplified by PCR ∼700-bp fragments from either end of the pm4 locus. The primers used to amplify the 5′-UTR 754-bp fragment encoding from −311 to 443 bp were the following: sense 5′-GCATGGTACCCCTTATTAAAGAGATTGGGAAGC-3′ and antisense 5′-GCATATCGATTTTCCTAATTCTGCAGTACC-3′, flanked with engineered KpnI and ClaI restriction sites (underlined). The 3′-UTR 680-bp fragment was amplified using the following primers: sense 5′-GCATGAATTCCAGGACAAATTGAAAATGCAG-3′ and antisense 5′-GCATCCGCGGATAAATTTCTTTAATCTTATGGC-3′. The PCR product contained 545 bp of coding sequence, extending 135 bp into the 3′-UTR and flanked by EcoRI and SacII restriction sites (underlined). These PCR products were directionally cloned into the pL0001 plasmid backbone using the restriction sites described above, placing the fragments into the construct as shown in Supplemental Figure S1C (see http://ajp.amjpathol.org), flanking the Toxoplasma gondii dihydrofolate reductase/thymidylate synthase (tgdhfr/ts) drug-selectable marker cassette. This plasmid was linearized by cleavage with ScaI, NaeI, and SapI, and the ∼6.5-kb fragment was purified by agarose gel electrophoresis.
For the generation of mutant lines 688, blood-stage parasites of cl15cy1 were transfected with 5 to 10 μg of gel-purified DNA linear fragments of pL1095, and mutant parasites were obtained by the standard method of drug (pyrimethamine) selection in mice.13 Pyrimethamine-resistant parasites were subsequently cloned by limiting dilution. The correct disruption of pm4 in two clones examined (688cl2 and 688cl3) was confirmed by the failure of PCR to amplify the expected 1122-bp from the central region of the pm4 locus using primer pair 1 + 2 (primer 1: 5′-TCCGAATATTTAACAATTCGTGC-3′ and primer 2: 5′-GTTTTTTGCAACTGCAAAACC-3′) under conditions for which this fragment was readily amplified from parental lines (Supplemental Figure S1D, see http://ajp.amjpathol.org). The expected integration into the pm4 locus was verified by successfully amplifying the predicted 5′ and 3′ boundary sequences using primer pairs 3 + 5 (primer 3: 5′-TTCCCTTGTGTCCTTTAAG-3′ and primer 5: 5′-CGCATTATAGAGTTCATTTTAC-3′) and 6 + 4 (primer 4: 5′-AAGCGGAGTTTATTGTCTGTC-3′ and primer 6: 5′-CACATAAAATGGCTAGTATGAATAG-3′) as shown in Supplemental Figure S1D (see http://ajp.amjpathol.org). Southern blot analysis of digested DNA from wild-type, 688cl2, and 688cl3 further confirmed the predicted integration event (data not shown).
For the generation of mutant line Δpm4+ blood-stage parasites of line 1037cl1 (wt+ parasites) were transfected, selected, and analyzed as described for line 688, resulting in the selection of a transgenic parasite line that expresses GFP-Luc and is deficient in PM4 expression. Two mutant clones of this line, 1092cl2 and 1092cl4, have been selected for further analysis.
The loss of PM4 expression was confirmed by Western analysis of total protein extracted from saponin-lysed blood-stage parasites. Samples were extracted in an equal volume of SDS sample buffer (100 mmol/L Tris-Cl, pH 6.8, 10% glycerol, 2% SDS, and 100 mmol/L 2-mercaptoethanol), boiled for 5 minutes, and separated by electrophoresis on 10% acrylamide gels (Bio-Rad Laboratories, Hercules, CA). After electrophoresis, proteins in the gel were transferred to a 0.2 μmol/L polyvinylidene difluoride membrane. The membrane was blocked for 24 hours (100 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 0.1% Tween 20, and 5% powdered milk) before incubation with primary antibody. Anti-PM4 antibodies were raised in a rabbit immunized with the predicted N-terminal peptide of the enzyme (KYEDSIELDQSLGLSC) cross-linked to carrier protein (keyhole limpet hemocyanin) and used at a dilution of 1:7,500 in blocking solution. Antibody bound to the membrane was visualized on X-ray film after incubation with a secondary antibody, goat anti-rabbit (horseradish peroxidase-conjugated), and reaction with the SuperSignal West Dura Extended Duration Substrate (Pierce Chemical, Rockford, IL). Equal loading of parasite protein samples was confirmed using anti-BiP antisera (1:10,000 dilution) provided by J. B. Adams and obtained from the Malaria Research and Reference Reagent Resource Center (MR4) (Manassas, VA), followed by incubation with goat anti-rat (horseradish peroxidase-conjugated) and developed with the chemiluminescent substrate, as above (Supplemental Figure S1E, see http://ajp.amjpathol.org).
Generation of Parasites Lines Expressing Luciferase
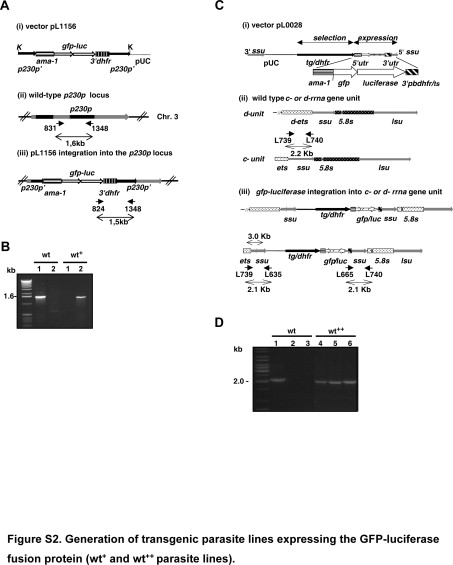
The transgenic parasite line 1037cl1 (wt+ parasites) contains the gfp-luc integrated into the p230p locus (PB000214.00.0) under the control of the schizont-specific ama1 promoter. The construct pL1156 is integrated by double crossover recombination and does not contain a drug-selectable marker gene. The DNA construct for the generation of this reporter line (Supplemental Figure S2A, see http://ajp.amjpathol.org) was made by replacing the α-tubulin II promoter of pL002414 with the ama1 promoter (EcoRV/BamHI fragment of pL0010). The ama1-gfpm3 (EcoRV/KpnI) fragment of this plasmid was subcloned in plasmid BSSK to create plasmid BSSKama1-gfp-3′utr. Finally, the eef1aa-gfp-3′utr cassette of plasmid pL0023 (pbGFP-Lucko230p-SMCON12) was replaced with the PstI/KpnI ama1-gfp-3′utr fragment of BSSKamaI-gfp-3′utr to obtain pL1141. Subsequently, the gfpm3-luc-3′utr (HpaI/KpnI) fragment of pL0028 (pPbgfp-lucSCH15) was introduced into pl1141 to create pL1156. The plasmids pL0024, pL0010, pL0023, and pL0028 can be obtained from MR4. Blood-stage parasites (cl15cy1) were transfected with 5 μg of linear fragments of KspI-digested plasmid pL1156,13 and transgenic parasites were selected by flow-sorting of GFP-expressing parasites as described.16 Flow-sorted, GFP-Luc-expressing parasites were subsequently cloned by limiting dilution. Correct integration of the construct into the p230p locus was analyzed by PCR using the following primers 831, 5′-CTTTATTTTTCAATTACCGCC-3′, 1348,13 and 824, 5′-CCAAGAAGGGCGGAAAGATC-3′, and by Southern analysis of restricted DNA (data not shown). Primers and sizes of the products are indicated in Supplemental Figure S2, A and B (see http://ajp.amjpathol.org). Correct timing of expression of GFP-Luc in schizonts was analyzed by determination of GFP expression during synchronized development of blood stages in live parasites using a fluorescence microscope (Leica DMRA HC “upright” microscope) as described.15
The transgenic parasite wt++ line (gfp-luc/cl2) contains the gfp-luc integrated into the c-/d-rrna gene unit under the control of the schizont-specific ama1 promoter. These parasites were generated using plasmid pL0028 (MRA-797, MR4)15 (Supplemental Figure S2C, see http://ajp.amjpathol.org). Correct integration of the construct was analyzed by diagnostic PCR as described previously17 (Supplemental Figure S2D, see http://ajp.amjpathol.org) and Southern analysis of restricted DNA (data not shown).
