

Abstract
Hippocampal sclerosis is a common pathological finding in patients with temporal lobe epilepsy, including children, but a causal relationship to early-life seizures remains in question. Neonatal status epilepticus in animals can result in neuronal death within the hippocampus, although macroscopic features of hippocampal shrinkage are not evident at adulthood. Here, we examined electrophysiological and pathological consequences of focally evoked status epilepticus triggered by intra-amygdala microinjection of kainic acid in postnatal day 10 rat pups. Neonatal status epilepticus resulted in extensive neuronal death in the ipsilateral hippocampal CA1 and CA3 subfields and hilus, as assessed by DNA fragmentation and Fluoro-Jade B staining 72 hours later. The contralateral hippocampus was not significantly damaged. Histopathology at P55/P65 revealed unilateral hippocampal sclerosis (grade IV, modified Wyler/Watson scale) comprising >50% CA1 and CA3 neuron loss and astrogliosis. Additional features included hydrocephalus ex vacuo, modest dentate granule cell layer widening, and altered neuropeptide Y immunoreactivity indicative of synaptic rearrangement. Hippocampal atrophy was also evident on magnetic resonance imaging. Depth electrode recordings at adulthood detected spontaneous seizures that involved the ipsilateral hippocampus and amygdala. A significant positive correlation was found between hippocampal pathology grade and both frequency and duration of epileptic seizures at adulthood. The current study demonstrates that experimental neonatal status epilepticus can result in classical unilateral hippocampal sclerosis and temporal lobe epilepsy.
Hippocampal sclerosis is the most common pathological finding in patients with temporal lobe epilepsy (TLE).1 Characteristics include gross atrophy, selective and asymmetric neuron loss within CA1, CA3 and dentate hilus, and gliosis.1–3 Additional features can include mossy fiber sprouting and granule cell layer dispersion.1,4
Hippocampal sclerosis is implicated as the cause of seizures and also associated cognitive deficits and seizure intractability, in patients with TLE.3,5–8 Hippocampal sclerosis is less common in children than adults with TLE, but may nevertheless be present in up to half or more patients.9–11 Pathology and neuroimaging studies have suggested seizures in children are the precipitant of hippocampal sclerosis, and hippocampal sclerosis the cause of TLE.10,12–17 Nevertheless, a causal link between early-life seizures, development of hippocampal sclerosis, and TLE in humans remains uncertain.
Animal models developed to address this question have demonstrated that hippocampal damage can result from early-life seizures.18 The immature rat brain is quite resistant to permanent seizure damage under 2 weeks of age, representing the time from newborn to 2 years of age in children.19,20 Possible mechanisms accounting for this have been reviewed elsewhere,21 and developmental age, route, and means of seizure induction all have strong influence on outcome. Prolonged seizures induced by hyperthermia in postnatal (P) day 10 rats cause only transient hippocampal injury and no permanent cell loss.22 Similarly, systemic kainic acid (KA) elicits seizures but transient, limited or no permanent hippocampal injury in rats younger than P18.23–25 In contrast, electrical stimulation of the perforant path in rat pups causes death of hilar and occasional pyramidal cells,26 and pilocarpine-induced status epilepticus in P12–14 rats causes moderate CA1 damage, minor or occasional CA3 cell death, but not hilar cell loss.27,28 Intrahippocampal or intraventricular KA injection in rat pups mainly damages the CA3 subfield.29–31 Nevertheless, despite showing neonatal rats are vulnerable to seizure damage, these models provide little evidence that gross hippocampal atrophy later develops in adolescence or adulthood.
The risk of epilepsy development after early-life status epilepticus in humans is estimated at 13 to 74%.32 Rates may be lower following febrile seizures.33 Immature rats subjected to status epilepticus develop TLE, with model-dependent prevalence rates of 0 to 42%.24,27,34–36 However, because neuronal death is not a consistent feature of the precipitating injury, a causal relationship between hippocampal pathology and epileptogenesis in these models is not supported.
The objective of our study was to investigate the outcome of status epilepticus induced by intra-amygdala KA in P10 rats. Focal generation of seizures in a brain region distant but nevertheless projecting to the hippocampus make this potentially relevant for addressing questions on the pathogenesis of hippocampal sclerosis. Our results indicate that neonatal status epilepticus in this model causes both unilateral hippocampal sclerosis and TLE.
Materials and Methods
Animal Model
Experiments were performed in accordance with protocols approved by the Legacy Institutional Animal Care and Use Committee and RCSI Research Ethics Committee. Animals were housed in a facility on a 12:12 light/dark cycle and provided access to food and water ad libitum. Seizure induction by intra-amygdala injection of KA was modified from Ben-Ari et al37 and experimental models developed by the authors.38,39 Male or female Sprague-Dawley rats (Harlan, Indianapolis, IN and Harlan, Bicester, UK) at P10 were used, with additional experiments using P9 and P11 pups, where indicated. On the day of the experiment, pups were removed from the dam, weighed, and anesthetized using isoflurane (5% induction, 1 to 2% maintenance in O2). Pups were placed in a stereotaxic frame equipped with a neonatal rat adaptor (David Kopf Instruments, Tujunga, CA), and body temperature was maintained at 37 ± 0.5°C by means of a homeostatic blanket (Harvard Apparatus, Boston, MA) and heat lamp. Following a midline incision, three partial burr holes were drilled bitemporally overlying the hippocampi and a third over frontal cortex (see Figure 1B). Cortical electroencephlogram (EEG) was recorded by means of skull-mounted recording electrodes (Plastics One, Roanoke, VA) using a Grass Comet digital EEG (Medivent, Lucan, Ireland). A fourth complete craniectomy was drilled for placement of a guide cannula. Coordinates for the guide cannula from Bregma were as follows: Anterior-posterior = −1.44 mm; Lateral = +2.85 mm. The guide cannula was lowered to the brain surface, and the entire assembly was fixed with dental cement. Anesthesia was discontinued, pups were placed in a warmed incubator, and electrodes were connected to the EEG via a swivel commutator. Following baseline EEG recordings up to 30 minutes, a 31-gauge internal cannula was inserted into the guide and lowered 3.5 mm below cortical surface to enable microinjection into the central and basolateral amygdala (Supplemental Figure S1A, see http://ajp.amjpathol.org). Pups received either 0.2 μl of vehicle (phosphate buffer, pH 7.4) or KA (0.1, 0.3, or 2 μg; Sigma-Aldrich, St. Louis, MO). EEG was routinely recorded up to 90 minutes, after which pups were returned to the dam. To establish the full duration of seizures in the model, an additional group of 0.1- and 2.0-μg KA-treated P10 pups were EEG-monitored for 4 hours after KA injection and again at 24 hours. Animals were euthanized at P13, P55, P65, or at adulthood (P120 to P150) (see Figure 1A).
Figure 1.
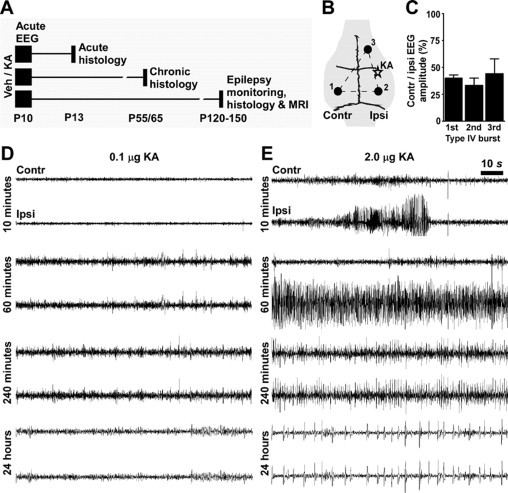
EEG and status epilepticus following intra-amygdala KA in P10 rats. A: Schematic of the experimental groups. Pups received intra-amygdala vehicle (veh) or KA at P10 and were analyzed at various times up to P150. B: Recording montage for acute bilateral EEG, depicting electrode placement over contralateral (contr. 1) and ipsilateral (ipsi. 2) cortex relative to the reference site (3). C: Graph showing lateralization of seizures in model as evidenced by lower EEG amplitude in contralateral recordings compared with ipsilateral signal strength for three sequential seizure events (averaged from n = 3 recordings). D and E: Representative EEG traces following intra-amygdala injection of 0.1 μg (D) and 2 μg (E) of KA, showing status epilepticus (continuous high-amplitude, high-frequency spiking) developing after injection of the high but not low dose of KA. Note seizure lateralization in E.
Acute EEG Analysis
Status epilepticus was defined as high-amplitude (>2 × baseline), high-frequency (>1 Hz) EEG spiking lasting at least 30 minutes. For assessment of seizure lateralization, spike amplitude was measured over 10-second excerpts in three consecutive events following injection of 2 μg of KA.
Hippocampal-Cortical and Amygdala-Cortical EEG with Video at Adulthood
Rats at P120 to P150 that had received intra-amygdala vehicle or KA (2 μg) at P10 were anesthetized and stereotaxically implanted with a twisted wire bipolar electrode (Plastics One) into either the dorsal hippocampus (coordinates: Anterior-posterior = −3.14 mm; Lateral = +1.5 mm; Vertical = −3.2 mm) or the amygdala at the level of dorsal hippocampus (coordinates: Anterior-posterior = −3.14 mm; Lateral = +4.6 mm; Vertical = −8.6 mm). Cortical EEG was recorded by means of two skull-mounted recording electrodes (Plastics One Inc) placed over the frontal cortex and cerebellum and secured with dental cement. Animals were allowed to recover for at least 24 hours before combined hippocampal-cortical or amygdala-cortical recordings using the Grass EEG system. Simultaneously, time-matched video was recorded using a Philips SPC 900NC PC camera. Each animal was recorded for 6 hours per day for up to 4 consecutive days. Video-EEG recordings were analyzed manually to determine the presence of spontaneous epileptic seizures and interictal events. Isolated high-amplitude spikes (>2 × baseline) with no abnormal behavioral correlate were classified as interictal events. Epileptic seizures were defined as high-frequency and high-amplitude EEG spiking lasting >5 seconds duration, according to similar definitions by others.35,36 Following detection of an electrographic event, video was analyzed for clinical features. Behavioral seizures were defined as behavior corresponding to Racine scale 3 or greater (score 3, bilateral forelimb clonus; score 4, forelimb clonus and rearing; score 5, forelimb clonus with rearing and loss of posture). Only events that fulfilled the electrographic and behavior criteria were counted as spontaneous seizures.
