

Abstract
Neuronal migration disorders are often identified in patients with epilepsy refractory to medical treatment. The prolonged or repeated seizures are known to cause neuronal death; however, the mechanism underlying seizure-induced neuronal death remains to be elucidated. An essential role of cyclin-dependent kinase 5 (Cdk5) in brain development has been demonstrated in Cdk5−/− mice, which show neuronal migration defects and perinatal lethality. Here, we show the consequences of Cdk5 deficiency in the postnatal brain by generating Cdk5 conditional knockout mice, in which Cdk5is selectively eliminated from neurons in the developing forebrain. The conditional mutant mice were viable, but exhibited complex neurological deficits including seizures, tremors, and growth retardation. The forebrain not only showed disruption of layering, but also neurodegenerative changes accompanied by neuronal loss and microglial activation. The neurodegenerative changes progressed with age and were accompanied by up-regulation of the neuronal tissue-type plasminogen activator, a serine protease known to mediate microglial activation. Thus age-dependent neurodegeneration in the Cdk5 conditional knockout mouse brain invoked a massive inflammatory reaction. These findings indicate an important role of Cdk5 in inflammation, and also provide a mouse model to examine the possible involvement of inflammation in the pathogenesis of progressive cognitive decline in patients with neuronal migration disorders.
Mammalian brains have a highly organized neocortical structure that forms the basis for higher cognitive functions. The proper development of the neocortex requires a series of steps that begins with migration of neurons from their birth place and initiation of neurite outgrowth, and ultimately leads to differentiation of neurons and the formation of connections with their appropriate targets.1 Recent studies have identified a large number of genes that are critical for these processes, and revealed that the mutations in these genes are associated with human neurological diseases.2,3 Neuronal migration disorders have been diagnosed with increasingly higher frequency in patients with epilepsy refractory to medical treatment and mental retardation.4 Structural abnormalities arising from neuronal migration disorders contribute to hyperexcitability in the brain, primarily through the creation of abnormal neuronal circuitry that results in an imbalance between excitatory and inhibitory synaptic transmission.5,6 The prolonged or repeated seizures are known to cause neuronal death, which may contribute to the progressive cognitive decline observed in some patients with epilepsy.7 Therefore, determining the molecular mechanism underlying these pathological processes is expected to have significant implications for the development of neuroprotective strategies in patients with neuronal migration disorders.
Cyclin-dependent kinase 5 (Cdk5) plays an important role in various aspects of cortical development, including neuronal migration, neurite outgrowth, and axonal pathfinding.8 Cdk5 is a member of the Cdk family of serine/threonine kinases. Unlike other Cdks that are major regulators of cell-cycle progression, Cdk5 is predominantly involved in phosphorylation of target proteins in neurons. Direct evidence of the involvement of Cdk5 in neuronal migration came from analysis of Cdk5−/− mice, which display perinatal lethality and extensive neuronal migration defects in the brain.9,10 The perinatal lethality of Cdk5−/− mice hampered our efforts to delineate the impact of Cdk5 deficiency on the postnatal brain. To circumvent this problem, we used the Cre-loxP recombination technique to generate a Cdk5 conditional knockout mouse (Cdk5 cKO). The Cdk5 gene was deleted in neurons of the forebrain by expression of cre recombinase, directed by the promoter of the CaMKII α gene encoding the α-subunit of the calcium/calmodulin-dependent kinase II (CaMKII). This strategy led to generation of viable mice with reduced expression of Cdk5 in the forebrain.
Here we report that elimination of neuronal Cdk5 from the developing forebrain caused complex neurological deficits, growth retardation, and early mortality in mice. Cdk5 cKO mice exhibit not only layering defects in the forebrain but also neurodegenerative changes that are accompanied by aberrant activation of astroglia and microglia. Our data suggest that cross talk between neurons and microglia may contribute to neurodegeneration in the brain with neuronal migration disorder.
Materials and Methods
Generation of CaMKIIcre-Directed Cdk5 Conditional Mutant Mouse
Mice carrying the floxed Cdk5 allele (flanked by two loxP motifs) were generated as described.11 The cre transgenic mice used in this study contained a cre transgene under the transcriptional control of the CaMKII α promoter, termed CaMKIIcre.12 The mouse Cdk5 locus on chromosome 5 is in close proximity to two neighboring genes, Accn3 and Slc4a2. Therefore, we considered the possibility that the expression of these genes might be affected by the deletion of their noncoding sequences during cre-mediated recombination (see Supplemental Figure S1 at http://ajp.amjpathol.org). To assess the possible effects of the deletion of these genes on the phenotype of Cdk5loxP/loxP; CaMKIIcre mice, termed Cdk5 cKO1 mice, we also generated Cdk5loxP/−; CaMKIIcre mice, termed Cdk5 cKO2 mice, in which the cre-mediated recombination would occur only in one allele without significantly altering the expression of the neighboring genes. These independent lines of Cdk5 cKO mice, Cdk5 cKO1 and Cdk5 cKO2 mice, were generated by crossing Cdk5loxP/loxP mice with Cdk5loxP/+; CaMKIIcre mice and with Cdk5+/−; CaMKIIcre mice, respectively.9,11,12 Genotypes of the mice were determined by PCR or Southern blot analysis, as described.11,13 Mice were housed under a 12 hours light/12 hours dark cycle. All of the investigations involving the mice were in compliance with the National Institutes of Health guidelines on the care and use of laboratory and experimental animals.