Growth and Multiplication of Δpm4 Blood Stages
The asexual multiplication rate in vivo, determined during the cloning procedure, is calculated as follows. The percentage of infected erythrocytes in mice injected with a single parasite is determined at days 8 to 11 by counting Giemsa-stained blood films. The mean asexual multiplication rate per 24 hours is then calculated, assuming a total of 1.2 × 1010 erythrocytes/mouse (2 ml of blood). The percentage of infected erythrocytes in mice infected with reference lines of the ANKA strain of P. berghei consistently ranges between 0.5 and 2% at day 8 after infection, resulting in a mean multiplication rate of 10 per 24 hours.18
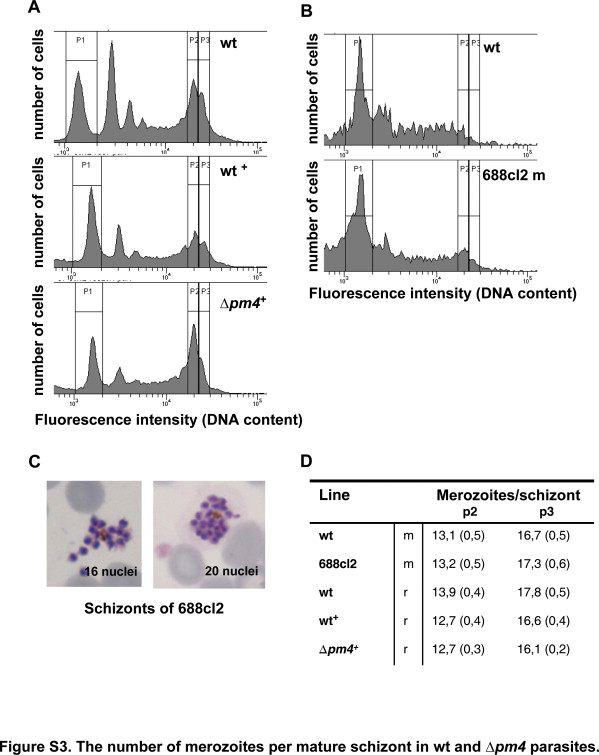
For determination of the number of merozoites per schizont, infected blood samples containing schizonts are stained with the DNA-specific, fluorescent dye Hoechst 33258 and analyzed by flow cytometry.18,19 Infected blood (1 to 3% parasitemia) is collected from Swiss-OF1 mice or Wistar rats by heart puncture and cultured overnight under standard culture conditions for collecting of P. berghei schizonts.20 After overnight culture the infected erythrocytes are separated from uninfected cells by Nycodenz density centrifugation as described13 and fixed in 0.25 (v/v) glutaraldehyde solution in PBS. These samples are stained with Hoechst 33258 at a concentration of 2 μmol/L for 1 hour at 37°C and analyzed with a FACScan (LSR II, Becton Dickinson, San Jose, CA). UV excitation of the Hoechst 33258 dye is performed with an argon ion laser (450/50 nm). The fluorescence intensity of a total of 50,000 cells per sample is measured, and data analysis is performed using CellQuest software (Becton Dickinson). The mean fluorescence intensity of free merozoites and/or ring-infected erythrocytes (first peak [P1] in the fluorescence histograms) (Supplemental Figure S3, see http://ajp.amjpathol.org) is proportional to the haploid DNA value18 and is set at 1. The number of merozoites per schizont is calculated by dividing the mean fluorescence intensity of mature schizonts (peaks P2 and P3 in the histograms) (Supplemental Figure S3, see http://ajp.amjpathol.org) by the mean fluorescence intensity of the merozoites/ring forms.18
The length of the asexual blood stage is determined in standard short-term cultures of synchronized P. berghei blood stages.18 In brief, cultured and purified schizonts, collected as described by Janse et al,13 are injected i.v. into the tail veins of mice. In these animals, merozoites invade within 4 hours after injection of the schizonts, giving rise to synchronized in vivo infections with a parasitemia of 0.5 to 3%, containing mainly (>90%) ring form parasites. At 2 to 4 hours after injection of the schizonts, infected blood is collected from the mice by heart puncture and incubated at a 1% cell density in complete culture medium (RPMI 1640 with 20% fetal calf serum) for a period of 24 hours at 37°C. At fixed time points after the start of the cultures, 1-ml samples are collected for analysis of the cell cycle by flow cytometry.18 Cells, fixed in 0.25 (v/v) glutaraldehyde solution in PBS, are stained with Hoechst 33258 at a concentration of 2 μmol/L for 1 hour at 37°C and analyzed with a FACScan (LSR II). UV excitation of Hoechst 33258 dye is performed with an argon ion laser (450/50 nm). The fluorescence intensity and size (forward/sideward scatter) of a total of 50,000 cells per sample are measured, and data analysis is performed using CellQuest software. No gate is set in the forward/sideward scatter for size selection of erythrocytes to include the small, free merozoites for analysis. The fluorescence intensity of infected erythrocytes is proportional to the DNA content of the parasites.19 The start of schizogony (the length of the G1 phase of trophozoites) is defined as the time point (hours postinvasion) at which the percentage of cells with more than the haploid/diploid DNA content (percentage of schizonts; gate P5 in Supplemental Figure S4, see http://ajp.amjpathol.org) had increased >5% compared with that at the previous time point.18 The time points at which the first schizonts are mature is defined as the time point (hours postinvasion) at which the percentage of free merozoites in gate P6 (Supplemental Figure S4, see http://ajp.amjpathol.org) had increased >5% compared with the previous time point.18
ECM Analysis
The development of cerebral complications was analyzed in several mouse strains that are susceptible to ECM (Swiss-OF1, Swiss-CD1, CBA/J, and C57BL/6). ECM in P. berghei infection is well characterized and is defined as the development of cerebral complications (drop in body temperature <34°C, paralysis, convulsions, and coma) at day 6 to 9 after infection of mice with 104 to 106 parasites.21,22 In this study mice were infected by i.p. injection of 105 to 106 infected erythrocytes (Δpm4-infected mice were also infected with a 10× higher dose than wild-type infected mice because of the lower multiplication rate of Δpm4 parasites). The onset of cerebral complications was determined by observation of several clinical signs such as ruffled fur, hunching, wobbly gait, limb paralysis, convulsion, and coma and by measuring the drop in body temperature at day 5 to 8 after infection at 6-hour intervals. The body temperature was measured using a laboratory thermometer (model BAT-12, Physitemp Instruments Inc., Clifton, NJ) with a rectal probe (RET-2) for mice. A drop in temperature below 34°C is indicative of cerebral complications,23 and experiments were terminated when the body temperature dropped below this value.
Sequestration of schizonts in whole bodies of live mice and isolated organs was visualized through imaging of luciferase-expressing, transgenic parasites with an intensified charge-coupled device photon counting video camera of the in vivo imaging system (IVIS 100, Xenogen Corporation, Alameda, CA) as described previously.15 Sequestration patterns were monitored in mice with synchronized infections. Synchronized infections (1 to 3% parasitemia) were established by injection of cultured, purified schizonts as described above. Imaging was performed between 17 and 25 hours after injection of schizonts. Imaging of individual organs, obtained by dissection from animals at 21 to 23 hours, was done as described previously.15 Imaging data were analyzed by using the programs LIVING IMAGE (Xenogen Corporation) and IGOR PRO (WaveMetrics, Lake Oswego, OR).
For histological analysis mice with asynchronous infection at day 7 postinjection were perfused first with PBS and then with 4% paraformaldehyde. The brains were removed and fixed with 4% paraformaldehyde at 4C° for 16 to 18 hours, rinsed in phosphate buffer for 24 hours, and cryoprotected with 10 and 30% sucrose each for 24 hours. The brains were divided into two hemispheres and frozen tissue was cut into 30-μm-thick longitudinal slices on a freezing cryotome. The sections were mounted on a slide and stained with H&E and analyzed with an upright Olympus epifluorescent/transmitted light microscope (magnification, ×400 or ×1000).
For the analysis of blood-brain barrier damage and endothelial integrity the mice with asynchronous infection at day 7 postinjection had been injected i.v. with 2% Evans Blue.24 Three hours after injection of the dye the mice were perfused through the heart with heparinized PBS, and subsequently the brain was removed24 and digital images were collected.
Bioluminescence analysis of brain from mice with asynchronous infection at day 7 postinjection was performed by an in vivo imaging system (IVIS 200, Xenogen) after extensive perfusion of the animals through the heart with heparinized PBS as described previously.15
Course of Δpm4 Infections, Drug Treatment, and Challenge with Wild-Type Parasites
The course of infection in BALB/c and NIH Swiss mice, which are not susceptible to ECM, was analyzed after either i.v. or i.p. inoculations of 107 parasitized erythrocytes. The parasitemia (% of infected erythrocytes) was determined by counting daily Giemsa-stained slides of tail blood. Challenge experiments were performed by i.v. injection of different doses (104 up to 107) of parasites: P. berghei (ANKA), Plasmodium yoelii 17X (MRA-426, MR4), and Plasmodium chabaudi ASS (MRA-429, MR4).
Protection immunity induced by drug treatment was analyzed in BALB/c mice injected i.p. with 107 wild-type parasites. Eighteen days postinfection, when the parasitemia was approximately 70 to 80% (as in Δpm4-infected animals), the mice were treated with 70 μg/ml pyrimethamine in the drinking water for 30 or 60 days. During this period the animals were monitored by Giemsa-stained slides to confirm the absence of parasites in the blood as in the Δpm4-infected animals that spontaneously cleared the infection. One week from the end of the drug treatment the mice were challenged i.v. with 107 wt++ parasites. Load and distribution of parasites were monitored by an in vivo imaging system (IVIS 200) as described above and by Giemsa-stained slides. Imaging data were analyzed as described above.