Neuropathology
Hippocampal sclerosis was assessed in P55 rats using a Wyler/Watson-adapted pathological grading system.40 Fresh-frozen whole brains were sectioned on a cryostat (12 μm) at the level of the dorsal hippocampus, as described previously.38,39 Next, sections were fixed in 10% formalin, dehydrated, and nissl-stained using cresyl violet. Demarcation of hippocampal boundaries was according to Lorente de No.41 Scoring was as follows: grade 0 = normal; I = slight (<10%) neuronal loss in CA3 or CA1; II = 10 to 50% neuronal loss in CA3 and/or CA1; III = >50% neuronal loss in CA3 with 10 to 50% neuronal loss in CA1; IV = >50% neuronal loss in CA3-CA1; and V = >50% neuronal loss in CA1-CA3 and damage to additional hippocampal regions, including dentate gyrus. Granule cell layer width was determined using Image J software, and granule cell counts were the average of five section lengths (100 μm) in two adjacent slices from n animals.
Immunohistochemistry and Neuronal Damage Detection
Immunohistochemistry and DNA fragmentation analysis was performed as previously described with modifications.38 After formalin fixation, sections were blocked in goat serum and incubated with specific antibodies against the following: glial fibrillary acidic protein (GFAP), neuropeptide Y (NPY) (Sigma-Aldrich), and NeuN (Chemicon International, Temecula, CA). Sections were washed in phosphate buffer and then incubated with fluorescein isothiocyanate-, Rhodamine (Jackson ImmunoResearch, West Grove, PA)-, AlexaFluor 488-, or AlexaFluor 568 (Molecular Probes, Eugene, OR)-conjugated secondary antibodies. DNA fragmentation was detected by terminal deoxynucleotidyl dUTP nick end labeling (TUNEL) according to manufacturer's recommendations (Roche Molecular Biochemicals, Indianapolis, IN). Sections were mounted in medium containing 4′,6 diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA) to stain nuclei. Fluoro-Jade B (FjB) staining was performed according to manufacturer's instructions (Chemicon International), as described previously.39 Sections were examined blinded to treatment, under a Nikon 2000s epifluorescence microscope fitted with a Hamamatsu Orca 285 camera (Micron Optical, Enniscorthy, Ireland) using excitation/emission wavelengths of 340/425 nm (blue), 500/550 nm (green), and 580/630 nm (red). For the hippocampal subfields, TUNEL, FjB, and NeuN counts were the mean of the total cell counts from the entire subfield from two adjacent sections of n animals, as described previously.38,39 Amygdala TUNEL, FjB, and neuron counts were the mean of four ×40 lens fields. Assignment of counts to specific subnuclei of the amygdala was not undertaken. To assess the proportion of dying cells with apoptotic morphology, mean counts from 100 cells on two adjacent hippocampal sections from seizure pups at 72 hours (n = 4) were examined for the presence of nuclear rounding and clumping of TUNEL-positive material.
Western Blotting
Western blotting was undertaken as described previously.38 Protein was extracted from hippocampi in lysis buffer containing a protease inhibitor mixture (Sigma-Aldrich) and 50-μg samples separated by 12 to 15% SDS-PAGE. Proteins were transferred to polyvinylidene difluoride membranes and incubated with specific antibodies against the following: α-tubulin and pan-14-3-3 (Santa Cruz Biotechnology, Santa Cruz, CA), NeuN (Chemicon International), and GFAP (Sigma-Aldrich). Protein levels were detected using chemiluminescence and semiquantitative densitometry undertaken on time-matched gels using AlphaEaseFC 4.0 software.
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) was performed using a 7T Bruker Biospec 70/30 system (Bruker Biospin, Ettlingen, Germany) with a 70-mm volume coil for transmission and an actively decoupled rat head surface coil for reception only (circular polarized). Rats were anesthetized with 5% isofluorane and maintained under sedation using 1.5% isofluorane. The animal's temperature (37 ± 1°C) and respiration were monitored for the duration of the experiment. High-resolution images were acquired using Rapid Acquisition with Relaxation Enhancement two-dimensional sequence with a Rapid Acquisition with Relaxation Enhancement factor of 8 and an echo time of 12.845 ms, resulting in an effective echo time of 38.5 ms (with a flip angle of 90° and 180° for the refocusing pulses). With an acquisition matrix of 256 × 256 and a field of view of 2.5 × 2.5 cm2, the pixel resolution was 98 μm/pixel. In the axial plane, 54 slices, each measuring 0.5 mm in thickness with no gaps, were acquired. Repetition time was 6676.9 ms, and six averages were used for a total measuring time of 21 minutes.
Data Analysis
All data are presented as the mean ± SEM. Statistical analysis was performed using analysis of variance with post hoc Fisher's protected least significant difference test and Student's t-test, as appropriate. Statistical analyses were performed using StatView software (SAS Institute, Cary, NC) and GraphPad Instat (San Diego, CA). Significance was accepted at P < 0.05.
Results
Focally Evoked Neonatal Status Epilepticus in P10 Rats
We first investigated the electrophysiological consequences of neonatal status epilepticus induced by intra-amygdala microinjection of KA in P10 rats. This model was selected because in adult rats it produces limbic status epilepticus, followed by classical unilateral hippocampal pathology and chronic TLE.37,42 P10 rats were selected for their hippocampal developmental age equivalence to that of a young human infant and the extensive knowledge of responses at this age to status epilepticus.19,20 However, EEG and histopathology responses to KA were similar in P9 and P11 rat pups (n = 4 per group given 2 μg; data not shown).
The surface EEG recording montage is depicted in Figure 1B. Epileptiform EEG was elicited in P10 rat pups in a dose-related manner following intra-amygdala microinjection of KA. Pups injected with 0.1 μg of KA developed mainly low-amplitude, high-frequency spiking that lasted several hours but that did not develop into status epilepticus (continuous high-amplitude, high-frequency spiking lasting at least 30 minutes) (Figure 1D). Behavioral features were mild and included brief episodes of masticatory movements, salivation, facial, and forelimb myoclonus. Recordings at 24 hours showed no abnormal behavior, and EEG features were unremarkable. Similar results were observed for 0.3 μg-injected pups (data not shown). In contrast, after the injection of pups with 2 μg of KA, initial low-amplitude, high-frequency EEG developed to high-amplitude, high-frequency epileptiform EEG seizures (Figure 1E). The pups displayed seizure-like behavior including initial masticatory movements and salivation, developing to periods of facial and forelimb myoclonus and progressing to the most commonly observed behavior of wild running, loss of posture, and swimming motions. The high-amplitude, high-frequency spiking in 2-μg KA-injected pups was sustained and corresponded with the wild running and swimming behaviors during recordings 1 to 2 hours after KA. Illustrating the KA dose relationship, analysis of this seizure EEG pattern during recordings from the period of 60 to 90 minutes after KA found duration correlated to KA dose (r = 0.7, P < 0.01, n = 14). Using the referential recording montage and analyzing the relative amplitudes of EEG spiking after 2 μg of KA recorded from the surface overlying the ipsilateral and contralateral cortex supported status epilepticus as focal with secondary generalization: contralateral EEG amplitude was lower (40 ± 3%) than ipsilateral amplitude (n = 3) in first-event polyspike seizures (Figure 1C). Analysis of subsequent epileptiform events showed the same degree of lateralization persisted (Figure 1C). By 4 hours after 2 μg of KA, status epilepticus had abated in all pups, with minimal behavioral and EEG displaying intermittent lower-amplitude, high-frequency spiking (Figure 1E). Recordings in pups 24 hours after 2 μg of KA showed residual spiking but normal behavior (Figure 1E).
Neonatal Status Epilepticus Results in Extensive and Permanent Unilateral Hippocampal Cell Death
Examination of acute pathology was undertaken 3 days following intra-amygdala injections by staining sections at the level of dorsal hippocampus for TUNEL and FjB, which label cells undergoing irreversible DNA fragmentation and cell death, respectively. Significant TUNEL and FjB staining was present within ipsilateral CA1 (Figure 2, A and B), CA3 (Figure 2, C and D), and the hilus (Figure 2, E and F) in pups that underwent status epilepticus induced by intra-amygdala KA (2 μg) compared with vehicle-injected and naive P13 pups. Regression analysis determined there was a positive correlation between high-amplitude, high-frequency seizure duration (in recordings up to 90 minutes post-KA) and CA3 TUNEL counts (r = 0.7, P < 0.01, n = 14), indicating damage was related to seizure duration. Although we focused on the dorsal hippocampus, damage was also present at the level of the ventral hippocampus after status epilepticus induced by 2 μg of KA in CA1, CA3, and the hilus (Supplemental Figure S1B, see http://ajp.amjpathol.org).
Figure 2.
