

Abstract
Purpose
The purpose of this study was to investigate the potential of a T-cell-related targeting method using a lentiviral vector-based gene delivery system.
Materials and Methods
A lentiviral vector system was constructed by co-incorporating an anti-CD3 antibody (OKT3) and a fusogen into individual viral particles. The incorporation of OKT3 and fusogen was analyzed using confocal microscopy and the in vitro transduction efficiency was evaluated using flow cytometry. Blocking reagents (ammonium chloride (NH4Cl) and soluble OKT3 antibody) were added into vector supernatants during transduction to study the mechanism of this two-molecule targeting strategy. To demonstrate the ability of targeted transduction in vivo, Jurkat.CD3 cells were xenografted subcutaneously into the right flank of each mouse and the lentiviral vector was injected subcutaneously on both sides of each mouse 8 hours post-injection. Subsequently, the reporter gene (firefly luciferase) expression was monitored using a noninvasive bioluminescence imaging system.
Results
By co-displaying OKT3 and fusogen on the single lentiviral surface, we could achieve targeted delivery of genes to CD3-positive T-cells both in vitro and in vivo.
Conclusions
These results suggest the potential utility of this engineered lentiviral system as a new tool for cell type-directed gene delivery.
Keywords: gene therapy, targeted gene delivery, lentiviral vectors, CD3 antigen
INTRODUCTION
T-cells are one type of white blood cells that play a pivotal role in eliciting immune responses to protect the body from infections and diseases. Efficient gene transfer into T-cells could allow for the potential treatment of numerous genetic disorders, such as arthritis and diabetes (1–5), and provide novel gene therapies for diseases such as cancer and immunodeficiency syndrome (6–10). Currently, ex vivo gene transfer protocols require enrichment and selection of large amount T-cells and these procedures are cumbersome and expensive. Thus, there is an increasing need to develop novel in vivo gene delivery system that could achieve efficient gene transfer, cell-specific targeting, and long-term stable expression.
To date, viruses are still the most efficient vectors for gene delivery. There have been many attempts to develop targeted gene delivery systems based on various viral vectors by altering or restricting the natural host range. Adenovirus and adeno-associated virus vectors generally exhibit better transduction efficiency, and therefore are one of the popular vector systems applied in the ongoing clinical investigations. However, their applications are limited due to the high prevalence of pre-existing immunity (11). The gamma-retroviral vector, a sub-family of retroviral vectors, can mediate the integration of the transgene into the host genome, and is therefore widely used for gene delivery when long-term transgene expression is important. One factor limiting the application of gamma-retroviral vectors is their inability to transduce non-proliferative cells (12). In contrast, the lentiviral vector, another family of retroviral vectors, is able to deliver genes into non-dividing and less proliferative cells (13), including naive T-cells (14), and have thus attracted more attention in the gene therapy field recently (15). Along the process of developing efficient lentiviral vectors, strategies have been devised to improve both their safety by separating necessary viral genome into multiple plasmids during viral vectors production (16), and their transduction titer by replacing the original envelope with other viral glycoproteins such as Vesicular Stomatitis virus glycoprotein (VSVG) (17).
Beyond pseudotyping retroviral vectors with other natural glycoproteins with broad tropisms, significant works have also been devoted to alter these proteins so that they can redirect the vectors to specific cell types (18–20). Taking advantage of the structural elasticity of many viral glycoproteins (21), cell-surface determinants such as single-chain antibodies, ligand peptides, growth factors, etc., (20, 22–25), have been inserted into the permissive sites of glycoproteins to guide these enveloped vectors to particular cells. Another popular method is the use of an antibody (26, 27) or an engineered protein (28, 29) as a “bridge molecule” that has two binding domains, one for the vector and the other for the target cells, to guide the vectors to intended cells. We and others have recently demonstrated a method that breaks the binding and fusion functions, which usually were provided by a single glycoprotein, into two distinct molecules and have found that retroviral vectors co-displaying these two molecules could achieve cell-specific targeting with reasonable efficiencies (30–32).
In this report, we further tested this two-molecule strategy by targeting lentiviral vectors to CD3-expressing T-cells by enveloping vectors with an anti-CD3 antibody (OKT3) and an engineered fusogen derived from Sindbis virus glycoprotein. We found that such a recombinant vector could specifically transduce not only CD3-expressing cell lines, but also human primary CD3-positive T-cells. This type of vectors was also able to preferentially deliver a reporter gene to a CD3-expressing cell line in an in vivo xenografted mouse model. The investigation of several fusogen variants demonstrated that the fusogen plays a key role in this targeting method and that mutations in the fusion loop region of the fusogen molecule could enhance the targeting efficiency.
MATERIALS AND METHODS
Construct preparation
To construct the plasmid for the expression of membrane-bound OKT3 (Fig. 1), the cDNA of the human kappa light chain constant region was PCR-amplified from a human IMAGE consortium clone (ATCC number: 10325172) using the forward primer, 5’–ATA AAC CGT ACG GTG GCT GCA CCA TCT GTC TTC–3’ (restriction site is underlined), and the backward primer, 5’–ATC GAT GTC GAC CTA ACA CTC TCC CCT GTT GAA GCT CTT TGT GAC–3’, and the resulting DNA was cloned into the downstream of the human CMV promoter in pBudCE4.1 (Invitrogen) to generate pAbhKL. The cDNA of the human gamma-1 heavy chain constant region including the transmembrane domain was PCR-amplified from a human IMAGE consortium clone (ATCC number: 7516296) using the forward primer, 5’–TCC TCA GCT AGC ACC AAG GGC CCA TCG GTC–3’, and the backward primer, 5’–CCG GCC AGA TCT CTA GGC CCC CTG TCC GAT CAT GTT CC–3’, and the resulting DNA was inserted into the downstream of the human EF1a promoter in pAbhKL to generate pAbhLH. The light chain (the forward primer, 5’–CAA ATT GTT CTC ACC CAG TCT CCA GCA ATC–3’, and the backward primer, 5’–AGC CAC CGT ACG GTT TAT TTC CAA CTT TGT CCC CGA GCC G–3’) and heavy chain (the forward primer, 5’–CAG GTC CAG CTG CAG CAG TCT GGG G–3’, and the backward primer, 5’–CTT GGT GCT AGC TGA GGA GAC TGT GAG AGT GGT GCC TTG GC–3’) variable regions of the OKT3 antibody were PCR-amplified from a mouse hybridoma cell line (ATCC number: CRL-8001), PCR-assembled with the human light chain and heavy chain leader peptide sequences, and the DNA products were inserted directly into the upstream of the corresponding constant regions in pAbhLH to form the construct OKT3 (Fig. 1). The construct of the lentiviral backbone plasmid FUGW was described previously (33). To construct pIgαβ (Fig. 1), the cDNAs of human Igα and Igβ proteins were cloned downstream of human CMV and EF1α promoters, respectively, in pBudCE4.1 (30).
Fig. 1.

