

Abstract
Transforming growth factor-β cooperates with oncogenic Ras to activate nuclear β-catenin during the epithelial to mesenchymal transition of hepatocytes, a process relevant in the progression of hepatocellular carcinoma (HCC). In this study we investigated the role of β-catenin in the differentiation of murine, oncogene-targeted hepatocytes and in 133 human HCC patients scheduled for orthotopic liver transplantation. Transforming growth factor-β caused dissociation of plasma membrane E-cadherin/β-catenin complexes and accumulation of nuclear β-catenin in Ras-transformed, but otherwise normal hepatocytes in p19ARF−/− mice. Both processes were inhibited by Smad7-mediated disruption of transforming growth factor-β signaling. Overexpression of constitutively active β-catenin resulted in high levels of CK19 and M2-PK, whereas ablation of β-catenin by axin overexpression caused strong expression of CK8 and CK18. Therefore, nuclear β-catenin resulted in dedifferentiation of neoplastic hepatocytes to immature progenitor cells, whereas loss of nuclear β-catenin led to a differentiated HCC phenotype. Poorly differentiated human HCC showed cytoplasmic redistribution or even loss of E-cadherin, suggesting epithelial to mesenchymal transition. Analysis of 133 HCC patient samples revealed that 58.6% of human HCC exhibited strong nuclear β-catenin accumulation, which correlated with clinical features such as vascular invasion and recurrence of disease after orthotopic liver transplantation. These data suggest that activation of β-catenin signaling causes dedifferentiation to malignant, immature hepatocyte progenitors and facilitates recurrence of human HCC after orthotopic liver transplantation.
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide and results in poor prognosis with 5-year survival rates of only 8.9% despite of treatment.1 Various risk factors such as viral infections, alcohol abuse, exposure to fungal hepatotoxins, or hereditary diseases can cause inflammation, fibrosis and cirrhosis, which finally lead to HCC.2 Major problems in combating HCC include diagnosis at advanced stages and thus few therapeutic options.3 Curative treatments involving tumor resection or orthotopic liver transplantation (OLT), which abolishes underlying cirrhosis, offer promising results but are limited to early stages.4 Since 1996, selection of HCC patients for OLT is restricted to those with few, small liver tumors, which excludes many patients at the time of diagnosis.5 This generates a need for novel parameters to assess risk of tumor recurrence, which are applicable even in patients that exceed the established criteria of tumor burden.6 Although clinicopathological features such as up-regulation of cytokeratin (CK)19 were found to be correlated with high risk of HCC recurrence after OLT, the underlying molecular and cellular mechanisms are still poorly understood.7
Molecular alterations responsible for HCC development and progression include 1) loss of tumor suppressors such as p53, retinoblastoma protein (Rb), p16INK4A or p14ARF; 2) activation of oncoproteins such as c-myc or c-Met; 3) activation and secretion of cytokines such as transforming growth factor (TGF)-β; and 4) alterations in the Wnt/Frizzled signaling leading to nuclear accumulation of β-catenin, which are found in 33 to 67% of HCC cases.8–11 While mutations of β-catenin are rather rare, other mechanisms activating Wnt signaling such as secretion of Frizzled7 contribute to nuclear accumulation of β-catenin and transcription of its target genes such as cyclin D1.10,12 Activation of β-catenin is essential for liver development as shown by deletion of β-catenin in gene-targeted mice, causing death around embryonic day 17 due to impaired liver cell proliferation and increased apoptosis.13 Furthermore, canonical Wnt/Frizzled signaling is connected to tumor progression and modulates differentiation and stemness of the intestinum.14 Noteworthy, nuclear β-catenin accumulation was strikingly demonstrated in cells of the invasive front and surrounding tissues in colon carcinomas.15 These data raise the question whether β-catenin-positive, migratory cancer stem cells (CSCs) are capable of causing tumor dormancy and distal metastases.
The epithelial to mesenchymal transition (EMT), a process particularly implicated in embryonic development, has increasingly been recognized as a crucial step in tumor progression and metastasis.16 Disruption of E-cadherin from the cell membrane as well as nuclear β-catenin accumulation are hallmarks of EMT as described in a variety of carcinoma models.15 Previous studies revealed numerous effectors of EMT of which TGF-β is a particularly potent one.16 In mouse models of hepatocellular EMT, TGF-β evades tumor-suppressive functions and induces a highly malignant, invasive phenotype of hepatocytes in collaboration with Ras/MAPK signaling, which is accompanied by autocrine TGF-β secretion.17,18 Interestingly, both urine and serum of patients with advanced liver tumors contain high TGF-β levels.19 In human HCC, EMT and thus invasiveness were also induced by the signal transducer and activator of transcription (STAT) 5b in cooperation with the hepatitis B oncoprotein HBX, or by TGF-β plus laminin-5.20,21 HCC cell lines having undergone EMT and expressing elevated levels of integrin-linked kinase were also largely resistant to inhibitors of the epidermal growth factor receptor.22 This is in line with observations that erlotinib or cetuximab showed only modest effects in HCC therapy, although they were highly effective in other cancer types.23
In this study, we show that the TGF-β-induced activation of nuclear β-catenin in neoplastic hepatocytes correlates with their transdifferentiation to early hepatocyte progenitors that abundantly express M2-PK and CK19, but lack epithelial CK8 and CK18. In human patients, primary HCC with high-level nuclear β-catenin expression showed disintegration of plasma membrane complexes of E-cadherin and β-catenin, suggesting EMT. Furthermore, nuclear β-catenin accumulation associated with enhanced vascular invasion and elevated recurrence of HCC after OLT.
Materials and Methods
Cell Culture
Epithelial MIM-R hepatocytes were generated by introducing a retroviral construct bicistronically expressing oncogenic v-Ha-Ras and green fluorescent protein into cultured, immortalized p19ARF−/− hepatocytes (MIM1-4).17 Retroviral transmission of MIM-R hepatocytes either with the constitutive active mutant β-catenin ΔGSK-KT3, its negative regulator axin or with Smad7 resulted in epithelial MIM-R-ABC, MIM-R-Axin, and MIM-R-S7 cells, respectively.24,25 Treatment of epithelial MIM-R cells with 1 ng/ml TGF-β1 (R&D Systems, Minneapolis, MN) over 14 days resulted in a stable fibroblastoid cell line, termed MIM-RT. MIM-R-S7 cells were termed MIM-RT-S7 after comparable TGF-β1 treatment. Fibroblastoid cells were grown on plastic, whereas epithelial cells required culture dishes coated with rat tail collagen, prepared as described.26 All cells were grown in RPMI 1640 plus 10% fetal calf serum and antibiotics at 37°C and 5% CO2, and were routinely screened for the absence of mycoplasma.