Histological Analyses
To examine cre-mediated recombination in the brain, CaMKIIcre transgenic mice were crossed with ROSA26-lacZ reporter mice.14 Brains obtained from their progenies were processed for X-gal staining, as described.15
For histological and immunohistochemical analyses, mice were anesthetized by intraperitoneal injections of avertin (250 mg/kg, Fluka, Milwaukee, WI). They were then perfused transcardially with 0.1 mol/L sodium phosphate buffer, pH 7.4, followed by Streck tissue fixative (Streck Laboratories, Inc., Omaha, NE), a non-crosslinking fixative. Dissected brains were kept in the fixative overnight at 37°C. The fixed brains were embedded in paraffin and cut into 5-μm-thick coronal or sagittal sections. Histological analyses were performed on the sections stained with cresyl violet or H&E. Immunohistochemistry was performed using either an avidin-biotin-peroxidase complex technique (Vector Laboratories, Burlingame, CA) with diaminobenzidine as a substrate, or immunofluorescence staining with Alexa Fluor 488- and 568-coupled secondary antibodies (Invitrogen, Carlsbad, CA). The following antibodies were used at the indicated dilutions: anti-NeuN (Chemicon International Inc., Temecula, CA; 1:1000); anti-glial fibrillary acidic protein (GFAP) (DAKO, Denmark; 1:4000); anti-F4/80 antigen (Serotec Ltd, Oxford, UK; 1:10); anti-Cux1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, 1:500); and anti-Ctip2 (Abcam Inc., Cambridge, MA, 1:500). The staining specificity of these antibodies was assessed by omission of the primary antibodies.
To detect neurodegeneration in the brain, coronal sections obtained from control and Cdk5 cKO1 mice at the ages of 2 to 3 months were stained with amino-cupric-silver as described.16 Animals were transcardially perfused with freshly prepared 4% paraformaldehyde in 0.1 mol/L cacodylate buffer, pH 7.4. The brains were removed and embedded in a gelatin matrix (NeuroScience Associates, Knoxville, TN). Free-floating sections (35 μm in thickness) were cut on a microtome and processed for staining.
Terminal dUTP Nick-End Labeling Analysis
Detection of 3′-OH termini of DNA strand breaks was performed on paraffin sections using an in situ cell death detection kit (Roche, Indianapolis, IN) following the recommendations of the manufacturer.
Western Blot Analyses
Preparation of brain lysates and Western blot analyses were performed as described previously.17 The primary antibodies were used at the indicated dilutions as follows: anti-Cdk5 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA; 1:2000), anti-neurofilament M (Chemicon International Inc., Temecula, CA; 1:10,000), anti-GFAP (Chemicon International Inc., Temecula, CA; 1:2000), and anti-actin (Sigma, St. Louis, MO; 1:2000) antibodies. Optical densities of the bands were quantified using an image analysis system with NIH Image software, version 1.62. To determine the levels of Cdk5, neurofilament-M, and GFAP protein, the data obtained with each antibody were normalized to the actin levels on the stripped and reprobed membranes. For reuse, the membranes were stripped for 20 minutes at 50°C in 63 mmol/L Tris-HCl, pH 6.8, containing 100 mmol/L 2-mercaptoethanol and 2% SDS.
Reverse Transcription-PCR
Total RNA was extracted from various regions of the mouse brains with TRIzol reagent (Invitrogen, Carlsbad, CA). Reverse transcription (RT)-PCR was performed using SuperScript First-Strand Synthesis System (Invitrogen, Carlsbad, CA) for generation of cDNA using 2 μg of total RNA in a 20 μl reaction. The following primers were used: for CD68, 5′- CCGAATCCTATACCCAATTCAG-3′ and 5′-AGCAGCCTGTAGCCTTAGAGAG-3′, generating a 372 bp product; for tumor necrosis factor (TNF)-α, 5′- AGCCGATTTGCTATCTCATACC-3′ and 5′- AGTACTTGGGCAGATTGACCTC-3′, generating a 183 bp product; for tissue-type plasminogen activator (tPA), 5′-GCCCTCTGGTGTGCATGATCAAT-3′ and 5′-TTCCAAAGCCAGACCTTCATCCTT-3′, generating a 364 bp product; for c-fos, 5′-CAACGCCGACTACGAGGCGTCAT-3′ and 5′-GTGGAGATGGCTGTCACCG-3′, generating a 189 bp product; and for glyceraldehyde-3-phosphate dehydrogenase, 5′-CCATCACCATCTTCCAGGAG-3′ and 5′-GCATGGACTGTGGTCATGAG-3′, generating a 322 bp product.
Plasminogen-Casein Zymography
Cryostat sections (12 μm in thickness) were obtained from mice that were transcardially perfused with ice-cold PBS, and analyzed for in situ proteinase activity as described.18 Briefly, 100 μl overlays consisting of 1% agarose, 1.7% boiled nonfat dry milk solution, and 30 μg/ml purified human plasminogen (Calbiochem, San Diego, CA) in PBS were applied to prewarmed tissue sections and spread evenly under 24 × 32 mm glass coverslips. The slides were incubated for 4 hours at 37°C in a humidified chamber. Identical experiments were performed with the overlay from which the plasminogen was omitted. Photographs were taken using dark-ground illumination. For SDS-polyacrylamide gel electrophoresis zymography, various regions of the brain were dissected from control and Cdk5 cKO1 mice, following perfusion with ice-cold PBS. Protein extraction and zymography were performed as described.19 Brain lysates were prepared in a lysis buffer consisting of 20 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, and 1% Nonidet-P40. Equal amounts of protein (25 μg) were loaded without reduction or heating onto 10% SDS polyacrylamide gels containing 20 μg/ml plasminogen and 0.4% boiled nonfat dry milk solution. Purified human tPA (0.5 ng, American Diagnostica Inc., Greenwich, CT) and brain lysates from tPA−/− mice were included as a positive control and a negative control, respectively. Following electrophoresis, the gels were soaked two times in 2.5% Triton-X 100 for 30 minutes to remove the SDS. Subsequently, the gels were placed in 0.1 mol/L glycine buffer, pH 8.0, and incubated for 12 hours at 37°C to allow proteolysis of the substrate in the gels. Finally, the gels were stained with 0.2% Coomassie Brilliant Blue-R250.
Results
Generation of Forebrain-Specific Cdk5 Knockout Mouse
We used the cre/loxP recombination technique to achieve selective inactivation of Cdk5 in the forebrain. When tested with a ROSA26-lacZ reporter mouse line,14 CaMKIIcre transgenic mice exhibited the cre-mediated recombination mainly in the forebrain (Figure 1, A and B), which was detected as early as E12.5 (data not shown). In contrast, few LacZ-positive cells were detected in the midbrain, hindbrain, and spinal cord. Both the Cdk5 cKO1 and Cdk5 cKO2 mice were born in an expected Mendelian frequency. To determine the amount of Cdk5 remaining in the brain after cre-mediated recombination, we performed Western blot analysis. Cdk5 protein levels in the forebrain of Cdk5 cKO1 and Cdk5 cKO2 mice were decreased to 30% to 55%, and to 5% to 30% in the control (Cdk5loxP/loxP) mice, respectively (Figure 1C), while only minor reductions were observed in the hindbrain and spinal cord (data not shown).
