

Abstract
Specifically expressed at intercellular adherens junctions of endothelial cells, VE-cadherin is a receptor that exhibits particular self-association properties. Indeed, in vitro studies demonstrated that the extracellular part of VE-cadherin elaborates Ca++-dependent hexameric structures. We hypothesized that this assembly could be at the basis of a new cadherin-mediated cell-cell adhesion mechanism. To verify this assumption, we first demonstrated that VE-cadherin can elaborate hexamers at the cell surface of confluent endothelial cells. Second, mutations were introduced within the extracellular part of VE-cadherin to destabilize the hexamer. Following an in vitro screening, three mutants were selected, among which, one is able to elaborate only dimers. The selected mutations were expressed as C-terminal Green Fluorescent Protein fusions in CHO cells. Despite their capacity to elaborate nascent cell-cell contacts, the mutants seem to be rapidly degraded and or internalized. Altogether, our results suggest that the formation of VE-cadherin hexamers protects this receptor and might allow the elaboration of mature endothelial cell-cell junctions.
Keywords: Adherens Junctions; metabolism; Animals; Antigens, CD; chemistry; genetics; metabolism; CHO Cells; Cadherins; chemistry; genetics; metabolism; Calcium; metabolism; Cell Adhesion; physiology; Cell Communication; Cricetinae; Cricetulus; Cross-Linking Reagents; pharmacology; Cytoplasm; metabolism; Dimerization; Endothelium, Vascular; cytology; metabolism; Fluorescent Antibody Technique; Green Fluorescent Proteins; genetics; metabolism; Immunoprecipitation; Intercellular Junctions; metabolism; Microscopy, Fluorescence; Mutation; genetics; Umbilical Veins; cytology; metabolism
Keywords: adhesion, cell-cell junctions, endothelium, hexamer, VE-cadherin
Introduction
Cadherins constitute a superfamily of glycoproteins that play an essential role in the control of cell polarity and tissue morphology (1). They mediate, via their ectodomains, Ca++-dependent homophilic interactions allowing the adhesion of cells expressing the same cadherin type. Recently, Nollet et al. (2) divided the superfamily of cadherins into five subfamilies, which can be distinguished according to their domain composition, genomic organization and protein sequences. Among these subfamilies classical type I cadherins and atypical type II cadherins are frequently found in adherens junctions and share a common modular organisation. Indeed, all the members of type I and II cadherins comprise five tandem extracellular modules designated EC1 to EC5, a transmembrane domain and a cytoplasmic tail. Mechanisms underlying the molecular structure of homophilic adhesive contacts between cadherins are controversial. According to the prevailing zipper model, cadherin molecules are thought to associate via both cis and trans dimers. It was proposed that lateral cis dimers are elaborated by molecules emerging from the same cell surface and require the insertion of tryptophan 2 from one molecule into a hydrophobic pocket on the adjacent molecule. These cis dimers are required for the elaboration of a second dimer interface that allows the anti-parallel alignment of cadherin molecules protruding from opposed cell surfaces (3). The participation of the tripeptide HAV within the N-terminal extracellular module EC1 of class I cadherins in the elaboration of these trans dimers is possible but not definitively assessed. Furthermore, the zipper model was recently contested by Boggon et al. (4) who established the tri-dimensional structure of the entire ectodomain of C-cadherin. In this structure, the above mentioned trytophane 2-mediated interface is involved in the maintenance of two adjoining cadherin molecules arranged in an anti-parallel fashion. According to Boggon et al. (4) and to the zipper model, only the N-terminal extracellular module EC1 is involved in cadherin trans interactions. On the contrary, Chappuis-Flament et al. (5) and Sivasankar et al. (6) established that the EC1 domain interacts with the EC4 or EC5 domain, indicating that multiple cadherin domains participate in the homophilic binding of type I cadherins.
To generate strong cell-cell adhesion, homophilic and homotypic interactions between the cadherin ectodomains must be strengthened by intracellular interactions involving the cytoplasmic tail of cadherins and allowing the attachment of cadherins to the cytoskeleton. This cytoplasmic domain exhibits highly conserved regions able to bind several common intracellular partners. Indeed, the C-terminus of the cytoplasmic domain of cadherins binds, in a mutually exclusive fashion, β-catenin or plakoglobin whereas the juxtamembrane domain binds the other catenin protein p120 (7). The anchorage of cadherins to the actin cytoskeleton is indispensable for the development of strong cell-cell adhesion (8). This connection is mediated by α-catenin which interacts, on one hand, with cadherin–bound β- or γ-catenins and, on the other, with the actin cytoskeleton, either directly or indirectly through linker proteins such as vinculin or α-actinin. Despite their physiological importance, little is known about the mechanisms regulating the dynamic rearrangement of the cadherin-catenin complex. Nevertheless, it was recently established that cadherins activate Rho family members (9) which, in turn, mediate the dynamic rearrangement of the actin cytoskeleton (10) and participate in the regulation of cadherin-mediated adhesion. Different cadherin-catenin complex interactions also constitute points of potential regulation for cadherin function (11,12). Catenin p120 emerges as a key regulator of cadherin function but its role remains elusive (13). Indeed, defective binding of p120 to the cytoplasmic tail of cadherins prevents the establishment of stable adhesive cell-cell contacts (14). More recent results established that p120 plays a critical role in the control of cadherin plasma membrane levels (15,16,17). Altogether, in spite of the key role of catenins in junction assembly and vascular organization, further studies are needed to understand how endothelial cells use these cytoskeletal linkages and these different signaling pathways to response to angiogenic and inflammatory processes.
VE-cadherin is specifically expressed at the adherens junction of endothelial cells (18) and was demonstrated to play fundamental roles in microvascular permeability, angiogenesis and cardiovascular physiology (19,20,21). Like other members of the family, VE-cadherin mediates calcium-dependent homophilic adhesion. On the one hand, VE-cadherin presents several commun features with the other cadherin family members but on the other hand, because of its endothelial cell selective expression, VE-cadherin seems to be a part of specific signaling pathways. The function and physiological role of VE-cadherin must be in particular adapted to the vascular endothelium in which it is expressed. Although VE-cadherin is classified as a type II cadherin, it shares week homology with the other type II cadherins. Moreover, the VE-cadherin amino acid sequence exhibits only 23% identity with that of classical type I cadherins. Type I cadherins have the HAV amino acid sequence in the cell adhesion recognition (CAR) sequence localized in the EC1 repeat, previously discussed for being involved in cell-cell adhesion. Interestingly, VE-cadherin lacks the HAV tripeptide and seems to have specific residues surrounding its CAR site. From these observations and although VE-cadherin possesses the typical cadherin modular organisation, this receptor may be considered as a particular cadherin with different functional and structural properties (22). Its peculiar characteristics are also reflected by its unique self-association properties. Although recent studies suggest that VE-cadherin homophilic binding occurs in a similar manner to that of classical cadherins, the mechanism is controversial and not well defined. Based on in vitro studies, we have recently demonstrated that the VE-cadherin ectodomain self-assembles into a Ca++-dependent hexamer (23). Multiple interactions, mainly involving the inter-module regions EC1-EC2 and EC3-EC4, promote the formation of a completely interdigitated hexameric configuration (24). This spatial organisation supports the novel mechanism for homophilic interactions of cadherins proposed by Zhu et al. (25), in which opposed cadherin ectodomain dimers are completely interdigitated to adhere in an antiparallel way.
In order to assess the role of the hexameric VE-cadherin assembly at the cell surface, we elaborated mutants unable to hexamerize. An in vitro selection, based on both the ability of the resulting mutated fragments to correctly fold and to loose the capacity to oligomerize as hexamers, identified three particular mutants, among which one oligomerizes as a dimer. The selected mutations were then expressed as C-terminally fused VE-cadherin-GFP constructs in CHO cells. In this study we show that the hexameric self assembly seems to play a role in the prevention of VE-cadherin internalization and/or degradation.