Subcutaneous and Orthotopic Tumor Formation
Cells were each trypsinized and washed with phosphate-buffered saline. Thereafter, 1 × 106 cells in 100 μl of Ringer's solution were subcutaneously injected each into three individual immunodeficient CB-17 SCID recipient mice as described.24 To induce orthotopic liver tumors, 5 × 105 cells in 20 μl of Ringer's solution were injected into the spleen of immunodeficient CB-17 SCID recipient mice.27 All injected cell types caused experimental tumors with an incidence of 100%. The experiments were performed twice using three mice per cell type and performed according to the Austrian guidelines for animal care and protection.
Immunohistochemical Analysis
Subcutaneous tumors and liver tissue were fixed in 4% phosphate-buffered paraformaldehyde and embedded in paraffin.24,25 Sections 4 μm thick were stained with hematoxylin and eosin (H&E) for standard microscopy. For immunohistochemistry, paraffin-embedded sections were treated with citric acid buffer (0.01 mol/L, pH 6.0) before staining with the following antibodies: anti-β-catenin (Transduction Laboratories, Lexington, UK), 1:100; anti-nondestructible, active β-catenin (Upstate, Lake Placid, NY), 1:100; anti-E-cadherin (Transduction Laboratories), 1:100; anti-CK8 (PROGEN, Heidelberg, Germany), 1:50; anti-CK18 (PROGEN), 1:50; anti-CK19 (University of Iowa, Iowa City, IA), 1:100; anti-M2-PK (Rockland, Philadelphia, PA), 1:100; anti-HNF-6 (Santa Cruz Biotechnology, Santa Cruz, CA), 1:100; anti-glutamine synthetase (Abcam, Cambridge, UK), 1:100. For staining with anti-green fluorescent protein antibody (Santa Cruz Biotechnology) at a dilution of 1:100, sections were pretreated with trypsin. After washing, sections were stained with corresponding, biotinylated secondary antibodies and visualization was performed with the Vectastain ABC kit using diaminobenzidine as substrate (Vector Laboratories, Burlingame, CA). Biotinylated antibodies against peanut agglutinin and Dolichos biflorus agglutinin (both Vector Laboratories) were each used at a dilution of 1:100.
HCC Tissue Array
The HCC tissue arrays used contained paraffin-embedded specimens of tumors and adjacent normal tissue collected from a total of 133 female and male HCC patients, all of which have undergone OLT for HCC at the Department of Transplantation Surgery, Medical University of Vienna, between 1982 and 2002.28 The anonymous pathology reports were obtained and the baseline characteristics of all 133 HCC cases analyzed in this study are outlined in Supplemental Table 1 at http://ajp.amjpathol.org. All histological specimens were reviewed for histological type and grade. Core biopsies with diameter of 0.6 mm were taken from each donor paraffin block, arrayed in triplicate, and 4-μm-thick sections cut from the array. After antigen retrieval in sodium citrate buffer (pH 6.0), sections were stained with the anti-active β-catenin antibody (Upstate) at a dilution of 1:100 or with anti-CK19 (Dako, Glostrup, Denmark), 1:100. Secondary antibody was used and visualization was performed with the Vectastain ABC kit using diaminobenzidine as substrate (Vector Laboratories). Evaluation of tissue arrays was performed by two independent researchers (G.Z. and M.M.) who were blinded regarding patient details. Immunostaining using the anti-active β-catenin antibody was scored on triplicate tissues by using the following arbitrary scale: no staining, low staining, medium staining, and high staining, judging both staining intensity of nuclei in β-catenin-positive tumor cells and percent nuclei positive in tumor tissues. Immunostaining using the anti-CK19 antibody was scored on triplicate tissues by evaluating the absence or the presence of staining in malignant hepatocytes.
Statistical Analysis
Nuclear β-catenin accumulation ordinal values were compared between groups of clinical features by means of the exact Jonckheere-Terpstra and Wilcoxon signed rank test in StatXact (StatXact Version 4.0, Cytel Software Corporation) and SAS (SAS. SAS/STAT User's Guide, Version 9, Cary, NC 27513: SAS Institute Inc., 2002–2003). P values are two-sided. P values below 0.05 were considered as statistically significant. The 95% confidence intervals for percentages were calculated in StatXact.
Results
Inhibition of TGF-β Signaling by Antagonistic Smad7 Abolishes EMT Signatures
Experimental HCC were generated in mice by orthotopic transplantation of green fluorescent protein-positive, oncogenic Ras-expressing hepatocytes (MIM-R), which display an epithelial organization. These liver tumors were compared with experimental HCC derived from fibroblastoid MIM-RT cells, in which EMT was induced by long-term TGF-β1 treatment of cultured MIM-R hepatocytes (see Supplemental Figure S1 at http://ajp.amjpathol.org).17
MIM-R tumors expressed a complex of the adherens junction proteins E-cadherin and β-catenin at the plasma membrane, while MIM-RT-derived tumors showed cytoplasmic expression or even loss of E-cadherin as well as delocalization of the plasma-membrane-bound β-catenin to cytoplasm and nucleus (Figure 1).24 However, liver tumors generated by MIM-R-S7 hepatocytes overexpressing the TGF-β signaling inhibitory protein Smad725 retained localization of E-cadherin/β-catenin complexes at the plasma membrane (Figure 1). Accordingly, subcutaneous tumors derived from MIM-RT-S7 cells, which failed to undergo EMT even after long-term TGF-β1 treatment,25 displayed intact E-cadherin/β-catenin complexes at cell borders (see Supplemental Figure S2 at http://ajp.amjpathol.org). These results demonstrate that TGF-β signaling is responsible for the dissociation of E-cadherin/ β-catenin complexes at cell boundaries in orthotopic tumors, a process indicating EMT.
Figure 1.
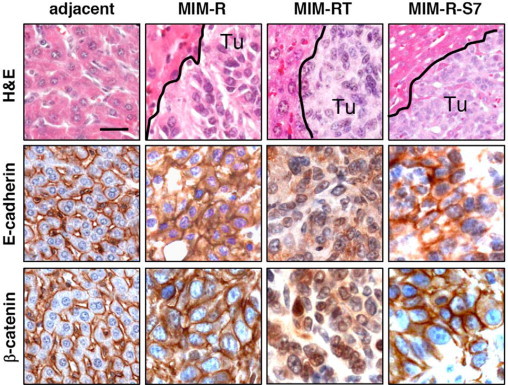
TGF-β-dependent EMT of hepatocytes associates with dissociation of E-cadherin/β-catenin complexes. Ras-transformed epithelial MIM-R hepatocytes, the same after EMT induced by TGF-β (MIM-RT cells), and MIM-R-S7 cells overexpressing Smad7, which disrupts TGF-β signaling, were transplanted into the spleen of immunocompromised SCID mice to generate orthotopic experimental HCC. The resulting liver tumors (Tu) and neighboring tissue from MIM-R tumors (adjacent) were collected after 20 days and processed for standard histology by H&E staining and for immunohistochemical staining by using antibodies against E-cadherin and total β-catenin (β-catenin). Black lines in upper panels indicate tumor-host borders. Scale bar = 25 μm.