Figure 1.
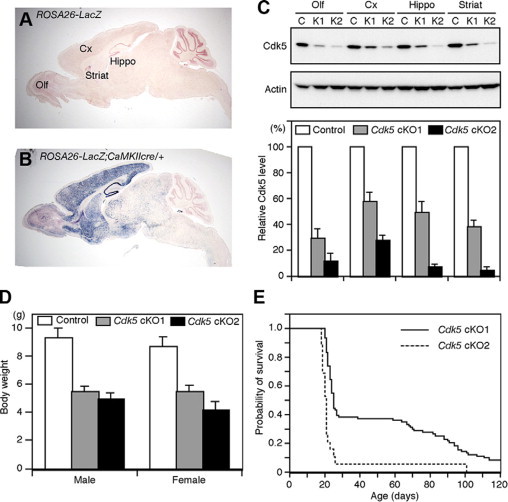
Generation of forebrain-specific Cdk5 knockout mouse. A and B: Cre-mediated recombination was examined by X-gal staining of sagittal sections obtained from 1-month-old ROSA26-lacZ reporter mice without (A), or with (B), the CaMKIIcre transgene. Olf, olfactory bulb; Cx, cerebral cortex; Hippo, hippocampus; Striat, striatum. C: Western blot showing a marked reduction of Cdk5 protein in the forebrain of Cdk5 cKO mice. C: control (Cdk5loxP/loxP) mice; K1, Cdk5 cKO1 (Cdk5loxP/loxP; CaMKIIcre) mice; K2, Cdk5 cKO2 (Cdk5loxP/-; CaMKIIcre) mice. In the bar graph, the Cdk5 levels relative to those of control mice are shown as means ± SEM (n = 3). D: Body weight of control, Cdk5 cKO1, and Cdk5 cKO2 mice at 20 days of age, showing growth retardation of Cdk5 cKO mice. E: Kaplan-Meier survival curves for Cdk5 cKO1 (n = 84) and Cdk5 cKO2 (n = 19) mice showing decreased viability. All of the control littermates survived beyond the observation period.
Loss of Neuronal Cdk5 from Developing Forebrain Caused Growth Retardation, Early Mortality, and Complex Neurological Deficits in Mice
Both the Cdk5 cKO1 and Cdk5 cKO2 mice were indistinguishable from the control littermates at birth, and their average body weight did not differ significantly from the controls by postnatal day (P) 5. However, a failure to thrive became apparent by P10. Both the Cdk5 cKO mice weighed as little as 50% to 60% of the weight of their control littermates (Figure 1D). The cause of this growth retardation remains unclear.
In addition to a failure to thrive, both Cdk5 cKO mice showed decreased viability after weaning (Figure 1E). Approximately 60% to 70% of Cdk5 cKO1 mice and 95% of Cdk5 cKO2 mice died within a week after weaning. The remaining 30% to 40% of the Cdk5 cKO1 mice died after 2 months of age, while some mutant mice survived beyond 4 months of age. Although the cause of the decreased viability of these mutant mice is not clear, we have observed some Cdk5 cKO1 mice that died immediately following spontaneous seizures.
Both the Cdk5 cKO1 and Cdk5 cKO2 mice displayed complex neurological deficits. One of the signs observed by P20 was dyskinesia of the limbs. When suspended by their tail, the mutant mice retracted their limbs toward their trunks in a dystonic fashion, rather than extending them as their control littermates did. Around the same age, the mutant mice developed tremors. We also noticed that the Cdk5 cKO1 mice developed seizures by 2 months of age, which were characterized by repetitive myoclonic jerks and generalized tonic seizures followed by loss of posture. Except for their earlier mortality, the phenotype of Cdk5 cKO2 mice was similar to that of Cdk5 cKO1 mice, suggesting that possible deletion of the neighboring genes, Accn3 and Slc4a2, in Cdk5 cKO1 mice does not affect their phenotype.
Brain Development in Cdk5 Conditional Mutant Mice
We reported earlier that Cdk5−/− mice displayed neuronal migration defects in the developing cerebrum and cerebellum, as well as in the hindbrain, including the facial nerve nucleus and inferior olive.9,20 To examine the effect of forebrain-specific reduction of Cdk5 expression on brain development, we compared the cytoarchitecture of the E18.5 brains of Cdk5 cKO1 mice with those of Cdk5+/+ (Figure 2, A,D,G,J) or Cdk5−/− mice using Nissl-stained sections. Cdk5 cKO1 mice displayed disruption of the laminar structure in the cerebral cortex (Figure 2C) and hippocampus (Figure 2F), similar to that of Cdk5−/− mouse brain (Figure 2, B and E), while some of the cortical neurons appeared to migrate normally, indicative of the presence of neurons that had escaped Cdk5 inactivation in the Cdk5 cKO1 forebrain. Cdk5−/− mice exhibited additional developmental abnormalities in the hindbrain, where the facial motor neurons did not migrate properly from the dorsal pons (Figure 2H). Furthermore, the inferior olive neurons were not aligned to form the characteristic structure of the inferior olive (Figure 2K). These abnormalities in the hindbrain were not observed in Cdk5 cKO1 mice (Figure 2, I and L), however, indicating that the CaMKIIcre-mediated recombination of the Cdk5 gene occurred specifically in the forebrain during brain development.
Figure 2.