Material and Methods
Construction of E. coli expression vectors
The mutated fragments VE-EC1-4Mi (i from 1 to 4) were elaborated from the pET expression vector containing the DNA sequence coding for VE-EC1-4 (amino acids 1-431) fused to an N-terminal methionine (23). The QuickChange site-directed mutagenesis kit (Stratagene Europe, Amsterdam, The Netherlands) was used to introduce amino acid substitutions.
Construction of mammalian expression vectors
The VE-Cad-GFP variants were elaborated from the pGFP expression vector containing the DNA sequence coding for VE-cadherin by using the Quick-Change site-directed mutagenesis kit (Stratagene). The resulting proteins VE-CadM1-GFP, VE-CadM2-GFP, VE-CadM3-GFP and VE-CadM4-GFP possessed the single E321/D, the double N98/A; D99/A, the triple D96/A; N98/A; D99/A and the double D320/A; E321/A amino acid substitutions, respectively, within the VE-cadherin ectodomain. The coding sequence of these VE-cadherin-GFP-derived constructs was verified by sequence analysis.
Production, purification and trypsin digestion of the mutated VE-EC1-4 fragments expressed in E. coli
The VE-EC1-4 mutants were produced and purified as previously described for the respective wild-type fragment (23). Proteolysis experiments were performed as previously described (24).
Cell culture and DNA transfection
Subconfluent CHO cells were transiently transfected using Exgen 500 (Euromedex, Mundolsheim, France) in serum-free Dulbecco’s modified Eagle’s medium (DMEM) (Life technologies, Cergy Pontoise, France) according to the supplier’s instructions. 3 hours later, the Exgen-containing medium was replaced by serum-free DMEM. Cells were then serum-starved for at least 10 hours in order to synchronize them in G1/G0 (26). After this starvation treatment, the transfected cells were grown in 10% bovine calf serum-containing DMEM. Immunoprecipitation and immunofluorescence experiments were performed at different times after serum-starvation arrest.
The EaHy926 cell line (referred to as EaHy throughout this manuscript) was a generous gift from C-J. S. Edgell (University of North Carolina, Chapel Hill, C., USA) (27).
Antibodies
The polyclonal Anti-Cad3 antibody, produced and purified as previously described (19), interacts with the extracellular part of VE-cadherin. The polyclonal Anti-GFP antibody was from Santa Cruz Biotechnology (California, USA) and the monoclonal antibodies directed against p120 and β-catenin were from Transduction Laboratories (Lexington Kentucky, USA). For immunofluorescence studies, the Alexa 350 and CY3 (Jackson Laboratory, Maine, USA) conjugated-antibodies were also used.
Analytical gel filtration chromatography
Analytical gel filtration experiments were performed on the various mutated fragments immediately following the concentration step or after a delay of 24 hours as previously described (23).
Cross-linking of the soluble VE-EC1-4M3 fragment
The recombinant fragment VE-EC1-4M3 (17 μM) was cross-linked with EDC [N-ethyl-3-(3-dimethylaminopropyl) carbodiimid from Sigma] at 20°C in MES [2-(N-morpholino) ethane sulfonic acid] buffer containing 5 mM Ca++. The molar ratio between the recombinant fragment and the cross-linker reagent was adjusted by varying the concentrations of the cross-linking reagent from 0 to 640 mM. The cross-linked fragments were analyzed by electrophoresis on 5–20 % gradient gels.
Cross-linking at the surface of both EaHy cells and transfected CHO cells
Cell surface cross-linking of confluent EaHy was performed with DTSSP. First, cell monolayers were washed with PBS then incubated with DTSSP and diluted in PBS containing an antiprotease cocktail (1 μg/ml leupeptin, 1 μg/ml aprotinin and one tablet of EDTA-free Complete, Amersham) for 80 min at 37°C. The cross-linking reaction was terminated by adding 1 M Tris, pH 7 and cooling the reaction on ice. Following washing, cell lysis was performed according to a protocol previously described (19). The cell extracts were then centrifuged at 13 000 × g for 20 min at 4°C. The supernatants were immediately analyzed by electrophoresis and Western blot.
Cross-linking of CHO cells transfected by VE-CadM3-GFP was also performed with DTSSP at three hours post serum starvation.
Immunofluorescence analysis and fluorescence microscopy
At different times after serum-starvation arrest, transfected CHO cells were fixed for 20 min at room temperature in PBS containing 3% (wt/vol) paraformaldehyde and permeabilized for 3 min at the same temperature in PBS containing 3% paraformaldehyde and 0.5% Triton X-100. Cells were sequentially incubated with different primary and secondary antibodies with extensive PBS washes after each antibody incubation. Preparations were then mounted using Mowiol (Sigma-Aldrich). The immunostained cells were examined after an overnight incubation, in the dark, at room temperature, using fluorescence microscopy. They were observed with an inverted microscope (IX70, Olympus) using a Metamorph acquisition system (Universal Imaging) and a cooled couple-charged device camera (MicroMAX-1300YHX ; Roper Scientific). For living cell experiments, an Olympus 100× Plan Apochromat objective (Ph3, 1.3 NA) or an Olympus 60× Plan Apochromat objective (Ph3, 1.4 NA) were used.
Immunoprecipitation
For immunoprecipitation experiments, transfected cells were lysed with 2 ml buffer E (10mM Tris pH 7.4 containing 5mM CaCl2, 150mM NaCl, 1% Triton X100, 1% NP40, 1μg/ml leupeptin, 1μg/ml aprotinin, 1mM PMSF, one tablet of EDTA-free Complete) for 20 min on ice and then incubated with 115 μg of the anti-VE-cadherin Anti-Cad3 antibody for 2 hours at 4°C. Immunological complexes were captured on 50 μl Protein A-Sepharose beads (Sigma) for 1h at 4°C and then washed four times with buffer E. Bound proteins were eluted from the beads by boiling in 100 μl of β mercaptoethanol-containing SDS buffer for 10 min and electrophoresed on 5% wide-format gels (gel dimensions: 1.5 mm thick, 160 × 170 mm, electrophoresis instrument: PROTEAN II xi from Biorad, Ivry/Seine, France) to improve separation. Transfer of proteins from wide-format gels was performed using a blotting cell with adapted specifications (Trans blot cell, from Biorad). Separated proteins were electroblotted onto reinforced membranes (OPTITRAN BA-S8S, Schleicher and Schuell, Dassel, Germany) which were blocked for 1h with 3 % gelatin-containing PBS. Nitrocellulose membranes were successively incubated with different antibodies using stripping buffer according to the supplier’s instructions (ECL kit, Amersham).
Immunocapture assay
To detect VE-cadherin fragments released from CHO cells transfected with the VE- cadherin-GFP variants, an ELISA was performed using the polyclonal antibody Anti-Cad3 biotinylated or not biotinylated to capture and detect the fragments of VE-cadherin as previously described (28).
Results
Selection of mutation-sites
Recently, we demonstrated that the extracellular part of VE-cadherin self-associates as a Ca++-dependent hexamer (23). The amino acids involved in stabilization of modules EC1 and EC2 were clearly identified in the X-ray structure of the two N-terminal extracellular domains of murine E-cadherin (29). These amino acids are conserved among cadherins as attested by the alignment of the extracellular parts of murine E- and human VE-cadherins (Figure 1A). They constitute the motif DXND which is found at EC1-EC2, EC2-EC3 and EC4-EC5 inter-module regions and is substituted by DXN/DE at the EC3-EC4 inter-module region in E- and VE-cadherin (Figure 1A).
Fig 1. Mutated VE-EC1-4 fragments.