Key constructs involved in targeting lentiviral vectors to T-cells. FUGW: a lentiviral backbone construct encoding green fluorescence protein (GFP) as a reporter gene controlled by a human ubiquitin-C promoter (Ubi). WRE: woodchuck responsive element; CMV enhancer: the enhancer element derived from human cytomegalovirus. OKT3: a construct encoding the membrane-bound human/mouse chimeric anti-CD3 antibody, as well as two promoters to drive the expression of the light and heavy chains of the antibody. CMV: human cytomegalovirus immediate-early gene promoter; EF1α: human elongation factor 1 α promoter; OKT3 Vκ: the variable domain of the kappa chain of the mouse OKT3 antibody; Human Cκ: the constant domain of the kappa chain of human immunoglobulin; OKT3 Vγ: the variable domain of the gamma chain of the mouse OKT3 antibody; Human Cγ: the constant domain of the gamma-1 chain of human immunoglobulin; TM: the transmembrane domain of the gamma-1 chain of human immunoglobulin; pA: polyadenylation signal. pIgαβ: a construct encoding the accessory proteins for human immunoglobulin, Igα and Igβ. Fusogen: a mutant fusogenic molecule derived from Sindbis virus glycoprotein. Indicated mutations were introduced to the fusion loop to generate the new variants of fusogen, SV2, SV3, and SV4.
The fusogen construct, SV1, was constructed in our laboratory. It was generated by PCR-amplification of the glycoprotein from a laboratory strain of Sindbis virus (NCBI accession number: J02363) and cloned into pcDNA3 (Invitrogen) under the control of the CMV promoter. Further manipulations, including the deletion of amino acids 61–64 of the E3 protein, insertion of a hemagglutinin epitope tag sequence (MYPYDVPDYA) between amino acids 71 and 74 of the E2 protein, and the mutation of amino acids 157KE158 into 157AA158 of the E2 protein, were performed to disrupt their binding ability to the heparin sulfate glycosaminoglycan structure (27, 30). Based on SV1, additional mutations at the fusion loop region of the E1 protein of the Sindbis virus glycoprotein were introduced by PCR to generate the fusogen variants, SV2, SV3 and SV4, respectively (Fig. 1). The integrity of these constructs was confirmed by DNA sequencing.
In order to construct the lentiviral backbone plasmid FULG (Fig. 8a), the P2A gene was synthesized (Operon Biotechnology), which contained a 2A cleavage sequence derived from Porcine Teschovirus (34), a furin cleavage sequence (35) and a flexible SGSG linker. P2A gene was first assembled with the cDNA of GFP to form P2A-GFP, and the resulting DNA was subsequently assembled with the cDNA of the firefly luciferase to generate Luc-P2A-GFP. Finally, the GFP in the FUGW construct was replaced with Luc-P2A-GFP to yield FULG.
Fig. 8.

Targeting Jurkat.CD3 cells in vivo using engineered lentiviral vectors. (a) Schematic representation of a biocistronic lentiviral backbone plasmid capable of delivering two reporter genes. Luc: firefly luciferase gene; P2A: a self-cleavage peptide sequence derived from Porcine teschovirus; GFP: green fluorescence protein. (b) Targeted delivery of reporter genes in vivo in a xenografted mouse model. Jurkat.CD3 cells (5 × 106) were injected subcutaneously on the right flank side of each mouse (RAG2−/−γc−/− female mice, 6–8 weeks). After 8 hours, the concentrated lentiviral vector (FULG/OKT3+SV3, 25 × 106 TU) was injected subcutaneously on both sides of each mouse. Six days later, firefly luciferase expression was imaged using a bioluminescence imaging system. FULG/VSVG was used as a non-targeting control. Two groups of experiments were conducted independently.
Cell lines
293T cells were maintained in DMEM medium (Sigma) and supplemented with 10% FBS, L-glutamine (10 ml/L), penicillin (100 units/ml), and streptomycin (100 units/ml). Jurkat.CD3, expressing the human CD3 antigen, was generated by transduction of a Jurkat cell line (J.RT3-T3.5, lacking the beta chain of the T-cell receptor, ATCC number: TIB-153) via a VSVG-pseudotyped lentiviral vector. The cDNAs of the alpha and beta chains of a human T-cell receptor were linked by a P2A sequence and the resulting genes were cloned downstream of the human ubiquitin-C promoter in the lentiviral backbone plasmid FUW to form FUW-TCR. The lentiviral vector FUW-TCR was pseudotyped with VSVG and used to transduce Jurkat cells. The resulting cells were cultured for two weeks and then subjected to Fluorescence activated cell sorter (FACS) sorting to obtain a uniform population of CD3-positive cells designated as Jurkat.CD3. Both Jurkat.CD3 and Jurkat cells were maintained in RPMI medium supplemented with 10% FBS (Sigma), L-glutamine (10 ml/L), penicillin (100 units/ml), and streptomycin (100 units/ml).
Viral vector production
A standard calcium phosphate precipitation method was employed to produce the various engineered lentiviral vectors in this study. 293T cells in a 6-cm plate (~ 90% confluence) were transiently transfected with the lentiviral backbone plasmid FUGW (5 µg), the OKT3 plasmid (2.5 µg), the pIgαβ plasmid (2.5 µg), the fusogen plasmid (2.5 µg), and other packaging plasmids (pMDLg/pRRE and pRSV-Rev). Four hours after transfection, the cell medium was replaced with pre-warmed fresh culture medium and the cells were incubated at 37°C and 5% CO2. The viral supernatants were harvested between 48 ~ 72 hours after the medium change. Viral supernatants were filtered through a 450-nm filter and used for transduction experiments. The fresh supernatants could also be concentrated by ultracentrifugation (Optima L-90K, Beckman Coulter) for 90 min at 25,000 × g and 4°C, and the resulting vector pellets were resuspended in cold filtered PBS.
Fluorescent labeling
For the detection of individual viral particle incorporating the antibody (OKT3) and fusogen, fresh viral supernatants were overlaid upon poly-L-lysine coated coverslips in a six-well culture dish and centrifuged at 3,700 × g and 4°C for 2 hours using a Sorval Legend RT centrifuge. The coverslips were rinsed with cold PBS twice and the adhered particles were immunostained by an Alexa594-conjugated anti-human IgG antibody (Molecular Probes), a biotin-conjugated anti-HA antibody (Miltenyi Biotec Inc.), and an anti-p24 antibody (NIH AIDS Research and Reference Reagent Program), and further incubated with FITC-conjugated streptavidin (BD Phamingen). The coverslips were mounted in Vectashield (an antifade mounting medium, Vector Laboratories). The images were acquired with a Zeiss LSM-510 laser scanning confocal microscope (Carl Zeiss) using a plan-apochromat 63×/1.4 oil immersion objective. The images were processed using the LSM 510 software version 3.2 SP2. For the detection of cell-virus binding, cells were incubated with lentiviral supernatants at 4°C for 30 minutes, and then spun down to a glass coverslip. The coverslips were rinsed with cold PBS twice and immunostained with the anti-p24 antibody.
Targeted transduction of cell lines in vitro
Target cells (Jurkat.CD3 or Jurkat) were seeded in a 24-well culture plate at the density of 0.2 × 106 per well. Spin-infection was conducted by adding the lentiviral supernatants (1 ml per well) and spinning at 2,500 rpm and 30°C for 90 min using a RT Legend centrifuge. Following the spin-infection, viral supernatants were replaced with fresh medium and were incubated at 37°C with 5 % CO2 for an additional 3 ~ 5 days. GFP expression was determined by FACS analysis. In order to determine the vector transduction titer, two-fold serial dilutions of the viral supernatants were added to the target cells. The titer was determined by the dilution range that exhibited a linear GFP expression response.
Transduction of human primary T-cells in vitro
Human peripheral blood mononuclear cells (PBMC) were activated for 48 hours in RPMI medium containing 10% FBS (Sigma), L-glutamine (10 ml/L), penicillin (100 units/ml), and streptomycin (100 units/ml), as well as 5 µg/ml phytohemagglutinin (PHA). After twice washes with PBS, PBMC were exposed to the targeting lentiviral vector FUGW/OKT3+SV3 and spin-infection was conducted for 90 min at room temperature. The cell medium was replaced with fresh culture medium supplemented with human recombinant IL-2 (10 µg/ml) and cultured for another 4 days at 37°C and 5% CO2. FACS analysis was used to measure the specific transduction of activated PBMC.