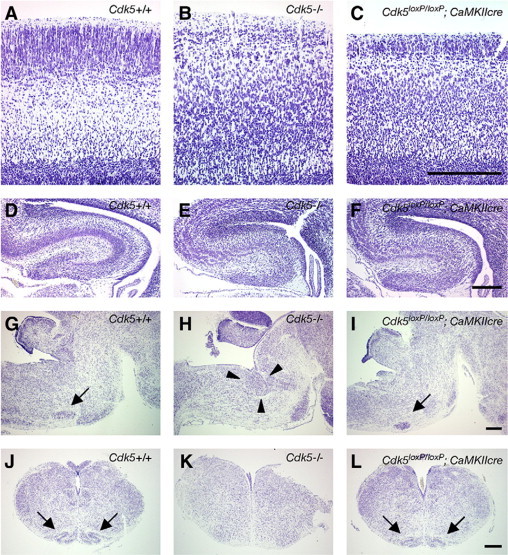
Forebrain-specific neuronal migration defects in the brain of Cdk5 conditional mutant (Cdk5 cKO1) mouse. Serial brain sections (5 μm in thickness) were obtained from E18.5 mouse embryos including wild-type (Cdk5+/+), Cdk5 knockout (Cdk5−/−), and Cdk5 cKO1 (Cdk5loxP/loxP; CaMKIIcre) mice, and stained with cresyl violet. A–C: Coronal sections of the cerebral cortex showing the layering defects in the Cdk5 cKO1 brain (C), which are similar to those in Cdk5−/− mouse brain (B). D–F: Coronal sections showing the disorganized hippocampus in the brain of Cdk5−/− (E) and Cdk5 cKO1 (F) mice. G–I: Sagittal sections of the hindbrain showing the ectopic facial motor neurons in Cdk5−/− mouse brain (H, indicated by arrowheads) and the normally located facial motor neurons in the wild-type brain (G, indicated by arrow) and Cdk5 cKO1 brain (I, indicated by arrow). J–L: Coronal sections of the medulla oblongata showing the inferior olive in the wild-type mice (J, indicated by arrows) and Cdk5 cKO1 mice (L, indicated by arrows), which is absent in Cdk5−/− mice (K). Scale bars = 250 μm.
Recent studies have identified many genes that define layer-specific identities within the neocortex.21 To examine the layer structure in the neocortex of Cdk5 cKO1 mice, we performed immunohistochemical staining of the mouse brains for the layer-specific markers: Cux1, a marker of layer II to IV, and Ctip2, a marker of layer V and VI. Consistent with the earlier report showing the inverted pattern of layer structure in the neocortex of Cdk5−/− mice,22 Cux1-immunopositive neurons were positioned below the Ctip2-immunopositive neurons in the Cdk5 cKO1 mouse brain (see Supplemental Figure S2 at http://ajp.amjpathol.org). This indicated that the later-born neurons stacked up in the neocortex in an inverted order.
Neurodegeneration in the Cdk5 Conditional Mutant Brain
Macroscopic inspection of the brains from 3-month-old Cdk5 cKO1 mice revealed a striking reduction in the size of the forebrain compared with control littermates (Figure 3, C and D), whereas the size of the E18.5 mutant brain did not differ from the controls (Figure 3, A and B). We examined the time-course of morphological changes in the mutant forebrain using H&E-stained coronal sections. Histological analysis revealed atrophic changes in the mutant forebrain that progressed with age, as evidenced by the progressive dilatation of the lateral ventricle (Figure 3, E–N). Amino-cupric silver staining revealed many silver-reactive fibers, reflecting axonal or dendritic degeneration in the neuropils of the cerebral cortex (Figure 3, O and P) and striatum (Figure 3, Q and R). This discovery indicated degenerative changes in the mutant forebrain. Consistent with these histological data, the number of terminal dUTP nick-end labeling-positive cells increased in the mutant forebrain (see Supplemental Figure S3 at http://ajp.amjpathol.org). To further examine if there was a loss of neurons in the mutant forebrain, we performed immunohistochemical staining of the mouse brains for NeuN, a marker for neurons. The density of NeuN-immunopositive cells appeared to decrease in the mutant forebrain compared with that of the control mice, suggesting neuronal loss in the mutant forebrain (Figure 4A). These results were corroborated by Western blot analysis for neurofilament-M, another marker for neurons. The levels of neurofilament-M decreased specifically in the mutant forebrain, while the decrease in the hippocampus was not statistically significant (Figure 4B). Neuronal loss is often accompanied by reactive astrogliosis. The specificity of the affected lesions in the mutant brain was also revealed by GFAP immunostaining (Figure 4C) and immunoblotting (Figure 4D). Thus the atrophic changes in the mutant forebrain likely resulted from neuronal loss that occurred as a consequence of the neurodegenerative process.
Figure 3.
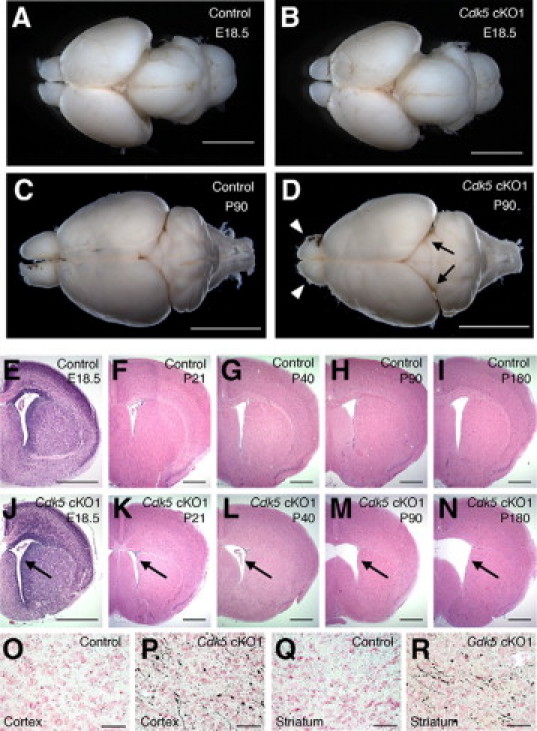
Neurodegeneration in the forebrain of Cdk5 conditional mutant (Cdk5 cKO1) mice. A–D: Dorsal view of the whole brains of 3-month-old mice (C and D) demonstrated the marked reduction in size of the olfactory bulb (indicated by arrowheads in panel D) and the shrinkage of the cerebral cortex (indicated by arrows in panel D) in Cdk5 cKO1 mice. Note that there were no differences in size between E18.5 control (A) and mutant (B) brains. E–N: Histological analysis of H&E-stained coronal sections revealed that the atrophic changes in the forebrain of Cdk5 cKO1 mice progressed with age, as evidenced by the progressive dilatation of the lateral ventricle (indicated by arrows). Note that E18.5 Cdk5 cKO1 brain (J) exhibited neuronal migration defects in the cerebral cortex, but not any atrophic changes. O–R: Amino-cupric silver staining on 2-month-old control (O and Q) and Cdk5 cKO1 (P and R) brains shows the silver-reactive cell bodies and fibers (axons or dendrites) in the cerebral cortex (P) and striatum (R) of Cdk5 cKO1 mice, indicative of neurodegenerative changes. Scale bars: 2 mm (A and B); 5 mm (C and D); 1 mm (E–N); 250 μm (O–R).