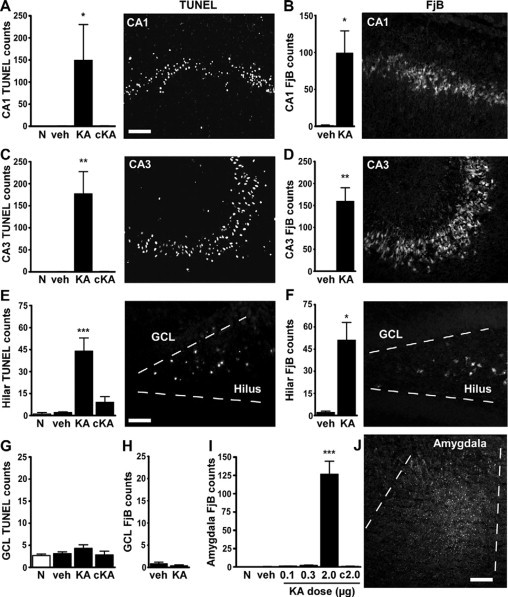
Permanent hippocampal damage 72 hours after neonatal status epilepticus in P10 rats. A–H: Quantification of ipsilateral TUNEL and FjB counts 72 hours after injection of vehicle (veh) or 2 μg of KA compared with age-matched naive (N) pups for CA1 (A and B), CA3 (C and D), hilus (E and F), and dentate granule cell layer (GCL) (G and H). Data from contralateral (c) fields are shown for 2-μg injected pups. Panels to the right of graphs are representative field views of TUNEL and FjB staining from 2-μg KA-injected pups at 72 hours. I: Quantification of FjB counts within the amygdala at the injection site for each dose tested. J: Representative photomicrograph of FjB staining within the ipsilateral amygdala at P13 in a 2-μg injected pup. Data are from n = 3–4 rats per group. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 compared with vehicle and naive pups. Scale bar in A–D, 100 μm; E and F, 50 μm; and J, 150 μm.
Systemic administration of the anticonvulsant lorazepam (6 mg/kg) 90 or 100 minutes after 2 μg of KA (n = 2–4 per group) reduced, although did not abolish seizure activity, on EEG (Supplemental Figure S1C, see http://ajp.amjpathol.org). Lorazepam-treated pups displayed ≥50% reduction in CA3 and CA1 cell death (data not shown).
TUNEL and FjB counts in the ipsilateral dentate granule cell layer of 2-μg injected pups were not different to controls (Figure 2, G and H). There was also no significant TUNEL or FjB staining in the contralateral hippocampal subfields of 2-μg KA-injected pups or in ipsilateral hippocampal subfields from pups given 0.1 or 0.3 μg of KA (Figure 2, A–H, and data not shown). Analysis of the ipsilateral amygdala at the site of microinjection revealed there was significant TUNEL (data not shown) and FjB staining in 2-μg injected pups compared with control and other KA doses (Figure 2, I and J).
Phenotype and Morphology of Degenerating Cells
Immunostaining of hippocampal sections determined 88.9 ± 3.2% of TUNEL-positive cells were immunoreactive for the neuronal nuclear protein NeuN (Supplemental Figure S1D, see http://ajp.amjpathol.org). Morphological features of apoptosis such as clumping of chromatin and apoptotic bodies were present in 3.8 ± 0.5% of TUNEL-positive cells (Supplemental Figure S1E, see http://ajp.amjpathol.org).
Neonatal Status Epilepticus Results in Unilateral Hippocampal Sclerosis
We next studied groups of rats at P55 and P65 (adolescence/early adulthood) to examine whether morphological characteristics of hippocampal sclerosis were present. Rats at P55 or P65 that underwent status epilepticus at P10 induced by 2 μg of KA displayed sclerosis of the ipsilateral hippocampus (Figure 3, A–C and E). There was no significant difference in the incidence or severity of hippocampal sclerosis between male and female rats given 2 μg of KA at P10 (data not shown). Morphological features comprised atrophy of the pyramidal cell layers and reduced overall size of the ipsilateral hippocampus (Figure 3, A–C and E). Hydrocephalus ex vacuo of the lateral ventricle ipsilateral to the side of KA injection was observed (Figure 3, A–C and E). Scoring of hippocampal sclerosis using a modified Wyler/Watson scale (see Materials and Methods) determined hippocampal sclerosis averaged grade IV (Figure 3G). Hippocampal sclerosis did not develop in pups given the lower KA doses, and neuropathologic scores were zero for hippocampi of P55 rats that received intra-amygdala vehicle at P10 and the contralateral hippocampus of 2-μg KA-injected animals (Figure 3, A–F, and data not shown).
Figure 3.
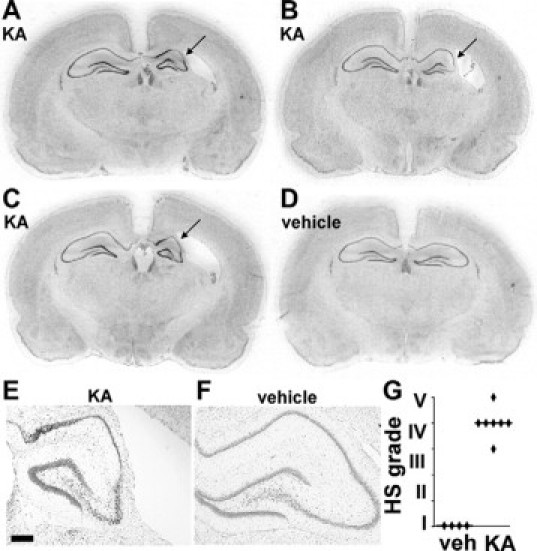
Unilateral hippocampal sclerosis at P55/P65 after neonatal status epilepticus in P10 pups. Representative nissl-stained sections from separate 2-μg KA-injected rats at P55 (A and B) and P65 (C), which underwent neonatal status epilepticus at P10. Note ipsilateral hippocampal sclerosis (arrows) and hydrocephalus ex vacuo of the lateral ventricle. D: Coronal section from a P55 rat that received intra-amygdala vehicle at P10. E and F: Representative photomicrographs (×2.5 lens) of ipsilateral hippocampus at P55 from rats that received intra-amygdala KA (2 μg) (E) and vehicle (F) at P10. Scale bar in E, 250 μm. G: Graph showing hippocampal sclerosis (HS) pathology scoring at P55 for rats given intra-amygdala vehicle (veh, n = 4) or KA (2 μg, n = 7) at P10.
Hallmarks of Hippocampal Sclerosis: Neuron Loss
Counts of NeuN-positive cells at the level of dorsal hippocampus were ∼40% of vehicle controls in the ipsilateral CA1 of P55 rats that were subject to neonatal status epilepticus at P10 (Figure 4A). In CA3, neuron counts were ∼20% of controls (Figure 4, B and C). Contralateral hippocampal CA1 and CA3 neuron counts at P55 in rats subject to neonatal status epilepticus at P10 were not different to controls (Figure 4, A and B). The extent of ipsilateral CA1 and CA3 neuron loss was similar in rats analyzed at P120 to P150 (data not shown). Neuron loss was also significant at adulthood within the ipsilateral hilus, where NeuN-positive counts in rats given 2 μg of KA at P10 were 42% (range, 13 to 91%) of vehicle controls (P < 0.05), whereas contralateral hilar neuron counts were not different to controls (data not shown). These data were supported by Western blot measurement of hippocampal levels of NeuN in separate rats at P65. NeuN levels at P65 in rats given 2 μg of KA at P10 were ∼50% of the vehicle control group (P < 0.001, n = 3 per group) (Figure 4D).
Figure 4.
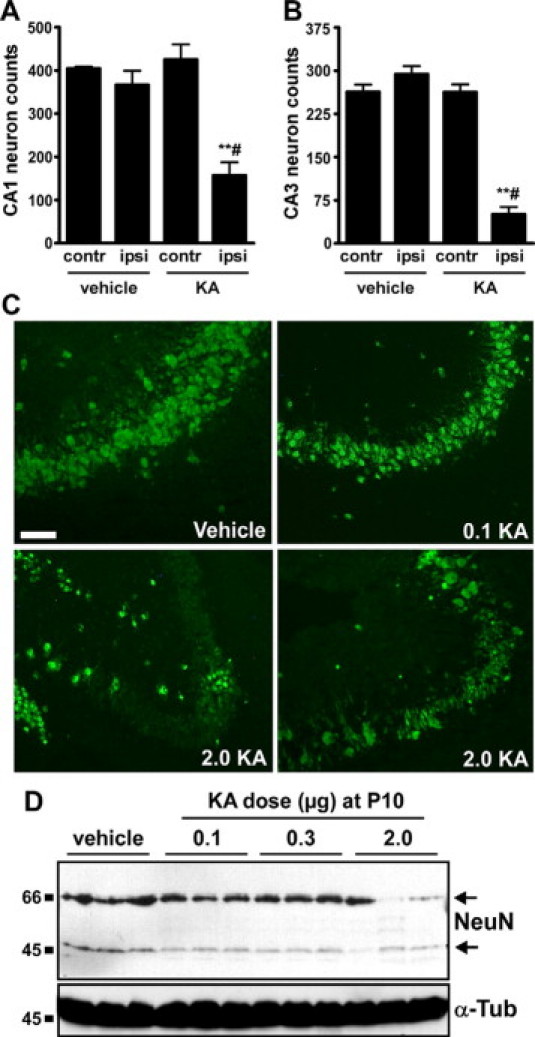
Hippocampal neuron loss at P55/65 following neonatal status epilepticus. A and B: Graphs depict NeuN-positive cell counts for contralateral (contr) and ipsilateral (ipsi) CA1 and CA3 subfields at P55 in rats given intra-amygdala vehicle or KA (2 μg) at P10 (n = 4–6 per group). Note significant loss of neurons in both subfields in rats subject to neonatal status epilepticus. ∗∗P < 0.01 versus ipsilateral vehicle control, #P < 0.05 versus contralateral KA. C: Fluorescence microscopy images (×20 lens) showing NeuN staining (green) of ipsilateral CA3 subfields of P55 rats that received intra-amygdala vehicle or KA (dose in micrograms) at P10. Note significant neuron loss in rats given 2 μg of KA. D: Western blot analysis at P65 of NeuN levels in hippocampal samples from vehicle- and KA-injected rats at P10 (n = 1 per lane). Note lower levels of NeuN protein in 2-μg KA-injected rats compared with other groups. α-Tubulin (α-Tub) is included as a protein loading control. Scale bar in C, 100 m.