Antibody competition and endosomal neutralization
A similar spin-infection protocol was used for this experiment. For the antibody competition assay, graded amounts of soluble OKT3 antibody or matched isotype antibody were added to Jurkat.CD3 cells before the addition of viral supernatants. For the neutralization experiment, different amounts of ammonium chloride (NH4Cl, dissolved in PBS) were added into viral supernatants during transduction. Viral supernatants were replaced with fresh medium after 90 min of spin-infection and cells were incubated for another 3 ~ 5 days before FACS analysis of GFP expression.
Targeted transduction of Jurkat.CD3 cells in vivo
Jurkat.CD3 cells (5 × 106) were injected subcutaneously into the right flank side of each mouse (RAG2−/−γc−/− female mice, 6–8 weeks). After 8 hours, the recombinant concentrated lentiviral vector FULG/OKT3+SV3 (25 × 106 TU, resuspended in 200 µl PBS) was injected subcutaneously on both sides of each mouse. As a non-targeting control, concentrated lentiviral vector FULG/VSVG was similarly injected into both sides of each mouse with an MOI = 5. Several days later, the reporter gene (firefly luciferase) expression was imaged using a bioluminescence imaging system (Xenogen IVIS 200). Images were analyzed by using Living Image 2.50.1 software. All injections were conducted using a 27G1/2 needle and 1 ml tuberculin slip tip syringe (BD Biosciences). This experiment was performed in two groups independently. All experiments were performed in accordance with the guidelines by the National Institute of Health and the University of Southern California on the Care and Use of Animals.
RESULTS
Construction and production of recombinant lentiviral vectors
Many viral surface glycoproteins are known to be responsible for both virus-cell binding and fusion with either cellular or endosomal membrane to release the viral genetic materials into the host cell cytoplasm (21). We were able to engineer lentiviral vectors in such a way that these two functions were separated and carried out by two distinct molecules on the surface of a single viral particle (30). In this engineered system, the initial cell-specific binding was directed by the antibody-antigen interaction and the subsequent fusion was induced by the surface-displayed fusogenic molecule (36).
To target CD3-positive T-cells, we constructed a membrane-bound human/mouse chimeric antibody derived from a mouse anti-human CD3 monoclonal antibody OKT3 (Fig. 1, designated as OKT3) (37, 38). The human CD3 antigen is a protein complex composed of four individual chains (CD3γ, CD3δ, and two chains of CD3ε). It is associated with the T-cell receptor (TCR) to support its surface expression and to participate in TCR signaling for the activation of T lymphocytes (39). This human CD3 antigen was used as the cell-surface receptor for engineered recombinant lentiviral vectors to target T-cells for the present study. In order to stably display the surface antibody to the cell surface and subsequently to the viral surface, we also constructed a plasmid capable of expressing the human immunoglobulin accessory proteins, Igα and Igβ (Fig. 1, designated as pIgαβ). The Igα and Igβ proteins are required to stably express the membrane-bound antibody molecules on the cell surface (40). This will allow the antibody to be incorporated onto the lentiviral vector surface.
Besides the antibody, another key molecule on the engineered vector surface is the fusogen. After endocytosis is induced by the binding of antibody-bearing vectors to cell-surface CD3, this fusogenic molecule will mediate fusion in the low pH endosomal environment to release the genetic contents of the vectors into the cytosol of cells. It is well-documented that Sindbis virus glycoprotein (SV) can pseudotype lentivirus (17) and that the fusion ability of SV is independent of receptor binding (26, 27). We and others have demonstrated that the binding-deficient and fusion-competent protein (SV1, Fig. 1) derived from SV could be effectively incorporated onto the lentiviral vector surface and serve as a fusogen to mediate transduction (27, 30). An intracellular trafficking study confirmed that the SV1 plays an important role in inducing fusion for targeting lentiviral vectors at the early endosome stage (36). We wanted to use this SV1 protein as a fusogen to test its ability to mediate lentiviral vector targeting to T-cells. A previous study in our laboratory also showed that the performance of the fusogen had a significant impact on the transduction efficiency of engineered viral vectors and that the introduction of mutations at the fusion loop of SV1 could enhance the targeting efficiency of vectors bearing anti-CD20 antibody to CD20-expressing cells (32, 41). These mutations on the E1 226 region of SV were originally identified by Kielian and coworkers, and they found that Sindbis virus incorporated with these mutations had an improved level of infectivity to cholesterol-depleted cells (42). We wanted to investigate the effect of these mutations on the ability of the fusogen variants (Fig. 1, designated as SV2, SV3, and SV4) to mediate engineered lentiviral vector targeting to T-cells.
Our targeting lentiviral vector, FUGW/OKT3+fusogen, co-expressed a membrane-bound anti-human CD3 antibody (OKT3) and one of fusogens (SV1, SV2, SV3 and SV4). Lentiviral vector pseudotyped with the VSVG envelope protein (FUGW/VSVG) was produced as non-targeting vector control. Lentiviral vectors displaying either antibody only (FUGW/OKT3) or fusogen only (FUGW/fusogen) were prepared for control purposes as well. FACS analysis of cells transfected by these various plasmids showed that almost all of the live cells had expression of the GFP reporter that was encoded in FUGW (Fig. 2a, upper). Among these GFP-positive cells, approximately 30% of the cells expressed both an OKT3 antibody and the SV1 fusogen (Fig. 2a, bottom), which likely produced functional FUGW/OKT3+fusogen vectors. As expected, FUGW/OKT only showed antibody expression and FUGW/Fusogen only showed fusogen expression on lentiviral vectors producing cells. Neither antibody nor fusogen expression was observed for FUGW/VSVG producing cells because of the absence of necessary antibody and fusogen plasmids. FACS analysis of 293T cells transfected with the various fusogens (SV1, SV2, SV3, and SV4) accompanied with either targeting antibody (OKT3) or control antibody (Ab) showed a similar level of surface expression of the fusogen (Fig. 2b), suggesting that they could be incorporated onto the lentiviral vector surface with similar efficiencies.
Fig. 2.

(a) Surface protein expression of 293T cells transfected to produce recombinant lentiviral vectors. 293T cells were transiently transfected with the lentiviral backbone plasmid FUGW, the antibody plasmid OKT3, the plasmid encoding antibody accessory proteins pIgαβ, the plasmid encoding a fusogen SV1, and other necessary packaging plasmids, to produce the targeting vector (FUGW/OKT3+fusogen). Co-transfection to produce vector bearing OKT3 only (FUGW/OKT3), bearing fusogen SV1 only (FUGW/fusogen), and bearing VSVG (FUGW/VSVG), were performed as controls. Two days post-transfection, GFP expression was analyzed by FACS (upper). Gating on the GFP-positive cells, the antibody and fusogen expressions were assessed by FACS staining using anti-human IgG and anti-HA tag antibodies, respectively (bottom). (b) FACS analysis of co-transfected vector-producing cells. 293T cells were transiently transfected with various plasmids as described above. Co-transfection using a non-relevant antibody lacking the binding specificity to CD3 (Ab), as well as co-transfection to produce vector bearing OKT3 only (FUGW/OKT3), and bearing VSVG (FUGW/VSVG), were performed as controls. Two days post-transfection, the antibody and fusogen expressions were measured by co-staining using anti-human IgG antibody and anti-HA tag antibody based on GFP positive cells by FACS.