Figure 4.
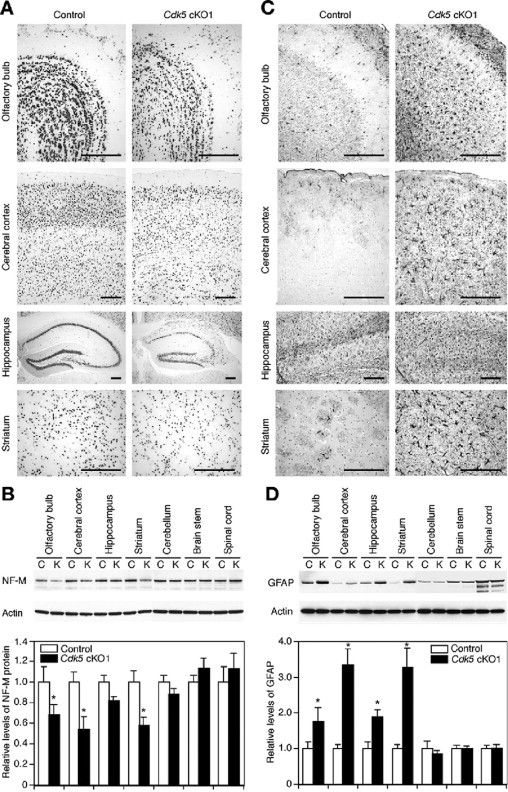
Neuronal loss and astrogliosis in the forebrain of Cdk5 conditional mutant (Cdk5 cKO1) mice. A: Immunostaining of 3-month-old mouse brains for NeuN, a marker for neurons, revealed a decreased density of NeuN-immunopositive cells in the olfactory bulb, cerebral cortex, hippocampus, and striatum of Cdk5 cKO1 mice, as compared with those of the control mice. B: Western blot analysis showed that the levels of neurofilament-M (NF-M), another marker for neurons, decreased in the forebrain of Cdk5 cKO1 mice, but not in the hindbrain and spinal cord. In the bar graph, quantitative results indicate the NF-M levels relative to those of the control mice and are shown as mean ± SEM (n = 3). Data are expressed in arbitrary units. The asterisk indicates P < 0.05, compared with the control mice (Mann-Whitney test). C: Immunostaining of 3-month-old mouse brains for glial fibrillary acidic protein (GFAP), a marker for astrocytes, revealed massive astrogliosis in the olfactory bulb, cerebral cortex, hippocampus, and striatum of Cdk5 cKO1 mice. D: Western blot analysis showed that the levels of GFAP increased in the forebrain of Cdk5 cKO1 mice, but not in the hindbrain and spinal cord. In the bar graph, quantitative results indicate the GFAP levels relative to those of the control mice and are shown as mean ± SEM. (n = 3). Data are expressed in arbitrary units. The asterisk indicates P < 0.05, compared with the control mice (Mann-Whitney test). Abbreviations in (B and D) are as follows: C, control (Cdk5loxP/loxP) mice and K, Cdk5 cKO1 (Cdk5loxP/loxP; CaMKIIcre) mice. Scale bars = 250 μm.
Microglial Activation in the Forebrain of Cdk5 Conditional Mutant Mice
Neurodegeneration is often accompanied by an inflammatory process that involves astroglial and microglial activation. In particular, microglial activation appears to play a pivotal role in the initiation and progression of neurodegenerative processes.23,24 We therefore examined if there was microglial activation in the mutant forebrain, in addition to astroglial activation. Activation of microglia was observed by immunohistochemistry with an antibody directed toward the F4/80 antigen, a surface glycoprotein known to be up-regulated on the activation of microglia.25 Intensely stained microglia were increased in the mutant forebrain, compared with that of the control mice (Figure 5A). This histological result was further corroborated by RT-PCR for CD68, which is specifically expressed by the macrophage/microglial cell lineage (Figure 5B).26 The levels of CD68 expression were approximately 1.5- to 3-fold higher in the mutant forebrain than in the control mouse brain (Figure 5B). Activated microglia have been shown to produce pro-inflammatory cytokines such as TNF-α.23,24 We found that TNF-α was expressed at higher levels in the mutant forebrain than in the brain of the control mice (Figure 5B). Therefore, Cdk5 deficiency led to activation of microglia in the forebrain, resulting in increased production of TNF-α.
Figure 5.
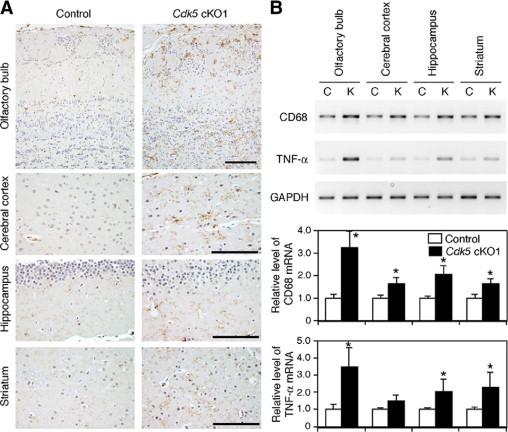
Aberrant activation of microglia in the forebrain of Cdk5 conditional mutant (Cdk5 cKO1) mice. A: Immunohistochemistry with anti-F4/80 antigen antibody revealed an increased number of activated microglia in the olfactory bulb, cerebral cortex, hippocampus, and striatum of 2- to 3-month-old Cdk5 cKO1 mice. Scale bars = 100 μm. B: RT-PCR on RNA extracted from 2- to 3-month-old mice revealed increased expression of CD68, a marker for microglia, and TNF-α, a cytokine produced by activated microglia, in the forebrain of Cdk5 cKO1 mice. C, control (Cdk5loxP/loxP) mice; K, Cdk5 cKO1 (Cdk5loxP/loxP; CaMKIIcre) mice. In the bar graphs, quantitative results indicate the levels of CD68 and TNF-α mRNA relative to those of the control mice, and are shown as mean ± SEM. (n = 3). Data are expressed in arbitrary units. *P < 0.05, compared with the control mice (Mann-Whitney test).