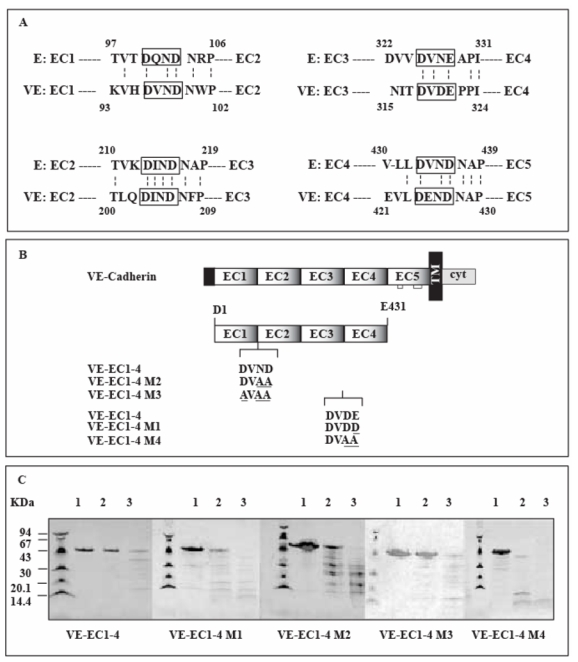
The extracellular region of cadherins consists of five homologous domains designated EC1 to EC5.
Panel A: Alignment of the extracellular parts of murine E- and human VE-cadherins
The four amino acid sequences linking the EC domains for mouse E. and human VE- cadherins are aligned. The conserved structural motifs between adjacent EC domains are boxed. Dotted vertical bars join amino acids conserved in both cadherin sequences. The numbers refer to the amino acids of E- and VE-cadherin according to the published sequences (GenBank P09803 and P33151 for mouse E- and human VE-cadherin, respectively)
Panel B : VE-cadherin mutants expressed in E. coli
VE-cadherin and its derived fragment VE-EC1-4 are schematically represented at the top. Mutations affecting conserved structural motifs, localized at the VE-EC1-4 inter-module sequences, are underlined. For comparison, the wild-type motifs from VE-cadherin are also mentionned. The name of each mutant is given at the left margin.
Panel C : Trypsin digestion of VE-cadherin mutants
Mutated VE-EC1-4 fragments were digested during 20 min at 22°C by trypsin using enzyme/fragment ratios of 0 (lane 1), 1.7 (lane 2), 13.6 (lane 3) U/μM. Molecular weight markers are at the left side of each gel. Trypsin-digested fragments were run through 8-25 % gels (Phast System, Amersham-Pharmacia). Following migration, proteins were detected by Coomassie blue staining.
In a previous study, we demonstrated that the four module-containing fragment VE-EC1-4 and the five module-containing fragment VE-EC1-5 possess the capacity to self-associate as hexamers (23). By contrast, fragments comprising one, two or three of the five extracellular modules of VE-cadherin do not associate or only associate as dimers (24). We also showed that the EC1-EC2 and the EC3-EC4 inter-module regions are critical for establishing VE-cadherin-mediated homophilic interactions. Consequently we attempted to destabilize the hexameric association by preferentially mutating amino acids within the motif DXN/DD/E in the EC1-EC2 and EC3-EC4 linker regions of the bacterially-expressed fragment VE-EC1-4. As shown in Figure 1B, the VE-cadherin DXN/DD/E motifs were altered by exchanging the asparagine and/or the negatively charged aspartic and glutamic acids with the uncharged amino acid alanine. These substitutions resulted in motifs with the structures DXAA or AXAA in the EC1-EC2 linker region of VE-EC1-4. Concerning the EC3-EC4 linker region, within the DXDE motif, the aspartic acid in position 320 was substituted by an alanine whereas the glutamic acid in position 321 was mutated to either an aspartic acid or an alanine.
Proteolytic experiments indicate that similar to wild type fragment, mutants M1, M2 and M3 are relatively resistant to trypsin digestion (Figure 1C). These results indicate that these fragments are correctly folded. In contrast to the mutants M1, M2 and M3, a fourth mutant, designated as VE-EC1-4 M4 appears to be almost completely digested by very low amounts of trypsin, probably because of a destabilization of its overall tri-dimensional structure (Figure 1C).
Oligomeric states of the mutated VE-EC1-4 fragments
The mutated VE-cadherin proteins, which have mutations in different calcium binding sites, now may have a much lower calcium binding capability. As it is known that cadherins need calcium to oligomerize and form cell-cell contacts, we verified whether amino acid substitutions described above prevent the oligomeric assembly of the VE-EC1-4 mutants by analyzing the capacity of the mutated fragments to self-associate by gel filtration chromatography. As illustrated in figure 2, the chromatographic profiles differ according to the mutations introduced. At 17 μM, wild-type VE-EC1-4 exhibited two peaks which corresponded to its monomeric (peak III) and hexameric forms (peak I) as previously described (23) (Figure 2A, left). The mutated fragment VE-EC1-4M1 also presented two peaks which possessed identical elution volumes to those observed for wild-type VE-EC1-4. This indicated that this mutant also self-associates as a hexamer. The shoulder of peak III in the chromatogram of mutant M1 might correspond to a monomer degradation product. The monomer/multimer composition at equilibrium can be estimated from the areas of peaks I and III of the chromatographic profiles. Using this method, it could be deduced from the chromatograms of Figure 2A that, at 17 μM, 91 % of VE-EC1-4 but only 40% of VE-EC1-4M1 are hexameric. With higher fragment concentrations (56 μM), these percentages increase (97 % for VE-EC1-4 and of 61% for VE-EC1-4M1, Figure 2A, right). The concentrations C50% for which 50% of the fragments are in their hexameric form were estimated as 1 μM for VE-EC1-4 (23) and 35 μM for VE-EC1-4M1. Despite the error introduced by the slight degradation of fragment VE-EC1-4M1, results seem to indicate that the single E321D substitution strongly diminishes the affinity of the hexameric self-association of VE-EC1-4.
Fig 2. Oligomeric states of the mutated VE-EC1-4 fragments.
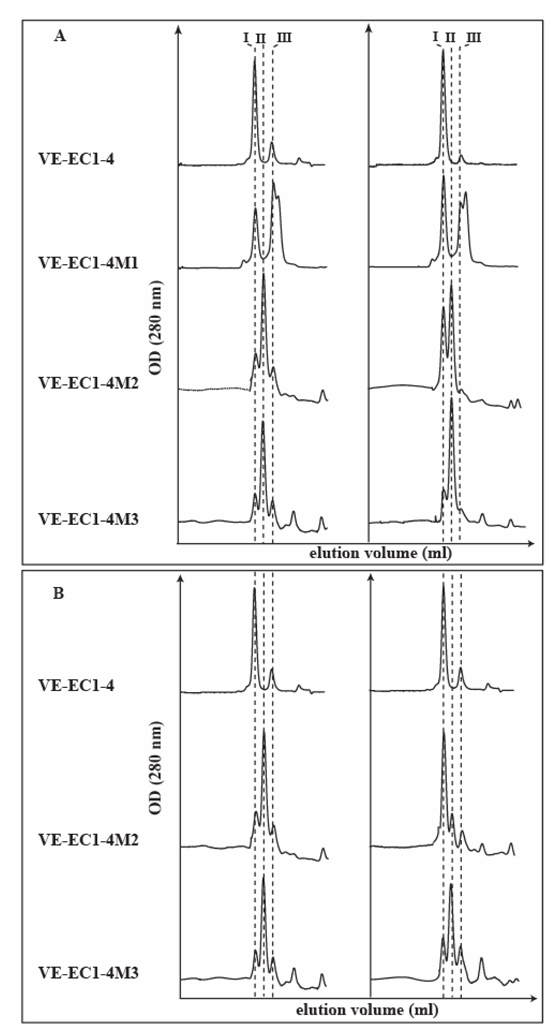
The oligomeric state of the fragments was studied by analytical gel filtration chromatography as previously described (Bibert et al. 2002). All the chromatographic runs were performed at 4°C.
Panel A : Impact of mutations on the molecular exclusion profiles of the VE-EC1-4 mutants.