Hallmarks of Hippocampal Sclerosis: Astrogliosis, NPY Changes, and Granule Cell Layer Widening
Next, we performed Western blot measurement of ipsilateral hippocampal levels of GFAP to determine whether sclerotic hippocampi featured astrogliosis, a common characteristic of hippocampal sclerosis. Hippocampal lysates from P65 rats that received intra-amygdala KA (2 μg) at P10 had dramatically elevated GFAP levels when compared with controls and to doses of KA that did not result in hippocampal sclerosis (P < 0.0001) (Figure 5A). Complementing these findings, tissue sections from sclerotic hippocampi at P55 displayed increased GFAP immunostaining within CA1 and CA3 compared with controls (Figure 5B and data not shown). This comprised a general increase in astrocyte numbers and widened/thickened astrocyte processes (Figure 5B).
Figure 5.
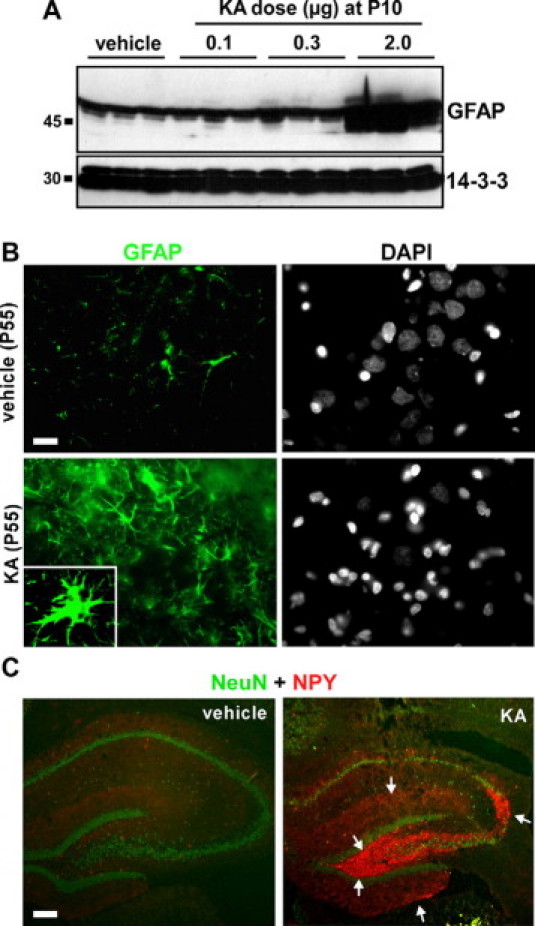
Hippocampal astrogliosis at P55/65 following neonatal status epilepticus. A: Western blot analysis at P65 of GFAP levels in hippocampal samples from rats given intra-amygdala vehicle or KA injection at P10 (n = 1 per lane). Note dramatically elevated GFAP in 2-μg KA-injected rats compared with other groups. 14-3-3 is included as a protein loading control. B: Fluorescence microscopy images (×40 lens) showing GFAP (green) staining of ipsilateral CA3 subfields of P55 rats that received either intra-amygdala vehicle or KA (2 μg) at P10. Note significant astrogliosis. Inset shows a higher power magnification of a single hypertrophic astrocyte. Nuclear morphology of cells is shown by 4′,6′-diamidino-2-phenylindole staining (DAPI) (gray). C: Representative photomicrographs showing hippocampus stained for NPY (red) and NeuN (green) at P55 in rats given either vehicle or KA (2 μg) at P10. NPY immunoreactivity demarcates hippocampal mossy fiber rearrangement (arrows). Scale bars in B, 12 μm; in C, 200 μm.
Hippocampal rearrangement of mossy fibers is a common feature of pathology in TLE patients and in animal models, although its function remains uncertain.43 NPY is expressed and released from the terminals of mossy fibers under these conditions and represents an immunohistochemical means of visualizing hippocampal rearrangement. NPY immunoreactivity in vehicle control rats was mainly present in isolated hilar cells, with some staining also detected in the outer part of the dentate molecular layer (Figure 5C). The KA-injected rats with hippocampal sclerosis at P55 displayed enhanced NPY immunoreactivity within the dentate molecular layer, hilus, and dendritic field of CA3 (Figure 5C).
Last, we investigated morphological changes to the dentate granule cell layer, which have been reported in experimental and human TLE.4,44 Rats at P55 that underwent 2 μg of KA-induced status epilepticus at P10 had a modest but significant widening of the dentate granule cell layer on the ipsilateral side compared with the contralateral side and when compared with the ipsilateral side of vehicle-injected controls (Figure 6, A, C, and D). Counts of granule neurons found no difference between ipsilateral or contralateral hippocampi for rats given vehicle or KA at P10 (Figure 6B). An ipsilateral difference in granule cell layer width at P55 was also evident in 2-μg KA-injected rats compared with rats that received 0.1 μg of KA (Figure 6E).
Figure 6.
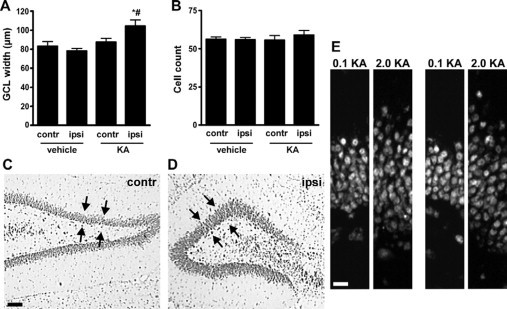
Granule cell layer widening at P55/65 following neonatal status epilepticus. A: Graph showing granule cell layer widths. Significant widening was detected in ipsilateral (ipsi) hippocampus of 2-μg KA-injected pups by adulthood (n = 4–6 per group). ∗P < 0.05 versus ipsilateral side of vehicle control; #P < 0.05 versus contralateral (contr) side of KA injected. B: Graph of granule cell layer counts from the same area show no differences between groups. C and D: Representative photomicrographs of the contralateral and ipsilateral dentate gyrus of a status epilepticus rat at P55 with widening of layer on ipsilateral side. Arrows highlight area of comparison. E: Representative photomicrographs of the upper blade of the ipsilateral dentate granule cell layer from 0.1- and 2-μg KA-injected rats at P55 at two different positions. Note difference in layer width despite numbers of cells being comparable. Scale bars in C, 100 μm; in E, 15 μm.
Amygdala (Injection Site) Pathology
Since KA (2 μg) injection at P13 caused cell death within the ipsilateral amygdala, we examined coronal brain sections at the corresponding level in P65 rats for evidence of neuronal loss. Counts of NeuN-positive cells within the injection site of the ipsilateral amygdala revealed lower numbers in KA-injected rats compared with vehicle-injected controls (Figure 7A). A small increase in GFAP immunoreactivity was also apparent in the KA-injected animals (Figure 7B).
Figure 7.
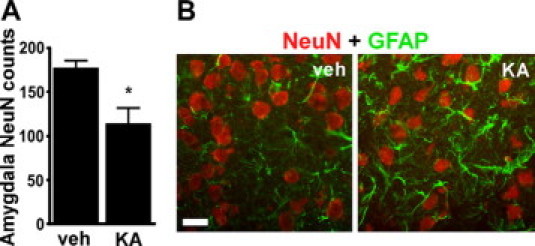
Amygdala injection site at P65. A: Graph showing NeuN counts within the ipsilateral amygdala of vehicle (veh)- and 2-μg KA-injected rats at P65 (n = 3–4 per group). ∗P < 0.05 compared with vehicle group. B: Representative double-label fluorescence immunostaining (×40 lens) of NeuN and GFAP in sections from P65 rats at the site corresponding to the intraamygala injection point at P10. Scale bar, 12 μm.
Neonatal Status Epilepticus Results in TLE at Adulthood
To determine whether neonatal status epilepticus leads to the development of TLE at adulthood, we undertook intermittent range-finding video and EEG monitoring of neonatal status epilepticus rats over several days at P55 (n = 5). Behavior consistent with epileptic seizures was occasionally observed in rats, which consisted of short episodes of facial stereotypies and head nodding. Such events were usually accompanied by brief epileptiform cortical EEG spiking, indicating seizures may be partial. However, secondarily generalized seizures with rearing and forelimb clonus were also observed. To fully evaluate the epileptic phenotype, additional rats subject to neonatal status epilepticus, and vehicle controls were instrumented for depth electrode recordings at the level of dorsal hippocampus with video at adulthood (P120 to P150; see Figure 1A). We first performed combined intrahippocampal-cortical EEG recordings (6 hours/day for 2 to 4 days). No epileptic seizure-like or epileptiform events were detected in rats that received intra-amygdala vehicle at P10 (n = 4) (Figure 8A). In contrast, electrographic seizures were detected in the ipsilateral hippocampus in 64% (7 of 11) of animals that received KA (2 μg) at P10. These were high-frequency events with typically increasing amplitude, usually followed by abrupt cessation or postictal depression (Figure 8, D and E). Seizures occurred with a mean incidence of 1.9 ± 0.2 per 6 hours recording session (range, 0 to 5). Mean spontaneous seizure duration was 201 ± 53 seconds (range, 6 to 1145 s). Corresponding cortical EEG typically displayed mild electrographic polyspike correlates of these hippocampal events when compared with background (Figure 8, D and E). The behavioral correlates of these seizures included head nodding, whole body clonus “jerk,” straub tail, unilateral and bilateral forepaw myoclonus, rearing, and loss of posture. Clinical symptoms typically began a few seconds before detection of hippocampal electrographic seizure EEG. Interictal events, characterized by low-frequency, high-amplitude spikes with no altered behavior were present in all of the animals that displayed spontaneous seizures and in three of four of the animals that did not display spontaneous seizures (Figure 8B). A subset of the rats (2 of 11) displaying temporal lobe seizures with limited cortical expression underwent occasional epileptic seizures characterized by high-amplitude, low-frequency spiking across cortical and hippocampal EEG with behavior characterized by intermittent immobility and myoclonic jerks (Figure 8C).