Imaging analysis of single viral particles and vector-cell binding
The confocal imaging method was used to directly visualize the incorporation of the antibody and fusogen on the surface of engineered lentiviral vectors. To avoid possible contamination of fluorescence signals from the FUGW construct in our imaging analysis, we used the FUW, which lacks a GFP reporter gene (Fig. 3a), as the transfer vector. Virus-producing 293T cells were transfected with various constructs for making lentiviral vectors. FUW/OKT3+SV1 is bearing both the OKT3 antibody and the SV1 fusogen, and FUW/VSVG is containing the VSVG envelope protein. Confocal images were acquired and fluorescent signals for p24, OKT3, and SV1 were detected in the green, red, and blue channels respectively. As shown in Fig. 3b, all three colors were observed for FUW/OKT+SV1 particles and a significant portion of the signals were co-localized in the merged image, indicating that OKT3 and SV1 could be co-incorporated into the same viral particle. Similar results were observed for vectors bearing the other mutant fusogens (SV2, SV3, and SV4) (data not shown). As expected, only green signals indicating viral particles were detected for the control FUW/VSVG vectors (Fig. 3b). The distribution of the p24 signals was further analyzed and the result could be fitted to a single Gaussian function (red dotted line in Figure 3c) with R2 = 0.98, suggesting that the fluorescence was emitted from single viral particles (35).
Fig. 3.
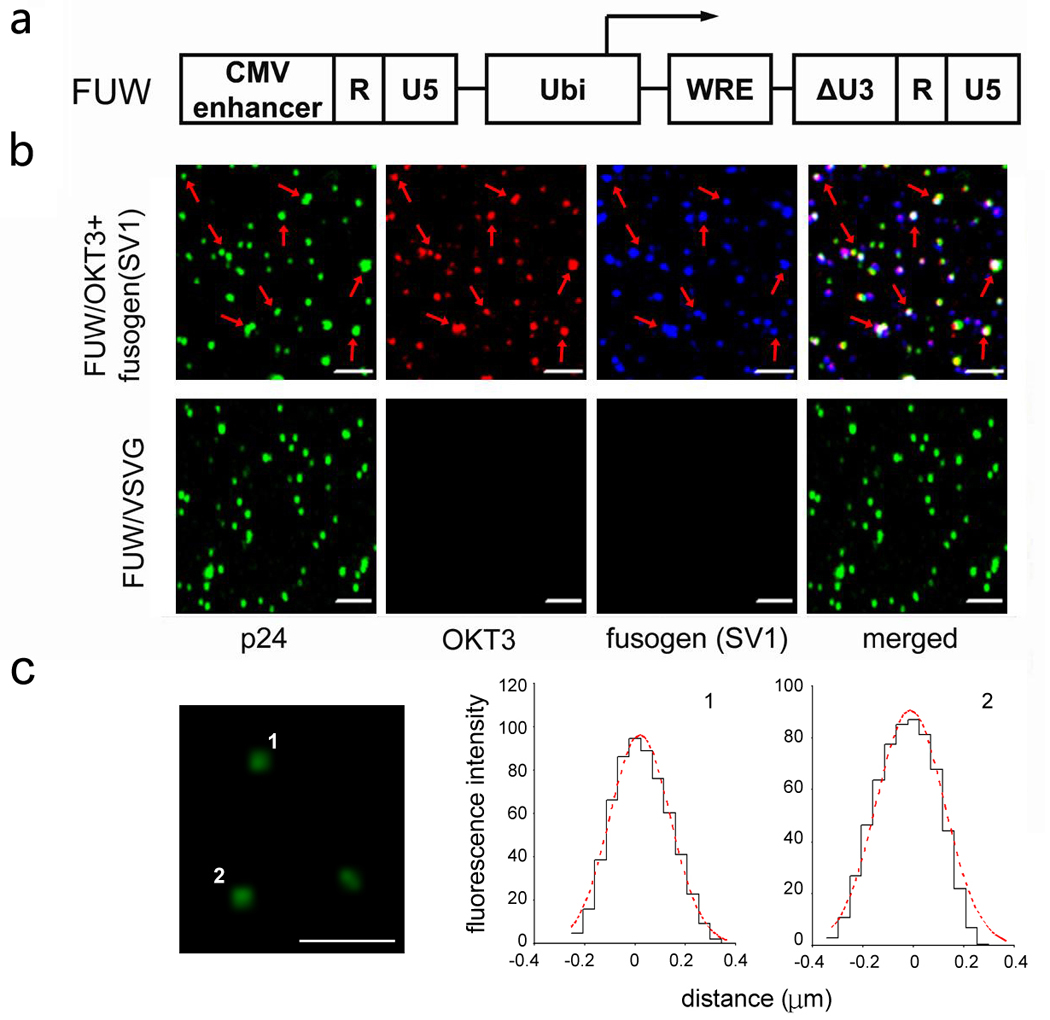
Confocal imaging analysis of the recombinant viral particles. (a) Schematic representation of the lentiviral backbone FUW, a similar construct to FUGW (see Fig. 1) except for the lack of a GFP reporter gene. (b) Confocal imaging analysis for the co-incorporation of antibody and fusogen on the same lentiviral particles. Indicated lentiviral supernatants, FUW/OKT3+fusogen and FUW/VSVG, were co-immunostained with antibody specific for OKT3 (red), antibody specific for the fusogen (blue), and antibody specific for the lentiviral capsid protein p24 (green). Overlapping green, red, and blue signals appear white in a merged image. The scale bar represents 2 µm. Images were acquired using a laser scanning confocal microscope (Zeiss LSM 510). (c) Confocal microscopy of p24-labeled lentivector. The relative fluorescence intensity distribution of two spots is shown. The distribution of the fluorescence fits to a Gaussian function. Scale bar represents 2 µm.
To demonstrate the specific binding between antibody-bearing vectors and target cells, we constructed a stable CD3-expressing cell line Jurkat.CD3 (Fig. 4a). Due to the lack of expression of the T-cell receptor beta chain, our parental Jurkat cells expressed a background level of CD3 (Fig. 4a&b). Immunostaining confirmed the expression of CD3 on Jurkat.CD3 cells (Fig. 4a). FACS analysis showed that Jurkat.CD3 cells expressed a physiologically relevant level of CD3, as compared to human primary T-cells (Fig. 4b). We used this Jurkat.CD3 cell line to investigate whether OKT3 on the lentiviral vector surface could mediate binding to target cells. The staining by p24-specific antibody clearly showed that there was binding between the Jurkat.CD3 cells and FUW/OKT3+SV1 (Fig. 4c). No binding was detected for Jurkat cells, suggesting that the binding of FUW/OKT3+SV1 to Jurkat.CD3 could be attributed to the CD3 antigen. When various lentiviral particles with different surface compositions (FUW/OKT3+SV1, FUW/OKT3, or FUW/SV1) were allowed to bind to Jurkat.CD3 cells, only the antibody-bearing particles (FUW/OKT3+SV1 and FUW/OKT3) could be readily detected at the target cell surface (Fig. 4d). This result further confirmed that the observed binding is dependent on the OKT3-CD3 interaction and is fusogen-independent.
Fig. 4.