Inhibition of TGF-β Signaling Abrogates Nuclear β-Catenin Localization
Immunohistochemical analysis of MIM-R liver tumor sections using an anti-active β-catenin antibody revealed clear nuclear staining in MIM-R hepatocytes,29 while adjacent tumor tissue was essentially negative (Figure 2A). MIM-RT-derived liver tumors showed even stronger nuclear accumulation of β-catenin in a high proportion of malignant hepatocytes, while inhibition of TGF-β signaling in MIM-R-S7 hepatocytes strongly reduced nuclear β-catenin staining (Figure 2A). Although this was observed in most MIM-R-S7-derived tumors (Figure 2B, upper panel), some nodules retained nuclear β-catenin with signal intensities much weaker than in MIM-RT tumors shown to have undergone EMT (Figure 2B, lower panel). Interestingly, nuclear β-catenin accumulation in MIM-R-S7-derived tumors occurred predominately at tumor-host borders (Figure 2, lower panels) suggesting a cross-talk with the tumor stroma. In conclusion, stabilization of nuclear β-catenin in liver tumors depends on both, TGF-β signaling and occurs in the context of EMT, but may also be associated with EMT-independent events since liver tumors generated by MIM-R hepatocytes also showed nuclear localization of β-catenin.
Figure 2.
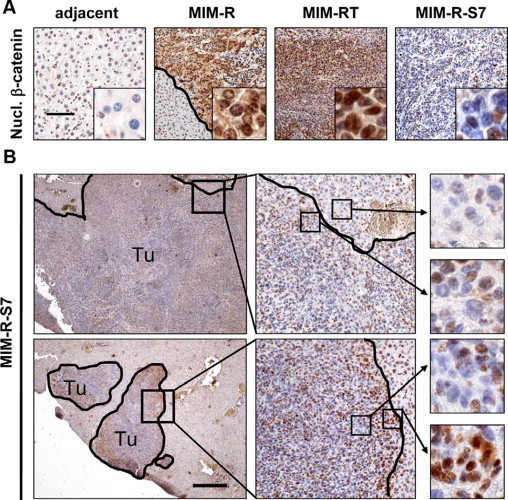
Accumulation of nuclear β-catenin is blocked by interference with TGF-β signaling. A: Experimental HCC generated by MIM-R, MIM-RT or MIM-R-S7 hepatocytes (Figure 1) were analyzed by immunohistochemical staining using an antibody against active β-catenin. “Adjacent” designates peritumoral tissue of MIM-R tumors. Insets: Part of panels at 10-fold higher magnification. Scale bar = 100 μm. B: Detailed analysis of MIM-R-S7-generated tumors stained as in A. Black lines indicate tumor-stroma border. Black boxes in left panels show location of fivefold magnified regions (middle panels). Boxed regions in upper middle panel show further fourfold magnifications of the stroma and the tumor (upper and lower right panels, respectively). Boxed regions in lower middle panel show low nuclear β-catenin levels in central tumor regions (upper small panel), whereas regions at the host-tumor border show intense staining for nuclear β-catenin (lower small panel). Tu, tumor. Scale bar = 500 μm.
Gain of Nuclear β-Catenin Induces Marker Proteins of Hepatic Progenitor Cells
Orthotopic liver tumors generated by MIM-R cells expressed cytokeratins CK8 and CK18, two markers of mature epithelial hepatocytes (Figure 3). A similar CK8/CK18 staining indicating a mature hepatocyte phenotype was observed in liver tumors derived from MIM-R-Axin hepatocytes, in which Wnt signaling and thus nuclear β-catenin accumulation is inhibited by axin.24 In contrast, liver tumors generated from fibroblastoid MIM-RT cells failed to express CK8 and CK18, suggesting a more immature phenotype (Figure 3). Accordingly, liver tumors of MIM-RT cells showed increased intrahepatic metastasis (data not shown). Overexpression of N-terminally truncated, constitutively active β-catenin in MIM-R cells (MIM-R-ABC) elicited a fourfold higher activity in β-catenin-responsive luciferase constructs and expression of known target genes of β-catenin such as c-myc and cyclin D1, as shown recently.24 In addition, the expression of glutamine synthetase, which represents a further target of β-catenin in hepatocytes,30 could be detected in MIM-R-ABC tumors but was hardly expressed in MIM-R-Axin tumors which are devoid of nuclear β-catenin (see Supplemental Figure S3 at http://ajp.amjpathol.org). Interestingly, those MIM-R-ABC cells also showed a suppressed CK8 and CK18 expression, although not to the same extent as observed in MIM-RT tumors (Figure 3). These data suggest that MIM-R tumors might contain rather well-differentiated hepatocytes, whereas tumors induced by MIM-RT cells that have undergone EMT lack hepatocytic differentiation.
Figure 3.
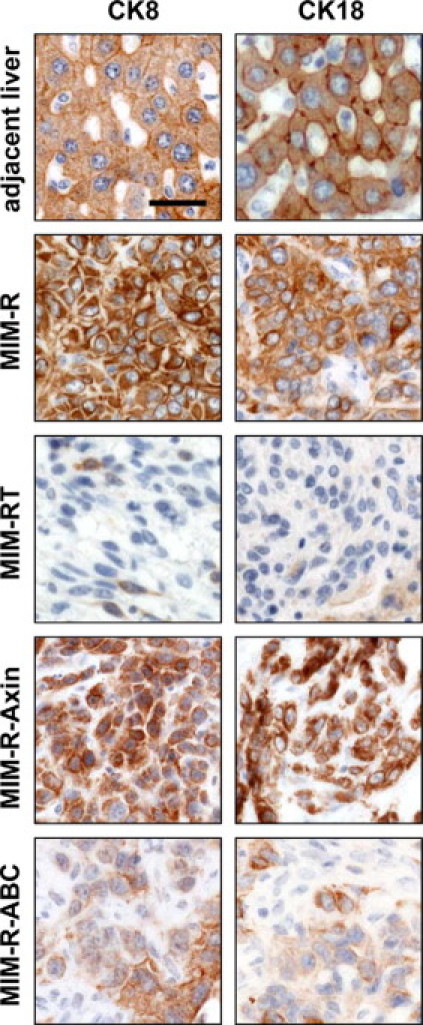
Activation of nuclear β-catenin in liver tumors by TGF-β or constitutive active β-catenin abolishes expression of the epithelial differentiation markers CK8 and CK18. The same cell types as in Figure 1 (MIM-R, MIM-RT) plus MIM-R cells with blocked (MIM-R-Axin) or constitutively active β-catenin signaling (MIM-R-ABC) were orthotopically transplanted into immunocompromised SCID mice. Green fluorescent protein-positive liver tumors were collected after 20 days and processed for immunohistochemistry using anti-CK8 or anti-CK18 antibodies. Scale bar = 25 μm.