Figure 8.
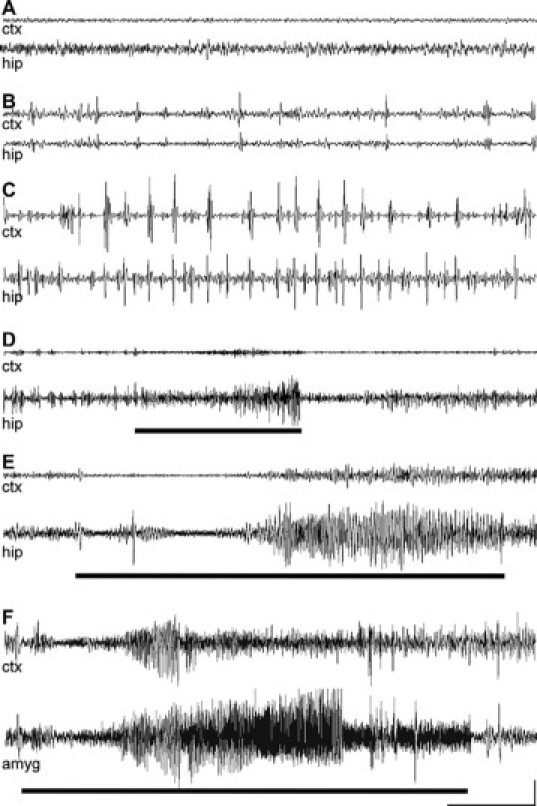
Spontaneous temporal lobe seizures at adulthood in rats subject to neonatal status epilepticus. A–E: Representative EEG traces from cortex (ctx) and hippocampal (hip) electrodes in adult rats following intra-amygdala injection at P10 of vehicle (A) or 2 μg of KA (B–E). A: Trace from a rat (P133) that received vehicle at P10. B: Interictal events associated with no alteration in behavior. C: High-amplitude, low-frequency electrographic events in hippocampus and cortex associated with periods of immobility and intermittent myoclonus. Brief (D) and prolonged (E) spontaneous electrographic seizures (high-amplitude, high-frequency spiking) recorded in the hippocampus. F: Representative spontaneous seizure captured in a P120 rat that received 2 μg of KA at P10 during combined intra-amygdala (amyg) and cortical EEG recordings. The bold lines represent the period of behavioral seizure (greater than Racine scale 3). Scale bar: voltage, 200 μV (A–F); time, 20 seconds (A–C) and 10 seconds (D–F).
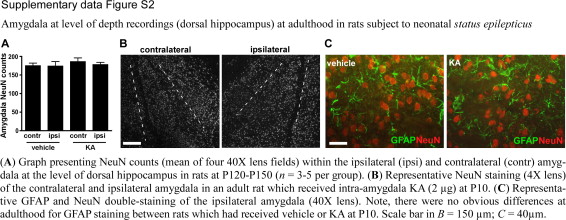
In similar experiments at adulthood with recording electrodes placed in the ipsilateral amygdala at the level of dorsal hippocampus and overlying cortex, spontaneous seizures were again detected in a majority of rats (three of four) that had received 2 μg of KA at P10 (Figure 8F). No seizures were detected during intra-amygdala recordings from adult rats that were given intra-amygdala vehicle at P10 (n = 2; data not shown). NeuN counts at P120 to P150 in the ipsilateral amygdala at the level of dorsal hippocampus were similar between KA- and vehicle-injected rats, and astrogliosis was not present (Supplemental Figure S2, see http://ajp.amjpathol.org).
Influence of Hippocampal Sclerosis on Epileptic Phenotype
Hippocampal sclerosis grading was undertaken after completion of video-EEG monitoring of animals at P120 to P150. Spearman rank correlation analysis determined that epileptic seizure frequency was positively correlated with hippocampal sclerosis grade (r = 0.83, P = 0.005) and that epileptic seizure duration was positively correlated with hippocampal sclerosis grade (r = 0.90, p = 0.0008). Frequency, but not duration, of spontaneous seizures was inversely correlated with hilar NeuN counts (r = −0.74, p < 0.05).
Detection of Hippocampal Sclerosis by MRI
Finally, we used MRI to visualize hippocampal sclerosis in rats at P125 to P150 that displayed spontaneous seizures (n = 4). Images were acquired in anesthetized rats using a high field (7 T) magnet. Anatomical scans identified unilateral hippocampal atrophy at P125 to P150 in rats given KA (2 μg) at P10 (Figure 9, A, B, and D). Follow-up pathology on a representative imaged animal confirming unilateral hippocampal atrophy is provided in Figure 9C.
Figure 9.
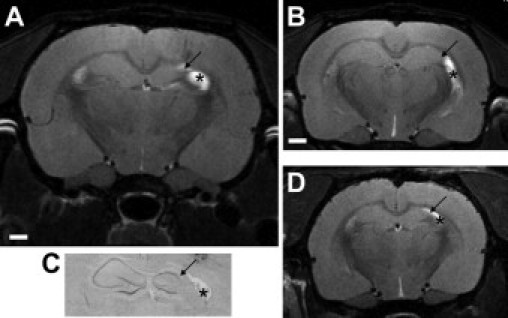
MRI confirmation of hippocampal sclerosis in epileptic rats at adulthood. A: MRI scan (7T) at the level of dorsal hippocampus of an epileptic rat at P150 that underwent neonatal status epilepticus, in which unilateral hippocampal sclerosis is evident (arrow). Asterisk denotes signal from hypertrophied ventrical. B: MRI from same epileptic rat in A at a more ventral level. C: Corresponding histology from the imaged animal with same features denoted. D: Imaged separate epileptic rat confirming interanimal similarity in sclerosis as resolved by MRI. Bar in A and B, 1 mm; in C and D, 0.75 mm.
Discussion
The present study demonstrates that intra-amygdala KA in neonatal rats causes status epilepticus and extensive acute unilateral hippocampal neurodegeneration. Significantly, rats developed unilateral atrophy of the hippocampus with attendant features of hippocampal sclerosis and spontaneous seizures involving the hippocampus. There is clinical data supporting a relationship between early-life seizures and development of hippocampal sclerosis and TLE.10,12–17 Causality is partly supported by animal data; with some exceptions, for example, systemic KA and prolonged seizures caused by hyperthermia, modest neuronal death is found in the immature rat hippocampus shortly after status epilepticus.26–28,31,45 However, normal hippocampal morphology or only minor atrophy is found at adulthood in these models, even when acute cell loss occurred. Hippocampal sclerosis, which refers to gross features of a hard shrunken hippocampus,1 has not been evident. The major finding in the present study was that neonatal rats of either gender subject to status epilepticus induced by intra-amygdala KA developed unilateral hippocampal sclerosis by adolescence/early adulthood. The ipsilateral hippocampus displayed extensive pyramidal neuron loss in both CA1 and CA3 subfields. Hilar neurons were also lost, whereas there was little ipsilateral cortical injury and no contralateral hippocampal pathology. Dentate granule neurons appeared spared, although these cells may die via an apoptotic process and be quickly removed,26 leading to underestimates of injury to this population. The pattern of neuron loss we observed is therefore similar, although not the same as classical hippocampal sclerosis in human TLE.1,2 The CA1 subfield is often the most damaged region in human hippocampal sclerosis and is preferentially damaged in immature rats after pilocarpine-induced status epilepticus.27,28 However, CA3 and hilar damage is common to human hippocampal sclerosis, whereas both CA3 and hilar cell loss are uncommon in the pilocarpine model in 2-week-old rats or younger.27,28,45 We did not see the bilateral damage reported following perforant pathway stimulation in immature rats,26 but the distribution of permanent damage is similar to that reported for infant monkeys after status epilepticus elicited by focal bicuculline, where there is hippocampal atrophy featuring CA1, CA3, and hilar neuron loss.46 This indicates potential clinical relevance, albeit recognizing there are more relevant neonatal seizure stimuli than KA, namely anoxia or pneumococcal meningitis.32,47 Other features of the model also support clinical relevance, including the activation of a temporal lobe structure that projects seizures to the hippocampus, avoidance of generalized effects of systemically administered convulsants on the brain and periphery, and use of P10 rats, which represent age equivalence to a term newborn or infant.19,20 The pathology also closely resembles the distribution of febrile seizure-induced transient hippocampal injury modeled by ∼20 minutes hyperthemic seizures in rats of the same age.22 Thus, distribution of damage, as well as EEG and behavior after KA, supports involvement of limbic structures. The explanation for the greater hippocampal damage in this amygdala onset model may be seizure intensity or duration differences or because of focal/intracerebral versus generalized/systemic onset seizures between this and other models.20,48 A secondary contribution of direct KA toxicity cannot be completely excluded. Both intraventricular and intrahippocampal KA cause permanent hippocampal damage in rat pups.29–31 However, pathology in those reports was mainly in the CA3 subfield. In contrast, we found damage to CA1 and CA3 was similar, and anticonvulsant administration could reduce seizures and cell death in the model. Acute cell death was also evident distant to the injection site within the ipsilateral ventral hippocampus. Taken together, the present study indicates that hippocampal damage from early-life seizures in rats can closely approximate the pattern and scale of hippocampal sclerosis seen in some patients.