Confocal imaging analysis of cell-virus binding. (a) Imaging detection of the surface expression of CD3 antigen on Jurkat.CD3 and Jurkat cells. CD3 was probed by immunostaining the cells with a mouse anti-human CD3 antibody and a secondary Cy5-conjugated anti-mouse antibody. The green color represents the CD3 on the cell surface and the blue color represents the cellular nucleus. (b) FACS analysis of the surface expression of CD3 on Jurkat, Jurkat.CD3, and human peripheral blood mononuclear cells (PBMC). (c) Imaging analysis of the lentiviral vector FUW/OKT3+SV1 binding to Jurkat.CD3 or Jurkat cells. For cell-virus binding, cells (2 × 106) were incubated with vector particles and were immunostained by antibody specific for the capsid protein p24 (green). The cellular nuclei were counterstained by 4'-6-diamidino-2-phenylindole (DAPI, blue). (d) Indicated lentiviral particles (FUW/OKT3+SV1, FUW/OKT3 or FUW/SV1), were incubated with Jurkat.CD3 cells and the same immunostaining as described in (c) was performed. The scale bar represents 5 µm.
Targeted transduction by recombinant lentiviral vectors in vitro
We studied the efficiency and specificity of engineered vectors in transducing CD3-expressing cells in vitro. Jurkat.CD3 cells were exposed to various viral supernatants and four days post-transduction, FACS analysis of the cells was performed to measure GFP transgene expression. As shown in Fig. 5a, lentiviral vectors bearing both OKT3 and one of four fusogens (FUGW/OKT3+fusogen) could efficiently transduce Jurkat.CD3 cells with more than 62% of them being GFP-positive. A low level background transduction (< 7%) was obtained when these vectors were used to transduce Jurkat cells. Similar background transductions were observed for the vectors bearing a non-relevant antibody (Ab) that is non-specific to CD3 and one of four fusogens (FUGW/Ab+fusogen) to transduce either Jurakt.CD3 or Jurkat cells. These background transductions were likely resulted from the residual binding function of fusogen to the heparin sulfate structure on the cell surface. Our data clearly showed that the engineered lentiviral vectors could target CD3-expressing cells and that the OKT3 on the vector surface played a key role in the success of targeted transduction. Although FUGW/OKT3 could bind to Jurkat.CD3 cells (Fig. 4d), it failed to transduce them and the background transduction efficiency was lower than the efficiency of targeting vectors transducing Jurkat cells. It further suggested that the fusogen is necessary for vector transduction and the background transduction is likely due to the remaining binding ability of fusogen.
Fig. 5.

Targeted transduction of lentiviral vectors in vitro. (a) Fresh unconcentrated lentiviral vectors either bearing an OKT3 antibody and a fusogen (FUGW/OKT3+fusogen), or bearing a non-relevant antibody (Ab, no specificity to CD3) and a fusogen (FUGW/Ab+fusogen), were added to transduce 0.2 × 106 Jurkat.CD3 cells or Jurkat cells. Transduction with lentiviral vectors bearing an OKT3 antibody only (FUGW/OKT3) or bearing the VSVG (FUGW/VSVG) were used as controls. Three days post-transduction, the transduction efficiency was measured by FACS analysis of the percentage of cells expressing GFP. Shaded column: transduction on Jurkat.CD3 cells; Open column: transduction on control Jurkat cells. (b) Specific viral titers for various engineered lentiviral vectors based on both Jurkat.CD3 and Jurkat cells. (c) Kinetics of the transgene expression in Jurkat.CD3 cells transduced with the targeting lentiviral vectors. Jurkat.CD3 cells were transduced with FUGW/OKT3+SV3 and continuously cultured for a month. GFP expression was determined by FACS at the indicated day.
Moreover, different transduction efficiencies were obtained for the four targeting lentiviral vectors bearing one of four different fusogens (FUGW/OKT3+fusogen (SV1-4)) in transducing Jurkat.CD3 cells. Among these vectors, FUGW/OKT3+SV3 gave the highest transduction efficiency (~80%). A two-fold dilution method was employed to titer these vectors based on both Jurkat.CD3 and Jurkat cells. Their titers are given in Fig. 5b. Because of its superior transduction efficiency, FUGW/OKT3+SV3 were chosen for the rest of investigations.
It is known that one of the advantages in using HIV-1-derived lentiviral vectors for gene delivery is in their ability to integrate genes of interest into the host genomes to permit long-term transgene expression. We monitored the GFP expression in Jurkat.CD3 cells transduced by FUGW/OKT3+SV3 to study the kinetics of transgene expression. The results showed that GFP expression was stable over a month during which the transduced cells were monitored (Fig. 5c).
Mechanistic study of targeted transduction of engineered vectors
The targeting lentiviral vectors in this study required two distinct proteins on the surface of virions to perform the binding and fusion functions, and both of them are important for efficient transduction. To confirm the role of the incorporated fusogen is to trigger fusion at the low pH environment of the endosomes, ammonium chloride (NH4Cl) with gradient concentrations were added into the FUGW/OKT3+SV3 supernatants during transduction to Jurkat.CD3 cells. GFP expression was quantified by FACS four days post-transduction and a decrease of transduction efficiency was observed in response to an increase in the concentration of NH4Cl (Fig. 6a), which is known to be able to neutralize the acidic endosomal environment (43). The addition of NH4Cl raised the pH at the fusion compartment for the engineered vector and resulted in the inhibition of transduction.
Fig. 6.

The effects of antibody competition and endosomal neutralization on lentiviral transduction in vitro. (a) Gradient concentrations of NH4Cl (dissolved in PBS) were pre-mixed with lentiviral supernatants (FUGW/OKT3+SV3) and added to Jurkat.CD3 cells for transduction. After replacement of the supernatant with fresh culture medium, the cells were incubated for another three days before FACS analysis of GFP expression. All of the data was normalized by the transduction efficiency compared to transduction without NH4Cl treatment. (b) Jurkat.CD3 cells were pre-incubated with gradient concentrations of soluble OKT3 antibody (OKT3) for 30 min, after which fresh unconcentrated FUGW/OKT3+SV3 supernatant (1 ml) was added for transduction. After replacement of the supernatant with fresh culture medium, the cells were incubated for additional three days before FACS analysis. The isotype-matched antibody was included in the experiment as a control.
To directly assess the role of the molecular interaction between OKT3 and CD3 in the process of targeted transduction, Jurkat.CD3 cells were exposed to FUGW/OKT3+SV3 supernatants in the presence of soluble OKT3 antibody. As shown in Fig. 6b, addition of this antibody markedly reduced the efficiency of transduction. This was likely due to molecular blocking by the soluble OKT3, which competed against the OKT3-bearing viral particles to bind to the CD3-expressing cells. The addition of an isotype control antibody had a minimal effect on transduction efficiency (Fig. 6b). These data both validated the hypothesis that membrane-anchored OKT3 plays a key role in directing engineered vectors to target cells and confirmed that CD3 is indeed the true receptor for the resulted transduction.
Targeted transduction of human primary T-cells by engineered vectors
An experiment was also performed to investigate the ability of the engineered lentiviral vectors to target human primary T-cells. The activated human peripheral blood mononuclear cells (PBMC) were transduced with FUGW/OKT3+SV3 at different multiplicity of infection (MOI). As shown in Figure 7a, about 1% GFP-positive cells were obtained at MOI=1 and all of them were CD3-postive T-cells. An increased dose of FUGW/OKT3+SV3 (MOI = 5) resulted in ~5.0% GFP-positive cells. Detail differences in the frequency and total functional GFP expression (iMFI) of populations of CD3+ and CD3− cells are shown in Fig. 7b. The integrated mean fluorescence intensity (iMFI), which was calculated by multiplying the percentage of positive cells with the mean fluorescence intensity (MFI), shows the same pattern as the frequency (Fig. 7b, right). PCR analysis of the genomic DNA extracted from these transduced T-cells confirmed the stable integration of the GFP gene (data not shown).