Contribution of Tissue-Type Plasminogen Activator to Microglial Activation
Microglia are sensitive to changes in their microenvironment and readily become activated in response to infections or injuries.23,24 We examined possible mechanisms underlying the aberrant activation of microglia in the forebrain of Cdk5 cKO1 mice. Taking the neuronal specificity of Cdk5 activity into consideration, we speculated that molecules secreted from Cdk5-deficient neurons might contribute to aberrant activation of microglia. It has been shown that microglial activation is induced by tPA, a serine protease that catalyzes the conversion of inactive plasminogen to the active protease plasmin.27 Neuronal tPA is induced and secreted in response to Ca2+ influx during events that increase neuronal activity, such as seizures and learning-related long-term potentiation.28 Furthermore, tPA is thought to mediate neurodegeneration, as evidenced by the observations that tPA−/− mice are resistant to neurotoxin-induced seizures and neurodegeneration.29 To test the possible contribution of tPA to microglial activation in the forebrain of Cdk5 cKO1 mice, we first examined plasminogen activator activity by in situ plasminogen-casein zymography. A striking increase in plasminogen activator activity was observed in the mutant forebrain compared with the brain of control mice (Figure 6, A–F). SDS- polyacrylamide gel electrophoresis/plasminogen-casein zymography further revealed that the tPA activity was increased in the mutant forebrain, but not in the hindbrain and spinal cord (Figure 6G).
Figure 6.
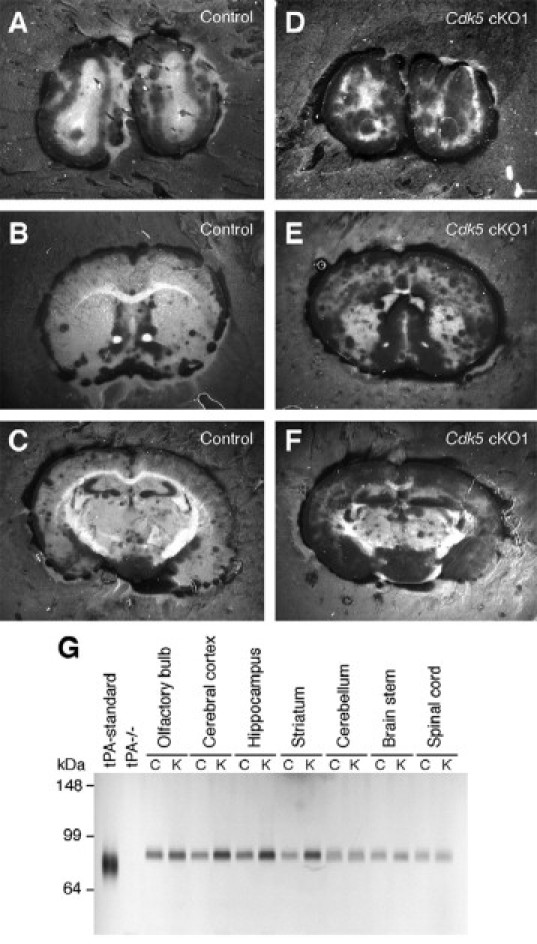
Tissue-type plasminogen activator (tPA) is up-regulated in the forebrain of Cdk5 conditional mutant (Cdk5 cKO1) mice. A–F: Localization of plasminogen activator activity (observed as dark zones) in the forebrain of control (A, B, and C) and Cdk5 cKO1 (D, E, and F) mice at the age of 3 months. Striking increases in plasminogen activator activity were observed in the olfactory bulb (D), cerebral cortex (E and F), ventral striatum (E), and CA3 region of the hippocampus (F) of Cdk5 cKO1 mice, as compared with those of the corresponding regions of control mice (A, B, and C). G: Brain region-specific activity of tPA measured by SDS-polyacrylamide gel electrophoresis/plasminogen-casein zymography using brain lysates from 3-month-old mice (n = 3). C, control (Cdk5loxP/loxP) mice and K, Cdk5 cKO1 (Cdk5loxP/loxP; CaMKIIcre) mice. Note the larger zones of proteolysis in the samples from the Cdk5 cKO1 forebrain, indicating the forebrain-specific increase of tPA activity in Cdk5 cKO1 mice.
To investigate the mechanism by which the tPA activity was increased in the forebrain of Cdk5 cKO1 mice, we examined tPA expression in the mutant forebrain at three different ages: at day 21 when the mutant mice had not yet developed seizures, and at 3- and 6-months when the mutant mice had developed seizures. RT-PCR revealed that tPA expression was increased in the 3- and 6-month-old mutant brain, but not in the day-21 mutant brain (Figure 7A). These changes in tPA expression correlated well with those of c-fos expression, which was used as an indicator of neuronal activity (Figure 7B). These data were consistent with the earlier observation that structural abnormalities arising from neuronal migration disorders contribute to hyperexcitability in the brain.5,6 Furthermore, the up-regulation of TNF-α, a cytokine produced by activated microglia, in the mutant forebrain temporally correlated with the up-regulation of tPA and c-fos (Figure 7C). Hyperexcitability in the mutant brain can cause a massive inflammatory reaction in an age-dependent manner, probably through the tPA induction, although the contribution of the other mechanisms cannot be excluded.
Figure 7.

Up-regulation of tPA in the forebrain of Cdk5 cKO1 mice with seizure activities is accompanied by the induction of TNF-α. A and B: RT-PCR revealed that tPA (A) and c-fos (B) expression were increased in the hippocampus of Cdk5 cKO1 mice with seizures at the ages of 3 and 6 months, but not of the seizure-free mutant mice at the age of day 21. C: These age-dependent changes in tPA and c-fos expression correlated well with those of TNF-α. The bar graphs indicate mRNA levels of each gene relative to those of the control mice, and are shown as mean ± SEM. (n = 3). Data are expressed in arbitrary units. *P < 0.05, compared with the control mice (Mann-Whitney test).