Splenectomy Experiments
For splenectomy, naïve and convalescent Δpm4cl6-infected BALB/c mice were anesthetized, and the spleen was removed.25 Two weeks after splenectomy the animals were infected with 107 wild-type parasites, and the parasitemia was determined by counting Giemsa-stained blood films of tail blood.
Passive Transfer of Immunoglobulin
IgGs were purified (Protein A antibody purification kit, PURE-1A, Sigma-Aldrich) using 1.5 ml of serum collected from BALB/c mice infected either with wild-type at day 18 after infection or with Δpm4cl6 parasites at day 18 and day 40 after injection (convalescent mice). Purified IgGs were injected i.p. into BALB/c mice for three consecutive days (−1, 0, and +1; 500-μg dose) relative to i.v. injection of 105 wt++ parasites. Parasite load and distribution in whole bodies of live mice were visualized through imaging of luciferase-expressing transgenic parasites at day 2 after the last immunoglobulin administration with an intensified-charge-coupled device photon counting video camera of the in vivo imaging system (IVIS 200) as described previously.15 In a second experiment 500 μg of the purified IgG was injected i.p. into BALB/c mice 2 days after i.v. injection of the wt++ parasite line. Parasite distribution in live mice was visualized through imaging of luciferase-expressing transgenic parasites 1 hour after the immunoglobulin administration. Imaging data were analyzed by using the programs LIVING IMAGE and IGOR PRO. Parasitemia was determined by daily analysis of Giemsa-stained blood films.
Statistical Analyses
We analyzed statistical significance by Student's t test with the GraphPad Prism software package and statistics for survival curves by MedCalc software.
Results
P. berghei Mutant Parasites Deficient in Expression of Plasmepsin 4
In this study several independent P. berghei mutant lines lacking PM4 (Δpm4 parasites) were generated by targeted disruption of the pm4 gene (Supplemental Table S1, see http://ajp.amjpathol.org). Two different DNA constructs have been used to disrupt pm4 through double crossover homologous recombination (see Materials and Methods). Correct disruption of the pm4 gene was confirmed by diagnostic PCR and Southern blot analysis (Supplemental Figure S1, see http://ajp.amjpathol.org). The absence of PM4 expression was shown by Northern analysis (data not shown) and Western analysis of blood-stage proteins (Supplemental Figure S1, see http://ajp.amjpathol.org). To monitor Δpm4 infections in live mice by real-time in vivo imaging we have introduced a gene encoding a GFP-luciferase fusion protein to serve as a reporter protein in wild-type parasites as well as in several Δpm4 parasite lines (Supplemental Figure S2 and Supplemental Table S1, see http://ajp.amjpathol.org). These luciferase-expressing parasites have been used to analyze parasite load and distribution and patterns of schizont sequestration by in vivo imaging as described previously.15
Modest Growth Delay of Blood Stages of Δpm4 Parasites
The in vivo asexual multiplication rate of Δpm4 parasites, defined as the fold increase in parasite numbers in mice during the first 8 to 11 days after infection with a single parasite, was reduced (Table 1). Our results showed a multiplication rate ranging from a 5.8- to a 7.0-fold increase per 24 hours in Δpm4 parasites compared with a consistent 10-fold increase in wild-type parasites.18 Flow cytometry analysis of cell cycle progression, following in vitro synchronized Hoechst 33258-stained blood-stage parasites,18 revealed that Δpm4 parasites have a prolonged cell cycle as shown by the delay in the onset of schizogony (length of the G1 phase) by approximately 3 hours and a similar delay in the production of mature schizonts (end of the S/M phase) (Table 1 and Supplemental Figure S4, see http://ajp.amjpathol.org). However, mature Δpm4 schizonts had normal numbers of daughter merozoites12–20 (Table 1; Supplemental Figure S3, see http://ajp.amjpathol.org). Interestingly, in synchronous cultures the development of Δpm4 schizonts seemed to be less “synchronous” than in the wild-type cultures. Analysis of Giemsa-stained samples from the cultures showed that 20 to 40% of the Δpm4 schizonts did not complete the cell division process within a 30-hour period compared with only 8 to 12% of wild-type parasites. A prolonged, less synchronized cell cycle in combination with a delayed and somewhat reduced schizont maturation process explain the observation of the slightly reduced multiplication rate in vivo. These results also demonstrate that the lysosomal plasmepsin, PM4, is not essential for either in vitro or in vivo intraerythrocytic development.
Table 1.
Growth Characteristics of Blood Stages of Wild-Type and Δpm4 Parasites
| Start of schizogony |
First mature schizonts |
Merozoites per schizont |
|||||
|---|---|---|---|---|---|---|---|
| Line | In vivo multiplication rate* | Hours† | Schizonts (%)† | Hours‡ | Merozoites (%)‡ | Gate P2§ | Gate P3§ |
| Wild-type | 10 (0) | 18 (0) | 14 (3) | ND | ND | 13.1 (0.5) | 16.7 (0.5) |
| Δpm4 (688cl2) | 5.8 (0.5) | 21 (0) | 15 (3) | ND | ND | 13.2 (0.5) | 17.3 (0.6) |
| wt+ (1037cl1) | 10 (0) | 19 (0) | 14 (2) | 23 (0) | 12 (2) | 12.7 (0.4) | 16.6 (0.4) |
| Δpm4+ (1092cl4) | 7.0 (0.8) | 22 (0) | 15 (3) | 26 (1) | 11 (3) | 12.7 (0.3) | 16.1 (0.2) |
Data are mean (SD).
ND, no data.
The multiplication rate per 24 hours is determined in mice infected with a single parasite.
The start of schizogony (S/M phase) was determined by flow cytometry (Supplemental Figure S4, see http://ajp.amjpathol.org) and is demonstrated by the presence of polyploid schizonts (cells with 3-16N present in P5 in Supplemental Figure S4, see http://ajp.amjpathol.org). The start of schizogony is defined as the time point (hours postinvasion) at which the percentage of cells with more than the haploid DNA content (% of schizonts; cells in gate P5 in Supplemental Figure S4, see http://ajp.amjpathol.org) had increased more than 5% compared with the averages of the previous time points. The percentage of schizonts in gate P5 is the mean of at least three measurements. Multiple measurements of blood samples of the haploid stages (rings and trophozoites) have shown that the percentage of cells in gate P5 has less than 2% variation.
The time point at which schizonts are mature is determined by flow cytometry as shown in Supplemental Figure S4 (see http://ajp.amjpathol.org) and is demonstrated by the presence of the haploid, free merozoites (gate P6 in Supplemental Figure S4, see http://ajp.amjpathol.org). This time point is defined as the time point (hours postinfection) at which the percentage of merozoites in P6 had increased more than 5% compared with the previous time point. The percentage of merozoites in gate P6 is the mean of at least three measurements.
The number of merozoites per schizonts is determined in cultured parasites by flow cytometry as described in Materials and Methods and shown in Supplemental Figure S3 (see http://ajp.amjpathol.org). The mean number of merozoites is shown for schizonts present in gate P2 and in gate P3 (Supplemental Figure S3, see http://ajp.amjpathol.org).
Δpm4 Parasites Have a Virulence-Attenuated Phenotype
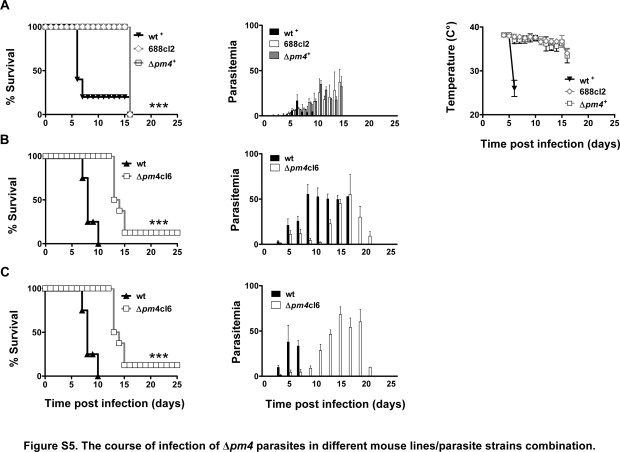
While studying the growth of Δpm4 parasites in vivo, we observed that they differed from wild-type parasites in terms of their lethality and the occurrence of ECM. To investigate this phenotype in more detail we studied Δpm4 infections in different mouse lines/strains that are known to be susceptible or resistant to ECM. Normally BALB/c and NIH Swiss mice do not develop ECM but die in the 3rd week from hyperparasitemia and severe anemia, whereas C57BL/6, Swiss-OF1, CBA/J, and Swiss-CD1 develop ECM at day 6 to 11 after infection.9,10 As expected, all BALB/c mice died between day 12 and 23 after wild-type infection with a parasitemia greater than 50%, within a time frame inversely correlated with the infection dose (ie, 105 to 107 parasites) (Figure 1A). Conversely, all Δpm4-infected BALB/c mice unexpectedly survived infection with a peak of parasitemia at day 16 to 22, which was rapidly cleared thereafter from the blood, resulting in undetectable parasitemia by microscopic analysis by day 25 (Figure 1A). NIH Swiss mice were also able to resolve the Δpm4 infections in a manner very similar to that in BALB/c mice (Figure 1B).
Figure 1.