To substantiate these findings, we analyzed various experimental liver tumors for the expression of M2-PK and CK19, two marker proteins for immature hepatic progenitors such as oval cells and for cholangiocytes. A positive correlation of these markers with nuclear accumulation of β-catenin would argue for the dedifferentiation of tumor cells to immature hepatocyte progenitors driven by Wnt/β-catenin signaling. Tumors derived from fibroblastoid MIM-RT cells and MIM-R-ABC cells displayed strong M2-PK and CK19 expression, while MIM-R-Axin tumors, in which β-catenin signaling is blocked, failed to express both markers similar to normal liver tissue adjacent to the tumors (Figure 4). Interestingly, MIM-R-derived tumors already expressed M2-PK and CK19. In addition, MIM-R-ABC generated tumors showed duct-like cell structures similar to the morphology exhibited in cholangiocarcinoma (see Supplemental Figure S1 at http://ajp.amjpathol.org). The strong staining for Dolichos biflorus agglutinin and peanut agglutinin and the absence of hepatocyte nuclear factor (HNF)-6 underlines the presence of duct-like cell structures in MIM-R-ABC-derived tumors, which nearly fails to occur in tumors generated from MIM-R-Axin cells (see Supplemental Figures S3 and S4 at http://ajp.amjpathol.org). These data suggest that malignant hepatocytes induced with TGF-β (MIM-RT) or expressing constitutive Wnt/β-catenin (MIM-R-ABC) might have the potential to dedifferentiate into hepatic progenitors.
Figure 4.
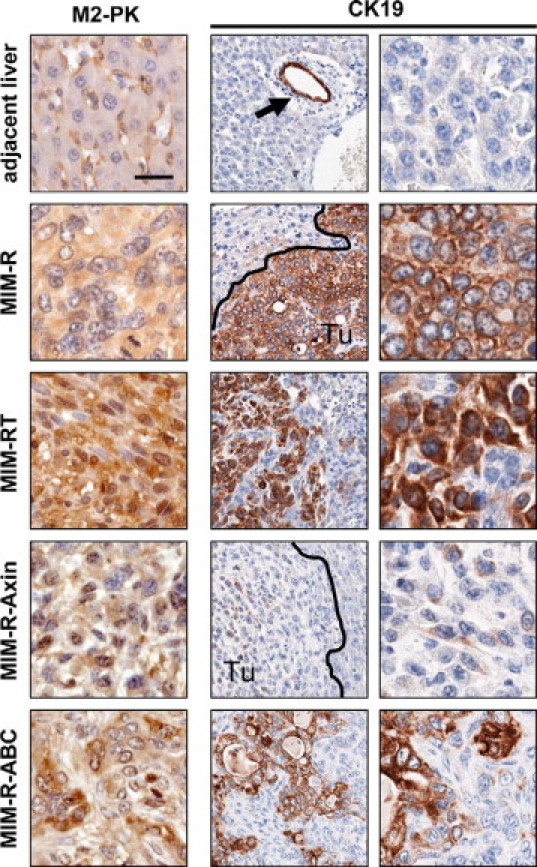
Nuclear β-catenin accumulation in liver tumor cells correlates with an immature hepatocyte progenitor phenotype. Orthotopic liver tumors generated by the same cell types as in Figure 3 were collected after 20 days and immunohistochemically stained with anti-M2-PK and anti-CK19 antibodies. Sections in middle panels show an overview of CK19 staining at lower magnification. Adjacent liver: Tissue surrounding MIM-R tumors. Arrow indicates bile duct in normal tissue. Black lines show tumor-host borders. Tu, tumor. Scale bar = 25 μm.
Loss or Redistribution of Plasma Membrane-Localized E-Cadherin Suggests EMT of Hepatocytes in Poorly Differentiated Human HCC
In an attempt to bridge human hepatocarcinogenesis to our mouse HCC models using normal murine hepatocytes with defined genetic alterations,24,27 we analyzed the epithelial organization of randomly selected primary human HCC by the immunolocalization of E-cadherin. Well differentiated human HCC of histological grade G131 showed plasma membrane expression of E-cadherin in 63% of cases (Figure 5A; Table 1), suggesting intact epithelial cell-cell communication of these human malignant hepatocytes. This finding was corroborated by semiquantitative evaluation of E-cadherin localization to plasma membrane, cytoplasm or its respective loss (Table 1). On the contrary, poorly differentiated human HCC (histological grade G3) frequently exhibited loss of plasma membrane E-cadherin expression (60% of HCC; Figure 5B; Table 1), which was retained at the membrane in only a minor proportion of HCC (Table 1; see Supplemental Table 2 at http://ajp.amjpathol.org). These data reveal that disruption of the E-cadherin/β-catenin complexes indicates changes of epithelial plasticity such as EMT of neoplastic hepatocytes in mouse (Figure 1) and human HCC (Figure 5B). Since β-catenin accumulation in cell nuclei of malignant hepatocytes is a strong, although not exclusive hallmark of EMT during hepatocarcinogenesis, the above observations predict that progression of human HCC involves nuclear β-catenin localization.
Figure 5.
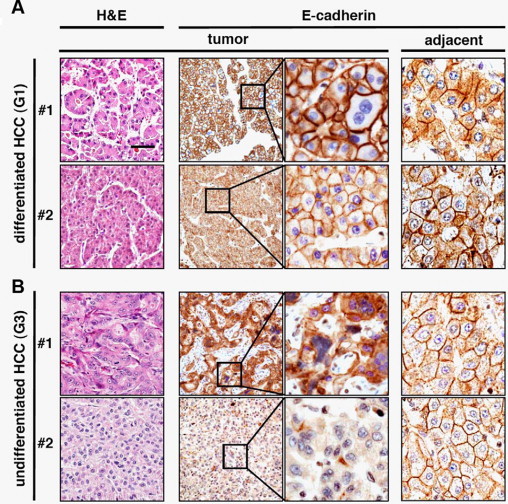
Poorly differentiated human HCC show cytoplasmic dislocation or loss of E-cadherin expression at plasma membrane, suggesting EMT. Human HCC samples were stained with H&E or anti-E-cadherin (E-cadherin) antibody. Two representative examples from both well-differentiated HCC (histological grading G1) (A) and poorly differentiated HCC tissues (histological grading G3) (B) are shown. The left panels show H&E staining. Black boxes in middle panels are regions depicted at fourfold magnification in right panels. E-cadherin is expressed at cell borders in HCC with G1 grade, whereas poorly differentiated HCC with G3 grade show either cytoplasmic delocalization or loss of E-cadherin. Hepatocytes of peritumoral tissues (adjacent) show plasma membrane-localized E-cadherin (right panels). Scale bar = 50 μm.
Table 1.