FPLC molecular exclusion profiles for VE-EC1-4 mutants were obtained at initial loading concentrations of 17 μM (left) or 56 μM (right).
Panel B : Kinetic study of the mutant association
The different mutants were concentrated at 17 μM and immediately analyzed by gel filtration chromatography (left). Then, they were stored at room temperature during 24 hours before being analyzed again by gel filtration chromatography (right).
The impact of double and triple mutations in the EC1-EC2 inter module region on the self-associating capacity of the fragments was more drastic than that of the single substitution in the EC3-EC4 intermodule region. Indeed, at 17 μM, the chromatographic profiles for purified VE-EC1-4M2 and VE-EC1-4M3 exhibited one major (peak II) and two minor peaks (peaks I and III) indicating that these fragments possess three different oligomeric states (Figure 2A, left side). By comparing the chromatographic profiles of the wild-type and the mutated fragments, it could be deduced that the minor peaks I and III correspond to the hexameric and the monomeric states, respectively. The major peak II should correspond to an intermediate oligomeric form. From the elution volume of peaks II, it could be assumed that this intermediate species might be a dimer, based on the calibration of the column (24). Cross-linking experiments using the homobifunctional cross-linking reagent EDC confirmed this assumption (Figure 3). Analysis by electrophoresis of the cross-linked VE-EC1-4M3 revealed a two-band pattern with apparent molecular weights compatible with the monomeric and the dimeric forms of VE-EC1-4M3.
Fig 3. Determination of the oligomeric state of VE-EC1-4M3 by cross-linking experiments.
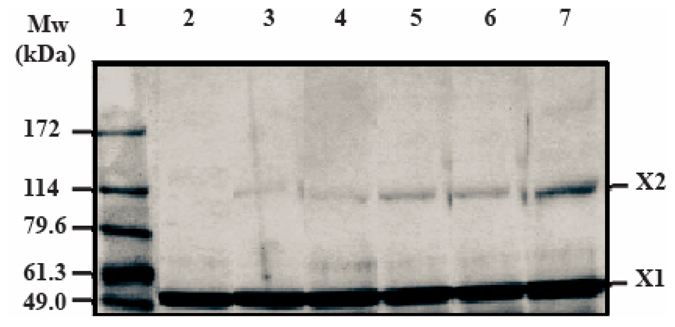
VE-EC1-4M3 (17 μM) was cross-linked using EDC at various concentrations: 0 mM (lane 2), 40 mM (lane 3), 80 mM (lane 4), 160 mM (lane 5), 320 mM (lane 6) and 640 mM (lane 7). Lane 1 : molecular weight markers. Cross-linked products were analyzed by SDS electrophoresis using a 5-20 % gradient gel. The oligomeric states of each cross-linked product are indicated on the right (X1: monomer; X2: dimer).
Increasing the fragment concentrations from 17 to 56 μM for the mutant VE-EC1-4M2 induced a significant increase of the percentage of hexamer (compare the corresponding chromatogram profiles at the left and right in Figure 2A). It could be deduced that an equilibrium exists between the dimeric and the hexameric forms of VE-EC1-4M2. By contrast, in similar conditions, the mutant VE-EC1-4M3 remains mainly dimeric.
Comparison of the gel filtration elution profiles obtained at 17 μM, immediately after the concentration step, and, after a delay of 24 hours at room temperature, showed an increase of the hexameric form for the mutant VE-EC1-4M2 (Figure 2B). Whereas the equilibrium between the monomeric and hexameric forms of wild-type VE-EC1-4 was rapidly established, the equilibrium existing between the three species of mutant VE-EC1-4M2 required several hours (Figure 2B). By contrast, the chromatographic profiles of mutant VE-EC1-4M3 changed only marginally with time. This indicated that addition of the mutation D96A to VE-EC1-4M2 generated a mutant, VE-EC1-4M3, which is trapped in a stabilized dimeric self- association (Figure 2B).
Altogether, the screening of the mutations allowed the characterization of three VE-EC1-4 mutants, which possess distinct oligomerization capacities. Whereas VE-EC1-4M1 self associates as a hexamer with a reduced affinity, VE-EC1-4M2 elaborates a dimeric form, at equilibrium with a hexameric form and VE-EC1-4M3 forms a stable dimer.
Expression of mutated VE-cadherin-GFP proteins at the CHO cell surface
The mutations described above were individually introduced in entire VE-cadherin proteins fused at their C-terminal part with Green Fluorescent Protein (VE-Cad-GFP) (Figure 4A). Three proteins were elaborated and designated as VE-CadM1-GFP, VE-CadM2-GFP, VE-CadM3-GFP (Figure 4A). For comparison, a fourth GFP-fused mutant, designated as VE-CadM4-GFP, was also generated by introducing the double substitutions in the EC3-EC4 inter region which were shown to disturb the stability of the recombinant fragment VE-EC1-4 in vitro (Figure 1C). Moreover, a mutated form of VE-cadherin lacking the cytoplasmic domain responsible for its interaction with β- and γ-catenins was also fused with GFP and named VE-Cadtr-GFP (30)(Figure 4A).
Fig 4. Expression of wild-type and mutated VE-cadherin-GFP variants in CHO cells.
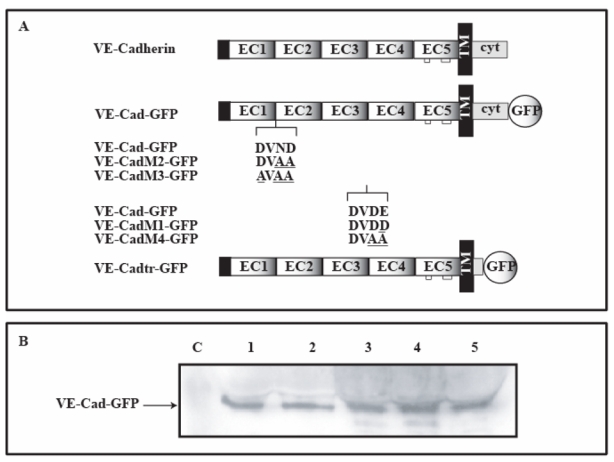
Panel A : Schematic representation of VE-cadherin-GFP proteins
Mutations described in Figure 1B were introduced into the entire VE-cadherin molecule fused at its C-terminus with GFP. Mutated amino acid residues are underlined. The names attributed to each protein construct are given at the left. VE-Cadtr-GFP possesses a truncated cytoplasmic domain unable to bind β- or γ-catenins.
Panel B: Immunoblot analysis of the cellular expression of the VE-cadherin-GFP constructs
CHO cells were transfected with cDNA constructs coding for the different VE-cadherin-GFP proteins. 3 hours after serum-starvation arrest, transfected CHO cells were lysed. Immunoblot analyses were then performed on cell lysates to verify expression of VE-Cad-GFP (lane 1), VE-Cadtr-GFP (lane 2), VE-CadM1-GFP (lane 3), VE-CadM2-GFP (lane 4), VE-CadM3-GFP (lane 5) using an anti-VE-cadherin antibody. No detectable level of endogenous VE- cadherin was observed in parental CHO cells (lane C).
CHO cells were transiently transfected with the expression vectors encoding the different VE-Cad-GFP variants. As illustrated in Figure 4B, Western blot analysis of total cell lysates, 3h post serum starvation arrest, by using an anti VE-cadherin antibody revealed a 130 kDa band for the wild-type molecule and the mutants VE-CadM1-GFP, VE-CadM2-GFP, VE-CadM3-GFP and VE-Cadtr-GFP whereas no band running 27 kDa lower on the gel, due to the absence of GFP fusion, was observed in untransfected CHO cells.
Oligomerization state of VE-cadherin at the surface of confluent endothelial cells
We then attempted to determine the oligomeric states of either the wild type or the mutated forms of VE-cadherin expressed at the cell surface.