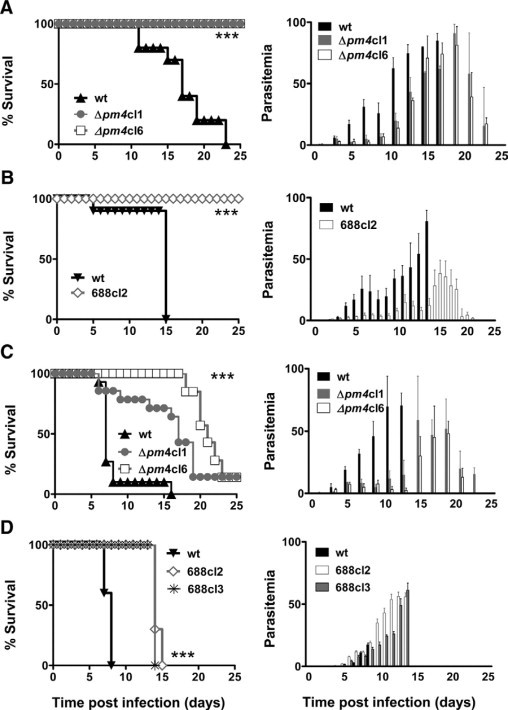
The course of infection of Δpm4 parasites in different mouse lines/parasite strains combinations. Cumulative death and parasitemia levels are shown in the left and right panels, respectively. A: BALB/c mice infected with 107 wild-type (wt) (n = 30) or Δpm4 parasites of lines Δpm4cl1 (n = 50) and Δpm4cl6 (n = 50). All wild-type infected mice died within the 3rd week after infection with high parasitemia, whereas all mice infected with Δpm4 parasites survived infection. B: NIH Swiss mice infected with 107 wild-type (cl15cy1) (n = 10) or Δpm4 parasites of line 688cl2 (n = 10). All NIH Swiss mice infected with wild-type (cl15cy1) parasites developed high parasitemia, and experiments were terminated on day 15 after infection at a parasitemia 80%. All mice infected with Δpm4 parasites after the peak of parasitemia were able to control the infection and completely cleared the parasites. C: C57BL/6 mice infected with 105 wild-type (n = 15) or Δpm4cl1 (n = 15) and Δpm4cl6 (n = 50) parasites Ninety percent of wild-type-infected C57BL/6 mice developed ECM at day 6 to 8 after infection. None of the mice infected with Δpm4 parasites showed signs of cerebral complications. Most Δpm4-infected mice (80%) developed high parasitemia and died in the 3rd week after infection. The surviving mice cleared the parasites. D: Swiss-OF1 mice infected with either 105 wild-type (cl15cy1) (n = 10) or Δpm4 parasites of lines 688cl2 (n = 13) or line 688cl3 (n = 6). All Swiss-OF1 mice developed ECM at day 6 to 8 after infection at a parasitemia ranging from 15 to 20%, whereas the mice infected with Δpm4 parasites did not show cerebral complications. These mice developed high parasitemia, and experiments were terminated between day 14 and 16 after infection at a parasitemia >30%. ***P < 0.0001.
When ECM-susceptible mice strains (C57BL/6, Swiss-OF1, CBA/J, and Swiss-CD1) were infected with Δpm4 parasites, symptoms of ECM were absent in all mice, whereas 80 to 100% of wild-type-infected mice died from ECM within 6 to 9 days after infection (Figure 1, C and D; Supplemental Figure S5, see http://ajp.amjpathol.or). The absence of ECM was not dependent on the dose of Δpm4 parasites inoculated. Mice inoculated with 105, 106, and 107 Δpm4 parasites consistently failed to develop ECM, whereas as few as 105 wild-type parasites were sufficient to cause ECM. In ECM-susceptible mice Δpm4 parasites caused prolonged infections and most mice (80 to 90%) died with a hyperparasitemia of more than 50% in the 3rd week without signs and symptoms of ECM (Figure 1, C and D; and Supplemental Figure S5, see http://ajp.amjpathol.org).
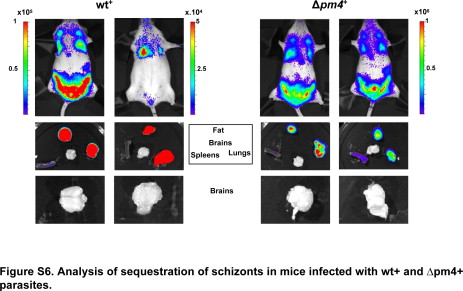
To further investigate the failure of Δpm4 parasites to cause ECM we analyzed schizont distribution in tissues and organs by real time in vivo imaging of schizonts. ECM-susceptible Swiss-OF1 mice were infected with parasites expressing the GFP-luciferase fusion gene under the control of the schizont-specific ama-1 promoter in either the wild-type or Δpm4 genetic background, termed wt+ or Δpm4+. The expression of luciferase under the schizont-specific ama-1 promoter allows the visualization of sequestered schizonts in live animals by in vivo imaging.15,26 Synchronized infections with wt+ or Δpm4+ parasites showed identical sequestration and distribution patterns in animals and in isolated organs. Similar to wt+, the Δpm4+ schizonts sequester mainly in the lungs, adipose tissue, and spleen (Figure 2, A–C).15 The timing of sequestration of Δpm4+ schizonts was delayed by a few hours (Figure 2A), in agreement with the prolonged cell cycle and delay in schizont maturation (Supplemental Figure S4, see http://ajp.amjpathol.org). Analysis of the brains of mice with synchronized infections showed the absence of schizont sequestration during the period of 20 to 24 hours after infection, both in Δpm4+- and in wt+-infected mice (Supplemental Figure S6, see http://ajp.amjpathol.org). In the above-mentioned studies the presence or absence of cerebral complications in mice was determined by the drop in body temperature and the presence of symptoms such as paralysis, convulsions, and coma. To more clearly establish that infections with Δpm4 parasites did not induce ECM, we examined the brain of mice with nonsynchronized wt+ and Δpm4+ infection by histology, by in vivo imaging, and by the Evans Blue dye extrusion method for the analysis of blood-brain barrier damage and endothelial integrity. At day 7 after infection, the brains of wt+-infected mice showed clear signs of cerebral hemorrhages and vascular permeability (Figure 3, A and B). In contrast, the brains of mice infected with 105 or 106 Δpm4+ parasites to reach parasitemias comparable to those in the wt+-infected mice did not show signs of inflammatory damage, and we found no evidence for vascular leakage (Figure 3, A and B). Using in vivo imaging we revealed a significantly higher parasite accumulation (ie, luciferase signals) in the brains of wt+-infected mice with cerebral complications at day 7 postinfection compared with the brains of Δpm4+-infected mice without ECM (Figure 3, C and D). The higher luciferase signals indicate an increased accumulation of parasites in the brain either as a result of accumulation of infected erythrocytes in tissues via hemorrhages or from increased specific adherence of schizonts as a result of inflammation as described previously.15,16 Because we imaged mice with asynchronous infections, no distinction can be made between these two possibilities because not only schizonts but also ring-infected forms contain luciferase activity as a result of carryover of the luciferase in ring forms from the schizonts.
Figure 2.
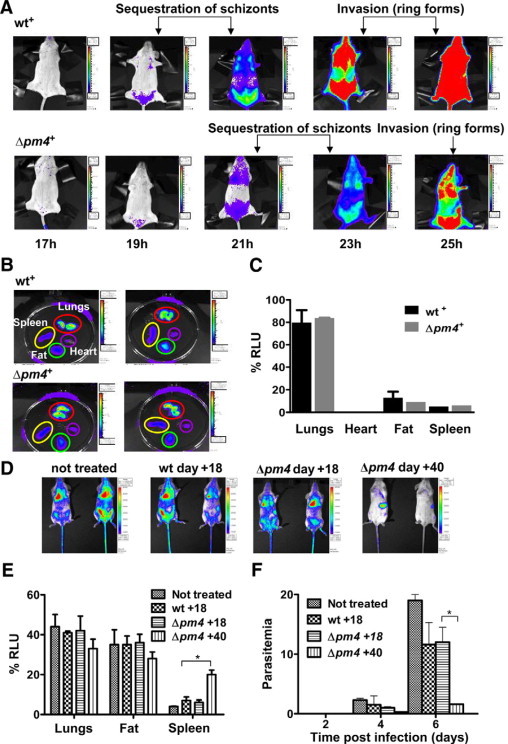
Distribution of luciferase expressing Δpm4+ and wt+ parasites in live mice as visualized by real time in vivo imaging through measurement of luciferase activity. A: A time course of an atypical distribution pattern of schizonts in mice with synchronous infections of Δpm4+ and wt+ parasites at different time points (hours) after the injection of purified merozoites. Schizont sequestration at 19 and 21 hours in wt+-infected mice (belly fat tissue, lungs, and spleen) and reinvasion of ring forms at 23 and 25 hours, resulting in bioluminescence in the whole body. In Δpm4+ parasites schizont sequestration and invasion start later (21 and 23 hours), but the distribution remains similar. B: Bioluminescence in organs of mice infected either with wt+ or Δpm4+ and isolated at 21 and 23 hours after infection, respectively. C: Quantification of luciferase signals in organs of mice infected with wt+ and Δpm4+ (six mice per group) showed no significant differences in parasite distribution. D: Parasite load and distribution in mice treated with IgG obtained from Δpm4 convalescent mice and infected with wt++ parasites. IgG purified from the serum of animals infected with Δpm4cl6 parasites at day 18 or 40 after injection were given to BALB/c mice at days −1, 0, and + 1 relative to the time of i.v. inoculation with wt++ parasites. As a control, mice were treated with IgG from wild-type infected mice obtained at day 18 postinfection. Nontreated: BALB/c mice infected with the wt++ parasite line only. Parasites were visualized at day 2 after infection of the mice with the wt++ parasites. E: Quantification of luciferase signals in organs of mice (six mice per group) treated with the different IgG samples shows a significant increase in parasite accumulation in the spleen of mice treated with IgG from convalescent animals 40 days postinfection. F: The course of the parasitemia in mice treated with the different IgG samples shows a significant reduction of the parasite load in mice treated with IgG that was collected from convalescent animals (day 40 after infection).*P < 0.05. RLU, relative light unit.