Discussion
Elimination of neuronal Cdk5 from the developing forebrain caused growth retardation, early mortality, and complex neurological deficits in mice. Characterization of Cdk5 cKO1 mice allowed us to define the consequences of Cdk5 deficiency in the postnatal brain. The forebrain of Cdk5 cKO1 mice showed neurodegenerative changes distinguished by neuronal loss and massive gliosis, in addition to disruption of the layered structure in the neocortex. Here, we discuss the mechanism underlying the deficits observed in Cdk5 cKO1 mice, and its impact on the pathogenesis of neurodegeneration.
Neurodegenerative changes in the Cdk5 cKO1 mouse brain were accompanied by a significant inflammatory process involving microglial and astroglial activation, leading to increased production of TNF-α. Although the inflammatory process has been considered to be secondary to the primary event of neuronal degeneration, mediators for inflammation such as TNF-α and other pro-inflammatory factors released by activated microglia can enhance neurodegeneration.24 Consistent with the notion that microglial activation plays a crucial role in neurodegenerative processes, inhibiting the microglial activation by infusion of macrophage/microglial inhibiting factor reduced excitotoxin-induced neurodegeneration.30 We examined the mechanism underlying aberrant activation of microglia in the forebrain of Cdk5 cKO1 mice, and identified tPA as a possible neuronal mediator contributing to microglial activation. tPA, which is secreted from neurons in response to elevation of intracellular Ca2+ levels,31 can activate microglia through interaction with annexin II, a cell-surface receptor on microglia.32 Overexpression of tPA in neurons has been shown to accelerate microglial activation in mice after induction of excitotoxicity.33 In contrast, mice lacking tPA showed very limited activation of microglia.29 In Cdk5 cKO1 mice, the expression of tPA was increased specifically in the forebrain where activated microglia were increased in number and produced TNF-α at higher levels. Furthermore, the temporal pattern of tPA expression correlated well with that of c-fos expression, which was used as an indicator of neuronal activity. Excessive neuronal activity causes an increase in the extracellular glutamate level, which activates glutamate receptors and leads to dramatic increases in intracellular Ca2+ levels.34 Thus it is possible that hyperexcitability in the brain with neuronal migration disorder might contribute to the tPA induction. We have generated an additional transgenic mouse, a Cdk5 cKO1 mouse in a tPA-null background. Our preliminary data revealed that a lack of tPA apparently diminished neuronal loss to some extent, and significantly reduced astrogliosis in the forebrain of the Cdk5 cKO1 mouse. These findings suggest the involvement of tPA-mediated inflammation in the pathogenesis of neurodegeneration (Utreras, Takahashi, and Kulkarni, unpublished).
There is another possible explanation for neurodegenerative changes observed in the Cdk5 cKO1 mouse forebrain. Cdk5 has been shown to play a role not only in neuronal migration but also in cell cycle regulation.35,36 Cdk5-deficient neurons fail to exit the cell cycle, leading to cell death.35 This observation is consistent with the recent reports demonstrating that the re-activation of cell cycle machinery in postmitotic neurons leads to their death both in vivo and in vitro.37,38 The role of Cdk5 in cell cycle inhibition involves the protein, but not its kinase activity. Thus, it has been proposed that Cdk5 may interact with other proteins in the nucleus to function as a cell cycle suppressor.35,36 This proposal is also supported by our earlier work demonstrating that cultured Cdk5-deficient neurons were more susceptible to apoptotic stimuli.39 However, other Cdk5 conditional knockout mice in which Cdk5 deletion was induced in the postnatal brain have not shown any abnormalities in the cytoarchitecture of the brain, nor have they shown any neurodegeneration. This suggests that a lack of Cdk5 protein is not sufficient to cause neuronal death in the postnatal brain.40,41 These observations indicate that the timing of Cdk5 deletion might be important for each phenotype, such as premature death and neurodegeneration. Therefore, neuronal migration defects observed in Cdk5 cKO1 mice in which Cdk5 was deleted as early as E12.5 might contribute to neurodegeneration in the forebrain.
While cellular alterations that are induced by seizures, such as neuronal loss and gliosis, are commonly known to occur in the human epileptic temporal lobe,34 more experiments will be needed to determine the degree to which inflammation contributes to the pathogenesis of neurodegeneration in the epileptic brain. Our data presented here may provide impetus for considering the development of anti-inflammatory therapies that can be used to avoid the progressive cognitive decline observed in some patients with neuronal migration disorders.
Acknowledgements
We thank Drs. Ioannis Dragatsis and Scott Zeitlin for providing the CaMKIIcre transgenic mice (R1ag#5), Dr. Kenneth Yamada and Dr. Yoshihiko Yamada for critical reading of the manuscript, and Harry Grant and Shelagh Powers for editorial assistance.