First, the endothelial cell line EaHy expressing wild-type VE-cadherin (27) was used in cross-linking experiments using the membrane-impermeable cross-linker DTSSP. Western blot analysis of DTSSP-incubated cell lysates revealed the formation of a multiple band pattern (Figure 5A). Due to the high VE-cadherin oligomer molecular weight, bands are quite weak and smeared but we could observe a different pattern which varied with the concentration of the cross-linking reagent, as previously observed for the fragment VE-EC1-4 (23). In the presence of 0.05 or 0.2 mM DTSSP, four bands could be detected (Figure 5A, lanes 1 and 2). For DTSSP concentrations ranging from 0.8 to 3.2 mM, six bands were obtained (Figure 5A, lanes 4–6). Because of the high molecular weight of VE-cadherin, we can not totally exclude the presence of additional multimeric species which could not be vizualized in gel. However, this result indicates that at the surface of endothelial cells, VE-cadherin does not associate as dimers. Moreover, this could reflect the oligomeric association found in solution with the recombinant fragment VE-EC1-4. By attributing a degree of oligomerization to each band, it was possible to relate the theoretical molecular weight (Mwth) to the electrophoretic mobility. A linear curve was obtained by plotting log Mwth against the corresponding electrophoretic mobility of each imunoblotted band (Figure 5C). This suggested that the six discrete bands corresponded to multimers containing one, two, three, four, five or six covalently cross-linked molecules of VE-cadherin from the bottom to the top of the gel. In fact, the five lower bands could correspond to partially cross-linked products whereas the upper one could correspond to the hexameric association found at the cell surface. Altogether, due to the low resolution of the gel, we can conclude that at the cell surface VE-cadherin aggegation state is at least trimeric and could be hexameric.
Fig 5. Oligomeric state of VE-cadherin at the surface of cells expressing VE-cadherin.
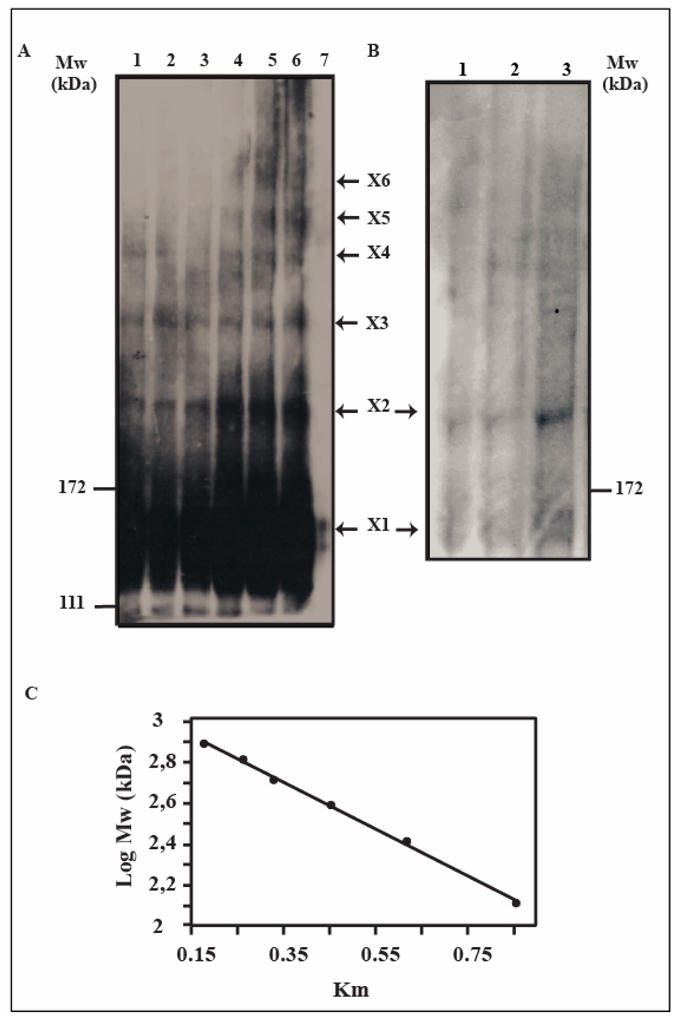
Panel A:Western blot analysis of EaHy cell surface-cross-linked VE-cadherin
EaHy cells were incubated with increasing concentrations of DTSSP (lanes 1: 0.05; 2: 0.2; 3: 0.4; 4: 0.8; 5: 1.6; 6: 3.2 mM of DTSSP). A control was obtained by incubating the cell monolayer with a dissociating Anti-VE-cadherin antibody before cross-linking with 3.2 mM of DTSSP (lane 7). Following cell lysis under non reducing conditions, lysates were electrophoresed on a 5% SDS polyacrylamide gel and subjected to Western blot analysis using a polyclonal antibody directed against EC3 VE-cadherin domain. The arrows at the right margin indicate the presence of VE-cadherin cross-linked products. The molecular weight of prestained marker proteins are indicated at the left.
Panel B: Western blot analysis of VE-cadherin cross-linked at the surface of VE-CadM3-GFP-transfected CHO cells.
VE-CadM3-GFP-transfected CHO cells were incubated with increasing concentrations of DTSSP (lanes 1: 0.8; 2: 1.6; 3: 3.2 mM of DTSSP). The lysates and the Western blot analysis were performed as previously described for EaHy cells (Panel A).
Panel C: Correlation between the migration coefficient and the molecular weight attributed to the immunoblotted bands.
Considering a 135 kDa molecular weight for VE-cadherin, the molecular weight of each electroblotted band was calculated from the hypothetical oligomeric state (X1: monomer, X2: dimer, X3: trimer, X4: tetramer, X5: pentamer, X6: hexamer). A linear correlation was then established between the migration coefficients Km and the molecular weight of the immunoblotted bands.
In a control experiment, the EaHy cell monolayer was cross-linked with DTSSP following a 2 hour-treatment with the Anti-VE cadherin antibody known to be able to disturb VE-cadherin-mediated homophilic interactions and cell-cell junctions (19). Following this treatment, only the band corresponding to the monomeric form of VE-cadherin was observed attesting that the upper bands effectively corresponded to multimeric forms of VE-cadherin (Figure 5A, lane 7). The faint intensity of this band may be due to the degradation of VE-cadherin once monomeric.
Secondly, to verify the oligomeric state of the mutant VE-CadM3-GFP at the cell surface, the corresponding transfected CHO cells were crosslinked using DTSSP, 3h post serum starvation arrest, the experimental conditions being identical to those used for the cell line EaHy (Figure 5B). Analysis by Western blot of the cell lysates revealed a single band. Based on the standard curve established previously connecting the migration coefficient and the VE-cadherin oligomer molecular weight, it can be concluded that this band corresponded to the dimeric form of VE-cadherin. This reflected the dimeric self-association of the recombinant fragment VE-EC1-4M3 found in vitro.
Expression of VE-cadherin-GFP variants at the surface of transfected CHO cells and recruitment of cytoplasmic partners
To verify whether VE-Cad-GFP proteins accumulated at cell-cell junctions, transfected cells were then imaged by GFP fluorescence (Figure 6A). At 3 h post serum-starvation arrest, the mutant proteins VE-CadM1-GFP, VE-CadM2-GFP, VE-CadM3-GFP and VE-Cadtr-GFP were mainly localized at cell-cell junctions similar to wild-type VE-Cad-GFP. This indicated that these mutated proteins were correctly transported to the cell surface. By contrast, as expected from our in vitro study, the mutant VE-CadM4-GFP was not able to be transported to the cell surface (data not shown). Numerous studies showed that cadherins, localized at the cell membrane, recruited, via their cytoplasmic tail, the intracellular partners p120 and β-catenins. To investigate whether the mutated VE-cadherin-GFP proteins, expressed at the CHO cell surface, were able to interact with catenins, junctional complexes were visualized at 3 h post serum-starvation arrest. Wild-type and mutated VE-cadherin-GFP molecules were observed using the intrinsic fluorescence of GFP whereas β-catenin and p120 were immuno-fluorescently detected (Figure 6A). For all mutants, both VE-cadherin-GFP and β-catenin colocalized at sites of cell-cell contacts, except for the mutant VE-Cadtr-GFP deleted for the β-catenin-binding domain. Similarly, wild type and mutated VE-cadherin-GFP and p120 exhibited an essentially identical distribution in areas of cell contact for all the variants studied.