Figure 3.
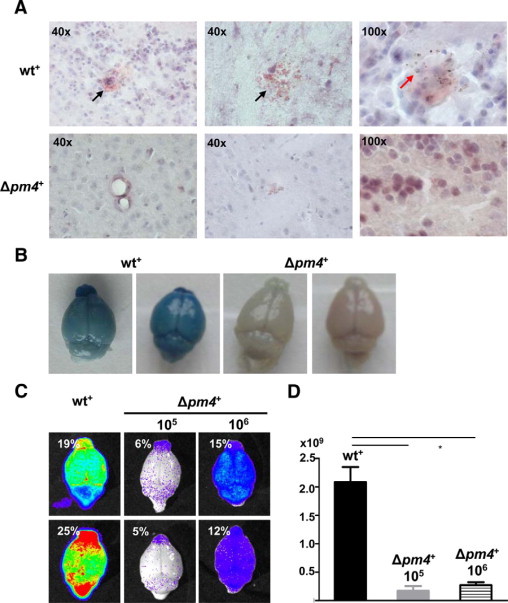
Analysis of cerebral complications in mice infected with wt+ and Δpm4+ parasites. A: Histological analysis of the brains of C57BL/6 mice with asynchronous infections at day 7 after injection of wt+ or Δpm4+ parasites. Longitudinal sections of the brain, stained with H&E, show extensive hemorrhagic areas in wt+-infected animals (black arrows), which are absent in Δpm4+-infected mice. In brains of wt+-infected mice, infected erythrocytes, recognized by pigment granules, were found as infiltrates in brain tissue (red arrow). B: Representative digital images of Evans Blue dye extrusion analysis of brains of wt+ or Δpm4+-infected C57BL/6 mice at day 7 postinfection. The blue staining of the brains shows vascular leakage in wt+-infected animals, which is absent in the brains of Δpm4+-infected mice. C: Bioluminescent images of brain isolated from C57BL/6 mice at day 7 after infection with 105 wt+ or 105 to 106 Δpm4+ parasites showing a significant higher parasite load (see D) in brains of wt+ than in brains of Δpm4+-infected mice as a result of differences in accumulation of infected erythrocytes in brain tissue. Percentages shown represent the parasitemias at the time of collection of the brains. D: Differences in luminescence signal between the brains of mice infected with either wt+ or Δpm4+ parasites (n = 6) showing the differences in parasite load. *P < 0.05.
Recovery from Δpm4 Infection Results in a Long-Lasting Malaria Immunity
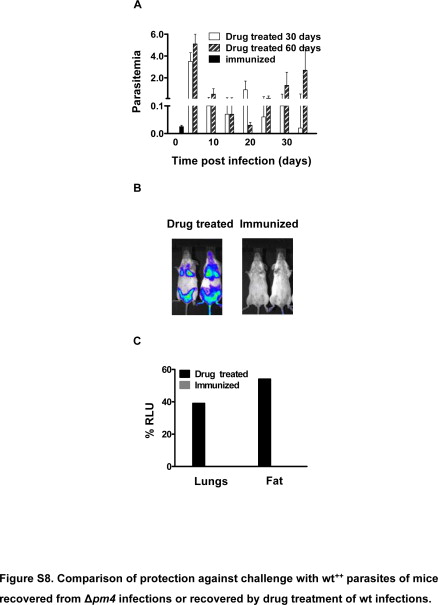
Attenuated pathogens are frequently used as vaccines, and, therefore, in this study, we tested whether the growth-attenuated Δpm4 parasites were able to elicit protective immunity. BALB/c, C57BL/6, and NIH Swiss mice, which had recovered from a single Δpm4 infection, were challenged with wild-type parasites. All convalescent mice of all strains tested were protected against P. berghei wild-type parasite challenge given at high doses (106 to 107 parasites). Challenged mice either failed to develop a detectable infection or showed a short-lasting, low-level parasitemia (<0.05%), which was usually cleared within 10 days (Table 2). The immune responses elicited by Δpm4 parasites were remarkably long-lasting. Up to 1 year after recovery from a Δpm4 infection, the mice were protected against challenge (Table 2). The mice were also protected against challenge with the closely related species P. yoelii. We inoculated BALB/c mice that had recovered 4 months earlier from a Δpm4 infection with P. yoelii-infected erythrocytes and found that all mice infected with 104 to 106 parasites were protected. Only after giving the highest challenge dose (107 parasites), did we observe a mortality rate of 50% (Table 2). NIH Swiss mice that had recovered from Δpm4 infection were also protected against challenge with P. yoelii. The challenge of these mice with P. chabaudi did not result in complete protection. However, compared with naïve mice the course of infection in Δpm4-recovered mice was significantly milder with lower parasitemias (Supplemental Figure S7, see http://ajp.amjpathol.org). In naïve mice a parasitemia greater than 50% was observed during the first wave of parasitemia, whereas in Δpm4-immunized mice the parasitemia remained below 10%. In addition, a clear recrudescent peak was absent. We also compared the protection elicited through infection with Δpm4 with that elicited by infection with wild-type followed by pyrimethamine drug treatment. Mice infected with wild-type parasites that were drug-treated developed a protective immunity against subsequent challenge with the wt++ parasite line although we observed substantial differences compared with Δpm4-induced protection (Supplemental Figure S8, see http://ajp.amjpathol.org). At day 30 and 60 after drug-induced recovery a challenge with wild-type parasites caused a mild but significant parasitemia (1 to 3%) that was several fold higher than that observed after challenging Δpm4-recovered mice (< 0.05%) (Supplemental Figure S8, see http://ajp.amjpathol.org). Furthermore, drug-treated mice failed to clear the infection for several weeks after challenge, whereas in Δpm4-recovered mice infections were cleared in less than 10 days. These results show that mice that are infected with Δpm4 attenuated parasites develop a stronger protective immune response than is induced in mice infected with wild-type parasites cleared by drug treatment.
Table 2.
Protection of Δpm4 Cured Mice against Challenge with Wild-Type Parasites of P. berghei and P. yoelii
| Mouse strain | Infected with/cured from | Challenged with | Time of challenge (days)* | Challenge dose (no. of parasites)† | No. protected/no. challenged |
|---|---|---|---|---|---|
| BALB/c | Δpm4cl6 | P. berghei | 60 | 107 | 30/30‡ |
| BALB/c | Δpm4cl6 | P. berghei | 120 | 107 | 30/30‡ |
| BALB/c | Δpm4cl6 | P. berghei | 180 | 107 | 20/20 |
| BALB/c | Δpm4cl6 | P. berghei | 270 | 107 | 9/9 |
| BALB/c | Δpm4cl6 | P. berghei | 360 | 107 | 7/7 |
| C57BL/6 | Δpm4cl6 | P. berghei | 120 | 107 | 10/10‡ |
| NIH Swiss | 688cl3 | P. berghei | 44/60 | 106 | 6/6 |
| BALB/c | Δpm4cl6 | P. yoelii (17X) | 120 | 107 | 5/10 |
| BALB/c | Δpm4cl6 | P. yoelii (17X) | 120 | 106 | 10/10 |
| BALB/c | Δpm4cl6 | P. yoelii (17X) | 120 | 105 | 10/10‡ |
| BALB/c | Δpm4cl6 | P. yoelii (17X) | 120 | 104 | 10/10‡ |
| NIH Swiss | 688cl3 | P. yoelii (17X) | 44 | 106 | 5/5 |
Number of days after recovery from infection with Δpm4cl6 or 688cl3.
Mice were challenged by i.v. injection of infected erythrocytes.
Three months after the initial challenge, 10 mice were rechallenged with 107 parasites and all mice remained protected.