Footnotes
Supported by the Division of Intramural Research of National Institute of Dental and Craniofacial Research, National Institutes of Health.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
{kind=link}
{kind=link}
{kind=link}
References
- 1.Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 2.Bielas S, Higginbotham H, Koizumi H, Tanaka T, Gleeson JG. Cortical neuronal migration mutants suggest separate but intersecting pathways. Annu Rev Cell Dev Biol. 2004;20:593–618. doi: 10.1146/annurev.cellbio.20.082503.103047. [DOI] [PubMed] [Google Scholar]
- 3.Mochida GH, Walsh CA. Genetic basis of developmental malformations of the cerebral cortex. Arch Neurol. 2004;61:637–640. doi: 10.1001/archneur.61.5.637. [DOI] [PubMed] [Google Scholar]
- 4.Guerrini R, Sicca F, Parmeggiani L. Epilepsy and malformations of the cerebral cortex. Epileptic Disord. 2003;Suppl 2:S9–S26. [PubMed] [Google Scholar]
- 5.Luhmann HJ, Karpuk N, Qü M, Zilles K. Characterization of neuronal migration disorders in neocortical structures. II. Intracellular in vitro recordings. J Neurophysiol. 1998;80:92–102. doi: 10.1152/jn.1998.80.1.92. [DOI] [PubMed] [Google Scholar]
- 6.Patel LS, Wenzel HJ, Schwartzkroin PA. Physiological and morphological characterization of dentate granule cells in the p35 knock-out mouse hippocampus: evidence for an epileptic circuit. J Neurosci. 2004;24:9005–9014. doi: 10.1523/JNEUROSCI.2943-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong M. Advances in the pathophysiology of developmental epilepsies. Semin Pediatr Neurol. 2005;12:72–87. doi: 10.1016/j.spen.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Smith DS, Greer PL, Tsai LH. Cdk5 on the brain. Cell Growth Differ. 2001;12:277–283. [PubMed] [Google Scholar]
- 9.Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pareek TK, Kulkarni AB. Cdk5, a journey from brain to pain: lessons from gene targeting. In: Tsai LH, Ip N, editors. Cdk5. Springer; Norwell MA: 2008. p. 211. [Google Scholar]
- 11.Hirasawa M, Ohshima T, Takahashi S, Longenecker G, Honjo Y, Veeranna, Pant HC, Mikoshiba K, Brady RO, Kulkarni AB. Perinatal abrogation of Cdk5 expression in brain results in neuronal migration defects. Proc Natl Acad Sci USA. 2004;101:6249–6254. doi: 10.1073/pnas.0307322101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis. 2000;26:133–135. doi: 10.1002/(sici)1526-968x(200002)26:2<133::aid-gene10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 13.Pareek TK, Keller J, Kesavapany S, Agarwal N, Kuner R, Pant HC, Iadarola MJ, Brady RO, Kulkarni AB. Cyclin-dependent kinase 5 modulates nociceptive signaling through direct phosphorylation of transient receptor potential vanilloid 1. Proc Natl Acad Sci USA. 2007;104:660–665. doi: 10.1073/pnas.0609916104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 15.Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A. Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281–292. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- 16.de Olmos JS, Beltramino CA, de Olmos de Lorenzo S. Use of an amino-cupric-silver technique for the detection of early and semiacute neuronal degeneration caused by neurotoxicants, hypoxia, and physical trauma. Neurotoxicol Teratol. 1994;16:545–561. doi: 10.1016/0892-0362(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi S, Saito T, Hisanaga S, Pant HC, Kulkarni AB. Tau phosphorylation by cyclin-dependent kinase 5/p39 during brain development reduces its affinity for microtubules. J Biol Chem. 2003;278:10506–10515. doi: 10.1074/jbc.M211964200. [DOI] [PubMed] [Google Scholar]
- 18.Sappino AP, Huarte J, Vassalli JD, Belin D. Sites of synthesis of urokinase and tissue-type plasminogen activators in the murine kidney. J Clin Invest. 1991;87:962–970. doi: 10.1172/JCI115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Chaudhry A, Chintala SK. Inhibition of plasminogen activation protects against ganglion cell loss in a mouse model of retinal damage. Mol Vis. 2003;9:238–248. [PubMed] [Google Scholar]
- 20.Ohshima T, Ogawa M, Takeuchi K, Takahashi S, Kulkarni AB, Mikoshiba K. Cyclin-dependent kinase 5/p35 contributes synergistically with Reelin/Dab1 to the positioning of facial branchiomotor and inferior olive neurons in the developing mouse hindbrain. J Neurosci. 2002;22:4036–4044. doi: 10.1523/JNEUROSCI.22-10-04036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 22.Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K. Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci. 1998;18:6370–6377. doi: 10.1523/JNEUROSCI.18-16-06370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 24.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 25.Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- 26.Inoue H, Sawada M, Ryo A, Tanahashi H, Wakatsuki T, Hada A, Kondoh N, Nakagaki K, Takahashi K, Suzumura A, Yamamoto M, Tabira T. Serial analysis of gene expression in a microglial cell line. Glia. 1999;28:265–271. doi: 10.1002/(sici)1098-1136(199912)28:3<265::aid-glia10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 27.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 29.Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- 30.Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8:19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- 31.Gualandris A, Jones TE, Strickland S, Tsirka SE. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci. 1996;16:2220–2225. doi: 10.1523/JNEUROSCI.16-07-02220.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siao CJ, Tsirka SE. Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J Neurosci. 2002;22:3352–3358. doi: 10.1523/JNEUROSCI.22-09-03352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siao CJ, Fernandez SR, Tsirka SE. Cell type-specific roles for tissue plasminogen activator released by neurons or microglia after excitotoxic injury. J Neurosci. 2003;23:3234–3242. doi: 10.1523/JNEUROSCI.23-08-03234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pitkanen A, Sutula TP. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002;1:173–181. doi: 10.1016/s1474-4422(02)00073-x. [DOI] [PubMed] [Google Scholar]
- 35.Cicero S, Herrup K. Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci. 2005;25:9658–9668. doi: 10.1523/JNEUROSCI.1773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Herrup K. Cdk5 and the non-catalytic arrest of the neuronal cell cycle. Cell Cycle. 2008;7:3487–3490. doi: 10.4161/cc.7.22.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ, Funk JO, Shapiro RA, Smith MA. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta. 2007;1772:467–472. doi: 10.1016/j.bbadis.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 38.Lee HG, Casadesus G, Nunomura A, Zhu X, Castellani RJ, Richardson SL, Perry G, Felsher DW, Petersen RB, Smith MA. The neuronal expression of MYC causes a neurodegenerative phenotype in a novel transgenic mouse. Am J Pathol. 2009;174:891–897. doi: 10.2353/ajpath.2009.080583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li BS, Zhang L, Takahashi S, Ma W, Jaffe H, Kulkarni AB, Pant HC. Cyclin-dependent kinase 5 prevents neuronal apoptosis by negative regulation of c-Jun N-terminal kinase 3. EMBO J. 2002;21:324–333. doi: 10.1093/emboj/21.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benavides DR, Quinn JJ, Zhong P, Hawasli AH, DiLeone RJ, Kansy JW, Olausson P, Yan Z, Taylor JR, Bibb JA. Cdk5 modulates cocaine reward, motivation, and striatal neuron excitability. J Neurosci. 2007;27:12967–12976. doi: 10.1523/JNEUROSCI.4061-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10:880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.