Fig 6. Cell-cell contacts initiated by expressed VE-Cad-GFP variants in CHO cells.
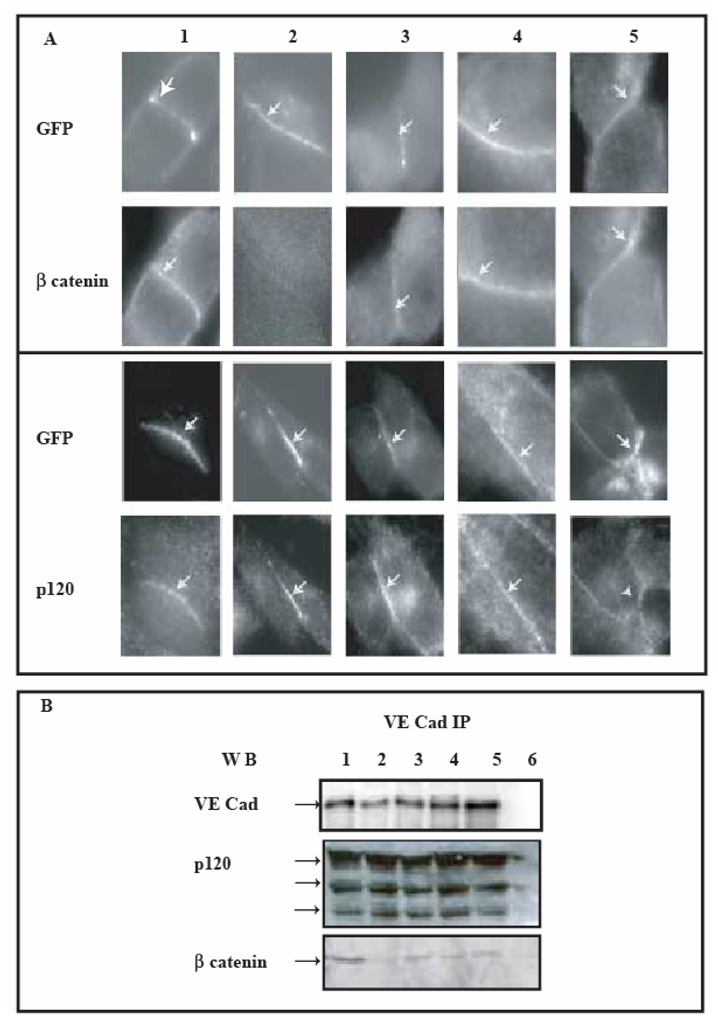
The ability of transfected cells to form a cadherin-based adhesion was assessed by immunofluorescence microscopy and immunoprecipitation methods. These experiments were performed 3h after serum-starvation arrest.
Panel A: Colocalization of VE-cadherin-GFP variants with G-catenin and p120 at intercellular junctions
VE-cadherin-derived proteins were observed using the intrinsic fluorescence of GFP whereas β-catenin and p120 were both immuno-fluorescently detected at the surface of transiently transfected CHO cells expressing VE-Cad-GFP: lane 1, VE-Cadtr-GFP: lane 2, VE-CadM1-GFP: lane 3, VE-CadM2-GFP: lane 4, VE-CadM3-GFP: lane 5. VE-cadherin-GFP variants and β-catenin or p120 colocalized, except for VE-Cadtr-GFP which failed to recruit β-catenin.
Panel B: Cytoplasmic partners of VE-cadherin-GFP variants
Lysates of CHO cells expressing VE-Cad-GFP (lane 1), VE-Cadtr-GFP (lane 2), VE-CadM1-GFP (lane 3), VE-CadM2-GFP (lane 4), VE-CadM3-GFP (lane 5) were immunoprecipitated with an Anti-VE-cadherin antibody. VE-cadherin immunoprecipitates were alternatively blotted with antibodies to either VE-cadherin, p120 and β-catenin. As a control, immunoprecipitation of untransfected CHO cell lysates was blotted in parallel (lane 6).
To directly determine whether VE-cadherin-GFP mutants were truly associated with catenins, VE-cadherin immunoprecipitates were blotted with antibodies to either β-catenin or p120 (Figure 6B). The results indicated that, at 3 h post serum-starvation arrest, the different VE-cadherin-GFP proteins were able to form complexes with the various isoforms of p120 expressed in CHO cells. In a similar way, they could associate with β-catenin, except for mutant VE-Cadtr-GFP.
It could be concluded that the formation of junctional complexes between cells was not disturbed by the mutations introduced within the extracellular domain of VE- cadherin and therefore does not depend on a hexamerically associated VE-cadherin.
Degradation and/or internalization of the VE-cadherin-GFP variants at the surface of transfected CHO cells
To study the fate of the mutated VE-cadherin-GFP variants, transfected CHO cells were observed 24h after serum-starvation arrest using the intrinsic fluorescence of GFP whereas VE-cadherin was immuno-fluorescently detected. (Figure 7A). After a 24h post-serum starvation arrest, cells expressing mutated VE-CadM1-GFP, VE-CadM2-GFP and VE-CadM3-GFP exhibited diffuse intracellular green fluorescence (Figure 7A, 3, 4 and 5). Moreover, immunofluorescent labeling of VE-cadherin showed only punctae near the nucleus. These results could possibly reflect an internalization of the mutated proteins (Figure 7A). By contrast, the wild type VE-Cad-GFP was concentrated at cell-cell junctions (Figure 7A, 1). Concerning the VE-Cadtr-GFP variant, in contrast to the three other VE-cadherin-GFP variants, this truncated mutant was able to react with the Anti-VE-cadherin antibody and was always concentrated at cell-cell junctions (Figure 7A, 2).
Fig 7. Impact of mutations on the stability of VE-cadherin-GFP variants.
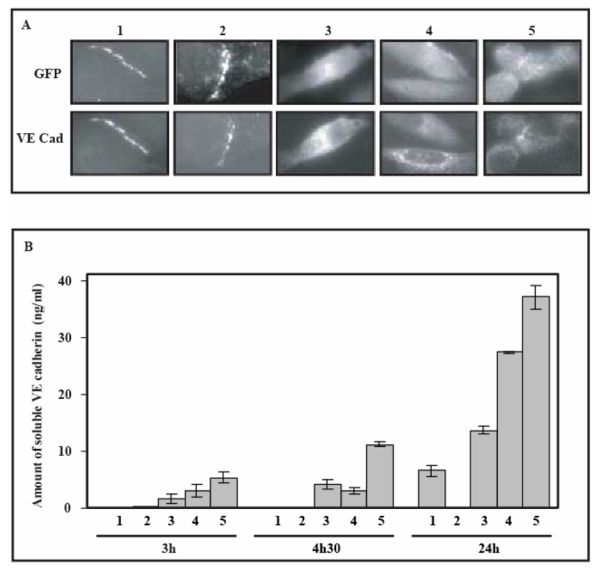
Panel A : Fluorescence microscopy of VE-cadherin mutants
24h post-serum starvation arrest, CHO cells expressing VE-Cad-GFP (1), VE-Cadtr-GFP (2), VE-CadM1-GFP ( 3), VE-CadM2-GFP ( 4), and VE-CadM3-GFP ( 5) were imaged for GFP fluorescence (top) and immunostained with an Anti-VE-cadherin antibody (bottom). Note that mutated VE-cadherin-GFP variants appeared as isolated cytoplasmic punctae excluded from intercellular contacts. By contrast, the wild-type and the truncated VE-cadherin-GFP co-localized at cell- cell junctions.