Protection Induced by Δpm4 Parasites Is Antibody- and Spleen-Mediated
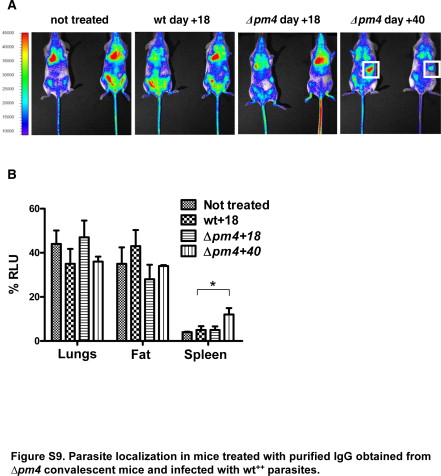
The nature of the immune responses elicited by Δpm4 parasites was investigated in BALB/c mice by passive antibody transfer experiments. Specifically, naïve mice that had received purified IgG from previously infected mice were inoculated with wt++ parasites to analyze the course of infection and parasite distribution by in vivo imaging. As expected, treatment with IgG collected from wild-type or Δpm4-infected animals at day 18 after infection, had no significant effect on the parasitemia and distribution of parasites (Figure 2, D–F). Treatment of mice with IgG collected from Δpm4 convalescent mice (day 40 after infection) had a strong effect on both parasite distribution and parasitemia (Figure 2E and F). Most wt++ parasites were removed from tissues/organs after the administration of IgG as shown by the strong decrease of luciferase activity throughout the whole body with the exception of the spleen in which luciferase activity was increased (Figure 2D). When animals were treated with IgG from convalescent mice during an ongoing wt++ infection, the parasitemia rapidly decreased, and parasites accumulated specifically in the spleen within 1 hour after injection of IgG (Supplemental Figure S9, see http://ajp.amjpathol.org). These results demonstrate that in P. berghei infection the spleen plays an essential role in antibody-mediated clearance of infected erythrocytes from the circulation. Rather than acting on parasite growth and/or erythrocyte invasion, antibodies seem to function by enhancing the clearance of infected erythrocytes by splenic macrophages. In addition, the observation that spleens from mice infected with Δpm4 parasites were on average twice the size of those from wild-type-infected mice (Figure 4A), emphasizing the role of the spleen in the resolution of Δpm4 infections. The role of the spleen was further investigated by infecting splenectomized BALB/c mice with Δpm4 parasites. These mice failed to clear Δpm4 parasites and showed recrudescent “waves” of parasitemia ranging between 15 and 50%, and more than 90% of the mice died between day 40 and 80 (Figure 4B). When convalescent mice were splenectomized followed by challenge with wild-type parasites, protection was also abrogated. The challenge of these animals with wild-type parasites resulted in infections with an oscillating parasitemia, ranging between 1 and 10% for a few days, followed by a rise of parasitemia to >50% that was invariably associated with death (Figure 4C).
Figure 4.
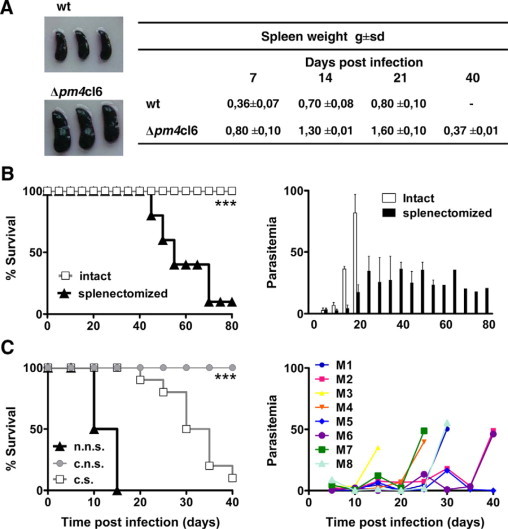
The role of the spleen in parasite clearance and protective immunity. A: Macroscopic observation of spleens from BALB/c mice infected with either 107 wild-type or Δpm4cl6 parasites. This analysis revealed that at 21 days postinfection the spleens of Δpm4-infected mice (lower panel) were much bigger than those from wild-type infected animals (upper panel). This observation was confirmed by comparing the spleen weight in either wild-type or Δpm4cl6-infected mice at different time points (7, 14, 21, and 40 days) after infection. Numbers represent the mean values ± SD of spleen weight of eight animals. B: Cumulative death (left panel) and levels of parasitemia (right panel) of either intact (gray) or splenectomized (black) BALB/c mice infected with 107 Δpm4cl6 parasites. ***P < 0.0001. C: Cumulative death (left panel) of splenectomized Δpm4 convalescent BALB/c mice (convalescent splenectomized [c.s.]) after a challenge with wild-type parasites. As a control Δpm4 convalescent intact BALB/c mice (convalescent not splenectomized [c.n.s.]) and naïve intact BALB/c mice (naïve not splenectomized [n.n.s]) were challenged with the same dose of wild-type parasites. Right panel: Time course of parasitemia in eight individual convalescent splenectomized BALB/c mice (M1–M8) after a challenge with wild-type parasites. None of the Δpm4 convalescent immune mice was able to control the parasitemia when challenged after splenectomy. All mice showed oscillating levels of parasitemia that increased sharply in a few days, leading to the death of the animals. ***P < 0.0001.
Discussion
We targeted for gene disruption in P. berghei its single blood-stage digestive vacuole aspartic protease, plasmepsin 4. Unexpectedly, despite the relatively minor parasite developmental consequences of the absence of PM4, we observed a marked effect on the virulence of these parasites both in ECM-resistant and ECM-susceptible mice. To date only a few mutant rodent parasites have been described in which disruption of a single parasite gene affects blood-stage growth in vivo. Recently, a mutant P. yoelii parasite that showed an attenuated phenotype in blood stages was generated by gene disruption.27 Disruption of the gene encoding purine nucleoside phosphorylase in the lethal P. yoelii YM strain resulted in impairment of parasite growth. Mice infected with this Δpnp parasite were able to clear the infection and induce protective immune responses. P. berghei parasites that lack one of the two identical gene copies encoding eEF1A18,28 showed a retarded proliferation and a prolonged cell cycle up to 20%, but these parasites were as virulent as wild-type parasites in mice and retained their ability to induce cerebral malaria. Therefore, the reduced growth rate alone does not explain the absence of ECM in Δpm4-infected mice and the attenuated virulence phenotype. Investigations in mouse models of malaria have shown that ECM is a complex neurological syndrome involving interactions between cytokines and the local cellular microenvironment.10,21,29 Recently, it has been demonstrated that regulatory T cells contribute to ECM by modulating immune responses, and depletion of these cells can protect mice from ECM.30 Although the role of host molecules in immunopathology (eg, interferon-γ, lymphotoxin-α, tumor necrosis factor-α, IL-10, and IL-12)21 is well documented, the parasite-derived molecules that trigger host responses leading to ECM pathology are essentially unknown. In P. falciparum infections, PfEMP1-mediated cytoadherence of schizonts in the brain has been proposed as an important parasite mediator of cerebral pathology.29 Although P. berghei schizonts also have a clear cytoadherence phenotype, they mainly sequester in the lungs, adipose tissue, and spleen and not in the brain vasculature. This suggests, as has been concluded previously, that cerebral pathology is unlinked to (CD36-mediated) sequestration.15 The Δpm4 parasites show a sequestration pattern comparable with that of wild-type parasites but fail to induce ECM, indicating the absence of an obvious link between ECM and cytoadherence in this experimental model. It has been suggested that parasite-derived “toxins” provide the essential trigger for the proinflammatory cytokine release that leads to ECM.29 For example, several studies have implicated glycosylphosphatidylinositol anchors in the induction of tumor necrosis factor production by macrophages and increase in inducible nitric oxide synthase synthesis.31,32 Furthermore, vaccination with the synthetic glycan moiety of glycosylphosphatidylinositol protected mice from ECM.33 In addition, the hemozoin pigment granule produced by Plasmodium has been reported to induce proinflammatory immune responses.34,35 Interestingly, P. falciparum mutants that lack all DV aspartic protease activity produce less hemozoin than wild-type parasites.4 The extent of hemozoin production in the Δpm4 parasites is not known, but the schizonts of these parasites seem to have smaller and more widely dispersed aggregates of hemozoin (C. J. Janse, unpublished observations). It is tempting to speculate that reduced hemozoin formation would prevent the induction of ECM. However, further studies are required to identify the critical parasite toxin that is missing in the Δpm4 parasites and is responsible for the “lack of ECM” phenotype.
The spontaneous recovery from an infection caused by Δpm4 parasites in BALB/c and NIH Swiss mice is accompanied with a long-lasting protective immunity against challenge with wild-type parasites. This phenotype shares some similarities with a virulence-attenuated P. berghei parasite (XAT) that was generated by radiation mutagenesis as well as the P. yoelii parasites lacking purine nucleoside phosphorylase.36,37 The XAT and Δpnp parasites were similarly characterized by an impaired growth rate in vivo, which also produced a self-resolving infection and induced a long-lasting immunity. Although XAT parasites demonstrated the possibility of generating virulence-attenuated parasites, they have been of limited use in understanding the molecular basis of virulence or immune mechanisms, leading to protective immunity. In XAT parasites the molecular basis of parasite attenuation is unknown and is difficult to disentangle from the numerous as yet uncharacterized DNA alterations induced by ionizing radiation. In contrast, Δpm4 and Δpnp parasites represent molecularly characterized experimental models; the further use of these parasites should provide more insights into parasite-derived factors that induce ECM as well mechanisms of protective immunity.