Several ancillary features of human hippocampal sclerosis were present in rats at adulthood in the model. This included unilateral hydrocephalus ex vacuo, presumably formed as a consequence of the atrophied hippocampus. Astrogliosis was also present in the sclerotic hippocampus, a stereotyped and proportional response to neuronal damage that may have a causal role in epileptogenesis.49 The changes to NPY expression and localization indicated possible axonal or synaptic reorganization. This may be consistent with mossy fiber sprouting seen in some immature seizure models.27,30,46 Of note, status epilepticus after pilocarpine in immature animals only results in mossy fiber sprouting at the age when vulnerability to seizure damage is manifest.27 Our data showed the changes were most prominent in CA3 and the hilus, suggesting neuronal death as a factor. Mossy fiber sprouting is implicated in the formation of epileptic circuits, although this has been questioned by short latent periods in some animal models and the occurrence of spontaneous seizures in animals without mossy fiber sprouting.50 Whether NPY changes also occur secondary to epileptic seizure effects is possible but not addressed presently. Granule cell layer widening was also evident in rats that developed hippocampal sclerosis after neonatal status epilepticus, and similar morphology is evident after status epilepticus in infant monkeys.46 This may indicate granule cell layer dispersion, which is sometimes present in hippocampi from patients with TLE,4 and a morphological feature possibly unique to epilepsy.51 However, the granule cell layer changes were less dramatic than the dispersion seen in certain adult models44 and an effect of compression-related changes in the present model cannot be excluded. Although the functional significance of granule cell layer and NPY changes are not fully understood, these data establish multiple pathological hallmarks of hippocampal sclerosis develop in this model.
Severance of afferent pathways to the hippocampus causes numerous cell and molecular alterations, including neuronal loss, synaptic rearrangement, and gliosis.52,53 Because the present model featured injury to the ipsilateral amygdala, input to the developing hippocampus is likely to be affected. However, major deafferentation-induced hippocampal atrophy can probably be excluded because i) lesions of the neonatal rat amygdala or entorhinal cortex by other methods do not result in hippocampal atrophy,54,55 and ii) the hippocampal neuron loss at adulthood was of similar extent to the degeneration already apparent at P13.
TLE develops in a subpopulation of children who experience early-life prolonged seizures,3 and this is also seen in rats subject to neonatal status epilepticus.47 However, TLE development is not more common in immature seizure models where minor permanent hippocampal injury is incurred than where absent.27,34–36,56 Thus, the influence of neuronal death for such epileptogenesis is questionable. In the present study, neonatal status epilepticus led to development of TLE. The hippocampus was recruited during these spontaneous seizures, although the site of ictogenesis may lie elsewhere because clinical signs of seizures often slightly preceded the electrographic bursts in the hippocampus. Similar minor temporal dissociation of behavior from hippocampal electrographic events has also been reported in rats that developed epilepsy after prolonged febrile seizures at P10.36 Although there was secondary generalization of some seizures, electrographic features on cortical EEG were typically subdued, which is noted in other models where seizures are limbic in semiology.36
The proportion of rats that developed epilepsy in our study was somewhat higher than those found in several previous models where damage was minimal.27,35,36,56 True epileptic seizure rates are almost certainly >64% because hippocampal epileptiform events were present in 91% of the rats, and our noncontinuous sampling was during the daytime, which can underestimate incidence.36 The duration of spontaneous seizures was also greater than those ictal manifestations reported in other models.35,36 Taken together, these data imply a significant influence of pathology on epileptic phenotype. Confirming this, we found both the incidence and duration of epileptic seizures was a function of hippocampal pathology: rats with greater hippocampal atrophy had more frequent and longer-lasting epileptic seizures.
The amygdala is often implicated in generation and spread of epileptic seizures in experimental and human TLE.57 Here, we detected amygdala involvement in spontaneous seizures at adulthood. Surprisingly, gross pathology was not present in epileptic rats at the site of these recordings (level of dorsal hippocampus), although amygdala damage was found at the injection point. Thus, in contrast to the hippocampus, a causal link between amygdala pathology and epileptogenesis is uncertain in this model. Indeed, the relationship between amygdala pathology and TLE remains incompletely understood.57 Notably, TLE can develop following neonatal seizures in models with transient but no permanent amygdala damage.36 Our findings also suggest spontaneous seizures are minimally or nonharmful to the amygdala. The rather modest pathology at the amygdala injection site at adulthood contrasts the hippocampal changes, supporting differences in vulnerability to KA/seizures and effectiveness of repair or proliferation mechanisms between these brain regions.37,58–61 Ultimately, multielectrode depth recordings, perhaps combined with tracking neuronal activation by molecular markers, may enable us to determine whether this or another region of the amygdala, the hippocampus, or other structure(s) is the site of ictogenesis.
If hippocampal or other damage and epileptic phenotype are causally related, this would support adjunctive neuroprotective treatment for early-life status epilepticus. Prompt seizure termination in patients is paramount during episodes of status epilepticus, but concerns remain over aggressive use of anticonvulsants in young children.62 In addition to respiratory depression, anticonvulsants may also induce apoptosis in the developing brain.63 These issues, and recent evidence that neuroprotection in adult mice subject to status epilepticus significantly reduced epileptic seizure number,64 strengthen a case for neuroprotection. A range of molecular targets may now warrant appraisal in the present model, and findings may also bear relevance to hippocampal sclerosis following early-life trauma-induced brain injury.65 Finding that hippocampal sclerosis was evident by MRI offers noninvasive approaches to monitoring pathogenesis and disease-modifying effects of potential treatments.
In summary, the present data demonstrate that experimental status epilepticus in neonatal rats can cause sufficient damage that the hippocampus develops a broad spectrum of pathological changes meeting a definition of hippocampal sclerosis. The rats also developed TLE. Although prolonged seizures in infants and children do not inevitably result in hippocampal sclerosis or TLE,3,33 the present animal data offer a potential ontogenic explanation for those clinical findings which have linked early-life seizures to development of hippocampal sclerosis and TLE in later life. These findings may also have clinical implications for treatment of status epilepticus in young children and infants and for approaches to mitigation of TLE.
Acknowledgements
We thank Jing Quan-Lan for technical support, and Ronan Conroy and Markus Rehm for statistical support. The authors also thank Jochen Prehn, Michael Farrell, Robert Meller and Roger Simon for scientific input and helpful discussion of the manuscript.
Footnotes
Supported by grants from the Health Research Board Ireland (RP/2007/37 and RP/2005/24), Science Foundation Ireland (04/IN3/B466 and 08/IN1/B1875), Wellcome Trust (GR076576MA), and the National Biophotonics and Imaging Platform Ireland.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
{kind=link}
Additional acute pathological and electrophysiological features of the model
(A) Amygdala tract following microinjection. Dotted circle approximates microinjection site. Schematic to the right is adapted from the Paxinos & Watson rat brain stereotaxic atlas and confirms region of delivery is the central amygdala (CeM) and adjacent basolateral amygdala (BLA). (B) Hippocampal damage after status epilepticus in P10 pups extends to the ventral hippocampus. Montage of 4X lens images at the level of the ipsilateral ventral hippocampus showing FjB staining 72 h after status epilepticus at P10. Note, damage is mainly within CA3 at this level but also present in CAl and hilus, while the granule cell layer (GCL) is not damaged. Panels to the left are 40X lens images of representative FjB staining. (C) Effect of lorazepam on seizure EEG. Representative EEG trace from a rat pup given 2 μg KA at PI 0 followed by lorazepam (6 mg/kg, intraperitoneal). Lorazepam reduced but did not fully abolish seizure activity. (D) Image (40X lens) of seizure-damaged (KA) ipsilateral CA3 at 72 h stained for the neuronal marker NeuN and TUNEL. Note majority of TUNEL-positive cells stain for NeuN. (E) Representative lOOX lens photomicrographs of individual CA3 TUNEL positive nuclei from KA-treated pups with features of apoptosis. Scale bars: B, 40 μm, D, 25 μm, E, 5 μm.
{kind=link}
Amygdala at level of depth recordings (dorsal hippocampus) at adulthood in rats subject to neonatal status epilepticus
(A) Graph presenting NeuN counts (mean of four 40X lens fields) within the ipsilateral (ipsi) and contralateral (contr) amygdala at the level of dorsal hippocampus in rats at PI20-PI50 (n = 3-5 per group). (B) Representative NeuN staining (4X lens) of the contralateral and ipsilateral amygdala in an adult rat which received intra-amygdala KA (2 μg) at P10. (C) Representative GFAP and NeuN double-staining of the ipsilateral amygdala (40X lens). Note, there were no obvious differences at adulthood for GFAP staining between rats which had received vehicle or KA at P10. Scale bar in B = 150 μm; C = 40μm.