Panel B : Detection of cleavage products of the extracellular part of VE-cadherin mutants by immuno capture analysis
Culture supernatants from CHO cells expressing VE-Cad-GFP (1), VE-Cadtr-GFP ( 2 ), VE-CadM1-GFP ( 3), VE-CadM2-GFP ( 4 ), VE-CadM3-GFP ( 5 ) were collected at different times after the serum-starvation arrest. Their contents in soluble VE-cadherin fragments were quantified using an immunocapture assay.
Moreover, on the other hand, to test additional proteolytic events occurring within the extracellular part of VE-cadherin, we developed an ELISA allowing the detection of soluble VE-cadherin fragments in the cell culture supernatants (Figure 7B). Effectively, some soluble fragments were detected in the supernatants of CHO cells expressing the different VE-cadherin-GFP mutants. In fact, the time course of the VE- cadherin degradation varied according to the nature of the mutants. Indeed, at 3h post serum starvation arrest, the extracellular part of mutants was not really degraded as attested by the amount of soluble fragments close to the background. By contrast, at 4h 30, VE-CadM3-GFP underwent substantial cleavages on its extracellular part. Moreover, 24h after serum-starvation arrest, high levels of soluble mutant-derived fragments were detected in the cell culture supernatants for the three variants (Figure 7B). Concerning the wild type VE-cadherin and VE-Cadtr-GFP variant, no soluble fragment were detected by this immuno-capture assay (Figure 7B, lanes 1 and 2 respectively).
These results were in good agreement with microscopy observations made previously on cells expressing the various VE-CadM-GFP mutants (Compare Figure 6 and Figure 7A).
Discussion
Recently, we demonstrated that the extracellular domain of VE-cadherin self-associates as a Ca-++dependent hexamer in solution (24,23). It remains to be shown whether these hexameric associations exist in vivo at the surface of confluent endothelial cells and participate in the elaboration of adherens junctions between endothelial cells.
In the present manuscript, we first established that VE-cadherin does not self-associate as a dimer at the surface of endothelial cells. This result is in contradiction with those obtained by Ahrens et al. (31) who showed that self-assembly of VE-cadherin follows a mechanism common to classical cadherins and self-associates as a dimer. To establish this result, the authors used a fusion between the ectodomain of VE-cadherin and the N-terminus of the trimeric coiled-coil domain of cartilage matrix protein. Divergence between Ahrens’ results and ours may be explained by the presence of the trimeric fusion protein which may not mimic the spatial organization of VE-cadherin molecules found at the endothelial cell surface.
We then inserted several mutations within the extracellular domain of VE-cadherin in order to destabilize its hexameric association. To facilitate the screening of the mutations, they were first introduced within the recombinant fragment VE-EC1-4 overlapping the four N-terminal extracellular modules of VE-cadherin. This fragment has previously been demonstrated to be able to self-associate as a hexamer in solution (23). Among twenty four mutations, only three were resistant to limited trypsin digestion. Thus, the single mutation E321/D within the EC3-EC4 inter-module region, the double mutation N98/A ; D99/A and triple mutation D96/A ; N98/A ; D99/A within the EC1-EC2 intermodule region did not disturb the overall tri-dimensional structure of VE-EC1-4 as attested by limited proteolysis resistance. As expected, they markedly affected the capacity of the fragments to oligomerize. Whereas the mutation E321/D causes an approximative 35 fold drop of the affinity to elaborate a hexamer, the triple mutant formed predominantly a dimer which did not evolve into a hexamer even after a one week incubation period. By contrast, the double mutant which also forms a dimer evolves rapidly into a hexamer. Altogether, our results demonstrate that intact EC1-EC2 and EC3-EC4 inter-module regions are required for elaborating efficient hexameric VE-cadherin self-association in vitro. They also show the existence of a dimeric intermediate, implicating the EC3-EC4 inter-module region, which can evolve into a hexameric assembly due probably to additional interactions, involving other EC or inter- module region.
To establish the in vivo participation of the VE-cadherin hexameric self-association in the elaboration of endothelial cell-cell junctions, the in vitro selected mutations were introduced within the extracellular part of VE-cadherin, C-terminally fused with GFP. The GFP fused-wild-type and -mutated VE-cadherin molecules were then expressed in CHO cells. For comparison, a fourth variant, VE-Cadtr-GFP, possessing a cytoplasmic tail deleted for the β-catenin-binding domain was also generated (32).
In transiently-transfected CHO cells, 3h post serum starvation, wild-type VE-cadherin–GFP was predominantly found at intercellular contact sites as previously described for the untagged VE- cadherin (18). Moreover, the different GFP-variants underwent the internal cellular quality control process and were transported to the cell surface where their localization is indistinguishable from that of the wild-type. At 3h post serum starvation arrest, they are also able to establish interaction with different partners such as p120 and β-catenin. By contrast, the mutant VE-CadM4-GFP exhibiting mutations D320/A; E321/A was not expressed at the cell surface reflecting the negative effect of these mutations on the stability of its tri-dimensional structure.
Introduction of the triple mutation abrogate the ability of VE- cadherin to elaborate hexameric complexes both in vitro and at the surface of CHO cells. The resulting triple mutant self-associates as dimers as established by cross-linking experiments. The fact that this triple mutant seems able to elaborate cell-cell contacts indicates that these dimeric structures possess an anti-parallel organization. Thus, on endothelial cells, cell-surface hexamers might also evolve from dimers which allow cell to adhere due to their anti-parallel orientation.
The mutant VE-Cadtr-GFP which possesses an intact extracellular domain and is deleted of the β-catenin binding site is able to fix p120 on its juxtamembrane region. Nevertheless, similar to wild type VE-Cad-GFP, this mutant is stable at least 24 h post serum starvation arrest. This correlates with the results obtained by Yap et al. (33) on a C-cadherin mutant deleted of the β-catenin binding domain which retains the capacity of wild type C-cadherin to promote adhesion of C cadherin expressing CHO cells. In contrast to VE-Cadtr-GFP, the three other variants lose their junctional localization between 3h and 24h after the serum starvation arrest and might become internalized and/or degraded. This seems to indicate that VE-Cadherin mutants, which can not self-associate as wild type VE-cadherin due probably to modified calcium affinity, are unable to promote stable cell-cell junctions.
It is difficult to speculate on the behaviour of VE-Cadherin mutants. PEST sequences, defined as sequences enriched with Pro, Glu or Asp and Ser or Thr residues (34), located close to or overlapping the β-catenin-binding domain of cadherins, may become exposed at the surface of mutant VE-cadherin molecules whereas they might be masked by the hexameric association. These PEST sequences should then target VE-cadherin molecules to the degradation pathway. According to Xiao et al. (35), VE-cadherin can be naturally internalized and processed through the endosomal-lysosomal pathway. Further experiments are needed to determine whether mutant VE-Cadherin degradation could be prevented by treating cells with chloroquine in order to inhibit lysosomal degradation or with proteasome inhibitors. Nevertheless, in our experiments, the rate of disapearance of VE-cadherin cell surface expression is strongly enhanced for the GFP-mutants when compared to that of the GFP-wild type protein. It is tempting to speculate that the mechanism driving these degradations is connected indirectly to the oligomeric state of VE-cadherin at the cell surface. The loss of the hexameric assembly results in a rapid internalization and/or proteolysis of VE-cadherin. The fact that mutants VE-CadM1-GFP and VE-CadM2-GFP which may have partially kept the capacity of the wild-type receptor to form hexameric structures are less cleaved than mutant VE-CadM3-GFP suggests that the hexamer self-assembly protects VE-cadherin from degradation. Comparatively, the VE-Cadtr-GFP mutant which possesses an intact extracellular domain able to hexamerize is not cleaved. Altogether, the results suggest that this protection against degradation may be assured by the compactness of the hexameric self-association of VE-cadherin.