Fig. 7.
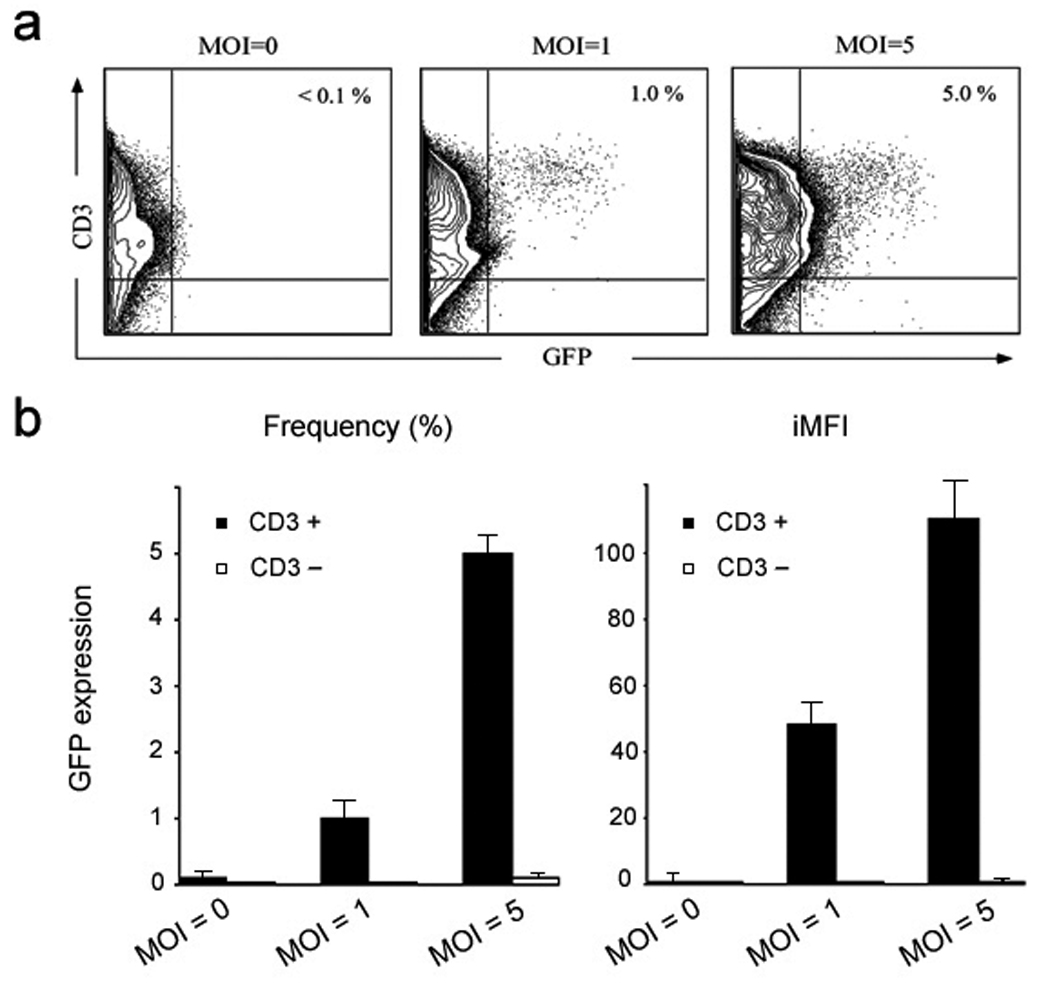
Targeted transduction of human primary T cells by the engineered lentiviral vectors. Human peripheral blood mononuclear cells (PBMC) were treated with phytohemagglutinin (PHA) for two days. The resulting cells were exposed to the lentiviral supernatant (FUGW/OKT3+SV3) at different multiplicity of infection (MOI). (a) The vector-treated cells were cultured for another 4 days with human recombinant interleukin 2 (IL-2) and analyzed by FACS for GFP expression. (b) The total frequency (based on live cells, left) and iMFI (right) for both CD3+ and CD3− cells.
Targeted transduction by engineered lentiviral vectors in vivo
An animal model was designed to examine the possibility of using our engineered lentiviral vectors to deliver genes in vivo in a cell-specific manner. For monitoring purposes, we made a biocistronic transfer vector, FULG, which is capable of expressing both firefly luciferase and GFP under the control of a human ubiquitin promoter (Fig. 8a). A self-cleavage peptide sequence derived from Porcine teschovirus was used to link these two genes to achieve efficient co-expression (34). This construct permitted us not only to evaluate the direct injection of vectors in mice non-invasively using a bioluminescence imaging technique (BLI), but also to analyze the cells isolated from injected animals in vitro using flow cytometry. Jurkat.CD3 cells (5 × 106) were first subcutaneously injected on the right flank side of the immunodeficient RAG2−/−γc−/− mice. Eight hours post-cell injection, concentrated targeting lentiviral vector (FULG/OKT3+SV3, 25 × 106 transduction units (TU), MOT = 5) was injected not only to the right flank site where the Jurkat.CD3 cells were xenografted, but also to the opposite left flank site where no cells were injected. As a non-targeting control, VSVG-pseudotyped lentiviral vector (FULG/VSVG (MOI = 5)) was applied to mice in the same way. The expression of the delivered reporter gene (firefly luciferase, encoded by FULG) was monitored and results were shown in Fig. 8b. Two groups of experiments were conducted independently. For the mouse treated with the FULG/OKT3+SV3 vector, a clear bioluminescence signal generated by firefly luciferase could be seen in the right flank side of the mouse where target Jurkat.CD3 cells were xenografted. On the left flank, which lacked the xenografted cells, no signal could be detected, although the same amount of FULG/OKT3+SV3 vector was injected. In contrast, administration of non-targeting FULG/VSVG with the same injection protocol generated bioluminescence signals on both sides of the mouse irrespective of whether the area was xenografted with Jurkat.CD3, confirming the broad tropism of VSVG-enveloped vectors. GFP expression was confirmed by FACS analysis of the Jurkat.CD3 cells harvested from the FULG/OKT3+SV3-injected mouse (data not shown).
DISCUSSION
Several viral vector systems are available for in vitro gene therapy, but the development of targeting vector for gene delivery both in vitro and in vivo appears to be much more difficult (20, 21). Such a targeting system requires the vector to be able to recognize a unique receptor on the surface of the target cells. Previously we showed that the co-incorporation of an antibody and a fusogen derived from a binding-deficient mutant Sindbis virus glycoprotein onto the same lentiviral particles could result in cell-specific transduction capabilities (30). This targeted transduction was accomplished through at least two molecular events: antibody-mediated binding at the target cell surface and fusogen-induced fusion at the low pH environment of the endosomal compartment of the target cell.
The results of this study demonstrated a potentially applicable way to engineer lentiviral vectors capable of delivering genes through the CD3 antigen, a marker that is uniquely expressed on T-cell surface. We constructed a membrane-bound human/mouse chimeric antibody (OKT3) specific for the human CD3 antigen to direct the vectors to T-cells. When this construct was used to co-transfect vector-producing 293T cells along with plasmids encoding antibody accessory proteins Igα and Igβ, a fusogen derived from the mutant Sindbis virus glycoprotein, a lentiviral backbone vector, as well as other standard packing plasmids, surface co-expression of OKT3 and fusogen was observed by FACS staining. Viral particles were harvested and subjected to direct visualization via confocal microscopy. Acquired images verified the co-incorporation of both proteins (OKT3 and fusogen) on the same viral particles. To study the specific binding of OKT3-bearing vectors to the desired cells, we constructed a Jurkat.CD3 cell line that constantly expressed the CD3 antigen as the target cells. Imaging analysis for vector-cell binding showed the CD3- and OKT3-dependent attachment of antibody-carrying viral particles to Jurkat.CD3 cells.