Localization of E-Cadherin in Human HCC
| HCC grading | E-cad at cell borders | Cytoplasmic E-cad | Loss of E-cad |
|---|---|---|---|
| G1 (n = 11) | 7/11 | 5/11 | 3/11 |
| G3 (n = 10) | 1/10 | 3/10 | 6/10 |
E-cad, E-cadherin; G1, differentiated HCC; G3, poorly differentiated HCC.
Nuclear β-Catenin Accumulation Predicts Vascular Invasion and Recurrence of Disease (ROD) in HCC Patients after OLT
To assess the role of nuclear β-catenin accumulation for HCC progression, we evaluated the expression of nuclear β-catenin by immunohistochemical analysis of human HCC tissue arrays covering a total number of 133 samples from patients scheduled for OLT. Noteworthy, 58.6% of HCC patients analyzed before OLT showed a medium to high nuclear β-catenin accumulation. To obtain more quantitative data, we discriminated between no, low, medium or high staining intensity (Figure 6A), indicating the degree of nuclear β-catenin stabilization. These data of nuclear β-catenin expression were then correlated with clinical parameters such as vessel invasion, cirrhosis, histological grading, metastasis, and ROD (see Supplemental Table 1 at http://ajp.amjpathol.org). Noteworthy, cirrhosis was not linked to nuclear stabilization of β-catenin as we found moderately higher levels of nuclear β-catenin in patients lacking cirrhosis (Table 2).
Figure 6.
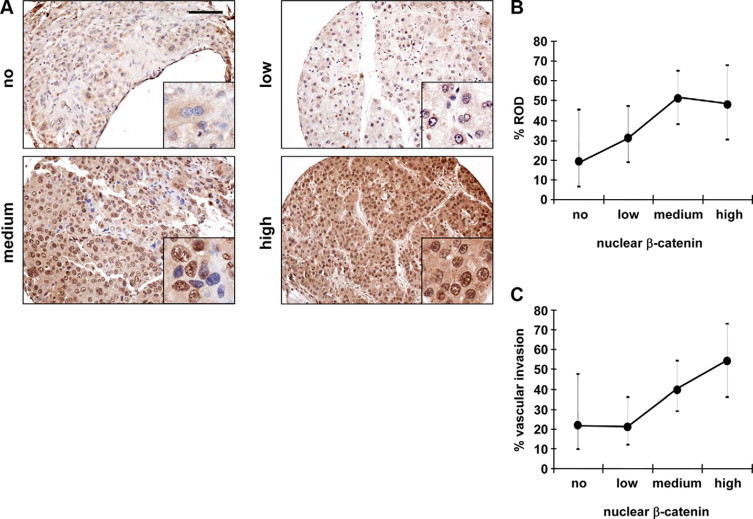
Nuclear β-catenin correlates with vascular invasion and tumor relapse in human HCC after OLT. Immunolocalization and staining intensity of samples from a human HCC tissue array (133 patients) was grouped into four arbitrary classes after staining with anti-activate β-catenin antibody. A: Representative examples for no (cytoplasmic β-catenin only), low (weak nuclear staining), medium (majority of nuclei strongly stained) or high nuclear β-catenin staining (all nuclei strongly stained) are shown. Insets: Selected regions at fivefold higher magnification. Scale bar = 100 μm. B and C: Nuclear β-catenin accumulation evaluated as shown in A associates with fostered recurrence of HCC (ROD) after OLT (B) and increased vascular invasion (C). Black circles show the percentages of patients positive for ROD (B) or vessel invasion (C) calculated from raw numbers of the different classes of nuclear β-catenin expression. Error bars = 95% confidence interval.
Table 2.
Nuclear β-Catenin Localization in Human HCC Facilitates ROD
| Nuclear β-catenin accumulation |
|||||
|---|---|---|---|---|---|
| Clinical features | No n (%) | Low n (%) | Medium n (%) | High n (%) | P |
| Vessel invasion | |||||
| Absent | 13 (14) | 32 (36) | 33 (37) | 12 (13) | 0.0018 |
| Present | 3 (7) | 7 (16) | 20 (47) | 13 (30) | |
| Cirrhosis | |||||
| No | 1 (6) | 1 (6) | 10 (63) | 4 (25) | 0.0396 |
| Yes | 15 (13) | 38 (32) | 43 (37) | 21 (18) | |
| Grading | |||||
| G1-2 | 14 (13) | 35 (32) | 45 (41) | 17 (15) | 0.0475 |
| G3 | 2 (9) | 4 (18) | 8 (36) | 8 (36) | |
| Metastasis | |||||
| Absent | 16 (13) | 36 (30) | 49 (41) | 20 (17) | 0.0530 |
| Present | 0 (0) | 3 (25) | 4 (33) | 5 (42) | |
| ROD | |||||
| No | 13 (16) | 27 (34) | 26 (33) | 13 (16) | 0.0142 |
| Yes | 3 (6) | 12 (22) | 27 (50) | 12 (22) | |
G, histological grade; no, low, medium or high, nuclear β-catenin immunoreactivity has exclusively been evaluated in hepatocytes of the HCC. Metastases include diseased lymph nodes as well as distal metastases. P < 0.05 was considered as statistically significant.
In line with the histological grading (G1–G3) before OLT, predicting good to bad prognosis,31 a large percentage of poorly differentiated HCC (G3) showed nuclear β-catenin at medium to high levels (each 36.4%), while no or low β-catenin levels were only found at 18.2% and 9.1%, respectively (Table 2). Noteworthy, the percentages of patients with G3 were calculated from raw numbers of the different classes of nuclear β-catenin expression. Correlation of nuclear β-catenin levels to metastasis was difficult, since metastases were very rare in the HCC analyzed due to the selection criteria of patients for OLT. Nevertheless, metastatic dissemination to lymph nodes or distal organs was slightly enhanced in HCC tissues exhibiting high levels of nuclear β-catenin stabilization (Table 2). Most importantly, we observed a highly significant increase of ROD in patients displaying elevated levels of nuclear β-catenin (Figure 6B; Table 2). Moreover, HCC showing medium and high levels of nuclear β-catenin exhibited not only high frequencies of HCC relapse (ROD; 50.9% and 48.0%, respectively), but also a striking increase in blood vessel invasion of tumor cells (37.7% and 52.0%, respectively; Figure 6C; Table 2). In addition, multivariable logistic regression models, which estimated the effect of β-catenin and the classical prognostic factors age and sex, revealed that nuclear β-catenin was significant with respect to vessel invasion and ROD, and thus coincided with results from univariate analyses shown in Table 2 (data not shown).