References
- 1.Mathern GW, Babb TL, Armstrong DL. Hippocampal Sclerosis In Epilepsy: A Comprehensive Textbook. In: Engel JJ, Pedley TA, editors. Lippincott-Raven Publishers; Philadelphia: 1997. p. 133. [Google Scholar]
- 2.Meldrum BS, Bruton CJ. Epilepsy. In: Adams JH, Duchen LW, editors. Oxford University Press; New York: 1992. p. 1246. [Google Scholar]
- 3.Guerrini R. Epilepsy in children. Lancet. 2006;367:499–524. doi: 10.1016/S0140-6736(06)68182-8. [DOI] [PubMed] [Google Scholar]
- 4.Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990;535:195–204. doi: 10.1016/0006-8993(90)91601-c. [DOI] [PubMed] [Google Scholar]
- 5.Davies KG, Hermann BP, Dohan FC, Jr, Foley KT, Bush AJ, Wyler AR. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res. 1996;24:119–126. doi: 10.1016/0920-1211(96)00008-3. [DOI] [PubMed] [Google Scholar]
- 6.Semah F, Picot MC, Adam C, Broglin D, Arzimanoglou A, Bazin B, Cavalcanti D, Baulac M. Is the underlying cause of epilepsy a major prognostic factor for recurrence? Neurology. 1998;51:1256–1262. doi: 10.1212/wnl.51.5.1256. [DOI] [PubMed] [Google Scholar]
- 7.Vossler DG, Kraemer DL, Knowlton RC, Kjos BO, Rostad SW, Wyler AR, Haltiner AM, Hasegawa H, Wilkus RJ. Temporal ictal electroencephalographic frequency correlates with hippocampal atrophy and sclerosis. Ann Neurol. 1998;43:756–762. doi: 10.1002/ana.410430610. [DOI] [PubMed] [Google Scholar]
- 8.Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. Lancet Neurol. 2004;3:663–672. doi: 10.1016/S1474-4422(04)00906-8. [DOI] [PubMed] [Google Scholar]
- 9.Kuzniecky R, Murro A, King D, Morawetz R, Smith J, Powers R, Yaghmai F, Faught E, Gallagher B, Snead OC. Magnetic resonance imaging in childhood intractable partial epilepsies: pathologic correlations. Neurology. 1993;43:681–687. doi: 10.1212/wnl.43.4.681. [DOI] [PubMed] [Google Scholar]
- 10.Mohamed A, Wyllie E, Ruggieri P, Kotagal P, Babb T, Hilbig A, Wylie C, Ying Z, Staugaitis S, Najm I, Bulacio J, Foldvary N, Luders H, Bingaman W. Temporal lobe epilepsy due to hippocampal sclerosis in pediatric candidates for epilepsy surgery. Neurology. 2001;56:1643–1649. doi: 10.1212/wnl.56.12.1643. [DOI] [PubMed] [Google Scholar]
- 11.Bocti C, Robitaille Y, Diadori P, Lortie A, Mercier C, Bouthillier A, Carmant L. The pathological basis of temporal lobe epilepsy in childhood. Neurology. 2003;60:191–195. doi: 10.1212/01.wnl.0000044055.73747.9f. [DOI] [PubMed] [Google Scholar]
- 12.Sager HJ, Oxbury JM. Hippocampal neuron loss in temporal lobe epilepsy: correlation with early childhood convulsions. Ann Neurol. 1987;22:334–340. doi: 10.1002/ana.410220309. [DOI] [PubMed] [Google Scholar]
- 13.Cendes F, Andermann F, Dubeau F, Gloor P, Evans A, Jones-Gotman M, Olivier A, Andermann E, Robitaille Y, Lopes-Cendes I, Peters T, Melanson D. Early childhood prolonged febrile convulsions, atrophy and sclerosis of mesial structures, and temporal lobe epilepsy: an MRI volumetric study. Neurology. 1993;43:1083–1087. doi: 10.1212/wnl.43.6.1083. [DOI] [PubMed] [Google Scholar]
- 14.Harvey AS, Grattan-Smith JD, Desmond PM, Chow CW, Berkovic SF. Febrile seizures and hippocampal sclerosis: frequent and related findings in intractable temporal lobe epilepsy of childhood. Pediatr Neurol. 1995;12:201–206. doi: 10.1016/0887-8994(95)00022-8. [DOI] [PubMed] [Google Scholar]
- 15.VanLandingham KE, Heinz ER, Cavazos JE, Lewis DV. Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol. 1998;43:413–426. doi: 10.1002/ana.410430403. [DOI] [PubMed] [Google Scholar]
- 16.Perez ER, Maeder P, Villemure KM, Vischer VC, Villemure JG, Deonna T. Acquired hippocampal damage after temporal lobe seizures in 2 infants. Ann Neurol. 2000;48:384–387. [PubMed] [Google Scholar]
- 17.Hermann BP, Seidenberg M, Bell B. The neurodevelopmental impact of childhood onset temporal lobe epilepsy on brain structure and function and the risk of progressive cognitive effects. Prog Brain Res. 2002;135:429–438. doi: 10.1016/S0079-6123(02)35040-4. [DOI] [PubMed] [Google Scholar]
- 18.Wasterlain CG, Niquet J, Thompson KW, Baldwin R, Liu H, Sankar R, Mazarati AM, Naylor D, Katsumori H, Suchomelova L, Shirasaka Y. Seizure-induced neuronal death in the immature brain. Prog Brain Res. 2002;135:335–353. doi: 10.1016/S0079-6123(02)35031-3. [DOI] [PubMed] [Google Scholar]
- 19.Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haut SR, Veliskova J, Moshe SL. Susceptibility of immature and adult brains to seizure effects. Lancet Neurol. 2004;3:608–617. doi: 10.1016/S1474-4422(04)00881-6. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Ari Y, Holmes GL. Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 2006;5:1055–1063. doi: 10.1016/S1474-4422(06)70626-3. [DOI] [PubMed] [Google Scholar]
- 22.Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nitecka L, Tremblay E, Charton G, Bouillot JP, Berger ML, Ben-Ari Y. Maturation of kainic acid seizure-brain damage syndrome in the rat. II. Histopathological sequelae. Neuroscience. 1984;13:1073–1094. doi: 10.1016/0306-4522(84)90289-6. [DOI] [PubMed] [Google Scholar]
- 24.Stafstrom CE, Thompson JL, Holmes GL. Kainic acid seizures in the developing brain: status epilepticus and spontaneous recurrent seizures. Brain Res Dev Brain Res. 1992;65:227–236. doi: 10.1016/0165-3806(92)90184-x. [DOI] [PubMed] [Google Scholar]
- 25.Chang D, Baram TZ. Status epilepticus results in reversible neuronal injury in infant rat hippocampus: novel use of a marker. Brain Res Dev Brain Res. 1994;77:133–136. doi: 10.1016/0165-3806(94)90220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson K, Holm AM, Schousboe A, Popper P, Micevych P, Wasterlain C. Hippocampal stimulation produces neuronal death in the immature brain. Neuroscience. 1998;82:337–348. doi: 10.1016/s0306-4522(97)00195-4. [DOI] [PubMed] [Google Scholar]
- 27.Sankar R, Shin DH, Liu H, Mazarati A, Pereira de Vasconcelos A, Wasterlain CG. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18:8382–8393. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nairismagi J, Pitkanen A, Kettunen MI, Kauppinen RA, Kubova H. Status epilepticus in 12-day-old rats leads to temporal lobe neurodegeneration and volume reduction: a histologic and MRI study. Epilepsia. 2006;47:479–488. doi: 10.1111/j.1528-1167.2006.00455.x. [DOI] [PubMed] [Google Scholar]
- 29.Cook TM, Crutcher KA. Intrahippocampal injection of kainic acid produces significant pyramidal cell loss in neonatal rats. Neuroscience. 1986;18:79–92. doi: 10.1016/0306-4522(86)90180-6. [DOI] [PubMed] [Google Scholar]
- 30.Leite JP, Babb TL, Pretorius JK, Kuhlman PA, Yeoman KM, Mathern GW. Neuron loss, mossy fiber sprouting, and interictal spikes after intrahippocampal kainate in developing rats. Epilepsy Res. 1996;26:219–231. doi: 10.1016/s0920-1211(96)00055-1. [DOI] [PubMed] [Google Scholar]
- 31.Montgomery EM, Bardgett ME, Lall B, Csernansky CA, Csernansky JG. Delayed neuronal loss after administration of intracerebroventricular kainic acid to preweanling rats. Brain Res Dev Brain Res. 1999;112:107–116. doi: 10.1016/s0165-3806(98)00161-8. [DOI] [PubMed] [Google Scholar]
- 32.Raspall-Chaure M, Chin RF, Neville BG, Scott RC. Outcome of paediatric convulsive status epilepticus: a systematic review. Lancet Neurol. 2006;5:769–779. doi: 10.1016/S1474-4422(06)70546-4. [DOI] [PubMed] [Google Scholar]
- 33.Chungath M, Shorvon S. The mortality and morbidity of febrile seizures. Nat Clin Pract Neurol. 2008;4:610–621. doi: 10.1038/ncpneuro0922. [DOI] [PubMed] [Google Scholar]
- 34.Anderson AE, Hrachovy RA, Antalffy BA, Armstrong DL, Swann JW. A chronic focal epilepsy with mossy fiber sprouting follows recurrent seizures induced by intrahippocampal tetanus toxin injection in infant rats. Neuroscience. 1999;92:73–82. doi: 10.1016/s0306-4522(98)00746-5. [DOI] [PubMed] [Google Scholar]
- 35.Kubova H, Mares P, Suchomelova L, Brozek G, Druga R, Pitkanen A. Status epilepticus in immature rats leads to behavioural and cognitive impairment and epileptogenesis. Eur J Neurosci. 2004;19:3255–3265. doi: 10.1111/j.0953-816X.2004.03410.x. [DOI] [PubMed] [Google Scholar]
- 36.Dube C, Richichi C, Bender RA, Chung G, Litt B, Baram TZ. Temporal lobe epilepsy after experimental prolonged febrile seizures: prospective analysis. Brain. 2006;129:911–922. doi: 10.1093/brain/awl018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben-Ari Y, Tremblay E, Ottersen OP. Injections of kainic acid into the amygdaloid complex of the rat: an electrographic, clinical and histological study in relation to the pathology of epilepsy. Neuroscience. 1980;5:515–528. doi: 10.1016/0306-4522(80)90049-4. [DOI] [PubMed] [Google Scholar]