The in vitro transduction study on both Jurkat.CD3 cells and human primary T-cells demonstrated the specific targeting and the long-term transgene expression capabilities of this engineered lentiviral vector. Vectors bearing both OKT3 and fusogen were able to achieve specific transduction to CD3-expressing Jurkat cells, while only background transduction was detected for vectors bearing both CD3-blind antibody and fusogen. The different transduction efficiencies of Jurkat and Jurkat.CD3 cells also suggest that the level of surface CD3 expression is correlated with the transduction efficiency of OKT3-bearing lentivectors. No transduction was detected for vectors displaying only one molecule (either OKT3 or a fusogen), indicating the requirement of both proteins for a successful transduction. Exposure of PHA-activated PBMC to FUGW/OKT3+SV3 resulted in preferential transduction of the CD3-positive T-cells, indicating that primary cells could be targeted by the engineered lentiviral vectors as well. We used a xenografted mouse model to test the ability of this vector system to deliver genes to CD3-expressing cells in vivo and found that a subcutaneous injection of concentrated targeting lentiviral vectors encoding firefly luciferase could preferentially modify grafted Jurkat.CD3 cells at the injection site.
In our lentiviral vector targeting system, the two separated molecules (OKT3 antibody and fusogen) are essential for inducing endocytosis and triggering fusion. To examine the transduction mechanism, we added ammonium chloride (NH4Cl) to the vector supernatants to neutralize the acidic environment of the target cell endosomes. The incorporated fusogen, which is known to require the low pH of the early endosomes to induce membrane fusion (36, 42), was responsible for the observed reduced infectivity. Soluble OKT3 antibody added into the vector supernatants during transduction had an inhibitory effect on the transduction, which was due to the competitive binding between the OKT3-bearing vector and the soluble OKT3 against the target cells. These assays confirmed the requirement of both molecules (OKT3 and fusogen) for targeting lentiviral vectors. Meanwhile, they shed some light on an infection process involving the specific binding of vectors to CD3, which induces the internalization of the vectors into the endosome, and the unfolding of fusion domain at the low pH condition to trigger fusion to release the viral core into the cytosol of the target cells.
A study by our laboratory found that the kinetics and trafficking properties of the fusogen could significantly influence the overall transduction behavior of the engineered lentiviral vectors (36). Kielian and her colleagues extensively studied the envelope glycoprotein of alphavirus (both Semliki Forest virus and Sindbis virus) and found that the amino acid sequences at the fusion loop region were pivotal for virus fusion (44). Their further study of Sindbis virus identified some key mutations on the glycoprotein that improved its infectivity towards cholesterol-depleted cells (42). We adapted these mutations into our original fusogen (SV1), which was essentially binding-deficient and fusion-competent version of the Sindbis virus glycoprotein (30) and generated additional three fusogens (SV2, SV3 and SV4). These fusogentic molecules exhibited enhanced abilities to mediate transduction of T-cells when paired with the OKT3 antibody. We suspect that, based on the available structural information on alphavirus glycoprotein (44, 45), the introduced mutations may either favor the structural reorganization of the E1 protein of the fusogen triggered by the low pH within the acidic endosome to form a trimer, which is a fusion-active configuration, or stabilize the fusion-competent configuration of the fusogen, or both. Experiments are underway to test this speculation by a liposome-based fusion assay described by Kielian and coworkers (46).
In summary, we demonstrate a method of targeting human T-cells by engineering lentiviral vectors to carry both an anti-CD3 antibody and a fusogen. Meanwhile, a more extensive characterization of this system, such as the number of OKT3 antibody and fusogen molecules needed on the vector surface in order to effectively achieve the modification of T-cells, is required for further optimization. We observed that it was amenable to alter the key domain of the fusogen to improve its ability to mediate vector transduction, thus a continuous effort to engineer the fusion domain may be a viable means to further improve the performance of this targeting method to T-cells. Ultimately, to target T-cells in vivo, the systemic delivery of the engineered vector is likely to be required and we will need to develop an assay to evaluate the feasibility of intravenous injection of this OKT3-bearing lentiviral vector to target T-cells in vivo.
ACKNOWLEDGEMENTS
We thank April Tai, Lili Yang and Steven Froelich for critical reading of the manuscript, and the USC Norris Center Cell and Tissue Imaging Core. This work was supported by a National Institute of Health grant. The following reagents was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: Monoclonal Antibody to HIV-1 p24 (AG3.0) from Dr. Jonathan Allan.
REFERENCES
- 1.Chernajovsky Y, Adams G, Triantaphyllopoulos K, Ledda MF, Podhajcer OL. Pathogenic lymphoid cells engineered to express TGF beta 1 ameliorate disease in a collagen-induced arthritis model. Gene Ther. 1997;4:553–559. doi: 10.1038/sj.gt.3300436. [DOI] [PubMed] [Google Scholar]
- 2.Aiuti A, Vai S, Mortellaro A, Casorati G, Ficara F, Andolfi G, Ferrari G, Tabucchi A, Carlucci F, Ochs HD, Notarangelo LD, Roncarolo MG, Bordignon C. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat. Med. 2002;8:423–425. doi: 10.1038/nm0502-423. [DOI] [PubMed] [Google Scholar]
- 3.Verhoeyen E, Dardalhon V, Ducrey-Rundquist O, Trono D, Taylor N, Cosset FL. IL-7 surface-engineered lentiviral vectors promote survival and efficient gene transfer in resting primary T lymphocytes. Blood. 2003;101:2167–2174. doi: 10.1182/blood-2002-07-2224. [DOI] [PubMed] [Google Scholar]
- 4.Drobyski WR, Morse HC, 3rd, Burns WH, Casper JT, Sandford G. Protection from lethal murine graft-versus-host disease without compromise of alloengraftment using transgenic donor T cells expressing a thymidine kinase suicide gene. Blood. 2001;97:2506–2513. doi: 10.1182/blood.v97.8.2506. [DOI] [PubMed] [Google Scholar]
- 5.Maurice M, Verhoeyen E, Salmon P, Trono D, Russell SJ, Cosset FL. Efficient gene transfer into human primary blood lymphocytes by surface-engineered lentiviral vectors that display a T cell-activating polypeptide. Blood. 2002;99:2342–2350. doi: 10.1182/blood.v99.7.2342. [DOI] [PubMed] [Google Scholar]
- 6.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, Binder GK, Slepushkin V, Lemiale F, Mascola JR, Bushman FD, Dropulic B, June CH. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA. 2006;103:17372–17377. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 9.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat. Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 10.Schumacher TN. T-cell-receptor gene therapy. Nat. Rev. Immunol. 2002;2:512–519. doi: 10.1038/nri841. [DOI] [PubMed] [Google Scholar]