In conclusion, our results establish that VE-cadherin hexamerization seems to play a role only for the maintenance of mature intercellular junctions while initial cell-cell contacts can be made without hexamerized VE-cadherin. These nascent contacts seem to be unstable and cannot evolve into functional adhesive complexes such as those present in mature tissue, suggesting that hexamerization might protect VE-cadherin against degradation.
Acknowledgments
We would like to thank Pr. Kaethi Geering for helpful comments on the manuscript. This work was supported by grants from the Association pour la Recherche sur le Cancer (ARC n° 9463 and 4447) and from the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC). BH is a recipient of a fellowship from ARP (Association de Recherche sur la Polyarthrite).
References
- 1.Takeichi M. Cadherin cell adhesion receptors as amorphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 2.Nollet F, Kools P, van Roy F. Phylogenetic analysis of the cadherin superfamily allows identification of six major subfamilies besides several solitary members. J Mol Biol. 2000;299:551–572. doi: 10.1006/jmbi.2000.3777. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, Hendrickson WA. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- 4.Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 5.Chappuis-Flament S, Wong E, Hicks LD, Kay CM, Gumbiner BM. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. J Cell Biol. 2001;154:231–243. doi: 10.1083/jcb.200103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sivasankar S, Gumbiner B, Leckband D. Direct measurements of multiple adhesive alignments and unbinding trajectories between cadherin extracellular domains. Biophys J. 2001;80:1758–1768. doi: 10.1016/S0006-3495(01)76146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol. 1994;14:8333–8342. doi: 10.1128/mcb.14.12.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- 9.Nakagawa M, Fukata M, Yamaga M, Itoh N, Kaibuchi K. Recruitment and activation of Rac1 by the formation of E-cadherin-mediated cell-cell adhesion sites. J Cell Sci. 2001;114:1829–1838. doi: 10.1242/jcs.114.10.1829. [DOI] [PubMed] [Google Scholar]
- 10.Lambert M, Choquet D, Mege RM. Dynamics of ligand-induced Rac1-dependent anchoring of cadherins to the actin cytoskeleton. J Cell Biol. 2002;157:469–479. doi: 10.1083/jcb.200107104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meigs TE, Fedor-Chaiken M, Kaplan DD, Brackenbury R, Casey PJ. Galpha12 and Galpha13 negatively regulate the adhesive functions of cadherin. J Biol Chem. 2002;277:24594–245600. doi: 10.1074/jbc.M201984200. [DOI] [PubMed] [Google Scholar]
- 12.Kaplan DD, Meigs TE, Casey PJ. Distinct regions of the cadherin cytoplasmic domain are essential for functional interaction with Galpha12 and beta-catenin. J Biol Chem. 2001;276:44037–44043. doi: 10.1074/jbc.M106121200. [DOI] [PubMed] [Google Scholar]
- 13.Vincent PA, Xiao K, Buckley KM, Kowalczyk AP. VE-cadherin:adhesion at arm’s length. Am J Physiol Cell Physiol. 2004;286:C987–C997. doi: 10.1152/ajpcell.00522.2003. [DOI] [PubMed] [Google Scholar]
- 14.Ireton RC, Davis MA, van Hengel J, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, Gilbert B, van Roy F, Reynolds AB. A novel role for p120 catenin in E-cadherin function. J Cell Biol. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Kojima S, Borisy GG, Green KJ. p120 catenin associates with kinesin and facilitates the transport of cadherin-catenin complexes to intercellular junctions. J Cell Biol. 2003;163:547–557. doi: 10.1083/jcb.200305137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huber AH, Stewart DB, Laurents DV, Nelson WJ, Weis WI. The cadherin cytoplasmic domain is unstructured in the absence of beta catenin. A possible mechanism for regulating cadherin turnover. J Biol Chem. 2001;276:12301–12309. doi: 10.1074/jbc.M010377200. [DOI] [PubMed] [Google Scholar]
- 17.Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco LP, Dejana E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol. 1992;118:1511–1522. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gulino D, Delachanal E, Concord E, Genoux Y, Morand B, Valiron MO, Sulpice E, Scaife R, Alemany M, Vernet T. Alteration of endothelial cell monolayer integrity triggers resynthesis of vascular endothelium cadherin. J Biol Chem. 1998;273:29786–29793. doi: 10.1074/jbc.273.45.29786. [DOI] [PubMed] [Google Scholar]
- 20.Dejana E, Bazzoni G, Lampugnani MG. Vascular endothelial VE.-cadherin: only an intercellular glue? Exp Cell Res. 1999;252:13–19. doi: 10.1006/excr.1999.4601. [DOI] [PubMed] [Google Scholar]
- 21.Hordijk PL, Anthony E, Mul FP, Rientsma R, Oomen LC, Roos D. Vascular-endothelial-cadherin modulates endothelial monolayer permeability. J Cell Sci. 1999;112:1915–1923. doi: 10.1242/jcs.112.12.1915. [DOI] [PubMed] [Google Scholar]
- 22.Shimoyama Y, Tsujimoto G, Kitajima M, Natori M. Identification of three human type-II classic cadherins and frequent heterophilic interactions between different subclasses of type-II classic cadherins. Biochem J. 2000;349:159–167. doi: 10.1042/0264-6021:3490159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Legrand P, Bibert S, Jaquinod M, Ebel C, Hewat E, Vincent F, Vanbelle C, Concord E, Vernet T, Gulino D. Self-assembly of the vascular endothelial cadherin ectodomain in a Ca2+-dependent hexameric structure. J Biol Chem. 2001;276:3581–3588. doi: 10.1074/jbc.M002667200. [DOI] [PubMed] [Google Scholar]
- 24.Bibert S, Jaquinod M, Concord E, Ebel C, Hewat E, Vanbelle C, Legrand P, Weidenhaupt M, Vernet T, Gulino-Debrac D. Synergy between extracellular modules of vascular endothelial cadherin promotes homotypic hexameric interactions. J Biol Chem. 2002;277:12790–12801. doi: 10.1074/jbc.M111597200. [DOI] [PubMed] [Google Scholar]
- 25.Zhu B, Chappuis-Flament S, Wong E, Jensen IE, Gumbiner BM, Leckband D. Functional analysis of the structural basis of homophilic cadherin adhesion. Biophys J. 2003;84:4033–4042. doi: 10.1016/S0006-3495(03)75129-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A. 1974;71:1286–1290. doi: 10.1073/pnas.71.4.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, Gulino-Debrac D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem. 2003;278:14002–14012. doi: 10.1074/jbc.M300351200. [DOI] [PubMed] [Google Scholar]
- 29.Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- 30.Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E. The molecular organization of endothelial cell to cell junctions: differential association of plakoglobin, beta-catenin, and alpha-catenin with vascular endothelial cadherin VE-cadherin. J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahrens T, Lambert M, Pertz O, Sasaki T, Schulthess T, Mege RM, Timpl R, Engel J. Homoassociation of VE-cadherin follows a mechanism common to “classical” cadherins. J Mol Biol. 2003;325:733–742. doi: 10.1016/s0022-2836(02)01286-x. [DOI] [PubMed] [Google Scholar]
- 32.Navarro P, Caveda L, Breviario F, Mandoteanu I, Lampugnani MG, Dejana E. Catenin-dependent and –independent functions of vascular endothelial cadherin. J Biol Chem. 1995;270:30965–30972. doi: 10.1074/jbc.270.52.30965. [DOI] [PubMed] [Google Scholar]
- 33.Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- 35.Xiao K, Allison DF, Kottke MD, Summers S, Sorescu GP, Faundez V, Kowalczyk AP. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. J Biol Chem. 2003;278:19199–19208. doi: 10.1074/jbc.M211746200. [DOI] [PubMed] [Google Scholar]