- 38.Henshall DC, Araki T, Schindler CK, Lan JQ, Tiekoter KL, Taki W, Simon RP. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. J Neurosci. 2002;22:8458–8465. doi: 10.1523/JNEUROSCI.22-19-08458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mouri G, Jimenez-Mateos E, Engel T, Dunleavy M, Hatazaki S, Paucard A, Matsushima S, Taki W, Henshall DC. Unilateral hippocampal CA3-predominant damage and short latency epileptogenesis after intra-amygdala microinjection of kainic acid in mice. Brain Res. 2008;1213:140–151. doi: 10.1016/j.brainres.2008.03.061. [DOI] [PubMed] [Google Scholar]
- 40.Watson C, Nielsen SL, Cobb C, Burgerman R, Williamson B. Pathological grading system for hippocampal sclerosis: correlation with magnetic resonance imaging-based volume measurements of the hippocampus. J Epilepsy. 1996;9:56–64. [Google Scholar]
- 41.Lorente de No R. I. Studies on the structure of the cerebral cortex. II. Continuation of the study of the ammonic system. J Psychol Neurol. 1934;46:143–177. [Google Scholar]
- 42.Tanaka S, Kondo S, Tanaka T, Yonemasu Y. Long-term observation of rats after unilateral intra-amygdaloid injection of kainic acid. Brain Res. 1988;463:163–167. doi: 10.1016/0006-8993(88)90541-0. [DOI] [PubMed] [Google Scholar]
- 43.Nadler JV, Tu B, Timofeeva O, Jiao Y, Herzog H. Neuropeptide Y in the recurrent mossy fiber pathway. Peptides. 2007;28:357–364. doi: 10.1016/j.peptides.2006.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heinrich C, Nitta N, Flubacher A, Muller M, Fahrner A, Kirsch M, Freiman T, Suzuki F, Depaulis A, Frotscher M, Haas CA. Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus. J Neurosci. 2006;26:4701–4713. doi: 10.1523/JNEUROSCI.5516-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cilio MR, Sogawa Y, Cha BH, Liu X, Huang LT, Holmes GL. Long-term effects of status epilepticus in the immature brain are specific for age and model. Epilepsia. 2003;44:518–528. doi: 10.1046/j.1528-1157.2003.48802.x. [DOI] [PubMed] [Google Scholar]
- 46.Jurgen Wenzel H, Born DE, Dubach MF, Gunderson VM, Maravilla KR, Robbins CA, Szot P, Zierath D, Schwartzkroin PA. Morphological plasticity in an infant monkey model of temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S70–S75. doi: 10.1111/j.1528-1157.2000.tb01560.x. [DOI] [PubMed] [Google Scholar]
- 47.Bender RA, Baram TZ. Epileptogenesis in the developing brain: what can we learn from animal models? Epilepsia. 2007;48(Suppl 5):2–6. doi: 10.1111/j.1528-1167.2007.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wasterlain CG. Invulnerability of the immature brain to seizures: do dogmas have nine lives? Epilepsy Curr. 2006;6:59–61. doi: 10.1111/j.1535-7511.2006.00099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boison D. The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol. 2008;84:249–262. doi: 10.1016/j.pneurobio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nissinen J, Halonen T, Koivisto E, Pitkanen A. A new model of chronic temporal lobe epilepsy induced by electrical stimulation of the amygdala in rat. Epilepsy Res. 2000;38:177–205. doi: 10.1016/s0920-1211(99)00088-1. [DOI] [PubMed] [Google Scholar]
- 51.Thom M, Martinian L, Williams G, Stoeber K, Sisodiya SM. Cell proliferation and granule cell dispersion in human hippocampal sclerosis. J Neuropathol Exp Neurol. 2005;64:194–201. doi: 10.1093/jnen/64.3.194. [DOI] [PubMed] [Google Scholar]
- 52.Kelley MS, Steward O. Injury-induced physiological events that may modulate gene expression in neurons and glia. Rev Neurosci. 1997;8:147–177. doi: 10.1515/revneuro.1997.8.3-4.147. [DOI] [PubMed] [Google Scholar]
- 53.Deller T, Haas CA, Frotscher M. Reorganization of the rat fascia dentata after a unilateral entorhinal cortex lesion: role of the extracellular matrix. Ann NY Acad Sci. 2000;911:207–220. doi: 10.1111/j.1749-6632.2000.tb06728.x. [DOI] [PubMed] [Google Scholar]
- 54.Bouwmeester H, Gerrits MA, Roozemond JG, Snapper J, Ronken E, Kruse CG, Westenberg HG, van Ree JM. Neonatal basolateral amygdala lesions affect monoamine and cannabinoid brain systems in adult rats. Int J Neuropsychopharmacol. 2007;10:727–739. doi: 10.1017/S1461145706007346. [DOI] [PubMed] [Google Scholar]
- 55.Harich S, Kinfe T, Koch M, Schwabe K. Neonatal lesions of the entorhinal cortex induce long-term changes of limbic brain regions and maze learning deficits in adult rats. Neuroscience. 2008;153:918–928. doi: 10.1016/j.neuroscience.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 56.Sankar R, Shin D, Mazarati AM, Liu H, Katsumori H, Lezama R, Wasterlain CG. Epileptogenesis after status epilepticus reflects age- and model-dependent plasticity. Ann Neurol. 2000;48:580–589. [PubMed] [Google Scholar]
- 57.Pitkanen A, Tuunanen J, Kalviainen R, Partanen K, Salmenpera T. Amygdala damage in experimental and human temporal lobe epilepsy. Epilepsy Res. 1998;32:233–253. doi: 10.1016/s0920-1211(98)00055-2. [DOI] [PubMed] [Google Scholar]
- 58.Wuerthele SM, Lovell KL, Jones MZ, Moore KE. A histological study of kainic acid-induced lesions in the rat brain. Brain Res. 1978;149:489–497. doi: 10.1016/0006-8993(78)90491-2. [DOI] [PubMed] [Google Scholar]
- 59.Ben-Ari Y, Tremblay E, Ottersen OP, Meldrum BS. The role of epileptic activity in hippocampal and “remote” cerebral lesions induced by kainic acid. Brain Res. 1980;191:79–97. doi: 10.1016/0006-8993(80)90316-9. [DOI] [PubMed] [Google Scholar]
- 60.Park JH, Cho H, Kim H, Kim K. Repeated brief epileptic seizures by pentylenetetrazole cause neurodegeneration and promote neurogenesis in discrete brain regions of freely moving adult rats. Neuroscience. 2006;140:673–684. doi: 10.1016/j.neuroscience.2006.02.076. [DOI] [PubMed] [Google Scholar]
- 61.Jung KH, Chu K, Lee ST, Kim JH, Kang KM, Song EC, Kim SJ, Park HK, Kim M, Lee SK, Roh JK. Region-specific plasticity in the epileptic rat brain: a hippocampal and extrahippocampal analysis. Epilepsia. 2009;50:537–549. doi: 10.1111/j.1528-1167.2008.01718.x. [DOI] [PubMed] [Google Scholar]
- 62.Abdel-Hamid HZ, Painter MJ. Approach to the management of neonatal status epilepticus. In: Wasterlain CG, Treiman DM, editors. The MIT press; Cambridge: 2006. p. 503. [Google Scholar]
- 63.Olney JW, Young C, Wozniak DF, Jevtovic-Todorovic V, Ikonomidou C. Do pediatric drugs cause developing neurons to commit suicide? Trends Pharmacol Sci. 2004;25:135–139. doi: 10.1016/j.tips.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 64.Jimenez-Mateos EM, Hatazaki S, Johnson MB, Bellver-Estelles C, Mouri G, Bonner C, Prehn JH, Meller R, Simon RP, Henshall DC. Hippocampal transcriptome after status epilepticus in mice rendered seizure damage-tolerant by epileptic preconditioning features suppressed calcium and neuronal excitability pathways. Neurobiol Dis. 2008;32:442–453. doi: 10.1016/j.nbd.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 65.Felderhoff-Mueser U, Ikonomidou C. Mechanisms of neurodegeneration after paediatric brain injury. Curr Opin Neurol. 2000;13:141–145. doi: 10.1097/00019052-200004000-00005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional acute pathological and electrophysiological features of the model
(A) Amygdala tract following microinjection. Dotted circle approximates microinjection site. Schematic to the right is adapted from the Paxinos & Watson rat brain stereotaxic atlas and confirms region of delivery is the central amygdala (CeM) and adjacent basolateral amygdala (BLA). (B) Hippocampal damage after status epilepticus in P10 pups extends to the ventral hippocampus. Montage of 4X lens images at the level of the ipsilateral ventral hippocampus showing FjB staining 72 h after status epilepticus at P10. Note, damage is mainly within CA3 at this level but also present in CAl and hilus, while the granule cell layer (GCL) is not damaged. Panels to the left are 40X lens images of representative FjB staining. (C) Effect of lorazepam on seizure EEG. Representative EEG trace from a rat pup given 2 μg KA at PI 0 followed by lorazepam (6 mg/kg, intraperitoneal). Lorazepam reduced but did not fully abolish seizure activity. (D) Image (40X lens) of seizure-damaged (KA) ipsilateral CA3 at 72 h stained for the neuronal marker NeuN and TUNEL. Note majority of TUNEL-positive cells stain for NeuN. (E) Representative lOOX lens photomicrographs of individual CA3 TUNEL positive nuclei from KA-treated pups with features of apoptosis. Scale bars: B, 40 μm, D, 25 μm, E, 5 μm.
Amygdala at level of depth recordings (dorsal hippocampus) at adulthood in rats subject to neonatal status epilepticus
(A) Graph presenting NeuN counts (mean of four 40X lens fields) within the ipsilateral (ipsi) and contralateral (contr) amygdala at the level of dorsal hippocampus in rats at PI20-PI50 (n = 3-5 per group). (B) Representative NeuN staining (4X lens) of the contralateral and ipsilateral amygdala in an adult rat which received intra-amygdala KA (2 μg) at P10. (C) Representative GFAP and NeuN double-staining of the ipsilateral amygdala (40X lens). Note, there were no obvious differences at adulthood for GFAP staining between rats which had received vehicle or KA at P10. Scale bar in B = 150 μm; C = 40μm.