- 11.Muller OJ, Kaul F, Weitzman MD, Pasqualini R, Arap W, Kleinschmidt JA, Trepel M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003;21:1040–1046. doi: 10.1038/nbt856. [DOI] [PubMed] [Google Scholar]
- 12.Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 14.Costello E, Munoz M, Buetti E, Meylan PR, Diggelmann H, Thali M. Gene transfer into stimulated and unstimulated T lymphocytes by HIV-1-derived lentiviral vectors. Gene Ther. 2000;7:596–604. doi: 10.1038/sj.gt.3301135. [DOI] [PubMed] [Google Scholar]
- 15.Kohn DB. Lentiviral vectors ready for prime-time. Nat. Biotechnol. 2007;25:65–66. doi: 10.1038/nbt0107-65. [DOI] [PubMed] [Google Scholar]
- 16.Aillesand LE, Naldini L. HIV-1-derived lentiviral vectors. Curr. Top. Microbiol. Immunol. 2002;261:31–52. doi: 10.1007/978-3-642-56114-6_2. [DOI] [PubMed] [Google Scholar]
- 17.Cronin J, Zhang XY, Reiser J. Altering the tropism of lentiviral vectors through pseudotyping. Curr. Gene Ther. 2005;5:387–398. doi: 10.2174/1566523054546224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandrin V, Russell SJ, Cosset FL. Targeting retroviral and lentiviral vectors. Curr. Top. Microbio. Immunol. 2003;281:137–178. doi: 10.1007/978-3-642-19012-4_4. [DOI] [PubMed] [Google Scholar]
- 19.Verhoeyenand E, Cosset FL. Surface-engineering of lentiviral vectors. J. Gene Med. 2004;6 Suppl 1:S83–S94. doi: 10.1002/jgm.494. [DOI] [PubMed] [Google Scholar]
- 20.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavillette D, Russell SJ, Cosset FL. Retargeting gene delivery using surface-engineered retroviral vector particles. Curr. Opin. Biotechnol. 2001;12:461–466. doi: 10.1016/s0958-1669(00)00246-9. [DOI] [PubMed] [Google Scholar]
- 22.Ager S, Nilson BH, Morling FJ, Peng KW, Cosset FL, Russell SJ. Retroviral display of antibody fragments; interdomain spacing strongly influences vector infectivity. Hum. Gene Ther. 1996;7:2157–2164. doi: 10.1089/hum.1996.7.17-2157. [DOI] [PubMed] [Google Scholar]
- 23.Marin M, Noel D, Valsesia-Wittman S, Brockly F, Etienne-Julan M, Russell S, Cosset FL, Piechaczyk M. Targeted infection of human cells via major histocompatibility complex class I molecules by Moloney murine leukemia virus-derived viruses displaying single-chain antibody fragment-envelope fusion proteins. J. Virol. 1996;70:2957–2962. doi: 10.1128/jvi.70.5.2957-2962.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chowdhury S, Chester KA, Bridgewater J, Collins MK, Martin F. Efficient retroviral vector targeting of carcinoembryonic antigen-positive tumors. Mol. Ther. 2004;9:85–92. doi: 10.1016/j.ymthe.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Funke S, Maisner A, Muhlebach MD, Koehl U, Grez M, Cattaneo R, Cichutek K, Buchholz CJ. Targeted cell entry of lentiviral vectors. Mol. Ther. 2008;16:1427–1436. doi: 10.1038/mt.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morizono K, Bristol G, Xie YM, Kung SK, Chen IS. Antibody-directed targeting of retroviral vectors via cell surface antigens. J. Virol. 2001;75:8016–8020. doi: 10.1128/JVI.75.17.8016-8020.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morizono K, Xie Y, Ringpis GE, Johnson M, Nassanian H, Lee B, Wu L, Chen IS. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 2005;11:346–352. doi: 10.1038/nm1192. [DOI] [PubMed] [Google Scholar]
- 28.Roux P, Jeanteur P, Piechaczyk M. A versatile and potentially general approach to the targeting of specific cell types by retroviruses: application to the infection of human cells by means of major histocompatibility complex class I and class II antigens by mouse ecotropic murine leukemia virus-derived viruses. Proc. Natl. Acad. Sci. USA. 1989;86:9079–9083. doi: 10.1073/pnas.86.23.9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boerger AL, Snitkovsky S, Young JA. Retroviral vectors preloaded with a viral receptor-ligand bridge protein are targeted to specific cell types. Proc. Natl. Acad. Sci. USA. 1999;96:9867–9872. doi: 10.1073/pnas.96.17.9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L, Bailey L, Baltimore D, Wang P. Targeting lentiviral vectors to specific cell types in vivo. Proc. Natl. Acad. Sci. USA. 2006;103:11479–11484. doi: 10.1073/pnas.0604993103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin AH, Kasahara N, Wu W, Stripecke R, Empig CL, Anderson WF, Cannon PM. Receptor-specific targeting mediated by the coexpression of a targeted murine leukemia virus envelope protein and a binding-defective influenza hemagglutinin protein. Hum. Gene Ther. 2001;12:323–332. doi: 10.1089/10430340150503957. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Zeigler L, Joo KI, Cho T, Lei Y, Wang P. Gamma-retroviral vectors enveloped with an antibody and an engineered fusogenic protein achieved antigen-specific targeting. Biotechnol. Bioeng. 2008;19:861–872. doi: 10.1002/bit.21903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 34.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single 'self-cleaving' 2A peptide-based retroviral vector. Nat. Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 35.Fang J, Qian J-J, Yi S, Harding TC, Tu GH, VanRoey M, Jooss K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat. Biotech. 2005;23:584–590. doi: 10.1038/nbt1087. [DOI] [PubMed] [Google Scholar]
- 36.Joo KI, Wang P. Visualization of targeted transduction by engineered lentiviral vectors. Gene Ther. 2008;15:1348–1396. doi: 10.1038/gt.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cosimi AB, Burton RC, Colvin RB, Goldstein G, Delmonico FL, LaQuaglia MP, Tolkoff-Rubin N, Rubin RH, Herrin JT, Russell PS. Treatment of acute renal allograft rejection with OKT3 monoclonal antibody. Transplantation. 1981;32:535–539. doi: 10.1097/00007890-198112000-00018. [DOI] [PubMed] [Google Scholar]
- 38.Cosimi AB, Colvin RB, Burton RC, Rubin RH, Goldstein G, Kung PC, Hansen WP, Delmonico FL, Russell PS. Use of monoclonal antibodies to T-cell subsets for immunologic monitoring and treatment in recipients of renal allografts. N. Engl. J. Med. 1981;305:308–314. doi: 10.1056/NEJM198108063050603. [DOI] [PubMed] [Google Scholar]
- 39.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 40.Reth M. Antigen receptors on B lymphocytes. Annu. Rev. Immunol. 1992;10:97–121. doi: 10.1146/annurev.iy.10.040192.000525. [DOI] [PubMed] [Google Scholar]
- 41.Lei Y, Joo I-I, Wang P. Enhancing Targeted Transduction of Enveloped Lentiviral Vectors by Engineering Fusogenic Molecules. 2009 doi: 10.1186/1754-1611-3-8. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu YE, Cassese T, Kielian M. The cholesterol requirement for sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 1999;73:4272–4278. doi: 10.1128/jvi.73.5.4272-4278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mellman I, Fuchs R, Helenius A. Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 1986;55:663–700. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- 44.Kielianand M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 2006;4:67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature. 2004;427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 46.Umashankar M, Sanchez-San Martin C, Liao M, Reilly B, Guo A, Taylor G, Kielian M. Differential cholesterol binding by class II fusion proteins determines membrane fusion properties. J. Virol. 2008;82:9245–9253. doi: 10.1128/JVI.00975-08. [DOI] [PMC free article] [PubMed] [Google Scholar]