We further evaluated the expression of CK19, already reported to correlate with high risk of HCC recurrence after OLT7 on the arrayed human HCC samples to address an association with nuclear β-catenin activation. Interestingly, a strong CK19-positive immunoreactivity significantly correlated with medium to high nuclear β-catenin expression (26.4% and 25.0%, respectively; see Supplemental Figure S5 and Supplemental Table 3 at http://ajp.amjpathol.org), confirming CK19 as marker for a highly malignant phenotype of HCC and corroborating findings described above for nuclear β-catenin. In contrast to nuclear β-catenin, CK19 showed no correlation with tumor grading, vessel invasion, or ROD but was significantly associated with lymph node or distal metastases (see Supplemental Table 3 at http://ajp.amjpathol.org). Therefore, the hyperproliferation of unorganized, solitary CK19-positive cells in the stroma close to the tumor border may suggest an invasion of malignant hepatocytes, which depends on the microenvironment (see Supplemental Figure S6; middle and lower panels, at http://ajp.amjpathol.org). In summary, these data suggest that CK19 associates with nuclear β-catenin activation and in addition, correlates with metastases.
Discussion
The role of nuclear β-catenin in the development, maintenance, and differentiation of normal and malignant liver progenitor cells is still poorly understood. The relevance and value of activated β-catenin for the prognosis of human HCC remains an open issue, since in-depth analysis is missing. In the present study we show that accumulation of nuclear β-catenin depends on TGF-β signaling in experimental murine liver tumors, resulting in the loss of epithelial markers and increased expression of hepatic progenitor markers. On the contrary, disruption of signaling along the TGF-β or Wnt/β-catenin pathways favors terminal differentiation of liver carcinoma cells. These mouse data were mirrored by a collection of human HCC samples, in which high nuclear β-catenin levels significantly associated with dedifferentiation, increased vessel invasion and recurrence of HCC after OLT.
To date, the most important parameter to select patients for OLT is still the size and number of tumors, since the Milan criteria defined in 1996 demand that only HCC patients with one or three nodules measuring 5 or up to 3 cm in diameter, respectively, are recommended for OLT.32 However, these Milan criteria are a continuing matter of debate, particularly as new clinicopathological and prognostic markers allowing a better selection for OLT have been defined, giving also those patients an access to transplantation who exceed the Milan criteria.6 CK19, a marker of both cholangiocytes (bile duct epithelial cells) and oval cells (bipotential hepatic progenitors) is an important example, since its enhanced expression correlated with a higher recurrence rate after OLT.7 Furthermore, Fiorentino et al33 proposed that a combination of low membranous E-cadherin, high proliferative MIB-1 index and nuclear β-catenin accumulation correlated with elevated relapse of HCC after OLT. However, only six of 83 patients with high nuclear β-catenin were reported in this study, a much lower percentage than usually observed.10 Our own studies found 58.6% of 133 HCC patient samples with high or medium nuclear β-catenin expression, which is in good agreement with previous investigations.10 Our collection of patient samples allowed us to demonstrate a close correlation of nuclear β-catenin accumulation and tumor recurrence after OLT, suggesting that high levels of nuclear β-catenin-positive tumor cells could provide an excellent prognostic marker for the relapse of HCC.
Importantly, the patient samples used in this study were assembled during 1982 and 2002. Therefore a large percentage of these samples from patients with OLT were collected before the introduction of Milan criteria in 199632 offering the unique chance to include various parameters of tumor progression in our analysis. Indeed, nuclear β-catenin was significantly associated with vascular invasion in advanced human HCC. This observation is in agreement we previous studies showing that nuclear β-catenin is associated with vascular invasion and frequently occurs in tumor cells of poorly differentiated HCC.10,30,34,35
Noteworthy, murine axin-overexpressing (MIM-R-Axin) tumor cells lacking nuclear β-catenin24 showed markers of differentiated hepatocytes such as CK8 and CK18 (Figure 3). Accordingly, inactivation of c-myc, which represents a target gene of Wnt/β-catenin signaling, led to the differentiation of HCC and expression of CK8.36,37 Furthermore, high levels of nuclear β-catenin were detected at the invasive front in colon cancer patients, suggesting that these nuclear β-catenin-positive cells are prone to metastasize.38 From these data we conclude that nuclear β-catenin, and most conceivably, its downstream target c-myc might be critically involved in the dedifferentiation and dissemination of malignant hepatocytes during tumor progression and metastasis. The expression of CK19, which correlates with nuclear β-catenin and was even detected in collectively and individually invading cells into the stroma, supports this hypothesis (see Supplemental Figure S6 at http://ajp.amjpathol.org).
TGF-β-dependent EMT has increasingly been recognized as an important mechanism of tumor progression by mediating enhanced migration, invasion and intravasation of various carcinomas such as those induced by oncogene-transformed breast or liver epithelial cells.39 Accordingly, up-regulation of Twist causing down-regulation of E-cadherin and EMT induction in human HCC, associated with elevated vascularization and metastasis.40 Furthermore, Mani et al41 described a connection between EMT and the occurrence of CSCs, since a CD44high/CD24low cell population could be linked to breast cancer stemness.41–43 In fact, various expression patterns of markers have been associated with CSCs, however, marker combinations unambiguously defining one specific CSC population are still missing. In liver cancer, CD90+/CD45− cells were designated as CSCs and found in tumors as well as blood samples of HCC patients.44 Furthermore, bipotent liver progenitors (oval cells) show expression of M2-PK, OV6 (mouse A6) as well as coexpression of CK18 and CK19, the latter typical for hepatocytes and cholangiocytes, respectively.45–48 Interestingly, our study revealed that hepatocytes from MIM-R-ABC-derived mouse tumors exhibited a high proportion of nuclear β-catenin-positive cells expressing M2-PK and CK19. This suggested that malignant hepatocytes can adopt an immature liver progenitor-like phenotype during progression, resembling neoplastic oval cells (Figure 4). Accordingly, tumors generated from MIM-R-ABC hepatocytes showed a duct-like morphology similarly to cholangiocarcinomas (see Supplemental Figures S1, S3, and S4 at http://ajp.amjpathol.org). In contrast, tumors lacking nuclear β-catenin displayed a differentiated HCC phenotype as shown by their expression of the mature hepatocyte markers CK8 and CK18.
Taken together, these data provide strong evidence that activated nuclear β-catenin is both necessary and sufficient for the malignant cell fate decision, since lack of nuclear β-catenin caused differentiation into mature hepatocytes while its presence induced retrodifferentiation into immature hepatocyte progenitors. It needs to be mentioned that the murine MIM hepatocytes used in this study were isolated from p19ARF knockout mice and transformed with oncogenic Ras. Although an influence of p19ARF (p14ARF in human) deficiency on hepatocyte differentiation cannot be entirely excluded, nontransformed p19ARF−/− behave like normal hepatocytes in every aspect tested.27 Importantly, there is no bias in tumorigenicity since hepatocytes immortalized via transgenic expression of the cytoplasmic domain of the Met receptor (termed MMH cells) execute tumor progression and EMT as observed in p19ARF null MIM hepatocytes.18,26 Since up to 20% of human HCC cases show p14ARF inactivation,49 a putative interplay of p14ARF and nuclear β-catenin in human HCC needs to be addressed in the future.