Through the combination of passive IgG transfer studies, in vivo imaging, and splenectomy experiments, we showed the critical role of the spleen in protective immunity induced by Δpm4 parasites. A marked decrease in circulating parasite load is directly associated with an increase in parasite accumulation in the spleen in mice treated with IgG from Δpm4 convalescent mice. Rather than directly acting on parasite growth and erythrocyte invasion, these observations indicate that protective antibodies seem to function mainly by redirecting infected erythrocytes to the spleen where they are removed from the blood. The importance of antiparasite antibodies for spleen-mediated elimination of blood stages and the corresponding protective immune responses has been indicated previously for both rodent and human malaria.24,38–41 Relatively little is known about the specific mechanisms that generate adaptive immunity in the spleen during a malaria infection. Our results indicate that Δpm4 parasites are suitable tools to investigate the specific immune mechanisms in the spleen crucial for the development of protective immunity. The knowledge from such studies might inform rational vaccine design and strategies that would reduce or abolish malarial pathology.
The limited success in generating protective immunity using so-called subunit vaccines, principally composed of one or a few parasite molecules (or indeed domains of molecules), has renewed an interest in use of whole attenuated parasite vaccines. Encouraging results have come from immunization with attenuated sporozoite vaccines both in mice and humans. These attenuated parasites, generated through irradiation42,43 and more recently by genetic modification44–48 or chemical attenuation49 are able to induce long-lasting sterile protective immunity against liver-stage infection. This study and the recent report on P. yoelii Δpnp mutants demonstrate that it is possible, by engineered inactivation of parasite proteins, to generate attenuated blood-stage parasites that are capable of inducing protective immunity against blood-stage infection. Such parasites should be powerful tools in elucidating parasite-derived factors that cause severe disease and should provide additional insight into factors that are required to induce protective immunity. Clearly, much is still to be understood, but the insights generated by virulence-attenuated parasites, such as the one described in this study, may help to assess the potential of generating malaria vaccine based on genetically attenuated blood stages.
Acknowledgements
MR4 provided plasmid pL0028, BiP antisera was contributed by John H. Adams, and P. chabaudi ASS was contributed by Wallace Peters and Brian L. Robinson. Jai Ramesar is acknowledged for the technical support with transfections and analysis of growth characteristics of the blood stage, Mario Rende and Anna Stabile for histological analyses, and Shahid Khan for stimulating discussions.
Footnotes
Supported by grants from the Italian Ministry of Research Programme for Relevant National Interest (2005065913_005) and for Basic Research Investments Programme (grant RBLA03C9F4_001), and the Wellcome Trust Functional Genomics Initiative (grant 66742 to A.P.W./C.J.J.). B.F.F. was supported by The Netherlands Organization for Scientific Research (ZonMw TOP grant 9120_6135). S.C. was supported by Fondazione Cassa di Risparmio di Perugia. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
R.S. and C.J.J. contributed equally to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Current address of A.P.W.: Division of Infection and Immunity, Institute of Biomedical Life Sciences & Wellcome Centre for Molecular Parasitology, Glasgow Biomedical Research Centre, University of Glasgow, Scotland.
Contributor Information
Roberta Spaccapelo, Email: roberta.spaccapelo@unipg.it.
John B. Dame, Email: damej@ufl.edu.
Web Extra Material
{kind=link}
Targeted gene disruption of the pm4 locus.
{kind=link}
Generation of transgenic parasites expressing the GFP-luciferase fusion protein (wt+ and wt++).
{kind=link}
The number of merozoites per mature schizont in control (wt, wt+) and in Δpm4 (688cl2, Δpm4+) infected erythrocytes.
{kind=link}
Cell cycle analysis measured by flow cytometry showing a delayed growth phenotype of the of Δpm4+ parasites.
{kind=link}
The course of infection of Δpm4 parasites in different mouse lines/parasite strains combinations.
{kind=link}
Analysis of sequestration of schizonts in mice infected with wt+ and Δpm4+ parasites.
{kind=link}
Challenge of P. berghei Δpm4 convalescent mice with wild type P. chabaudi parasites.
{kind=link}
Comparison of protection against challenge with wt+ parasites of mice recovered from Δpm4 infections or recovered by drug cure of wt infections.
{kind=link}
Parasite localization in mice treated with purified IgG obtained from Δpm4 convalescent mice and infected with wt++ parasites.
Genetically modified parasite lines generated.
References
- 1.Coombs GH, Goldberg DE, Klemba M, Berry C, Kay J, Mottram JC. Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 2001;17:532–537. doi: 10.1016/s1471-4922(01)02037-2. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R, Liu J, Beatty W, Pelosof L, Klemba M, Goldberg DE. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc Natl Acad Sci USA. 2002;99:990–995. doi: 10.1073/pnas.022630099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dame JB, Yowell CA, Omara-Opyene L, Carlton JM, Cooper RA, Li T. Plasmepsin 4, the food vacuole aspartic proteinase found in all Plasmodium spp. infecting man. Mol Biochem Parasitol. 2003;130:1–12. doi: 10.1016/s0166-6851(03)00137-3. [DOI] [PubMed] [Google Scholar]
- 4.Bonilla JA, Bonilla TD, Yowell CA, Fujioka H, Dame JB. Critical roles for the digestive vacuole plasmepsins of Plasmodium falciparum in vacuolar function. Mol Microbiol. 2007;65:64–75. doi: 10.1111/j.1365-2958.2007.05768.x. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc Natl Acad Sci USA. 2006;103:8840–8845. doi: 10.1073/pnas.0601876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omara-Opyene AL, Moura PA, Sulsona CR, Bonilla JA, Yowell CA, Fujioka H, Fidock DA, Dame JB. Genetic disruption of the Plasmodium falciparum digestive vacuole plasmepsins demonstrates their functional redundancy. J Biol Chem. 2004;279:54088–54096. doi: 10.1074/jbc.M409605200. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Gluzman IY, Drew ME, Goldberg DE. The role of Plasmodium falciparum food vacuole plasmepsins. J Biol Chem. 2005;280:1432–1437. doi: 10.1074/jbc.M409740200. [DOI] [PubMed] [Google Scholar]
- 8.Bonilla JA, Moura PA, Bonilla TD, Yowell CA, Fidock DA, Dame JB. Effects on growth, hemoglobin metabolism and paralogous gene expression resulting from disruption of genes encoding the digestive vacuole plasmepsins of Plasmodium falciparum. Int J Parasitol. 2007;37:317–327. doi: 10.1016/j.ijpara.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Lou J, Lucas R, Grau GE. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin Microbiol Rev. 2001;14:810–820. doi: 10.1128/CMR.14.4.810-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Souza JB, Riley EM. Cerebral malaria: the contribution of studies in animal models to our understanding of immunopathogenesis. Microbes Infect. 2002;4:291–300. doi: 10.1016/s1286-4579(02)01541-1. [DOI] [PubMed] [Google Scholar]
- 11.Carvalho TG, Menard R. Manipulating the Plasmodium genome. Curr Issues Mol Biol. 2005;7:39–55. [PubMed] [Google Scholar]
- 12.Janse CJ, Franke-Fayard B, Mair GR, Ramesar J, Thiel C, Engelmann S, Matuschewski K, van Gemert GJ, Sauerwein RW, Waters AP. High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol Biochem Parasitol. 2006;145:60–70. doi: 10.1016/j.molbiopara.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Janse CJ, Ramesar J, Waters AP. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc. 2006;1:346–356. doi: 10.1038/nprot.2006.53. [DOI] [PubMed] [Google Scholar]
- 14.Kooij TW, Franke-Fayard B, Renz J, Kroeze H, van Dooren MW, Ramesar J, Augustijn KD, Janse CJ, Waters AP. Plasmodium berghei α-tubulin II: a role in both male gamete formation and asexual blood stages. Mol Biochem Parasitol. 2005;144:16–26. doi: 10.1016/j.molbiopara.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Franke-Fayard B, Janse CJ, Cunha-Rodrigues M, Ramesar J, Buscher P, Que I, Lowik C, Voshol PJ, den Boer MA, van Duinen SG, Febbraio M, Mota MM, Waters AP. Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc Natl Acad Sci USA. 2005;102:11468–11473. doi: 10.1073/pnas.0503386102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janse CJ, Franke-Fayard B, Waters AP, Ramesar J, Tomas AM, van der Wel AM, Thomas AW. Selection by flow-sorting of genetically transformed, GFP-expressing blood stages of the rodent malaria parasite, Plasmodium berghei. Nat Protoc. 2006;1:614–623. doi: 10.1038/nprot.2006.88. [DOI] [PubMed] [Google Scholar]