Tumor dormancy, eg, due to resting carcinoma cells in the bone marrow, is a major issue of debate, addressing the prominent influence of the microenvironment on cell fate.50 Accordingly, Brabletz et al38 showed that cells with nuclear β-catenin accumulation prevailed at the invasive front of colon cancers while being essentially absent in the tumor center. Likewise, TGF-β and/or Wnt ligands secreted by the tumor-stroma and activating nuclear β-catenin accumulation, may mediate cell plasticity and EMT in HCC. Accordingly, Wnt5a and Wnt11 were remarkably elevated in malignant murine hepatocytes that underwent EMT in response to TGF-β in cell culture (data not shown). In addition, hepatic myofibroblasts vastly contribute to tumor progression by secreting TGF-β and by causing accumulation of nuclear β-catenin in malignant hepatocytes at the invasive front.26,51 Although hepatic myofibroblasts abundant in fibrotic and cirrhotic livers most likely produce these cytokines, we could not find a correlation between cirrhosis and nuclear β-catenin-positive HCC (Table 2). Thus, other mechanisms, eg, direct involvement of inflammatory cells, might critically contribute to the cytokine and chemokine production that triggers dedifferentiation and EMT.
In summary, our data demonstrate the relationship between markers of immature hepatic progenitors, ie, CK19 and M2-PK, and nuclear β-catenin accumulation after EMT in murine liver tumors. In mouse, nuclear β-catenin accumulation was strongly dependent on TGF-β signaling, which remains to be confirmed in human HCC. Importantly, our results suggest that circulating hepatic CSCs that express activated nuclear β-catenin may repopulate the new organ after OLT, leading to HCC recurrence. Further investigations of hepatic CSCs and their response to microenvironmental signals may uncover further markers allowing to better select patients for OLT and limit the problem of relapsing disease.
Acknowledgements
We thank Prof. Lukas Kenner for critical reviewing of immunohistochemical data.
Footnotes
Supported by the Austrian Science Fund, FWF, grant P19598-B13 (to W.M.), and the Austrian Science Fund, FWF, Special Research Program SFB F28 (to H.B. and W.M.).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Web Extra Material
Baseline characteristics of 133 HCC patients
Localization of E-cadherin in human HCC
CK19 associates with nuclear β-catenin accumulation in human HCC
References
- 1.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 2.Friedman SL. Mechanisms of disease: mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1:98–105. doi: 10.1038/ncpgasthep0055. [DOI] [PubMed] [Google Scholar]
- 3.Sangiovanni A, Del Ninno E, Fasani P, De Fazio C, Ronchi G, Romeo R, Morabito A, De Franchis R, Colombo M. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology. 2004;126:1005–1014. doi: 10.1053/j.gastro.2003.12.049. [DOI] [PubMed] [Google Scholar]
- 4.El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology. 2008;134:1752–1763. doi: 10.1053/j.gastro.2008.02.090. [DOI] [PubMed] [Google Scholar]
- 5.Freeman RB., Jr Transplantation for hepatocellular carcinoma: The Milan criteria and beyond. Liver Transpl. 2006;12:S8–S13. doi: 10.1002/lt.20936. [DOI] [PubMed] [Google Scholar]
- 6.Ishizaki Y, Kawasaki S. The evolution of liver transplantation for hepatocellular carcinoma (past, present, and future) J Gastroenterol. 2008;43:18–26. doi: 10.1007/s00535-007-2141-x. [DOI] [PubMed] [Google Scholar]
- 7.Durnez A, Verslype C, Nevens F, Fevery J, Aerts R, Pirenne J, Lesaffre E, Libbrecht L, Desmet V, Roskams T. The clinicopathological and prognostic relevance of cytokeratin 7 and 19 expression in hepatocellular carcinoma. A possible progenitor cell origin. Histopathology. 2006;49:138–151. doi: 10.1111/j.1365-2559.2006.02468.x. [DOI] [PubMed] [Google Scholar]
- 8.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 9.Tannapfel A, Busse C, Weinans L, Benicke M, Katalinic A, Geissler F, Hauss J, Wittekind C. INK4a-ARF alterations and p53 mutations in hepatocellular carcinomas. Oncogene. 2001;20:7104–7109. doi: 10.1038/sj.onc.1204902. [DOI] [PubMed] [Google Scholar]
- 10.Lee HC, Kim M, Wands JR. Wnt/Frizzled signaling in hepatocellular carcinoma. Front Biosci. 2006;11:1901–1915. doi: 10.2741/1933. [DOI] [PubMed] [Google Scholar]
- 11.Rossmanith W, Schulte-Hermann R. Biology of transforming growth factor beta in hepatocarcinogenesis. Microsc Res Tech. 2001;52:430–436. doi: 10.1002/1097-0029(20010215)52:4<430::AID-JEMT1028>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 12.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan X, Yuan Y, Zeng G, Apte U, Thompson MD, Cieply B, Stolz DB, Michalopoulos GK, Kaestner KH, Monga SP. Beta-catenin deletion in hepatoblasts disrupts hepatic morphogenesis and survival during mouse development. Hepatology. 2008;47:1667–1679. doi: 10.1002/hep.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 15.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells-an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 16.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 17.Fischer AN, Herrera B, Mikula M, Proell V, Fuchs E, Gotzmann J, Schulte-Hermann R, Beug H, Mikulits W. Integration of Ras subeffector signaling in TGF-{beta} mediated late stage hepatocarcinogenesis. Carcinogenesis. 2005:931–942. doi: 10.1093/carcin/bgi043. [DOI] [PubMed] [Google Scholar]
- 18.Gotzmann J, Huber H, Thallinger C, Wolschek M, Jansen B, Schulte-Hermann R, Beug H, Mikulits W. Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-beta1 and Ha-Ras: steps towards invasiveness. J Cell Sci. 2002;115:1189–1202. doi: 10.1242/jcs.115.6.1189. [DOI] [PubMed] [Google Scholar]
- 19.Reiss M. Transforming growth factor-beta and cancer: a love-hate relationship? Oncol Res. 1997;9:447–457. [PubMed] [Google Scholar]
- 20.Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 with transforming growth factor-beta1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129:1375–1383. doi: 10.1053/j.gastro.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 21.Lee TK, Man K, Poon RT, Lo CM, Yuen AP, Ng IO, Ng KT, Leonard W, Fan ST. Signal transducers and activators of transcription 5b activation enhances hepatocellular carcinoma aggressiveness through induction of epithelial-mesenchymal transition. Cancer Res. 2006;66:9948–9956. doi: 10.1158/0008-5472.CAN-06-1092. [DOI] [PubMed] [Google Scholar]
- 22.Fuchs BC, Fujii T, Dorfman JD, Goodwin JM, Zhu AX, Lanuti M, Tanabe KK. Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res. 2008;68:2391–2399. doi: 10.1158/0008-5472.CAN-07-2460. [DOI] [PubMed] [Google Scholar]
- 23.Zhu AX, Stuart K, Blaszkowsky LS, Muzikansky A, Reitberg DP, Clark JW, Enzinger PC, Bhargava P, Meyerhardt JA, Horgan K, Fuchs CS, Ryan DP. Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer. 2007;110:581–589. doi: 10.1002/cncr.22829. [DOI] [PubMed] [Google Scholar]
- 24.Fischer AN, Fuchs E, Mikula M, Huber H, Beug H, Mikulits W. PDGF essentially links TGF-beta signaling to nuclear beta-catenin accumulation in hepatocellular carcinoma progression. Oncogene. 2007;26:3395–3405. doi: 10.1038/sj.onc.1210121. [DOI] [PubMed] [Google Scholar]
- 25.Mikula M, Proell V, Fischer AN, Mikulits W. Activated hepatic stellate cells induce tumor progression of neoplastic hepatocytes in a TGF-beta dependent fashion. J Cell Physiol. 2006;209:560–567. doi: 10.1002/jcp.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, Jechlinger M, Waerner T, Weith A, Beug H, Mikulits W. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene. 2006;25:3170–3185. doi: 10.1038/sj.onc.1209083. [DOI] [PubMed] [Google Scholar]
- 27.Mikula M, Fuchs E, Huber H, Beug H, Schulte-Hermann R, Mikulits W. Immortalized p19ARF null hepatocytes restore liver injury and generate hepatic progenitors after transplantation. Hepatology. 2004;39:628–634. doi: 10.1002/hep.20084. [DOI] [PubMed] [Google Scholar]
- 28.Sieghart W, Fuereder T, Schmid K, Cejka D, Werzowa J, Wrba F, Wang X, Gruber D, Rasoul-Rockenschaub S, Peck-Radosavljevic M, Wacheck V. Mammalian target of rapamycin pathway activity in hepatocellular carcinomas of patients undergoing liver transplantation. Transplantation. 2007;83:425–432. doi: 10.1097/01.tp.0000252780.42104.95. [DOI] [PubMed] [Google Scholar]
- 29.Lahsnig C, Mikula M, Petz M, Zulehner G, Schneller D, van Zijl F, Huber H, Csiszar A, Beug H, Mikulits W. ILEI requires oncogenic Ras for the epithelial to mesenchymal transition of hepatocytes and liver carcinoma progression. Oncogene. 2009;28:638–650. doi: 10.1038/onc.2008.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cieply B, Zeng G, Proverbs-Singh T, Geller DA, Monga SP. Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology. 2009;49:821–831. doi: 10.1002/hep.22695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou L, Rui JA, Wang SB, Chen SG, Qu Q, Chi TY, Wei X, Han K, Zhang N, Zhao HT. Outcomes and prognostic factors of cirrhotic patients with hepatocellular carcinoma after radical major hepatectomy. World J Surg. 2007;31:1782–1787. doi: 10.1007/s00268-007-9029-z. [DOI] [PubMed] [Google Scholar]
- 32.Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, Montalto F, Ammatuna M, Morabito A, Gennari L. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med. 1996;334:693–699. doi: 10.1056/NEJM199603143341104. [DOI] [PubMed] [Google Scholar]
- 33.Fiorentino M, Altimari A, Ravaioli M, Gruppioni E, Gabusi E, Corti B, Vivarelli M, Bringuier PP, Scoazec JY, Grigioni WF, D'Errico-Grigioni A. Predictive value of biological markers for hepatocellular carcinoma patients treated with orthotopic liver transplantation. Clin Cancer Res. 2004;10:1789–1795. doi: 10.1158/1078-0432.ccr-1149-3. [DOI] [PubMed] [Google Scholar]
- 34.Endo K, Ueda T, Ueyama J, Ohta T, Terada T. Immunoreactive E-cadherin, alpha-catenin, beta-catenin, and gamma-catenin proteins in hepatocellular carcinoma: relationships with tumor grade, clinicopathologic parameters, and patients' survival. Hum Pathol. 2000;31:558–565. doi: 10.1053/hp.2000.6683. [DOI] [PubMed] [Google Scholar]
- 35.Tien LT, Ito M, Nakao M, Niino D, Serik M, Nakashima M, Wen CY, Yatsuhashi H, Ishibashi H. Expression of beta-catenin in hepatocellular carcinoma. World J Gastroenterol. 2005;11:2398–2401. doi: 10.3748/wjg.v11.i16.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 37.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH, Felsher DW. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 38.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–158. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 39.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 40.Niu RF, Zhang L, Xi GM, Wei XY, Yang Y, Shi YR, Hao XS. Up-regulation of Twist induces angiogenesis and correlates with metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2007;26:385–394. [PubMed] [Google Scholar]
- 41.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 43.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 44.Yang ZF, Ngai P, Ho DW, Yu WC, Ng MN, Lau CK, Li ML, Tam KH, Lam CT, Poon RT, Fan ST. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology. 2008;47:919–928. doi: 10.1002/hep.22082. [DOI] [PubMed] [Google Scholar]
- 45.Oertel M, Shafritz DA. Stem cells, cell transplantation and liver repopulation. Biochim Biophys Acta. 2008;1782:61–74. doi: 10.1016/j.bbadis.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–3822. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 47.Apte U, Thompson MD, Cui S, Liu B, Cieply B, Monga SP. Wnt/beta-catenin signaling mediates oval cell response in rodents. Hepatology. 2008;47:288–295. doi: 10.1002/hep.21973. [DOI] [PubMed] [Google Scholar]
- 48.Parent R, Marion MJ, Furio L, Trepo C, Petit MA. Origin and characterization of a human bipotent liver progenitor cell line. Gastroenterology. 2004;126:1147–1156. doi: 10.1053/j.gastro.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 49.Anzola M, Cuevas N, Lopez-Martinez M, Saiz A, Burgos JJ, Martinez de Pancorboa M. P14ARF gene alterations in human hepatocellular carcinoma. Eur J Gastroenterol Hepatol. 2004;16:19–26. doi: 10.1097/00042737-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Alix-Panabieres C, Riethdorf S, Pantel K. Circulating tumor cells and bone marrow micrometastasis. Clin Cancer Res. 2008;14:5013–5021. doi: 10.1158/1078-0432.CCR-07-5125. [DOI] [PubMed] [Google Scholar]
- 51.van Zijl F, Mair M, Csiszar A, Schneller D, Zulehner G, Huber H, Eferl R, Beug H, Dolznig H, Mikulits W. Hepatic tumor-stroma crosstalk guides epithelial to mesenchymal transition at the tumor edge. Oncogene. 2009;28:4022–4033. doi: 10.1038/onc.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Baseline characteristics of 133 HCC patients
Localization of E-cadherin in human HCC
CK19 associates with nuclear β-catenin accumulation in human HCC