- 17.Franke-Fayard B, Trueman H, Ramesar J, Mendoza J, van der Keur M, van der Linden R, Sinden RE, Waters AP, Janse CJ. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Janse CJ, Haghparast A, Speranca MA, Ramesar J, Kroeze H, del Portillo HA, Waters AP. Malaria parasites lacking eef1a have a normal S/M phase yet grow more slowly due to a longer G1 phase. Mol Microbiol. 2003;50:1539–1551. doi: 10.1046/j.1365-2958.2003.03820.x. [DOI] [PubMed] [Google Scholar]
- 19.Janse CJ, Van Vianen PH. Flow cytometry in malaria detection. Methods Cell Biol. 1994;42:295–318. doi: 10.1016/s0091-679x(08)61081-x. [DOI] [PubMed] [Google Scholar]
- 20.Janse CJ, Waters AP. Plasmodium berghei: the application of cultivation and purification techniques to molecular studies of malaria parasites. Parasitol Today. 1995;11:138–143. doi: 10.1016/0169-4758(95)80133-2. [DOI] [PubMed] [Google Scholar]
- 21.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 22.Engwerda C, Belnoue E, Gruner AC, Renia L. Experimental models of cerebral malaria. Curr Top Microbiol Immunol. 2005;297:103–143. [PubMed] [Google Scholar]
- 23.Curfs JH, van der Meide PH, Billiau A, Meuwissen JH, Eling WM. Plasmodium berghei: recombinant interferon-γ and the development of parasitemia and cerebral lesions in malaria-infected mice. Exp Parasitol. 1993;77:212–223. doi: 10.1006/expr.1993.1078. [DOI] [PubMed] [Google Scholar]
- 24.Benchenane K, Berezowski V, Fernandez-Monreal M, Brillault J, Valable S, Dehouck MP, Cecchelli R, Vivien D, Touzani O, Ali C. Oxygen glucose deprivation switches the transport of tPA across the blood-brain barrier from an LRP-dependent to an increased LRP-independent process. Stroke. 2005;36:1065–1070. doi: 10.1161/01.STR.0000163050.39122.4f. [DOI] [PubMed] [Google Scholar]
- 25.Eling WM. Chronic, patent Plasmodium berghei malaria in splenectomized mice. Infect Immun. 1982;35:880–886. doi: 10.1128/iai.35.3.880-886.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franke-Fayard B, Waters AP, Janse CJ. Real-time in vivo imaging of transgenic bioluminescent blood stages of rodent malaria parasites in mice. Nat Protoc. 2006;1:476–485. doi: 10.1038/nprot.2006.69. [DOI] [PubMed] [Google Scholar]
- 27.Ting LM, Gissot M, Coppi A, Sinnis P, Kim K. Attenuated Plasmodium yoelii lacking purine nucleoside phosphorylase confer protective immunity. Nat Med. 2008;14:954–958. doi: 10.1038/nm.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tewari R, Ogun SA, Gunaratne RS, Crisanti A, Holder AA. Disruption of Plasmodium berghei merozoite surface protein 7 gene modulates parasite growth in vivo. Blood. 2005;105:394–396. doi: 10.1182/blood-2004-06-2106. [DOI] [PubMed] [Google Scholar]
- 29.Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. 2005;5:722–735. doi: 10.1038/nri1686. [DOI] [PubMed] [Google Scholar]
- 30.Amante FH, Stanley AC, Randall LM, Zhou Y, Haque A, McSweeney K, Waters AP, Janse CJ, Good MF, Hill GR, Engwerda CR. A role for natural regulatory T cells in the pathogenesis of experimental cerebral malaria. Am J Pathol. 2007;171:548–559. doi: 10.2353/ajpath.2007.061033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boutlis CS, Riley EM, Anstey NM, de Souza JB. Glycosylphosphatidylinositols in malaria pathogenesis and immunity: potential for therapeutic inhibition and vaccination. Curr Top Microbiol Immunol. 2005;297:145–185. doi: 10.1007/3-540-29967-x_5. [DOI] [PubMed] [Google Scholar]
- 32.Riley EM, Wahl S, Perkins DJ, Schofield L. Regulating immunity to malaria. Parasite Immunol. 2006;28:35–49. doi: 10.1111/j.1365-3024.2006.00775.x. [DOI] [PubMed] [Google Scholar]
- 33.Schofield L, Hewitt MC, Evans K, Siomos MA, Seeberger PH. Synthetic GPI as a candidate anti-toxic vaccine in a model of malaria. Nature. 2002;418:785–789. doi: 10.1038/nature00937. [DOI] [PubMed] [Google Scholar]
- 34.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med. 2005;201:19–25. doi: 10.1084/jem.20041836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci USA. 2007;104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waki S, Tamura J, Imanaka M, Ishikawa S, Suzuki M. Plasmodium berghei: isolation and maintenance of an irradiation attenuated strain in the nude mouse. Exp Parasitol. 1982;53:335–340. doi: 10.1016/0014-4894(82)90076-5. [DOI] [PubMed] [Google Scholar]
- 37.Yoshimoto T, Yoneto T, Waki S, Nariuchi H. Interleukin-12-dependent mechanisms in the clearance of blood-stage murine malaria parasite Plasmodium berghei XAT, an attenuated variant of P. berghei NK65. J Infect Dis. 1998;177:1674–1681. doi: 10.1086/515301. [DOI] [PubMed] [Google Scholar]
- 38.Quinn TC, Wyler DJ. Resolution of acute malaria (Plasmodium berghei in the rat): reversibility and spleen dependence. Am J Trop Med Hyg. 1980;29:1–4. doi: 10.4269/ajtmh.1980.29.1. [DOI] [PubMed] [Google Scholar]
- 39.Weiss L. Mechanisms of splenic control of murine malaria: cellular reactions of the spleen in lethal (strain 17XL) Plasmodium yoelii malaria in BALB/c mice, and the consequences of pre-infective splenectomy. Am J Trop Med Hyg. 1989;41:144–160. doi: 10.4269/ajtmh.1989.41.144. [DOI] [PubMed] [Google Scholar]
- 40.Engwerda CR, Beattie L, Amante FH. The importance of the spleen in malaria. Trends Parasitol. 2005;21:75–80. doi: 10.1016/j.pt.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 41.Engwerda CR, Good MF. Interactions between malaria parasites and the host immune system. Curr Opin immunol. 2005;17:381–387. doi: 10.1016/j.coi.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman SL, Goh LM, Luke TC, Schneider I, Le TP, Doolan DL, Sacci J, de la Vega P, Dowler M, Paul C, Gordon DM, Stoute JA, Church LW, Sedegah M, Heppner DG, Ballou WR, Richie TL. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J Infect Dis. 2002;185:1155–1164. doi: 10.1086/339409. [DOI] [PubMed] [Google Scholar]
- 43.Wykes M, Good MF. A case for whole-parasite malaria vaccines. Int J Parasitol. 2007;37:705–712. doi: 10.1016/j.ijpara.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Mueller AK, Labaied M, Kappe SH, Matuschewski K. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature. 2005;433:164–167. doi: 10.1038/nature03188. [DOI] [PubMed] [Google Scholar]
- 45.Mueller AK, Camargo N, Kaiser K, Andorfer C, Frevert U, Matuschewski K, Kappe SH. Plasmodium liver stage developmental arrest by depletion of a protein at the parasite-host interface. Proc Natl Acad Sci USA. 2005;102:3022–3027. doi: 10.1073/pnas.0408442102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Dijk MR, Douradinha B, Franke-Fayard B, Heussler V, van Dooren MW, van Schaijk B, van Gemert GJ, Sauerwein RW, Mota MM, Waters AP, Janse CJ. Genetically attenuated, P36p-deficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proc Natl Acad Sci USA. 2005;102:12194–12199. doi: 10.1073/pnas.0500925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aly AS, Mikolajczak SA, Rivera HS, Camargo N, Jacobs-Lorena V, Labaied M, Coppens I, Kappe SH. Targeted deletion of SAP1 abolishes the expression of infectivity factors necessary for successful malaria parasite liver infection. Mol Microbiol. 2008;69:152–163. doi: 10.1111/j.1365-2958.2008.06271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silvie O, Goetz K, Matuschewski K. A sporozoite asparagine-rich protein controls initiation of Plasmodium liver stage development. PLoS Pathog. 2008;4:e1000086. doi: 10.1371/journal.ppat.1000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purcell LA, Yanow SK, Lee M, Spithill TW, Rodriguez A. Chemical attenuation of Plasmodium berghei sporozoites induces sterile immunity in mice. Infect Immun. 2008;76:1193–1199. doi: 10.1128/IAI.01399-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Targeted gene disruption of the pm4 locus.
Generation of transgenic parasites expressing the GFP-luciferase fusion protein (wt+ and wt++).
The number of merozoites per mature schizont in control (wt, wt+) and in Δpm4 (688cl2, Δpm4+) infected erythrocytes.
Cell cycle analysis measured by flow cytometry showing a delayed growth phenotype of the of Δpm4+ parasites.
The course of infection of Δpm4 parasites in different mouse lines/parasite strains combinations.
Analysis of sequestration of schizonts in mice infected with wt+ and Δpm4+ parasites.
Challenge of P. berghei Δpm4 convalescent mice with wild type P. chabaudi parasites.
Comparison of protection against challenge with wt+ parasites of mice recovered from Δpm4 infections or recovered by drug cure of wt infections.
Parasite localization in mice treated with purified IgG obtained from Δpm4 convalescent mice and infected with wt++ parasites.
Genetically modified parasite lines generated.